Introduction
Ovarian cancer is the second most common
gynecological cancer and currently ranks as one of the leading
causes of cancer-related deaths among women worldwide (1,2).
Epithelial ovarian cancer (EOC) is the most common and lethal type
of the disease, which accounts for approximately 85–95% of all
ovarian cancer cases. EOC treatment commonly results in a low
response rate to surgical resection, radiotherapy, chemotherapy and
poor prognosis with a median survival of <5 years in
approximately 30% of patients (3,4). The
high morbidity and mortality rates are contributed to the fact that
patients may appear asymptomatic in the early stages; therefore,
70–75% of patients in the advanced stages are often diagnosed after
the cancer has circulated throughout the abdominal cavity (5,6).
Therefore, a rapidly expanding understanding of the molecular
mechanisms underlying the pathogenesis of EOC, especially the
mechanisms that affect OC cell invasive, has the potential to
strongly influence treatment outcomes for this devastating
disease.
The histone H3 lysine 36 demethylase enzyme (KDM2A)
has been previously shown to demethylate histone H3K36, contain an
F-box domain, a JmjC domain, a CxxC zinc finger domain, a PHD
domain and 3 leucine-rich repeat elements belonging to the KDM gene
family (7–9). KDM2A was first discovered in kidney
COS-7 cells exhibiting robust histone demethylase activity
(10). KDM2A was found to be highly
expressed in lung (11), pancreatic
cancer (12), colorectal
adenocarcinoma (13), breast
(14), gastric cancer (15) and certain blood diseases (16), which have been reported to be
related to tumor aggressiveness and metastasis, chemotherapy
resistance and poor prognosis. This protein has been linked to many
signaling pathways, which promote cancer metastasis and malignancy.
Wagner et al showed that KDM2A promoted lung tumorigenesis
via the ERK1/2 signaling pathway (11). Chen et al reported that KDM2A
promoted angiogenesis and stemness by upregulating Jagged1
(17). However, the role of KDM2A
and its underlying mechanism still remain unclear in EOC
proliferation, migration and metastasis. In the present study, we
demonstrated that KDM2A is overexpressed in EOC and that KDM2A
promoted EOC progression and induced EMT. Furthermore, KDM2A
influenced the biological behaviors previously mentioned by
regulating the PI3K/AKT/mTOR pathway. These findings suggest that
KDM2A may serve as a potential therapeutic target for the clinical
management of EOC.
Materials and methods
Bioinformatic analysis
Gene profiling data of ovarian normal surface
epithelia and ovarian cancer epithelial samples were downloaded
from the GEO dataset (http://www.ncbi.nlm.nih.gov/geo). GSE14407 dataset was
selected for bioinformatic analysis (18). The differential analysis was
performed using R package ‘limma’ (19). Differential genes obtained from
GSE14407 were visualized using the R package ‘pheatmap’ (https://CRAN.R-project.org/package=pheatmap) and
‘ggplot2’ (20). RNA sequencing
data of KDM2A was achieved from the TCGA data portal (https://cancergenome.nih.gov/), containing 374 ovarian
cancer samples. Corresponding clinical data were also downloaded
and filtered out for useful information. Kaplan-Meier survival
curves were conducted to assess the prognostic value of KDM2A using
the R package ‘survival’ (https://CRAN.R-project.org/package=survival).
Ovarian cancer tissue samples
Human specimens were obtained between 01 January
2005 and 31 December 2011 from 27 patients, with a mean age of 46
years (range, 18–73 years), who underwent primary tumor resection
at Renmin Hospital of Wuhan University (Wuhan, China). Among the 27
cases, the specimen groups consisted of EOC (n=9), borderline
ovarian tumors (n=9) and normal ovary tissues (n=9). All specimens
were confirmed by at least 2 pathologists. In the present study,
the patients accepted no chemotherapy or radiotherapy before
surgery. Before conducting our scientific investigation, consent
was obtained from all the patients and the study was approved by
the Ethics Committee of Wuhan University (Wuhan, China).
Immunohistochemistry
The immunohistochemical analysis of KDM2A expression
in human EOC was performed as previously described (21). The rate of KDM2A-positive cells was
scored semi-quantitatively in each section.
Cell culture and reagent
The human OC cell lines A2780 and SKOV3 were
obtained from the State Key Laboratory of Molecular Biology,
Institute of Biochemistry and Cell Biology, Shanghai Institutes for
Biological Sciences, Chinese Academy of Sciences (Shanghai, China).
The cells were respectively cultured in MEM/F12 and RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS) (both from
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 1%
penicillin and 1% streptomycin (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) in a CO2 incubator under
standardized conditions. Antibodies KDM2A (cat. no. ab191387),
GAPDH (cat. no. ab181602), and β-actin (cat. no. ab8227) were
obtained from Abcam plc. (dilution 1:500; Cambridge, UK).
Antibodies phospho-PI3K (cat. no. sc4257), PI3K (cat. no. sc4292),
phospho-Akt (cat. no. sc4060), Akt (cat. no. sc4691), phospho-mTOR
(cat. no. sc5536), mTOR (cat. no. sc2983), MMP2 (cat. no. sc10736),
MMP9 (cat. no. sc10737), Bcl-2 (cat. no. sc2872), Bax (cat. no.
sc14796), E-cadherin (cat. no. sc14472), N-cadherin (cat. no.
sc13116) and vimentin (cat. no. sc5741) were purchased from Cell
Signaling Technology (dilution 1:500; Danvers, MA, USA). LY294002
was reported as the inhibitor of the PI3K, which was purchased from
Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China) (22).
Establishment of the stable
KDM2A-knockdown cell line Transfection
In order to knockdown the expression of endogenous
KDM2A, a lentivirus containing an shRNA sequence targeting KDM2A
was designed and synthesized by Shanghai GenePharma Co., Ltd.
(Shanghai, China). The shRNA sequence was as follow:
GGTGGGCAGTAGGAATCAA. The cells were seeded at ~1.0×105
cells/well into 6-well plates and cultured at 37°C overnight under
standard conditions. After 50% confluence was reached, the number
of cells in a well was counted using a hemocytometer. KDM2A shRNA
was transfected into the cells in Opti-MEM (Invitrogen; Thermo
Fisher Scientific, Inc.) at a multiplicity of infection (MOI) of 10
[MOI = transducing units per cell (TU) number/cells], according to
the manufacturer's instructions. The culture medium was replaced
after a 24-h incubation. A total of 48 h after transfection, the
cells were observed and photographed under a fluorescence
microscope. After successful transfection, the shRNA sequence was
stably expressed. Untransfected cells were used as a blank control,
while cells transfected with scrambled shRNA were considered as the
negative control (shControl).
