Introduction
The resistance of cancer cells appears to be a major
issue in current oncology treatment, as it hinders successful
treatment and worsens the prognosis of oncology patients. The
mechanisms involved in cancer cell resistance also reveal a broad
network of interactions at the molecular level and confirm the
great complexity of the human organism. As such, further
investigation that may utilize the acquired knowledge to predict
and successfully treat different diseases, including cancer, is
necessary. The goal of scientists should include the blockage of
tumor resistance and improvement of therapeutic options,
consequently improving patient prognosis. Several clinical studies
have shown a poor prognosis of cancer patients in the case of
recombinant erythropoietin (EPO) administration (1,2). This
growth hormone has a wide application mostly in the amelioration of
anemia that accompanies chemotherapy. Its action is mediated
through the EPO receptor (EPOR), and stimulation of EPOR in the
cell leads to the activation of different signaling pathways
followed by the activation of transcription factors participating
in the regulation of many cellular processes. These effects are
mitogenic, antiapoptotic, and anti-inflammatory, among other cell
protective effects (3). Although it
has long been assumed that EPO only acts on blood-forming cells,
its action has also been demonstrated in the non-hematopoietic
environment, including nerve, retinal, myocardial, and other cells,
where the presence of EPOR has been detected (4). Furthermore, EPOR expression has also
been identified in several types of cancer cell lines (5,6). In light
of the presence of EPOR, activation of EPO/EPOR signaling pathways,
and a broad scale of potential effects in the tumor environment,
the adverse outcomes of the abovementioned clinical trials can be
explained. In contrast, functional EPORs have not been concisely
shown to exist in human tumor cell lines (7). Supportive EPO therapy is currently used
in cancer patients treated with paclitaxel, which belongs to the
group of unique mitotic inhibitors, taxanes (8). Their usage shows great promise in
anticancer therapy due to their antiproliferative and
antiangiogenic actions observed together with the antimetastatic
activity on cancer cells. Although taxanes are assumed to have high
potential in the treatment of various types of cancer, they have
not been shown to be effective in every case (9). While Larsson et al (10) demonstrated a correlation between EPOR
and both the estrogen receptor (ER) and progesterone receptor (PR)
in breast cancer cells, Volgger et al (11) found a positive correlation between the
EPOR/ER/PR status and higher local cancer recurrence. In this
regard, EPOR silencing was found to reduce the proliferation of
both EPOR- and ERα-positive breast cancer cells but not
ERα-negative cells (12). Based on
our previous study (13) utilizing
EPOR-overexpressing and ERα-negative mammary adenocarcinoma RAMA
37-28 cells, we investigated the role of EPOR in the sensitivity
and/or resistance of these cells to chemotherapeutic agent
paclitaxel. The role of EPO in the proliferation and the apoptosis
of RAMA 37-28 cells under both control and paclitaxel conditions
was also investigated.
Materials and methods
Cell lines and cell culture
Rat mammary adenocarcinoma cell line RAMA 37 and its
clone RAMA 37-28 stably transfected with human EPOR were cultivated
in RPMI-1640 medium (Gibco/Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Gibco/Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin
(both from Invitrogen/Thermo Fisher Scientific, Inc.). Cells were
maintained at 37°C in a constant atmosphere of 5% CO2,
21% O2, 74% N2 and 95% humidity.
Therapeutic agents
Epoetin α (EPO) (Binocrit; Sandoz) and paclitaxel
(Ebewe Pharma) were purchased commercially. Both agents were stored
at 4°C. EPO was provided as a 40,000 IU/ml solution and was diluted
to make a final concentration of 10 IU/ml. Paclitaxel was provided
as a 7 mM solution and was diluted at a ratio 1:69 to construct a
0.1 mM working solution before the preparation of a final
concentration of 200 nM. Both EPO and paclitaxel solutions were
prepared fresh before use.
Proliferation assays
The proliferation assays were carried out using a
clonogenic assay and Incucyte ZOOM system (Essen BioScience).
