Introduction
In the last few years, increased evidence strongly
indicates that altered microRNA (miRNA or miR) expression can play
a significant role in cancer development and progression depending
on the tissue type and specific target (1). miRNAs are small non-coding regulatory
RNAs (19–25 nucleotides) that bind to specific sites of their
target genes and regulate post-transcriptional gene expression
(2). To date, the mechanisms
regulating normal miRNA expression and triggering their
deregulation in malignant diseases are poorly understood. The
primary control of miRNA expression occurs at the transcriptional
level. miRNA genes are frequently located in intergenic regions,
being independently transcribed, or are situated within exonic and
intronic regions and may share a promoter with the host gene,
although intragenic miRNA-specific promoters have also been
reported (3,4). miRNA genes may be regulated through
epigenetic mechanisms, such as specific histone modifications
and/or DNA methylation of CpG islands in promoter regions, or by
regions that are located next to miRNA genes (4). This regulation is tightly controlled and
has a great impact on the establishment and maintenance of cell
identity, since miRNAs, together with transcription factors, are
major regulators of cell phenotype (5). Furthermore, it has been proposed that
small subsets of miRNAs are capable of discriminating cell lines
and tissues, reinforcing the impacts of these molecules on cell
type-specific networks (6).
Burkitt lymphoma (BL) is a highly aggressive B-cell
malignancy that accounts for 40% of non-Hodgkin's lymphomas in
children and adolescents (7). Three
clinical variants of BL, including endemic, sporadic and
HIV-associated BL, have been described. Virtually all endemic BL
cases are EBV-associated, whereas less than 30% of sporadic and
30–40% of AIDS-related BLs are EBV-positive (8). The tumour is characterized by the
presence of chromosomal translocations, mainly t(8;14) or, less
frequently, t(2;8) or t(8;22), involving the c-MYC oncogene
and immunoglobulin genes [IGH (q32), IGK (p12), and
IGL (q11), respectively] leading to the constitutive
activity of the c-MYC promoter (9–11). Recent
studies have revealed recurrent mutations in the TCF3, a direct
target CCND3. In addition, CCND3 mutations were frequent in
sporadic BL (38%) and HIV-associated BL (67%) but not endemic BL
(1.8%), indicating a distinct genetic pathogenesis among BL
subtypes (12,13). Emerging evidence indicates that
abnormal modulation of mRNA transcription via miRNAs regulated by
MYC may be a significant event in BL pathogenesis (14). In this context, a signature of miRNAs,
including molecules regulated by MYC, has been proposed to
differentiate BL from diffuse large B-cell lymphoma (DLBCL), with
miR-29b being downregulated in BL (15,16). This
observation, together with the association between miR-29 and
histone marks, that are indicative of typical enhancers and
super-enhancers (5), indicates that
the deregulation of miR-29 family members may play a role in BL
pathogenesis. The miR-29 family has been described as a suppressor
of tumours regulating multiple oncogenic pathways in diverse types
of cancer (17–19). MYC also acts in the epigenetic
suppression of miR-29 by inducing histone deacetylation and histone
trimethylation in B-cell lymphomas (20). Additionally, DNA methylation has been
associated to transcription regulation of numerous cancer-related
genes including certain miRNAs such as miR-34b, miR-124a, miR-200,
among others (4).
In the present study, the contribution of DNA
methylation to miR-29 silencing was underlined in BL cell lines and
tumour samples, targeting both promoters and enhancers using
pyrosequencing quantitative assays. In addition, the current status
of the interplay between MYC and miR-29 regulation was reviewed,
highlighting the potential role of EBV-miRNAs in miR-29 regulation
for BL pathogenesis.
Materials and methods
Cell lines and treatments
The BL cell lines: Raji (EBV+) obtained
from the American Type Culture Collection (ATCC), Daudi
(EBV+) and BL41 (EBV−) kindly provided by Dr
Boulanger (Greehey Children's Cancer Research Institute, The
University of Texas Health Science Center, San Antonio, TX, USA),
Namalwa (EBV+) kindly provided by Dr Favaro (Laboratório
de Biologia Molecular e Celular, Hemocentro, Unicamp, Campinas, São
Paulo, Brazil) and Ramos (EBV−) provided by Professor
Andrei Thomas-Tikhonenko (Department of Pathobiology, School of
Veterinary Medicine, University of Pennsylvania, Philadelphia,
Pennsylvania, USA) were cultured accordingly (21). Mycoplasma contamination PCR and short
tandem repeat (STR) PCR were performed to confirm that the cell
lines were mycoplasma-negative and to verify their genotypes. For
the experiments, cells were treated with DNMT inhibitor
[5-aza-2′-deoxycytidine (decitabine); Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany] at 1.0, 0.5, 0.25 and 0.125 µM for 24 or 72 h.
The cells were collected and evaluated for DNA methylation and
protein expression.
Patients
Formalin-fixed paraffin-embedded tumour samples from
10 patients with pediatric BL were selected for the analysis. The
study was approved by the Brazilian National Cancer Institute
Ethics Research Committee (CEP Registration no. 18/09). The
diagnosis of BL was confirmed according to the criteria described
by the 2008 World Health Organization (WHO) classification for
hematopoietic diseases (22).
Portions of these data have been previously published (23).
DNA isolation and bisulfite conversion
from cell lines and tissues of patients
Genomic DNA was extracted with a QIAamp DNA FFPE
Tissue kit (Qiagen, Inc.) from a tumour block fixed in 10% neutral
buffered formaline and embedded in paraffin (FFPE) using sections
of 10 µm. The average period of fixation was 24 h at room
temperature. DNA from cells lines was extracted using the QIAamp
DNA kit (Qiagen, Inc.) and quantified on a NanoDrop 1000 (Thermo
Fisher Scientific, Waltham, MA, USA). Bisulfite conversion of 500
ng of genomic DNA was performed using the EpiTect Bisulfite Kits
(Qiagen, Inc.) according to the manufacturer's protocol.
Pyrosequencing assay
The previously bisulfite-converted DNA was amplified
using Platinum Taq DNA Polymerase (Thermo Fisher Scientific, Inc.)
in a 50-µl final volume according to the manufacturer's protocol.