Cell viability assay
Cell Counting Kit-8 (CCK-8; purchased from Dojindo
Laboratories, Kumamoto, Japan) was used to determine the inhibitory
effect of the silencing of KDM2A on the proliferation of A2780 and
SKOV3 cell lines according to the manufacturer's instructions.
Cells were seeded at ~10.0×103 cells/well into 96-well
plates with 10% FBS and cultured for 24, 48 and 72 h. Then, 10 µl
of CCK-8 was added and subsequently incubated for 1 h at 37°C. The
absorbance value (OD) at 450 nm of every well was measured with a
spectrophotometric plate reader. Assays were performed in
triplicate on 3 independent experiments.
Colony formation assay
An appropriate number of cells were plated on a
6-well plate (500 cells/2 ml/well) and cultured in 2 ml medium with
10% FBS. Then, cells were changed every 3 days with 10%
mycoplasma-free FBS for 2 weeks until the cells in the plates had
formed colonies that were of substantially right size (50
cells/colony or greater). The cells were fixed with 2 ml 75%
ethanol at room temperature for 15 min, and then stained with 0.5%
crystal violet. The cloning efficiency was calculated by dividing
the number of wells containing proliferating cells with the total
number of cell-plated wells.
Flow cytometric analysis of apoptosis
with PE/7-ADD staining
For apoptosis analysis, cells were treated for 48 h.
The cells were suspended with 100 µl of 1X binding buffer (0.1 mM
HEPES/NaOH, 1.4 M NaCl, 25 mM CaCl2 and pH 7.4) and
stained with 5 µl of PE Annexin V and 5 µl of 7-amino-actinomycin
(7-ADD) for 15 min at room temperature and then 400 µl 1X binding
buffer was added to each tube (Annexin V-PE Apoptosis Detection
kit; BD Pharmingen; BD Biosciences, San Diego, CA, USA). Analysis
of the results was carried out by BD FACSAria (BD Biosciences,
Franklin Lakes, NJ, USA). Data were quantified using FlowJo
Software (FlowJo, LLC, Ashland, OR, USA).
Hoechst staining
The 3 groups of A2780 and SKOV3 cells were fixed
using methanol acetic acid for 15 min and then stained with Hoechst
33342 for 5 min. Being washed twice with PBS, the cells were
immediately photographed under an Olympus BX51 inverted microscope
at ×400 magnification (Olympus Corp., Tokyo, Japan).
Wound healing assay
Untransfected and transfected cells were seeded at
5.0×105 cells/well in 6-well plates and cultured
routinely. After reaching 90% confluence, the cell monolayer was
scratched with a sterile pipette tip. After washing 3 times with
PBS for 5 min each to clear the floating cells, 1.5 ml MEM/F12 and
RPMI-1640 medium supplemented with 1% FBS were added into each
well. Photographs were taken by an Olympus BX51 inverted microscope
at ×100 magnification (Olympus Corp.) at 0, 24 and 48 h after
scratching. Results were indicated as the relative width of
scratch-the distance migrated relative to the original scratched
distance. The experiment was conducted 3 times.
Cell invasion assay
The invasive ability of cells was measured using the
Corning® Matrigel® Basement Membrane Matrix
(cat. no. 356234; Corning Inc., Corning, NY, USA) and a 24-well
Transwell chamber (Corning Inc.) according to the manufacturer's
protocol. The number of cells that passed through an 8-µm
polycarbonate membrane was calculated. The polycarbonate surface of
each chamber was covered with 50 µl Matrigel (1:8 dilution) to
create an artificial basement membrane. Cells were cultured at 37°C
in FBS-free MEM/F12 and RPMI-1640 medium for 24 h. After serum
starvation, the cells were seeded at 1×105 cells/well in
the upper Transwell chamber, which contained ~200 µl serum-free
medium. The lower chamber was filled with 500 µl of the medium
supplemented with 10% FBS. After an incubation of 48 h at 37°C, the
chambers were fixed at room temperature with paraformaldehyde for
30 min. Cells that had attached to the upper surface of the
chambers were removed with a sterile cotton swab, and cells that
adhered to the lower surface were stained with 0.1% crystal violet
(Guangfu Institute of Superfine Chemical Industry, Tianjin, China)
for 20 min at room temperature. The numbers of stained cells were
counted using an inverted microscope (Olympus IX 70–142; Olympus
Corp.) in 8 random fields. The experiment was repeated 3 times.
Western blot analysis
Cell lysates containing equal amounts of protein was
subjected to 12% SDS-PAGE and then transferred to 0.45 µm
Immobilon-P transfer membranes (EMD Millipore, Billerica, MA, USA)
by electroblotting. Membranes were blocked at room temperature for
1 h in TBST (50 mmol/l Tris-HCL, pH 7.6, 150 mmol/l NaCl and 0.1%
Tween-20) containing 5% non-fat dry milk and then incubated with
primary antibody at 4°C overnight. The membranes were then
incubated with appropriate anti-rabbit secondary antibody (dilution
1:10,000; Sigma-Aldrich; Merck KGaA) for 1 h at room temperature
and visualized with a chemiluminescence substrate kit
(Pierce™ ECL Western Blotting Substrate; Thermo
Scientific Fisher, Inc.). The intensity of the target protein bands
was normalized to the loading control, GAPDH/β-actin.
Statistical analysis
Statistical analyses were performed using SPSS v.
17.0 software (SPSS, Inc., Chicago, IL, USA). Quantitative data are
expressed as the mean ± standard deviation (SD). The log-rank test,
one-way ANOVA and and the Tukey's test were used. The statistically
significant difference was indicated in the figures and legends as
follows: *P<0.05, **P<0.01, ***P<0.001.