Clonogenic assay
Both the RAMA 37 and its clone RAMA 37-28 cell lines
were seeded on 6-well plates overnight. EPO (10 IU) and/or
paclitaxel (200 nM) was then added to the relevant groups of wells.
Cells that survived a 72-h incubation period with paclitaxel alone
or with the addition of EPO were seeded on other 6-well plates in
the amount of 1,000 cells/well and then incubated for 10 days. A
similar procedure was adopted in the case of control groups. After
10 days, the medium was aspirated and colonies were stained by
methylene blue (500 µl/well) at 1% concentration. The number of
colonies was determined using Clono Counter software (14). The results were analyzed statistically
using GraphPad Prism 5.01 software (GraphPad Software, Inc.).
Incucyte ZOOM system
The Incucyte ZOOM system allowed the monitoring of
the proliferation of both control and treated/experimental groups
in a defined environment of a standard incubator. Simplistically
this system presents a microscope gantry placed in a humified
incubator, and a networked external controller hard-drive made it
possible to gather images and process experimental data.
Experimental groups included controls (without the addition of any
therapeutic agents), cells with EPO alone or in combination with
paclitaxel, and cells with paclitaxel alone. All experimental
groups were seeded in a 96-well plate in hexaplicates at a
concentration of 3,000 cells/well. Cell proliferation was monitored
for 120 h using the Incucyte ZOOM system placed in an incubator.
The experiment was initiated by seeding 100 µl of cell
suspension/well. Cells were left to adhere and 24 h after seeding,
another 100 µl of culture medium with or without therapeutic agents
was added. Experiments were replicated three times. Images of the
wells of the 96-well plate were collected every 2 h by IncuCyte
ZOOM 4× objective (Nikon Plan Apo Lambda 4×/0.20; cat. no. 4466)
IncuCyte ZOOM integrated software (Essen Bioscience) was used to
analyze the results.
EPOR silencing
Cells seeded on 96-well plates were treated with 2
µM of on-targeting siRNA (ON-TARGETplus SMARTpool Human EPOR siRNA;
Dharmacon). Experimental group of RAMA 37-28 cells treated with
non-targeting siRNA (ON-TARGETplus Non-targeting Control siRNA;
Dharmacon) at the concentration of 2 µM served as the negative
control. Untreated RAMA 37-28 cells served as the controls. Three
independent experiments, including samples in triplicates (96-well
format), were conducted. Briefly, 100 µl of cell suspension/well
with cells at the concentration of 3,000/well was used. Cells were
diluted in antibiotic-free complete medium (with the supplement of
10% FBS) and incubated at 37°C overnight. siRNA stock solution (100
µM) was prepared in 1X siRNA buffer and the concentration of siRNA
was verified using UV spectrophotometry at 260 nm. Resuspended
siRNA was stored at −20°C, and each diluted siRNA at the
concentration of 2 µM was prepared freshly before use. The
appropriate volume of 2 µM siRNA and the appropriate volume of
DharmaFECT transfection reagent (Dharmacon) in the 2 separate tubes
were diluted with serum-free and antibiotic-free medium according
to the manufacturer's instructions. The contents of each tube were
mixed gently by pipetting up and down and subsequently incubated
for 5 min at room temperature (RT). They were then merged into 1
tube and further incubated for 20 min at RT. Afterward, a
sufficient amount of antibiotic-free complete medium was added to
the mix for the desired volume of the transfection medium. Culture
medium from the wells of the 96-well plates was removed and
replaced by 100 µl of the appropriate transfection medium. After 48
h of cell incubation, the transfection medium was replaced with
complete medium with or without the addition of therapeutic agents
paclitaxel and/or EPO (200 µl/well). Incucyte monitoring of the
cells was stopped after 144 h from cell seeding. Incucyte ZOOM
integrated software (Essen Bioscience) was used to analyze the
results.
Both transfection procedures, as well as the
treatment of cells with or without therapeutic agents, were
conducted before western blot analyses using a 6-well plate format.
In this case, RAMA 37 and RAMA 37-28 cells were stimulated with
paclitaxel for 2 different periods (15 min or 24 h) and lysed
afterward to perform western blot analysis.