The primers utilized were designed using PyroMark Assay Design
Software 2.0.2 (Qiagen, Inc.): primer 1, 29b2/c gene
forward, AGAAGGTAGGGTTGTAAGGA and reverse,
AAATCCCCACTCTCTAACCTATCTTTAT; primer 2, 29b2/c gene forward,
TAGTAAATATATAAGTGGGGGAAGAAGGGG and reverse,
TATCAAAACCAAAAACCTCTAAATAACC; primer 1, 29a/b1 gene forward,
GTTTTTTAGAGAGTTTTGGGTTGTT and reverse,
CCTAAAACAAAATCCCTACAAATTTTCA. The fragments amplified with these
primer sets contained 5, 6 and 5 CpG sites, respectively. All
reverse primers were biotinylated. The sequences for analysis were
localized to two regions: One in the promoter of the
miR-29b2/c gene (Chromosome 1:207823418-207824167) and the
other within the promoter flank region of the miR-29b2/c
gene (Chromosome 1:207823405-207824179). Another sequence for
analysis was located within the enhancer of the miR-29a/b1
gene (Chromosome 7:130897044-130897608). Two percent agarose gel
electrophoresis was used to confirm DNA amplification. Biotinylated
PCR products in a total volume of 40 µl were immobilized on
streptavidin-coated Sepharose beads (GE Healthcare). Pyrosequencing
was performed using PyroGold PyroMark Q96 reagents in the PyroMark
Q96 ID (both from Qiagen, Inc.). The percentage of each CpG site
was generated automatically using the PyroMark Q96 software
(version1 0.6).
miRNA extraction from BL cell lines
and real-time quantitative PCR (qRT-PCR)
Total RNA (including the miRNA) from BL41 and Raji
cells was extracted using TRIzol™ reagent (TRIzol™; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
concentration and quality of the extracted material was determined
with the 260/280 ratio using NanoDrop 1000 (Thermo Fisher
Scientific Inc.). The miRNA expression analyses were assessed by
primer-specific TaqMan® MicroRNA Reverse Transcription
(Applied Biosystems; Thermo Fisher Scientific, Inc.) and
hsa_miR-29a (ID 2112), hsa_miR-29b (ID 413), hsa_miR-29c (ID 587)
and RNU6B (NR_002752) were examined using the StepOne™
System (Applied Biosystems; Thermo Fisher Scientific, Inc.).
RNU6B was used to normalize the miRNA expression levels.
Thermocycling conditions consisted of incubation at 50°C for 2 min
and 95°C for 10 min, followed by 40 cycles of denaturation at 95°C
for 15 sec, and annealing and extension at 60°C for 1 min. To
calculate the relative expression, the 2−ΔΔCq method was
applied (24). Duplicate reactions
were performed in all PCR assays.
Western blotting
Western blot assays were performed as previously
described (23), using the following
antibodies: Anti-c-Myc (dilution 1:250; clone no. 9E10; Calbiochem;
cat. no. OP10), anti-DNMT1 (dilution 1:1,000; clone no. H-300;
Santa Cruz Biotechnology, Inc; cat. no. SC-13032) and DNMT3B
(dilution 1:1,000, clone no. 52A1018, Imgenex; cat. no. IMG-184A).
β-actin (dilution 1:1,000; clone B-6; cat. no. A5441) and HSC-70
(dilution 1:1,000; cat. no. sc-7298; both from Santa Cruz
Biotechnology, Inc.) antibodies were used as loading controls.
Mouse and rabbit secondary antibodies (dilution 1:10,000; cat. no.
A9169) were purchased from GE Healthcare. The blots were developed
using the c-Digit imaging system (LI-COR Biosciences) and analysed
by Image Studio Lite version 3.1.
Transfection of an inhibitor of
EBV-miR-BART6-5p
The Cy3-labbed control siRNA (Ambion; Thermo Fisher
Scientific, Inc.) was used to monitor the efficiency of the
transfection assay with Lipofectamine 2000 or RNAiMAX (both from
Invitrogen; Thermo Fisher Scientific, Inc.). Raji cells
(1×105) were transfected with Cy3-labeled siRNA (200
nM), washed with phosphate-buffered saline and analysed by a CyAn
ADP analyzer flow cytometer and Summit v4.3 software (both from
Beckman Coulter, USA). Next, Raji cells were transfected with 100
nM of an inhibitor of BART6-5p antagomir (cat. no. 4464084) or the
mimic negative control (cat. no. 4464058) (Life Technologies;
Thermo Fisher Scientific, Inc.) utilizing the method derived from
Mazzoccoli et al (21). Then,