Results
KDM2A is upregulated and associated
with poor survival in EOC
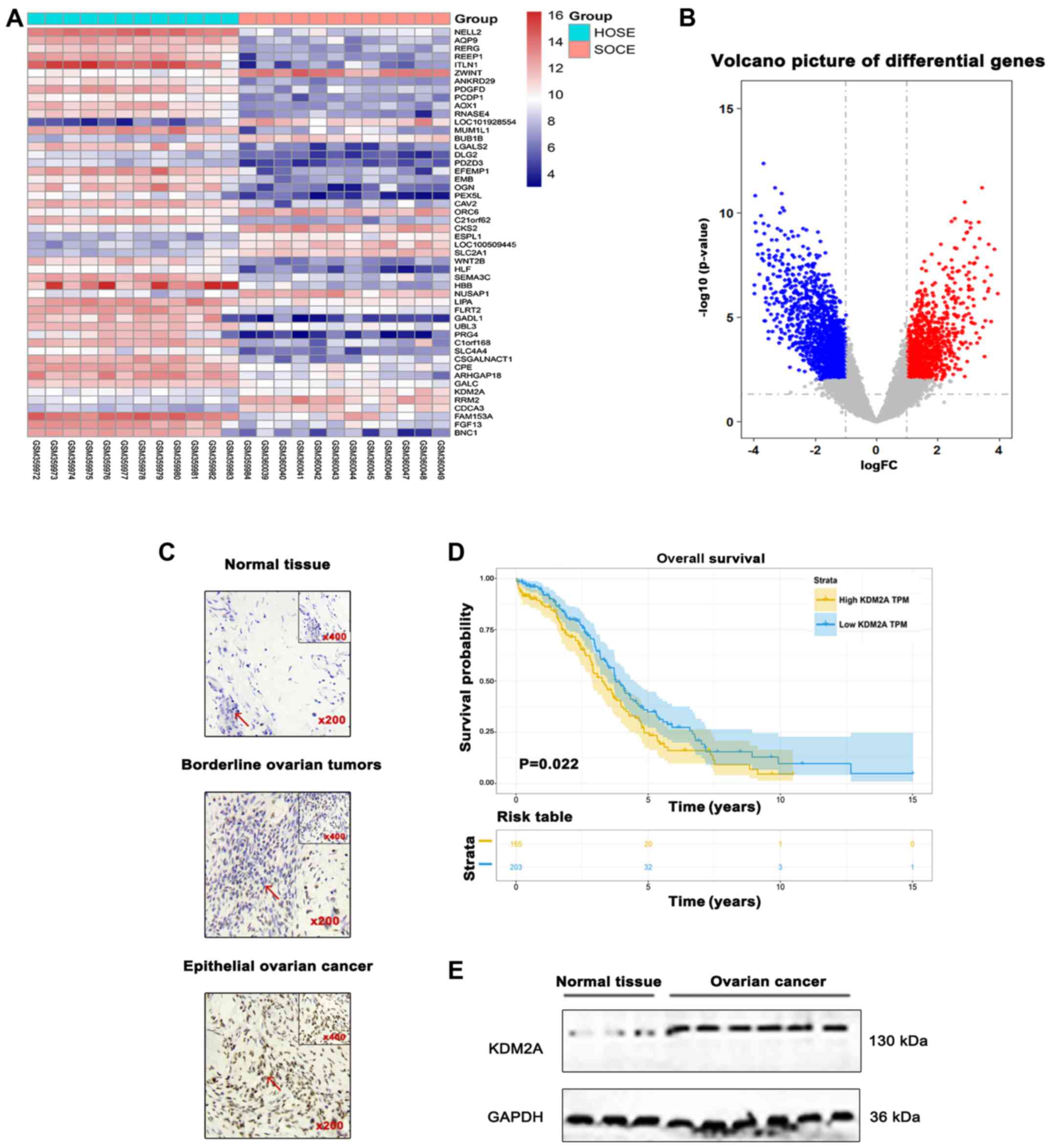
Differential analysis was performed for the GSE14407
dataset. The results showed that expression of KDM2A was
significantly upregulated in 12 serous ovarian cancer epithelial
samples (SOCE) compared to that noted in the 12 healthy ovarian
surface epithelial samples (HOSE) (Fig.
1A and B). Differences in the immunoreactivity of KDM2A
staining were observed in EOCs, borderline epithelial ovarian
tumors, and normal epithelial ovary tissues, predominantly
expressed in the nucleus and light cytoplasmic staining. EOCs
showed strong positive staining, while borderline epithelial
ovarian tumors and normal ovary tissues showed low-level expression
(Fig. 1C). Kaplan-Meier survival
analysis suggested that patients with high KDM2A expression had a
poor prognosis in overall survival (Fig. 1D). In addition, the expression level
of KDM2A was higher in 7 fresh EOC tissues compared with that in 3
fresh normal ovary tissues (Fig.
1E).
Assessment of KDM2A expression after
shRNA transfection
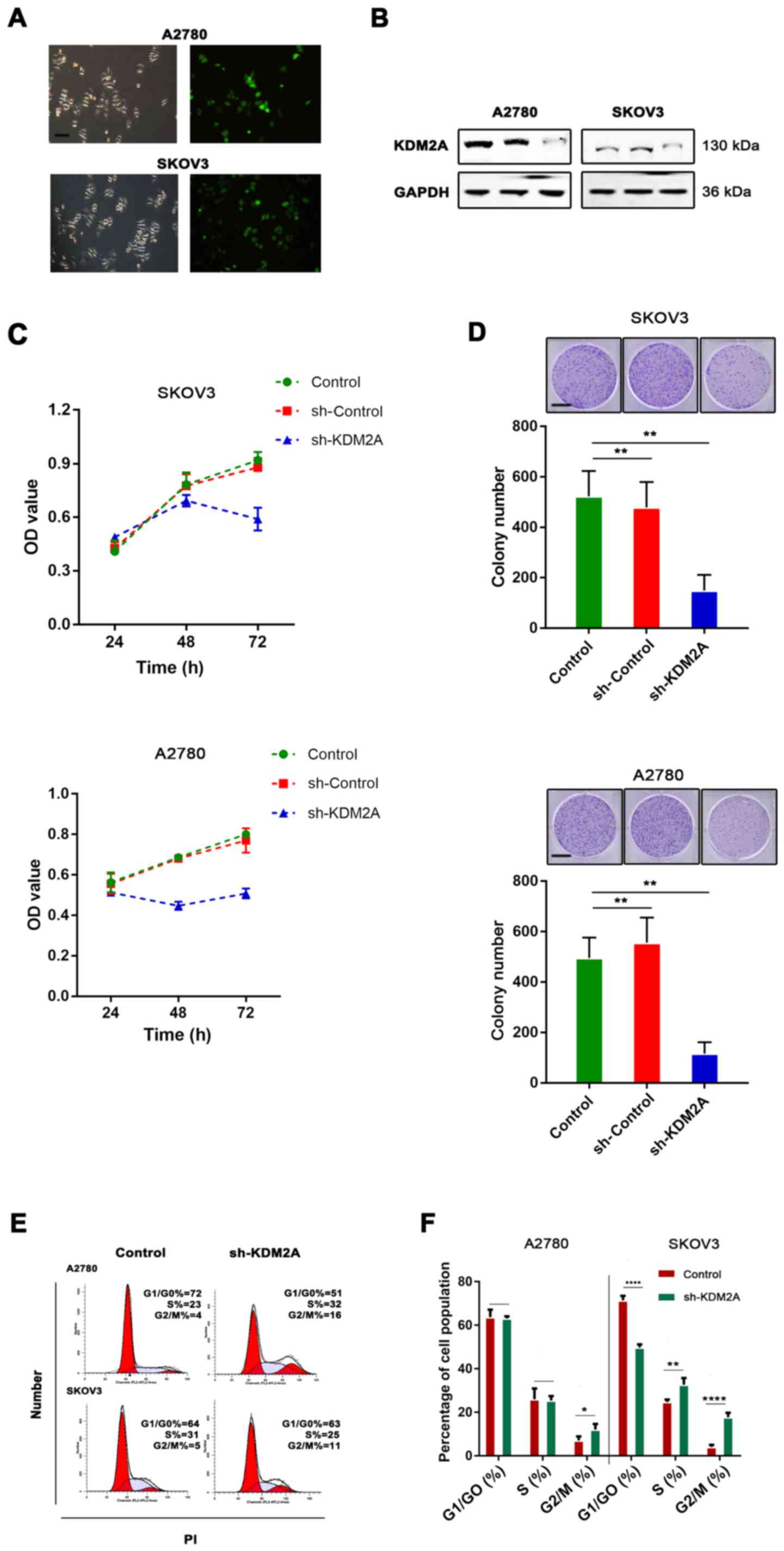
To test the function of KDM2A in EOCs, shRNA
transfection was used for KDM2A knockdown. The knockdown efficiency
was assessed by western blot analysis. The results showed that the
expression of KDM2A was significantly decreased after shRNA-KDM2A
transfection in human ovarian cancer cell lines SKOV3 and A2780
(Fig. 2A and B).