Western blot analysis
Western blot analysis was carried out according to
the generally accepted protocol. Specifically, cells were lysed
using lysis buffer in the presence of protease and phosphatase
inhibitor cocktail (Thermo Fisher Scientific, Inc). Protein samples
were separated by 12% SDS-PAGE and electrotransferred to
polyvinylidene difluoride membranes (Bio-Rad). The following
primary antibodies: anti-p-ERK1/2 (cat. no. 9101S), anti-p-AKT
(cat. no. 9271S), anti-STAT5 (cat. no. 9363S), anti-ERK1/2 (cat.
no. 9102S), anti-AKT (cat. no. 9272S), anti-BAX (cat. no. 2772S),
anti-BCL-XL (cat. no. 2762S), anti-caspase 3 (cat. no. 14220S),
anti-β-actin (cat. no. 3700S) (all from Cell Signaling Technology,
Inc.; 1:1,000 dilution), anti-p-STAT5 (cat. no. 50095; Temecula;
1:1,000 dilution), anti-EPOR (A82; Amgen; 1:2,000 dilution), and
HRP-conjugated secondary antibodies (Pierce Chemical; 1:2,000
dilution), were used for detection. β-actin antibodies were used as
controls for equal protein loading. The visualization was performed
using the ECL Western blotting substrate (Thermo Fisher Scientific,
Inc.) and Biomax imaging film (Kodak) or ChemiDoc XRS+ Imaging
system (Bio-Rad). The films were scanned with the GS-800 Calibrated
Densitometer, and the quantification was performed using Image J
software version 1.52 (NIH; National Institutes of Health,
Bethesda, MD, USA). The results are shown as the mean densities
from 3 independent experiments.
Apoptosis assays
Annexin V and caspase 3/7
activity
Cells seeded on a 96-well plate (3,000 cells/well in
100 µl/well) in triplicates were allowed to adhere overnight.
Annexin V reagent (Incucyte Annexin V Green Reagent for Apoptosis;
Essen Bioscience, final dilution of 1:200) or Caspase 3/7 reagent
(Incucyte Caspase 3/7 Green Apoptosis Reagent; Essen Bioscience,
final dilution of 1:1,000) were added together with paclitaxel
and/or EPO 72 h after cell seeding and EPOR silencing (experimental
groups with siRNA and nt siRNA against EPOR). The rate of
activation of Annexin V and both caspase 3/7 in cells was monitored
with the Incucyte ZOOM system every 1 h after treatment of the
cells. Plates were pre-warmed prior to data acquisition to avoid
condensation and expansion of the plate, which would hinder
autofocus. The maxima of excitation and emission were 490/515 nm
and 500/530 nm for Annexin V and caspase 3/7, respectively. Images
of wells of the 96-well plate were collected by Nikon 20×
objective. Incucyte ZOOM integrated software (Essen Bioscience) was
used to minimize background fluorescence and quantify fluorescent
objects.
Statistical analysis
The data were statistically analyzed using ANOVA
followed by Tukey's multiple comparison tests in ORIGIN analysis
software (OriginLab Co., Northampton, MA, USA). The results were
considered significant at the probability level P<0.05 and
P<0.01.
Results
The effects of different concentrations of
paclitaxel on the response of rat mammary adenocarcinoma RAMA 37
and RAMA 37-28 cells were monitored by MTT assay (data not shown).
From MTT cell survival plots, we determined the concentration of
the paclitaxel drug (200 nM), which inhibited the cell survival of
both cell lines by 50% (IC50). This concentration was
used in other analyses.