the cells were treated with decitabine (1.0 µM) and after 72 h, the
cells were harvested, and miRNA was extracted and quantified as
aforementioned.
Statistical analysis
Comparisons among groups were performed using
Kruskal-Wallis and Dunnett's post hoc test. P-values <0.05 were
considered to indicate a statistically significant difference, and
analysed using GraphPad Prism software (PRISM 5.0; GraphPad
Software Inc.).
Results
miR-29a/b1 and miR-29b2/c genes are
epigenetically silenced by methylation at promoter and enhancer
regions in BL cells
To confirm previous research that revealed
methylation in miR-29a/b1 and miR-29b2/c by MSP assays in BL cell
lines, the miR-29 promoter and enhancer regions were explored using
the pyrosequencing method for investigation of BL tumour samples.
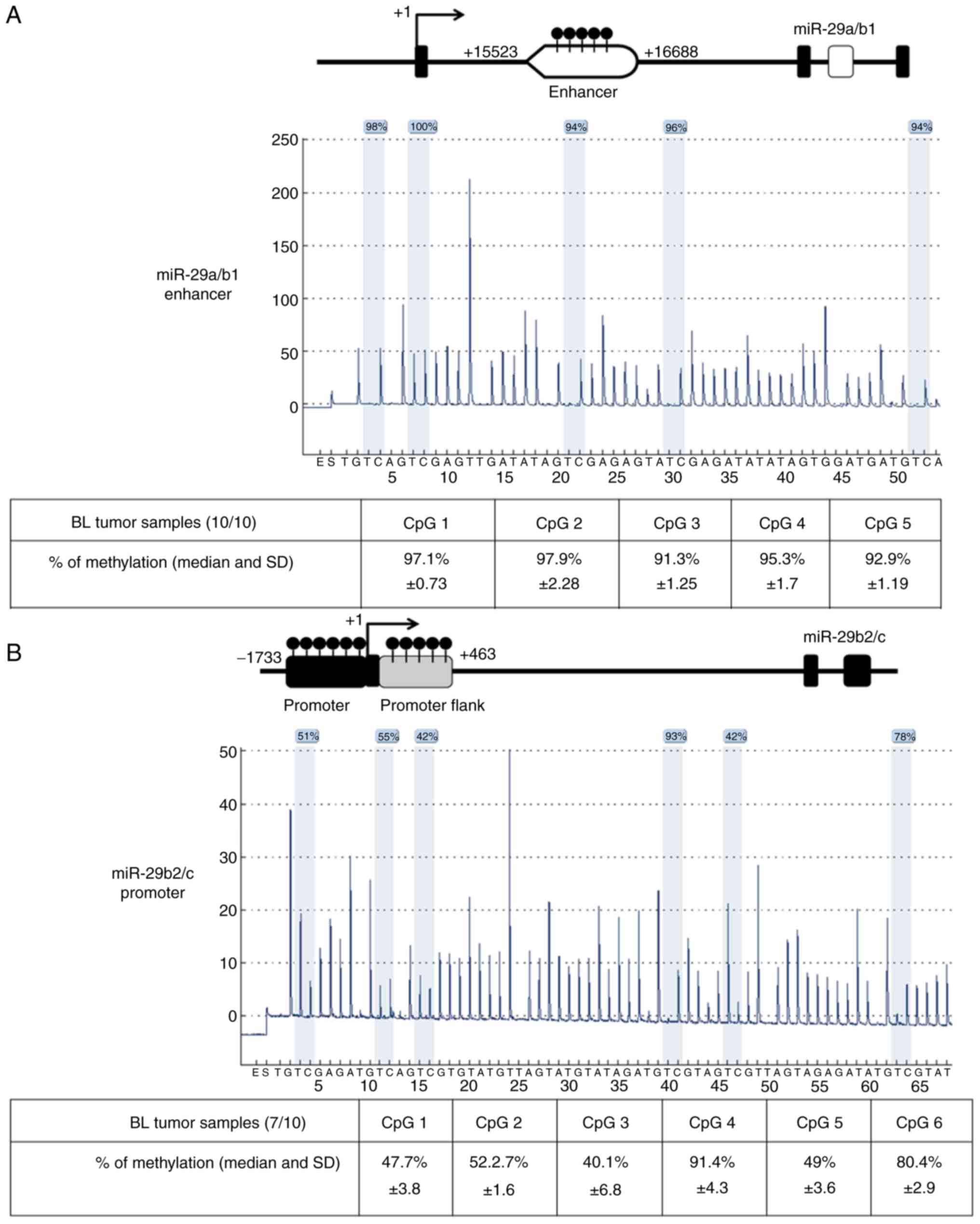
Data of BL patients are described in Table I. Quantitative pyrosequencing
methylation analysis of five CpG sites for miR-29a/b1 enhancer
region and six CpG sites for miR-29b2/c promoter region
demonstrated high levels of DNA methylation in 10/10 and 7/10 BL
tumour samples, respectively as depicted (Fig. 1). The mean of methylation levels in
CpG sites was 92.8% in miR-29a/b1 enhancer and 64% in miR-29b2/c
promoter in BL tumour samples. Additionally, the high methylation
level observed in BL tumour samples could explain the low miR-29s
levels reported previously by our group (23). In Raji and BL41 cell lines the results
also revealed mean methylation levels of 96 and 99% in CpG sites
located in miR-29a/b1 enhancer and 89 and 91% in miR-29b2/c
promoter flanking regions, respectively (Fig. S1A and C). Notably, the mir-29b2/c
promoter from Raji cells revealed lower mean methylation levels
(63%) than in BL41 cells (92%) at six evaluated CpG sites (Fig. S1B). Thus, using BL tumour tissues
analysed by pyrosequencing, it was concluded that silencing by
methylation can work together with other mechanisms to potentiate
the suppression of miR-29 in BL cells (20).
 | Table I.Clinical and biological features of
BL patients. |
Table I.
Clinical and biological features of
BL patients.
| Patient | Sex/Age | Initial site of
disease | Stage | LDH (U/l) | EBV (ISH) | Follow-up |
|---|
| 1 | F/2 | Abdomen | III | 394 | Positive | Alive |
| 2 | M/5 | Abdomen | III | 595 | Positive | Dead |
| 3 | M/6 | Abdomen | III | 510 | Positive | Alive |
| 4 | M/3 | Abdomen and
testicle | III | 845 | Positive | Dead |
| 5 | M/5 | Abdomen | III | 929 | Positive | Alive |
| 6 | F/6 | Abdomen | III | 536 | Negative | Alive |
| 7 | M/9 | Cervical mass | IV | 2,438 | Negative | Dead |
| 8 | M/11 | Abdomen | III | 1,205 | Positive | Alive |
| 9 | M/5 | Cervical mass | I | 453 | Positive | Alive |
| 10 | M/2 | Abdomen | III | 621 | Positive | Alive |
The treatment of BL cells with
DNA-demethylating agent decitabine reverses methylation and induces
miR-29 expression
Since tumour suppressive miRNAs are inactivated in
cancer cells by aberrant DNA methylation at promoter regions,
resulting in suppression, the effects of demethylating agent
decitabine was investigated on the miR-29 methylation and
expression levels in BL cells. After 24 h with 1.0 µM decitabine,
the miR-29 promoter and enhancer regions were demethylated in both
BL cell lines. In BL41 cells, the mean of cells that were
methylated in CpG sites dropped 22% in the miR-29a/b1 enhancer, 21%
in the miR-29b2/c promoter and 56% in the miR-29b2/c promoter flank
in comparison to the control. As for Raji cells, the mean of
methylation levels in CpG sites dropped 40% in the miR-29a/b1
enhancer, 21% in the miR-29b2/c promoter and 20% in the miR-29b2/c
promoter flank in comparison to the control (Fig. S1). Similar results were observed at
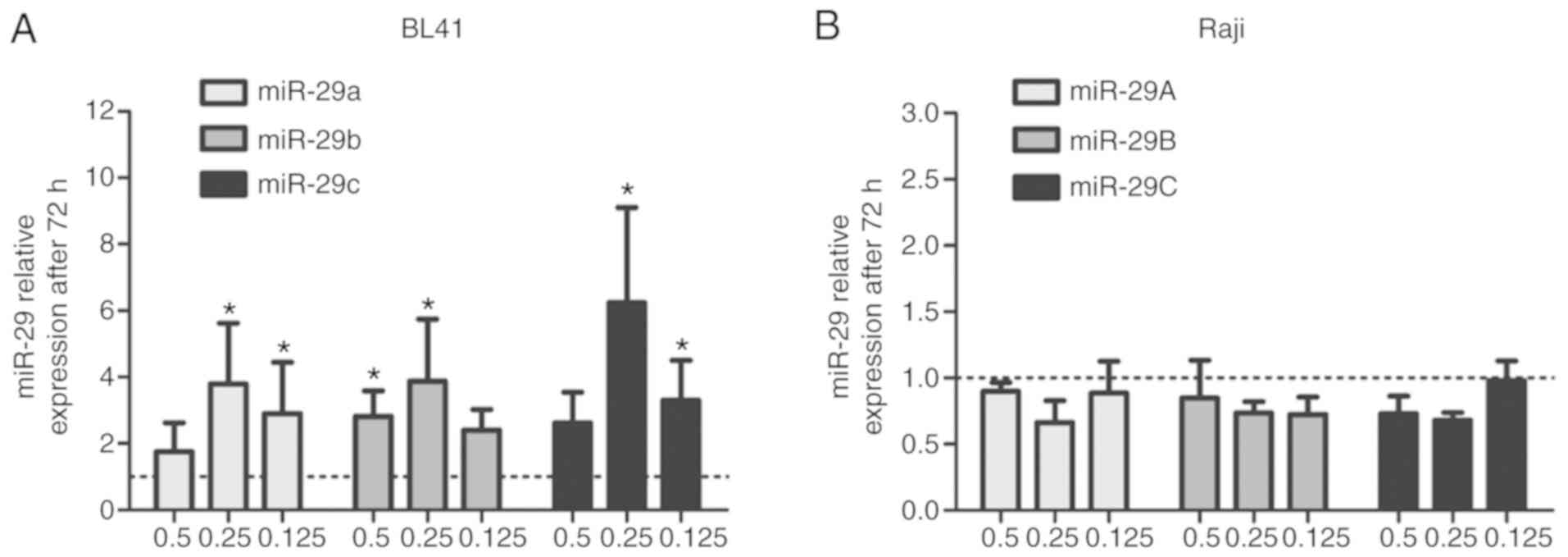
72 h, even using lower decitabine concentrations (Fig. S2). In addition, the miR-29 expression
levels were upregulated by decitabine treatments (0.5, 0.25 and
0.125 µM) at 72 h in BL41 cells (Fig.