Silencing of KDM2A reduces
proliferation and delays G2 phase progression
We found that KDM2A inhibition in A2780 and SKOV3
cells prevented proliferation. We tested our hypothesis to confirm
whether cell viability and the colony-forming capabilities were
determined by cell proliferation. As shown in Fig. 2C, CCK-8 was used to measure the
viability of EOC cells transfected with sh-KDM2A. The viability of
the sh-KDM2A cells was significantly lower than that for cells
transfected with the sh-Control and the control group. The colony
formation assay indicated that the colony-forming capabilities of
the SKOV3 and A2780 cells were significantly impaired due to KDM2A
depletion (Fig. 2D). To explore
whether the effects of KDM2A depletion on cell behaviors were
related to cell cycle distribution, we performed flow cytometric
analysis. The evidence showed that the cell cycle was related to
the proliferative capabilities of cells; therefore, the effect of
KDM2A in EOC was further investigated. The percentage of G2/M phase
cells was increased after silencing of KDM2A (Fig. 2E and F). These results demonstrated
cell cycle arrest in the G2/M transition due to KDM2A
knockdown.
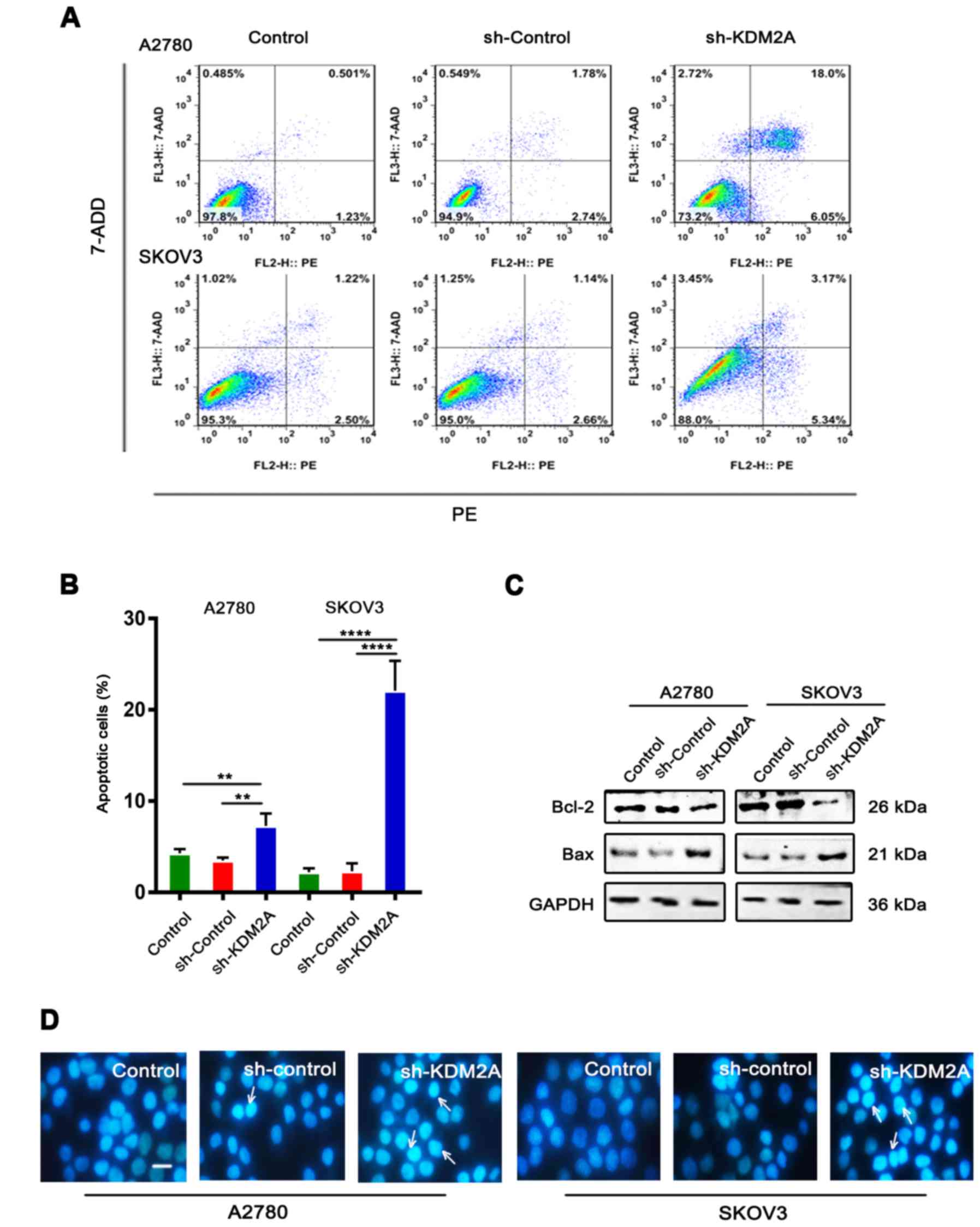
Downregulation of KDM2A stimulates
cell apoptosis
Flow cytometric analysis with PE/7-ADD staining
showed that the early apoptotic cell population in the sh-KDM2A
group was higher than that in the other 2 groups in the 2 cell
lines (Fig. 3A and B). Moreover,
western blot analysis showed that the expression level of Bcl-2 was
downregulated, while the expression level of Bax was upregulated in
the same conditions (Fig. 3C).
Hoechst 33342 staining showed that the sh-KDM2A group had a greater
number of apoptotic cells compared to the number of apoptotic cells
in the 2 control groups (Fig. 3D).
Additionally, cells of the sh-KDM2A group showed marked
morphological changes in the nuclear size than cells of the control
groups.
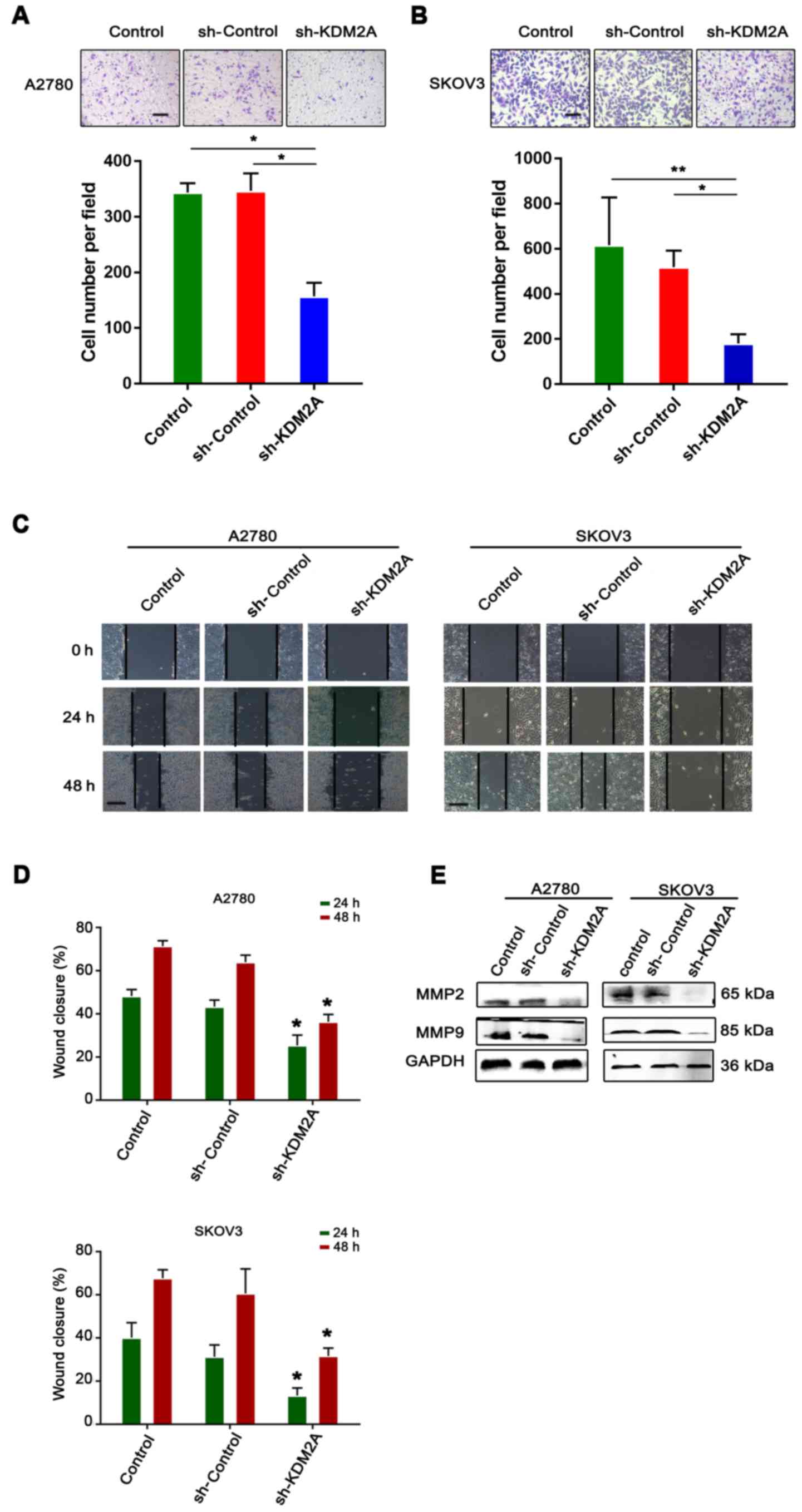
Inhibition of KDM2A attenuates tumor
migration and invasion in vitro
The serum-stimulated Matrigel invasion assay
demonstrated that the percentages of human ovarian cancer cells
that migrated through the polycarbonate membrane in the sh-KDM2A
transfection group were significantly lower than those in the
control groups (Fig. 4A and B).
Therefore, downregulation of KDM2A decreased cell invasion of the
human ovarian cancer cell lines after transfection.
The monolayer wound healing assay was used to assess
cell migration. The transfected cells showed a significant
deceleration in closure of the wound scratch width after 24 and 48
h compared with the sh-Control and control groups (Fig. 4C and D).
To study the effect of KDM2A depletion on cellular
response, the expression of MMP2 and MMP9 were measured in SKOV3
and A2780 cell lines. As shown in Fig.
4E, the 2 transfected proteins, showed decreased expression in
the sh-KDM2A group compared to the negative control and the control
group.
KDM2A promotes epithelial-mesenchymal
transition (EMT) via the PI3K/AKT/mTOR signaling pathway using the
LY294002 in EOC cells
Since EMT has been increasingly recognized as a
crucial event in cancer metastasis, we measured the expression of
EMT markers and EMT transcription factors before/after KDM2A
knockdown by western blotting. The expression level of E-cadherin
was markedly upregulated, while those of N-cadherin and vimentin
were significantly decreased in A2780 sh-KDM2A and SKOV3 sh-KDM2A
cells compared with the sh-Control and the control cells (Fig. 5A).