2A) but not in Raji cells (Fig.
2B). Similar results only for the high-decitabine concentration
were reported previously by our group using a non-quantitative
methylation-specific PCR assay (MSP) (21).
DNA methyltransferase inhibitor,
decitabine, downregulates MYC protein levels in a dose-dependent
manner
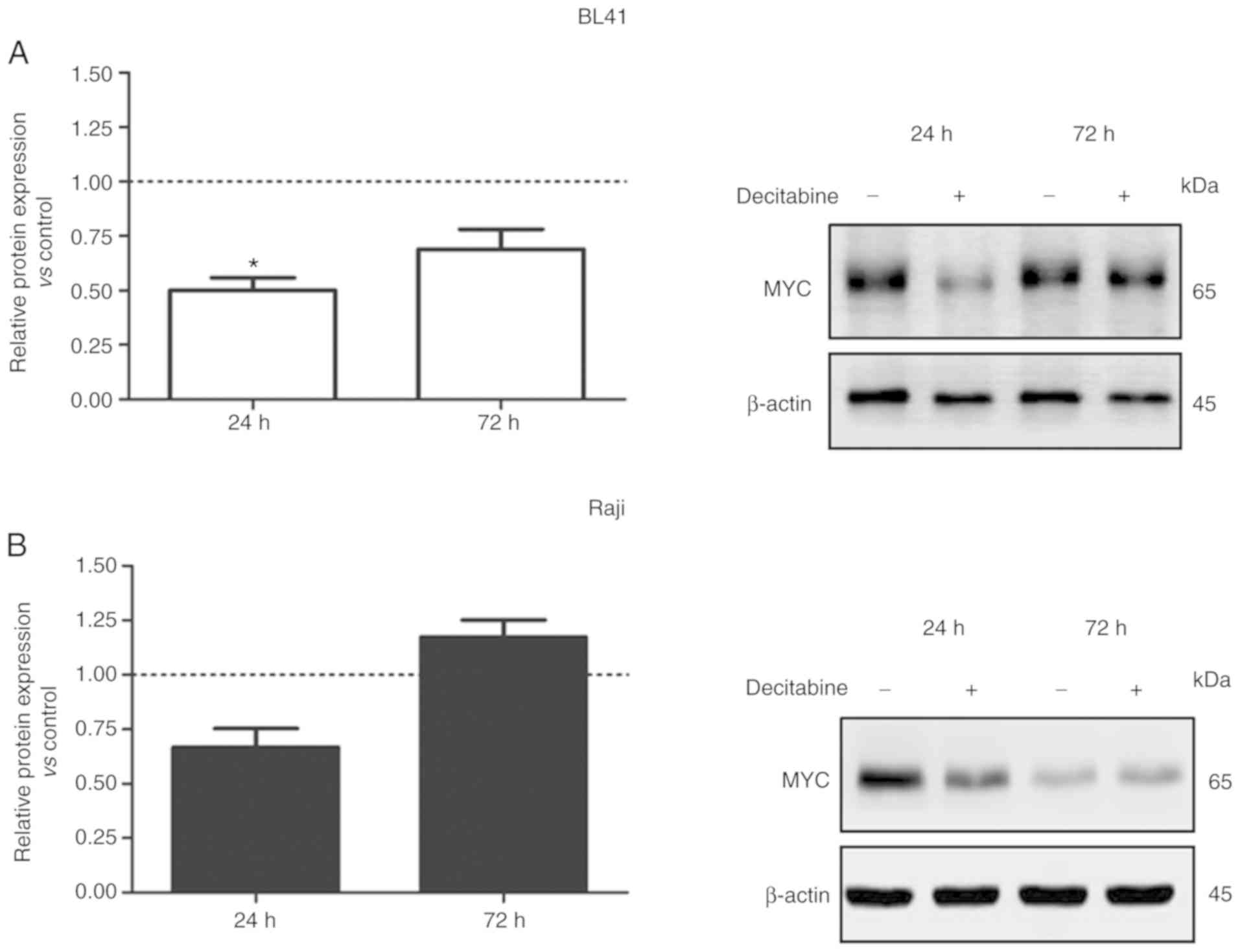
Given that MYC is involved in miR-29 regulation, we
further evaluated the effect of decitabine on MYC protein
expression. Upon 24 and 72 h of decitabine treatment (1.0 µM), MYC
expression levels were reduced in BL41 cells (Fig. 3A). Notwithstanding the variations
observed on MYC expression levels in the control (without treatment
of decitabine), these variations were expected due the prolonged
cell culture (24 to 72 h), since the medium was not replaced during
the assay. The cell cycle status and the concentration of fetal
bovine serum, for example, may contribute to the stabilization of
mRNA and protein levels. In addition, a slight decrease in the
level of β-actin was detected in the presence of decitabine.
However, this variation did not have impact on MYC expression
analysis (Fig. 3A). A similar result
was observed in Raji cells at 24 h after decitabine treatment
(Fig. 3B). These data indicated that
demethylation induced by decitabine may affect other genes or
miRNAs that target the MYC regulatory network (25). However, lower doses of decitabine
reduced DNMT1 expression in BL41 and Raji cells with no effects on
MYC protein levels (Fig. 3C and
D).
EBV-miR-BART6-5p inhibition modulates
miR-29 expression in BL cells treated with decitabine
It has been reported that EBV regulates the
expression of non-coding RNA in gastric carcinoma (GC). The
methylation of both viral and host DNA is one of the major
mechanisms involved in EBV-associated GC tumour development
(26). These studies indicate that
EBV infection alters the host miRNA profile. In our study, despite
the demethylation of miR-29 promoter CpG sites observed in Raji, an
EBV-positive cell line, an increase in the expression levels was
not detected (Fig. 2B). Therefore,
whether the observed lack of miR-29 expression could be related to
the expression of EBV-miRNAs was investigated since it has been
reported that an EBV-miRNA, miR-BART6-5p, suppresses Dicer
expression by binding to four target sites that are present within
the 3′-UTR of human Dicer (27).