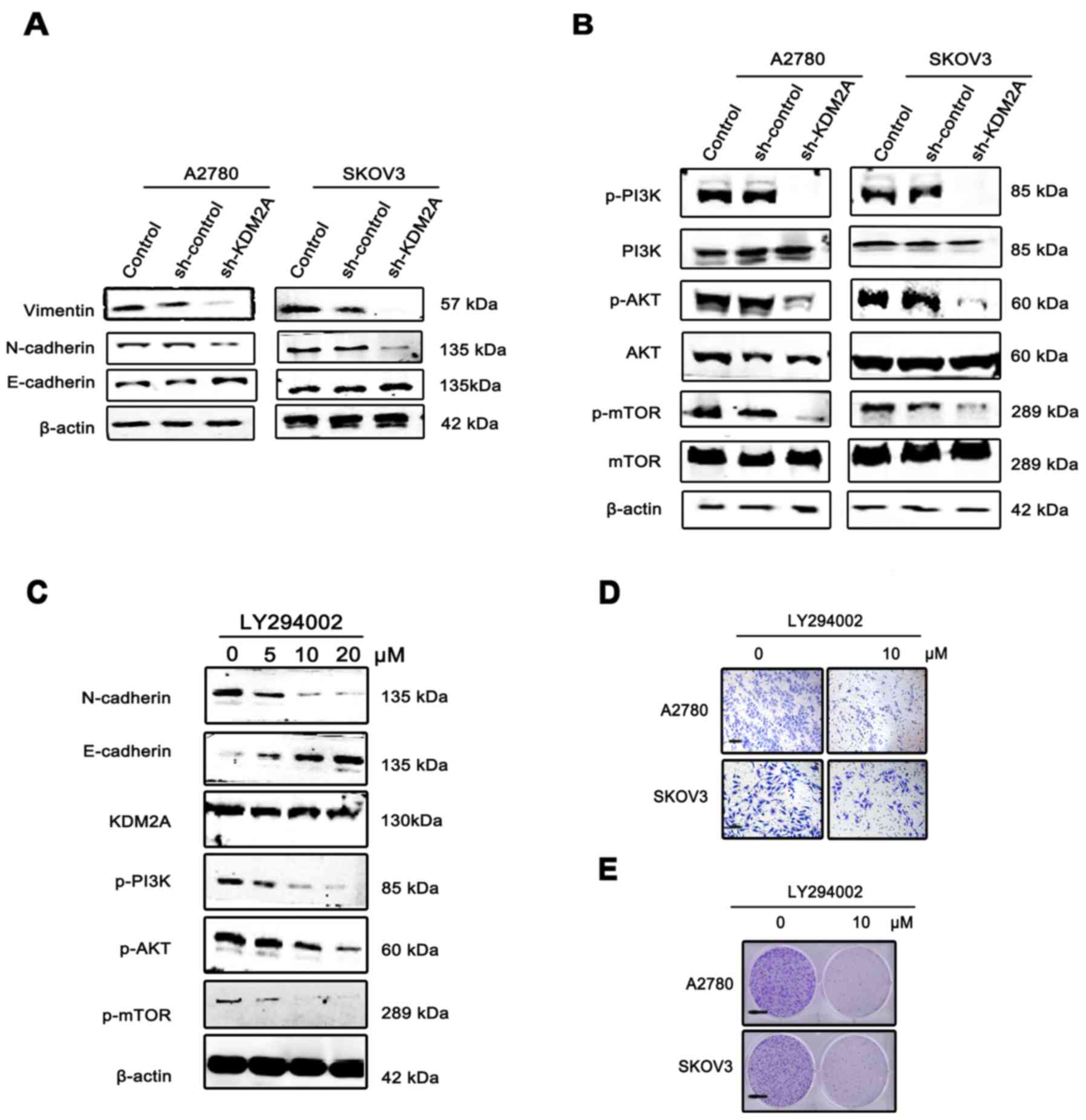 | Figure 5.KDM2A facilitates cancer cell EMT via
the PI3K/AKT/mTOR pathway. (A) Expression of E-cadherin, N-cadherin
and vimentin, EMT phenotype markers, in KDM2A-knockdown cells and
the 2 control groups. (B) Activation of the PI3K/AKT/mTOR pathway
in the 3 groups. (C) Expression of KDM2A, the EMT phenotype and
proteins of the PI3K/AKT/mTOR pathway in A2780 cells were
administered gradient concentrations of the PI3K inhibitor
(LY294002, 0, 5, 10 and 20 µmol/l). (D) The migration ability of
SKOV3 and A2780 cells gradually decreased with 10 µmol/l LY294002.
Scale bar, 50 µm. (E) LY294002 inhibited the colony formation of
SKOV3 and A2780 cells. Fewer colonies were formed in the treated
group compared with the control group. EMT, epithelial-mesenchymal
transition. Scale bar, 100 µm. |
To elucidate the molecular mechanism involved in
KDM2A-mediated EMT in EOC cells, we analyzed the key components of
the PI3K/AKT/mTOR signaling pathway. Western blot analysis showed
that KDM2A knockdown decreased the expression of phospho (p)-PI3K,
p-AKT and p-mTOR compared to the control groups (Fig. 5B). Using the PI3K signaling pathway
inhibitor, LY294002 (0, 5, 10 and 20 µmol/l), we treated the A2780
and SKOV3 cells for 48 h. The results showed that inactivation of
the PI3K signaling pathway led to suppression of EMT in a
dose-dependent manner, while the expression level of KDM2A was not
significantly changed (Fig. 5C).
The migration ability of SKOV3 and A2780 cells gradually decreased
with 10 µmol/l LY294002 (Fig. 5D).
In addition, LY294002 inhibited the colony formation of SKOV3 and
A2780 cells. Fewer colonies were formed in the treated group
compared with the control group (Fig.
5E).
Discussion
In the present study, our results indicated the
critical roles of KDM2A in epithelial ovarian cancer (EOC). The
significant increase of KDM2A expression in EOC was determined by
the GEO database. Similarly, immunohistochemical analysis and
western blot analysis confirmed the overexpression of KDM2A in EOC
patients compared with the borderline ovarian tumor and normal
ovary tissues. Moreover, the Kaplan-Meier estimate showed that
KDM2A expression was negatively associated with patient survival,
indicating that higher KDM2A expression was correlated with lower
overall survival rates of patients with EOC.
To further examine the role of KDM2A in EOC, we
investigated the effect of KDM2A on the viability of EOC cells
in vitro. The results showed that knockdown of KDM2A
expression in EOC cells significantly decreased cell proliferation
and efficiency of colony formation. In agreement with our findings,
KDM2A functions as an oncogene in several types of cancer and
several cancer cell lines possess high-level expression of KDM2A
(23). Many researchers have
concluded that KDM2A may be a promising target in anticancer
therapeutics. Huang et al showed that forced expression of
KDM2A increased the growth and motility of gastric cancer cells by
downregulating PDCD, a known tumor suppressor (24). Kong et al reported that
miR-29b and KDM2A are involved in the proliferation and metastasis
of gastric cancer cells (15).