Then, miR-BART6-5p expression was silenced and the effect of
decitabine on miR-29 expression levels was examined. The
transfection efficiency assay was confirmed in the Raji cell line
using Cy3-labeled control siRNA as revealed in Fig. S3.
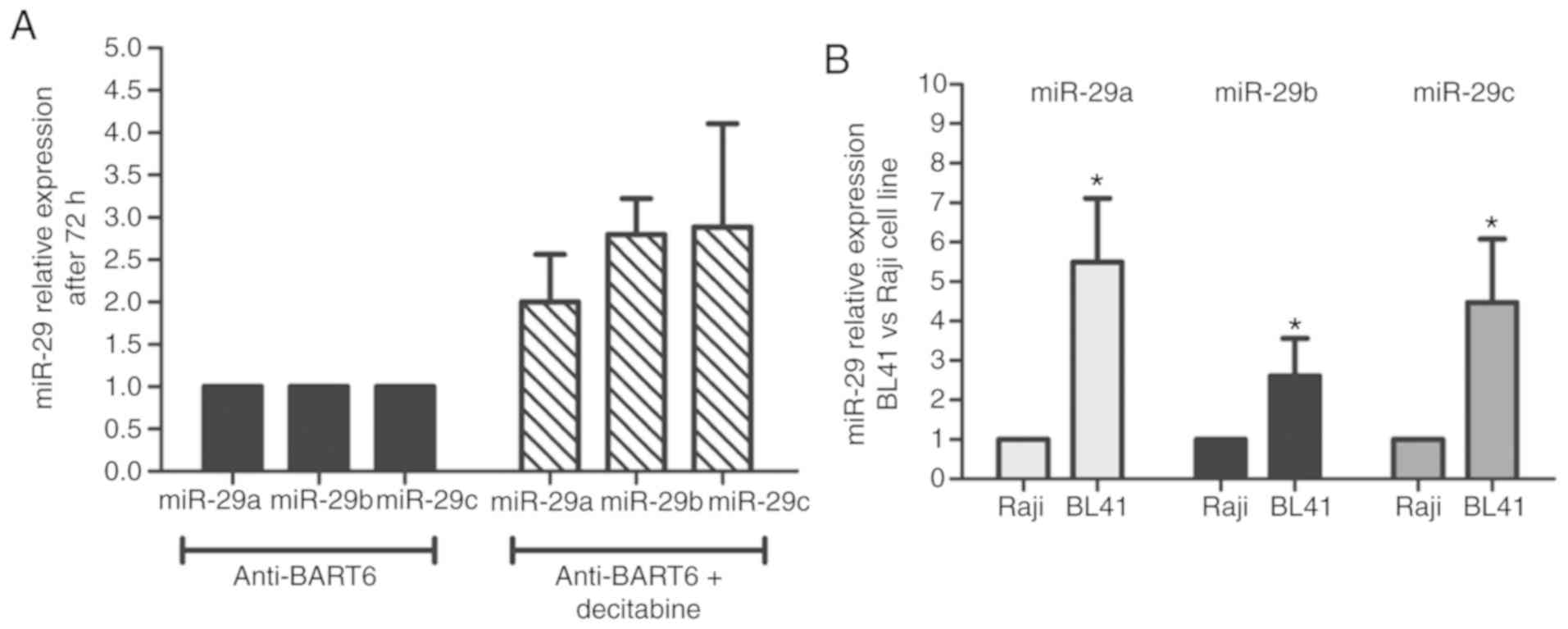
Suppression of miR-BART6-5p by antagomir followed by
decitabine treatment was associated with an increase of miR29a/b/c
expression levels in comparison to only antagomir (Fig. 4A). There was no difference between the
mimic negative control and non-transfected cells regarding miR-29
expression (data not shown). In addition, the basal miR-29
expression levels in the BL41 cell line were increased in
comparison to Raji cells, supporting the hypothesis of a
differential miR-29 regulatory mechanism between EBV-positive and
EBV-negative BL cells (Fig. 4B). The
analysis of additional BL cell lines (EBV-positive and negative)
indicated the same differential expression levels of miR-29a and
miR-29c (Fig. S4).
Discussion
Recent studies have revealed the numerous roles of
miRNAs underscoring the biological significance of their
alterations in cancer development (1,28). The
miR-29 family of miRNAs has been identified to function as a group
of tumour suppressors (29,30) in several cancer types via inhibition
of targets related to proliferation, migration and invasion
(31,32). In addition, the miR-29 family members
(miR-29a/b/c) are also termed ‘epi-miRs’ due to their role in the
regulation of epigenetic modifiers such as enzymes involved in DNA
methylation and demethylation [DNA methyltransferases [DNMTs] and
ten-eleven translocation (TET) enzymes] (4). Recently, our group demonstrated that the
transfection of miR-29s modulated DNA methyltransferase 3B in BL
cells. Moreover, overexpression of miR-29a/b/c resulted in the
inhibition of other targets that are associated with cell cycle
control, proliferation, and apoptosis (21,23).
miR-29s were previously reported to be regulated by MYC
contributing to tumorigenesis (33).
The protein c-MYC has a critical role in different cellular
processes and its protein stability is tightly regulated (34,35). In
BL, the chromosomal translocation places its promoter next to the
immunoglobulin enhancers, increasing the transcription of the mRNA,
followed by protein translation (9–11).
MYC promotes B-cell lymphomagenesis through a
complex interplay with several signalling pathways, including miRNA
regulation (36–39). Among the several MYC-regulated miRNAs,
it was revealed that MYC may suppress the expression of miR-29s
through the recruitment of the HDAC3/EZH2 co-repressor complex to
the promoter region of miR-29 genes (20). EZH2 and HDCA3 are part of the Polycomb
repressive complex 2 (PRC2), which specifically trimethylates
histones at K27, resulting in epigenetic silencing. Although MYC
activity can mediate the suppression of miR-29 through HDAC and
EZH2 recruitment, other mechanisms can work together to potentiate
suppression. In fact, more than one mechanism of epigenetic
regulation can cooperate and enhance regulation (28). Notably, it is worth considering the
promoter tissue miRNA-specificity regarding aberrant miRNA cancer
methylation (28,40). Approximately 45.5% of miRNA genes have
been shown to be methylated in at least two cancer types (41). Epigenetic silencing by promoter
hypermethylation of miRNAs with tumour suppressor activities has
been described in different types of cancers, being associated with
aberrant miRNA expression (41,42).
Therefore, miRNA methylation in cancer has been investigated by
pharmacological unmasking experimental assays using a DNA
methyltransferase inhibitor, 5-aza-2′-deoxycytidine (5-aza-CdR, DAC
or decitabine), and histone deacetylase (HDAC) inhibitors, as well
as double knockout models for DNA methyltransferase enzymes DNMT1
and DNMT3B (43,44). Given the regulatory role of MYC in
miR-29 expression, methylation was assessed as an additional
mechanism that may contribute to miR-29 silencing in BL tumour
samples. In the previous study we used MSP assays to reveal the
miR-29 promoter and enhancer gene methylation in BL cell lines
(21). The MSP assay has known
disadvantages such as the use of relative comparisons instead of
absolute quantification (45,46). Using quantitative bisulfite
pyrosequencing assays, methylation was investigated in CpG sites
located in the promoter and enhancer regions of miR-29a/b/c in BL
tumour samples. It was demonstrated that BL cell lines as well as
BL tumours exhibit hypermethylation of miR-29a/b1 enhancer
and miR-29b2/c promoter regions.
It has been proposed that the determination of cell
identity during development is highly associated with enhancer
usage, which is locally regulated by DNA methylation (47). Active enhancers are characterized by a
higher density of transcription factor binding sites, DNA
hypomethylation, a high level of H3K27ac and the binding of a
mediator (48). However, enhancers
not only play a role in early lineage commitment but also regulate
the expression of cell type-specific genes in differentiated cells,
such as macrophages and B cells (48). Other key determinants of cell fate
include transcription factors and miRNAs, enclosing a complex
regulatory network where these different factors interact with each
other (5). It is not surprising that
these networks are often dysregulated in cancer, a condition in
which cell identity and commitment to tissue homeostasis is lost.
Notably, differential expression of key transcription factors, such
as MYC overexpression in BL (9–11), of
miRNAs, as exemplified by the upregulation of miR-21 and
downregulation of let-7 in breast cancer (49), and alterations in enhancer methylation
have been revealed in tumours (50).
In the present study, we propose a complex network that may play a
role in BL pathogenesis, in which MYC overexpression, a common
feature of this neoplasia, leads to DNMT1 and DNMT3B upregulation
that is associated with miR-29 silencing by methylation of promoter
and enhancer regions.
The MYC/miR-29/DNMT3B circuit was identified by El
Baroudi et al as a feed-forward loop in which a master
transcription factor regulates a miRNA and, together with it, a set
of protein coding genes that are targeted by the same miRNA (Type
II) (51). Moreover, these feedback
regulations point to complex networks between miRNAs and epigenetic
machinery enhancing the human gene regulatory network. Notably, MYC
can lead to epigenetic deregulation through induction of DNMT1 and
DNMT3B transcription. The transcriptional regulation of DNMT3B by
MYC during B-cell lymphomagenesis has been recently demonstrated
in vivo (52). It is
noteworthy to mention that a previous study of our group
demonstrated overexpression of DNMT3B in ~86% of BL tumour samples
(23). Additionally, it was also
described by Poole et al that MYC binds to the DNMT1 and
DNMT3B promoters in BL-like cells and deregulates the expression of
both DNMT1 and DNMT3B. Moreover, the knockdown of endogenous MYC
leads to diminished DNMT3B expression levels in human BL cell
lines. Thus, a model was proposed where MYC additionally controls
DNA methylation through overexpression of DNMT1 and DNMT3B during
lymphomagenesis (53).
It has also previously revealed that miR-29a/b/c
expression is downregulated in BL tumour samples (23). Since miRNA expression can be lost due
to methylation, as demonstrated in the present study in BL cell
lines, 10 BL tumour samples were screened for methylation on
miR-29a/b1 and miR-29b2/c. Methylation in CpG sites located in the
promoter and enhancer regions was also confirmed in tumour tissues.
These findings suggest a relationship between downregulation of
tumour-suppressive miR-29a/b/c, overexpression of DNMT1 and DNMT3B
and hypermethylation in miR-29a/b1 and miR-29b2/c in
BL cells.
Another layer of complexity in miR-29 regulation was
revealed in our study using an EBV-positive BL cell line (Raji).
Regarding methylation of the promoter, promoter flanking region,
and enhancer regions of miR-29, it was observed in the Raji cells
that although treatment with lower concentrations of decitabine
decreased the methylation levels of evaluated CpG sites, it did not
increase miR-29 expression. One possible explanation would be
related to EBV, which has been reported to decrease the levels of
miRNAs including miR-29 in peripheral blood-derived B-cells
infected in vitro with EBV (54). EBV proteins have also been reported to
contribute to methylation, in which the EBV proteins EBNA3A/C bind
next to the BIM gene promoter, followed by CpG methylation
(55). Although EBV latency may vary
among BL EBV-positive cell lines, miRNA production is a common
feature among the different EBV latency types (56). Amongst miRNAs, the EBV-BART cluster
comprised of several miRNAs has been revealed to regulate both EBV
and host protein expression (57). It
has been reported that EBV-miRNA-BART prevents apoptosis by
inhibiting caspase-3 and enhances cell growth of EBV-infected
B-cells. Thus, EBV maintains lymphomas via its miRNAs (58). Notably, BART6 decreases DICER1
expression, which is involved in miRNA maturation in the cell
cytoplasm (27). In the present
study, after transfection with an antagomir to specifically inhibit
BART6-5p, the treatment with decitabine resulted in increased
miR-29 expression. The results indicated a role for EBV miRNAs in
the complex miR-29 regulation of BL tumours associated with EBV
adding a new layer of complexity in miR-29 regulation in BL
tumours.
Furthermore, it was demonstrated for the first time
by pyrosequencing of multiple CpG sites that BL tumour samples
present high levels of methylation in both promoter and enhancer
regions. Although the idea of a functional assay to evaluate the
activity of the enhancer would be relevant, Suzuki et al
(5) have shown that the region
analysed by us in miR-29a/b1 sequence (chr7:
130,582,141-130,582,237, genome version GRCh37/hg19) flanks a
typical enhancer region (chr7:130582783-130585367, genome version
GRCh37/hg19) identified in CD19-positive primary cells, and
encompasses a super-enhancer region (chr7: 130577042-130754905,
genome version GRCh37/hg19) in CD20-positive cells. The same
authors have also revealed that enhancers and super-enhancers are
cell type-specific and their activity has a great impact on cell
fate, which could be mediated by the regulation of master miRNAs.
Therefore, the miR-29a/b1 region analysed in our study is
considered as specifically relevant for our model on BL.
In summary, the miR-29a/b1 and miR-29b2/c genes have
methylated CpG sequences at promoter and enhancer regions that may
contribute to the regulation of the expression of miR-29s in BL
tumours. The findings indicate interplay between MYC and miR-29
regulation, highlighting the potential role of EBV-microRNAs in
miR-29 regulation for BL pathogenesis. On the other hand, studies
focusing on EBV-miRNAs in lymphomas are limited to cell lines.