Consistent with this finding, KDM2A showed a similar effect on cell
apoptosis in this study. We showed that downregulation of KDM2A
decreased cell migration and invasion in vitro, suggesting
that KDM2A promotes EOC tumor metastasis. Hematogenous
intravasation and extravasation are less accepted as a mechanism of
EOC metastasis. Instead, when cells reach a threshold value,
limited by epithelial-mesenchymal transition (EMT) or EMT-like
events and growth, a primary OC would passively shed cells from
their original location, leading to spheroid formation and
formation of peritoneal implantation metastasis (6,25,26).
EMT is a morphological change of cells or tissues
that lose their epithelial characteristics and gain mesenchymal
properties. EMT is the mechanism that has been well-characterized
for the transition of early-stage ovarian cancers to invasive and
metastatic malignancies, promoting the aggressiveness of ovarian
cancers and obtaining the characteristics of stem cells after a
loss of cell polarity and cell-cell adhesion (27–29).
Cells undergoing EMT exhibit loss of E-cadherin and increased
N-cadherin or vimentin expression, leading to tumor cell invasion
and metastasis. E-cadherin is responsible for cell-cell adhesion
(30,31); its loss is associated with tumor
invasion, metastasis and poor prognosis (32,33).
In addition, N-cadherin or vimentin controls tumor progression
(34,35). There is sufficient evidence
supporting the opinion that the development of EMT in ovarian
cancer causes aggressive phenotypes that may promote metastasis
(34). Liu et al
demonstrated that β-hCG-depleted SKOV3 cells had increased
E-cadherin and decreased vimentin and N-cadherin expression
(36). In addition, Colomiere et
al demonstrated that expression of mesenchyme-associated
N-cadherin and vimentin was consistent with the change in
fibroblast-like morphology and migratory phenotype (37). Unsurprisingly, these results
confirmed our hypothesis. In this study, we demonstrated that
silencing of KDM2A suppressed the EMT process by increasing the
expression level of the epithelial marker, E-cadherin, while
reducing the mesenchymal markers, N-cadherin and vimentin in
vitro. These data suggest that KDM2A promotes cancer cell
invasion and migration via activation of EMT.
EMT is regulated by various signaling pathways,
including the Wnt/β-catenin, Notch, TGF-β and PI3K/AKT signaling
pathways (38). Activation of the
PI3K/AKT pathway is universal in many malignancies and is
associated with carcinogenesis of all ovarian cancer subtypes
(39,40). In this study, silencing of KDM2A
downregulated levels of phospho-PI3K, phospho-AKT and phospho-mTOR,
leading to inhibition of EOC progression. Furthermore, the pathway
was inactivated in SKOV3 cells with the use of a PI3K inhibitor,
resulting in EMT inhibition. Thus, we provide evidence that KDM2A
may promote EMT through activation of the PI3K/AKT/mTOR pathway in
EOC cells.
In summary, the present study demonstrated several
roles of KDM2A in EOC. First, KDM2A was overexpressed in EOC and
KDM2A has potential as a prognostic marker of EOC. Second,
silencing of KDM2A was able to suppress proliferation, migration,
invasion and EMT, as well as promote apoptosis in EOC.
Additionally, we gained insight into the potential mechanism and
showed that KDM2A regulated EMT and the PI3K/AKT/mTOR signaling
pathway involved in metastasis. Although there were some notable
discoveries in this study, many limitations still exist. One is PCR
and animal experiments were not implemented. Although some
researchers have confirmed changes of KDM2A expression in other
tumors in vivo and in vitro (11), the results may be different. We
demonstrated the effects of PI3K inhibitors in ovarian cancer, but
we did not observe the consequence after the treatment of AKT and
mTOR inhibitors. Despite these limitations, these results show that
KDM2A may serve as a therapeutic target for clinical treatments of
EOC patients. Finally, evidence suggests a correlation between EMT
and the characteristics of embryonic neural cells, emphasizing the
significance of the epigenetic modification enzyme enhancer
(41,42). Thus, KDM2A could cause neural
development and this behavior may be associated with
tumorigenesis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the correspongding author upon reasonable
request.
Authors' contributions
DHL, JY, LH and LKG conceived and designed the
study. DHL, JM and YL prepared the experimental materials and
performed the in vitro assays. JC, JMT and MH performed the
bioinformatics study and the data interpretation. DHL, LH, JMT and
STL performed the statistical analysis. DHL, LH and MH wrote the
manuscript. STL, YL, MH and LH reviewed and edited the manuscript.
All authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Before conducting the scientific investigation, the
consent was obtained from all the patients and the study was
approved by the Ethics Committee of Wuhan University (Wuhan,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Auersperg N: Ovarian surface epithelium as
a source of ovarian cancers: Unwarranted speculation or
evidence-based hypothesis? Gynecol Oncol. 130:246–251. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Holschneider CH and Berek JS: Ovarian
cancer: Epidemiology, biology, and prognostic factors. Semin Surg
Oncol. 19:3–10. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marsden DE, Friedlander M and Hacker NF:
Current management of epithelial ovarian carcinoma: A review. Semin
Surg Oncol. 19:11–19. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lengyel E: Ovarian cancer development and
metastasis. Am J Pathol. 177:1053–1064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi Y and Whetstine JR: Dynamic regulation
of histone lysine methylation by demethylases. Mol Cell. 25:1–14.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Klose RJ and Zhang Y: Regulation of
histone methylation by demethylimination and demethylation. Nat Rev
Mol Cell Biol. 8:307–318. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Faundes V, Newman WG, Bernardini L, Canham
N, Clayton-Smith J, Dallapiccola B, Davies SJ, Demos MK, Goldman A,
Gill H, et al: Histone lysine methylases and demethylases in the
landscape of human developmental disorders. Am J Hum Genet.
102:175–187. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsukada Y, Fang J, Erdjument-Bromage H,
Warren ME, Borchers CH, Tempst P and Zhang Y: Histone demethylation
by a family of JmjC domain-containing proteins. Nature.