Animal models for EBV-associated carcinogenesis are required to
address the regulatory network involving EBV-miRNAs.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
INCT for Cancer Control: CNPq 573806/2008-0/FAPERJ
E26/170.026/2008; FAPERJ E-26/110.375/2014; SWISS-BRIDGE
Foundation, sub-project 1B/2014-2018. LM and MCR were supported by
grants from the Brazilian National Cancer Institute (INCA), Health
Ministry.
Availability of data and materials
The datasets generating during this study are
included within the article; data not shown is available from the
corresponding author upon request.
Authors' contributions
CEK and LM devised the study concept. LM and MCR
performed the experiments and generated and analysed the data. LM,
MCR, SCSL, and CEK interpreted the data and drafted the manuscript.
CEB revised the BL cases and performed FISH assays. CEK and SCSl
obtained funding and supervised the study. All authors critically
reviewed and approved the final version of the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The present study was approved by the Brazilian
National Cancer Institute Ethics Committee (registration number
18/09) in accordance with the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that that they have no competing
interests.
References
|
1
|
Esteller M: Epigenetic changes in cancer.
F1000 Biol Rep. 3:92011. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ying SY, Chang CP and Lin SL:
Intron-mediated RNA interference, intronic microRNAs, and
applications. Methods Mol Biol. 629:205–237. 2010.PubMed/NCBI
|
|
4
|
Moutinho C and Esteller M: MicroRNAs and
epigenetics. Adv Cancer Res. 135:189–220. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Suzuki HI, Young RA and Sharp PA:
Super-enhancer-mediated RNA processing revealed by integrative
MicroRNA network analysis. Cell. 168:1000–1014.e15. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Landgraf P, Rusu M, Sheridan R, Sewer A,
Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M,
et al: A mammalian microRNA expression atlas based on small RNA
library sequencing. Cell. 129:1401–1414. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hochberg J, Waxman IM, Kelly KM, Morris E
and Cairo MS: Adolescent non-Hodgkin lymphoma and Hodgkin lymphoma:
State of the science. Br J Haematol. 144:24–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brady G, Macarthur GJ and Farrell PJ:
Epstein-Barr virus and Burkitt lymphoma. Postgrad Med J.
84:372–377. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dalla-Favera R, Bregni M, Erikson J,
Patterson D, Gallo RC and Croce CM: Human c-myc onc gene is located
on the region of chromosome 8 that is translocated in Burkitt
lymphoma cells. Proc Natl Acad Sci USA. 79:7824–7827. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taub R, Kirsch I, Morton C, Lenoir G, Swan
D, Tronick S, Aaronson S and Leder P: Translocation of the c-myc
gene into the immunoglobulin heavy chain locus in human Burkitt
lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci USA.
79:7837–7841. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Willis TG and Dyer MJ: The role of
immunoglobulin translocations in the pathogenesis of B-cell
malignancies. Blood. 96:808–822. 2000.PubMed/NCBI
|
|
12
|
Schmitz R, Young RM, Ceribelli M, Jhavar
S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, et
al: Burkitt lymphoma pathogenesis and therapeutic targets from
structural and functional genomics. Nature. 490:116–120. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sander S, Calado DP, Srinivasan L, Köchert
K, Zhang B, Rosolowski M, Rodig SJ, Holzmann K, Stilgenbauer S,
Siebert R, et al: Synergy between PI3K signaling and MYC in Burkitt
lymphomagenesis. Cancer Cell. 22:167–179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Oduor CI, Kaymaz Y, Chelimo K, Otieno JA,
Ong'echa JM, Moormann AM and Bailey JA: Integrative microRNA and
mRNA deep-sequencing expression profiling in endemic Burkitt
lymphoma. BMC Cancer. 17:7612017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lenze D, Leoncini L, Hummel M, Volinia S,
Liu CG, Amato T, De Falco G, Githanga J, Horn H, Nyagol J, et al:
The different epidemiologic subtypes of Burkitt lymphoma share a
homogenous micro RNA profile distinct from diffuse large B-cell
lymphoma. Leukemia. 25:1869–1876. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hezaveh K, Kloetgen A, Bernhart SH,
Mahapatra KD, Lenze D, Richter J, Haake A, Bergmann AK, Brors B,
Burkhardt B, et al: Alterations of microRNA and microRNA-regulated
messenger RNA expression in germinal center B-cell lymphomas
determined by integrative sequencing analysis. Haematologica.
101:1380–1389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu K, Liu L, Zhang J, Wang Y, Liang H,
Fan G, Jiang Z, Zhang CY, Chen X and Zhou G: miR-29b suppresses the
proliferation and migration of osteosarcoma cells by targeting
CDK6. Protein Cell. 7:434–444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kwon JJ, Factora TD, Dey S and Kota J: A
systematic review of miR-29 in cancer. Mol Ther Oncolytics.
12:173–194. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Garzon R, Heaphy CE, Havelange V, Fabbri
M, Volinia S, Tsao T, Zanesi N, Kornblau SM, Marcucci G, Calin GA,
et al: MicroRNA 29b functions in acute myeloid leukemia. Blood.
114:5331–5341. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang X, Zhao X, Fiskus W, Lin J, Lwin T,
Rao R, Zhang Y, Chan JC, Fu K, Marquez VE, et al: Coordinated
silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic
target of histone modification in aggressive B-Cell lymphomas.
Cancer Cell. 22:506–523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mazzoccoli L, Robaina MC, Apa AG, Bonamino
M, Pinto LW, Queiroga E, Bacchi CE and Klumb CE: miR-29 silencing
modulates the expression of target genes related to proliferation,
apoptosis and methylation in Burkitt lymphoma cells. J Cancer Res
Clin Oncol. 144:483–497. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Campo E, Swerdlow SH, Harris NL, Pileri S,
Stein H and Jaffe ES: The 2008 WHO classification of lymphoid
neoplasms and beyond: Evolving concepts and practical applications.
Blood. 117:5019–5032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Robaina MC, Mazzoccoli L, Arruda VO, Reis
FR, Apa AG, de Rezende LM and Klumb CE: Deregulation of DNMT1,
DNMT3B and miR-29s in Burkitt lymphoma suggests novel contribution
for disease pathogenesis. Exp Mol Pathol. 98:200–207. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guan H, Xie L, Klapproth K, Weitzer CD,
Wirth T and Ushmorov A: Decitabine represses translocated MYC
oncogene in Burkitt lymphoma. J Pathol. 229:775–783. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shinozaki-Ushiku A, Kunita A, Isogai M,
Hibiya T, Ushiku T, Takada K and Fukayama M: Profiling of
virus-encoded MicroRNAs in Epstein-Barr Virus-associated gastric
carcinoma and their roles in gastric carcinogenesis. J Virol.