439:811–816. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wagner KW, Alam H, Dhar SS, Giri U, Li N,
Wei Y, Giri D, Cascone T, Kim JH, Ye Y, et al: KDM2A promotes lung
tumorigenesis by epigenetically enhancing ERK1/2 signaling. J Clin
Invest. 123:5231–5246. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nalla AK, Williams TF, Collins CP, Rae DT
and Trobridge GD: Lentiviral vector-mediated insertional
mutagenesis screen identifies genes that influence androgen
independent prostate cancer progression and predict clinical
outcome. Mol Carcinog. 55:1761–1771. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cao LL, Du C, Liu H, Pei L, Qin L, Jia M
and Wang H: Lysine-specific demethylase 2A expression is associated
with cell growth and cyclin D1 expression in colorectal
adenocarcinoma. Int J Biol Markers. 1:17246008187640692018.
|
|
14
|
Chen JY, Luo CW, Lai YS, Wu CC and Hung
WC: Lysine demethylase KDM2A inhibits TET2 to promote DNA
methylation and silencing of tumor suppressor genes in breast
cancer. Oncogenesis. 6:e3692017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kong Y, Zou S, Yang F, Xu X, Bu W, Jia J
and Liu Z: RUNX3-mediated up-regulation of miR-29b suppresses the
proliferation and migration of gastric cancer cells by targeting
KDM2A. Cancer Lett. 381:138–148. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu L, Li Q, Wong SH, Huang M, Klein BJ,
Shen J, Ikenouye L, Onishi M, Schneidawind D, Buechele C, et al:
ASH1L links histone H3 lysine 36 dimethylation to MLL leukemia.
Cancer Discov. 6:770–783. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen JY, Li CF, Chu PY, Lai YS, Chen CH,
Jiang SS, Hou MF and Hung WC: Lysine demethylase 2A promotes
stemness and angiogenesis of breast cancer by upregulating Jagged1.
Oncotarget. 7:27689–27710. 2016.PubMed/NCBI
|
|
18
|
Bowen NJ, Walker LD, Matyunina LV, Logani
S, Totten KA, Benigno BB and McDonald JF: Gene expression profiling
supports the hypothesis that human ovarian surface epithelia are
multipotent and capable of serving as ovarian cancer initiating
cells. BMC Med Genomics. 2:712009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ito K and Murphy D: Application of ggplot2
to Pharmacometric Graphics. CPT Pharmacometrics Syst Pharmacol.
2:e792013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu J, Meng Y, Zhang Z, Yan Q, Jiang X, Lv
Z and Hu L: MARCH5 RNA promotes autophagy, migration, and invasion
of ovarian cancer cells. Autophagy. 13:333–344. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Wu T, Wang Y, Yang L, Hu C, Chen L
and Wu S: γ-Glutamyl cyclotransferase contributes to tumor
progression in high grade serous ovarian cancer by regulating
epithelial-mesenchymal transition via activating PI3K/AKT/mTOR
pathway. Gynecol Oncol. 149:163–172. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang Y, Qian X, Shen J, Wang Y, Li X, Liu
R, Xia Y, Chen Q, Peng G, Lin SY and Lu Z: Local generation of
fumarate promotes DNA repair through inhibition of histone H3
demethylation. Nat Cell Biol. 17:1158–1168. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang Y, Liu Y, Yu L, Chen J, Hou J, Cui
L, Ma D and Lu W: Histone demethylase KDM2A promotes tumor cell
growth and migration in gastric cancer. Tumour Biol. 36:271–278.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Naora H and Montell DJ: Ovarian cancer
metastasis: Integrating insights from disparate model organisms.
Nat Rev Cancer. 5:355–366. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shield K, Ackland ML, Ahmed N and Rice GE:
Multicellular spheroids in ovarian cancer metastases: Biology and
pathology. Gynecol Oncol. 113:143–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kotiyal S and Bhattacharya S: Breast
cancer stem cells, EMT and therapeutic targets. Biochem Biophys Res
Commun. 453:112–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van Roy F and Berx G: The cell-cell
adhesion molecule E-cadherin. Cell Mol Life Sci. 65:3756–3788.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Christofori G and Semb H: The role of the
cell-adhesion molecule E-cadherin as a tumour-suppressor gene.
Trends Biochem Sci. 24:73–76. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Berx G, Cleton-Jansen AM, Nollet F, de
Leeuw WJ, van de Vijver M, Cornelisse C and van Roy F: E-cadherin
is a tumour/invasion suppressor gene mutated in human lobular
breast cancers. EMBO J. 14:6107–6115. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pećina-Slaus N: Tumor suppressor gene
E-cadherin and its role in normal and malignant cells. Cancer Cell
Int. 3:172003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Duran GE, Wang YC, Moisan F, Francisco EB
and Sikic BI: Decreased levels of baseline and drug-induced tubulin
polymerisation are hallmarks of resistance to taxanes in ovarian
cancer cells and are associated with epithelial-to-mesenchymal
transition. Br J Cancer. 116:1318–1328. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cheng YC, Tsao MJ, Chiu CY, Kan PC and
Chen Y: Magnolol inhibits human glioblastoma cell migration by
regulating N-cadherin. J Neuropathol Exp Neurol. 77:426–436. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu N, Peng SM, Zhan GX, Yu J, Wu WM, Gao
H, Li XF and Guo XQ: Human chorionic gonadotropin β regulates
epithelial-mesenchymal transition and metastasis in human ovarian
cancer. Oncol Rep. 38:1464–1472. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Colomiere M, Ward AC, Riley C, Trenerry
MK, Cameron-Smith D, Findlay J, Ackland L and Ahmed N: Cross talk
of signals between EGFR and IL-6R through JAK2/STAT3 mediate
epithelial-mesenchymal transition in ovarian carcinomas. Br J
Cancer. 100:134–144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cheaib B, Auguste A and Leary A: The
PI3K/Akt/mTOR pathway in ovarian cancer: Therapeutic opportunities
and challenges. Chin J Cancer. 34:4–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sain N, Krishnan B, Ormerod MG, De Rienzo
A, Liu WM, Kaye SB, Workman P and Jackman AL: Potentiation of
paclitaxel activity by the HSP90 inhibitor
17-allylamino-17-demethoxygeldanamycin in human ovarian carcinoma
cell lines with high levels of activated AKT. Mol Cancer Ther.
5:1197–1208. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cao Y: Tumorigenesis as a process of
gradual loss of original cell identity and gain of properties of
neural precursor/progenitor cells. Cell Biosci. 7:612017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang Z, Lei A, Xu L, Chen L, Chen Y,
Zhang X, Gao Y, Yang X, Zhang M and Cao Y: Similarity in
gene-regulatory networks suggests that cancer cells share
characteristics of embryonic neural cells. J Biol Chem.
292:12842–12859. 2017. View Article : Google Scholar : PubMed/NCBI
|