89:5581–5591. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Iizasa H, Wulff BE, Alla NR, Maragkakis M,
Megraw M, Hatzigeorgiou A, Iwakiri D, Takada K, Wiedmer A, Showe L,
et al: Editing of Epstein-Barr virus-encoded BART6 microRNAs
controls their dicer targeting and consequently affects viral
latency. J Biol Chem. 285:33358–33370. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Malumbres M: miRNAs and cancer: An
epigenetics view. Mol Aspects Med. 34:863–874. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fabbri M, Ivan M, Cimmino A, Negrini M and
Calin GA: Regulatory mechanisms of microRNAs involvement in cancer.
Expert Opin Biol Ther. 7:1009–1019. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao JJ, Lin J, Lwin T, Yang H, Guo J,
Kong W, Dessureault S, Moscinski LC, Rezania D, Dalton WS, et al:
microRNA expression profile and identification of miR-29 as a
prognostic marker and pathogenetic factor by targeting CDK6 in
mantle cell lymphoma. Blood. 115:2630–2639. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kinoshita T, Nohata N, Hanazawa T, Kikkawa
N, Yamamoto N, Yoshino H, Itesako T, Enokida H, Nakagawa M, Okamoto
Y and Seki N: Tumour-suppressive microRNA-29s inhibit cancer cell
migration and invasion by targeting laminin-integrin signalling in
head and neck squamous cell carcinoma. Br J Cancer. 109:2636–2645.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nishikawa R, Chiyomaru T, Enokida H,
Inoguchi S, Ishihara T, Matsushita R, Goto Y, Fukumoto I, Nakagawa
M and Seki N: Tumour-suppressive microRNA-29s directly regulate
LOXL2 expression and inhibit cancer cell migration and invasion in
renal cell carcinoma. FEBS Lett. 589:2136–2145. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chang TC, Yu D, Lee YS, Wentzel EA, Arking
DE, West KM, Dang CV, Thomas-Tikhonenko A and Mendell JT:
Widespread microRNA repression by Myc contributes to tumorigenesis.
Nat Genet. 40:43–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sears RC: The life cycle of C-myc: From
synthesis to degradation. Cell Cycle. 3:1133–1137. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Majello B and Perini G: Myc proteins in
cell biology and pathology. Biochim Biophys Acta. 1849:467–468.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F,
Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC, et al: Global
mapping of c-Myc binding sites and target gene networks in human B
cells. Proc Natl Acad Sci USA. 103:17834–17839. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Meyer N and Penn LZ: Reflecting on 25
years with MYC. Nat Rev Cancer. 8:976–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim JW, Mori S and Nevins JR: Myc-induced
microRNAs integrate Myc-mediated cell proliferation and cell fate.
Cancer Res. 70:4820–4828. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tao J, Zhao X and Tao J: c-MYC-miRNA
circuitry: A central regulator of aggressive B-cell malignancies.
Cell Cycle. 13:191–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Baer C, Claus R and Plass C: Genome-wide
epigenetic regulation of miRNAs in cancer. Cancer Res. 73:473–477.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Strmsek Z and Kunej T: MicroRNA silencing
by DNA methylation in human cancer: A literature analysis.
Noncoding RNA. 1:44–52. 2015.PubMed/NCBI
|
|
42
|
Lopez-Serra P and Esteller M: DNA
methylation-associated silencing of tumor-suppressor microRNAs in
cancer. Oncogene. 31:1609–1622. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lujambio A and Esteller M: CpG island
hypermethylation of tumor suppressor microRNAs in human cancer.
Cell Cycle. 6:1455–1459. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Saito Y and Jones PA: Epigenetic
activation of tumor suppressor microRNAs in human cancer cells.
Cell Cycle. 5:2220–2222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tost J and Gut IG: DNA methylation
analysis by pyrosequencing. Nat Protoc. 2:2265–2275. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Colella S, Shen L, Baggerly KA, Issa JP
and Krahe R: Sensitive and quantitative universal Pyrosequencing
methylation analysis of CpG sites. Biotechniques. 35:146–150. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Perino M and Veenstra GJ: Chromatin
control of developmental dynamics and plasticity. Dev Cell.
38:610–620. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Whyte WA, Orlando DA, Hnisz D, Abraham BJ,
Lin CY, Kagey MH, Rahl PB, Lee TI and Young RA: Master
transcription factors and mediator establish super-enhancers at key
cell identity genes. Cell. 153:307–319. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Elghoroury EA, ElDine HG, Kamel SA,
Abdelrahman AH, Mohammed A, Kamel MM and Ibrahim MH: Evaluation of
miRNA-21 and miRNA let-7 as prognostic markers in patients with
breast cancer. Clin Breast Cancer. 18:e721–e726. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Karpinski P, Pesz K and Sasiadek MM:
Pan-cancer analysis reveals presence of pronounced DNA methylation
drift in CpG island methylator phenotype clusters. Epigenomics.
9:1341–1352. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
El Baroudi M, Corà D, Bosia C, Osella M
and Caselle M: A curated database of miRNA mediated feed-forward
loops involving MYC as master regulator. PLoS One. 6:e147422011.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sabò A, Kress TR, Pelizzola M, De Pretis
S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et
al: Selective transcriptional regulation by Myc in cellular growth
control and lymphomagenesis. Nature. 511:488–492. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Poole CJ, Zheng W, Lodh A, Yevtodiyenko A,
Liefwalker D, Li H, Felsher DW and van Riggelen J: DNMT3B
overexpression contributes to aberrant DNA methylation and
MYC-driven tumor maintenance in T-ALL and Burkitt's lymphoma.
Oncotarget. 8:76898–76920. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Godshalk SE, Bhaduri-McIntosh S and Slack
FJ: Epstein-Barr virus-mediated dysregulation of human microRNA
expression. Cell Cycle. 7:3595–3600. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Paschos K, Smith P, Anderton E, Middeldorp
JM, White RE and Allday MJ: Epstein-barr virus latency in B cells
leads to epigenetic repression and CpG methylation of the tumour
suppressor gene Bim. PLoS Pathog. 5:e10004922009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Price AM and Luftig MA: To be or not IIb:
A multi-step process for Epstein-Barr virus latency establishment
and consequences for B cell tumorigenesis. PLoS Pathog.
11:e10046562015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Klinke O, Feederle R and Delecluse HJ:
Genetics of Epstein-Barr virus microRNAs. Semin Cancer Biol.
26:52–59. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Vereide DT, Seto E, Chiu YF, Hayes M,
Tagawa T, Grundhoff A, Hammerschmidt W and Sugden B: Epstein-Barr
virus maintains lymphomas via its miRNAs. Oncogene. 33:1258–1264.
2014. View Article : Google Scholar : PubMed/NCBI
|