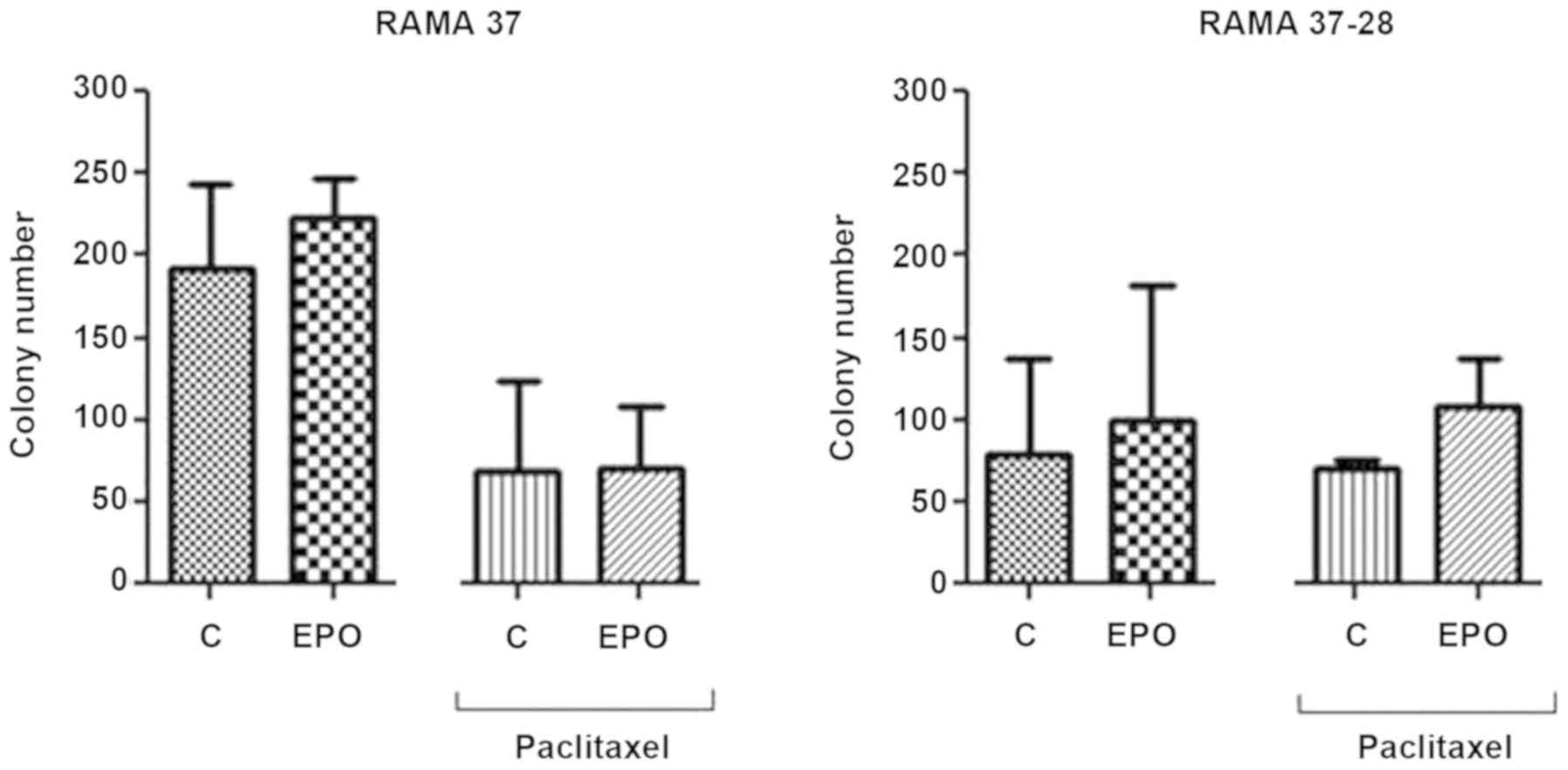
Clonogenic assay
Clonogenic assay was first used to evaluate the
proliferation of the EPOR-overexpressing RAMA 37-28 cells compared
to the parental RAMA 37 cells in control and paclitaxel conditions
(Fig. 1). In this regard, the effect
of single EPO or its combination with paclitaxel was also studied.
Although we observed the overall slower proliferation of RAMA 37-28
cells compared to RAMA 37 cells, the colony number was decreased
only slightly after paclitaxel treatment in the EPOR-overexpressing
RAMA 37-28 cells as opposed to the parental RAMA 37 cells. In
addition, EPO in combination with paclitaxel stimulated (protected)
RAMA 37-28 cell proliferation.
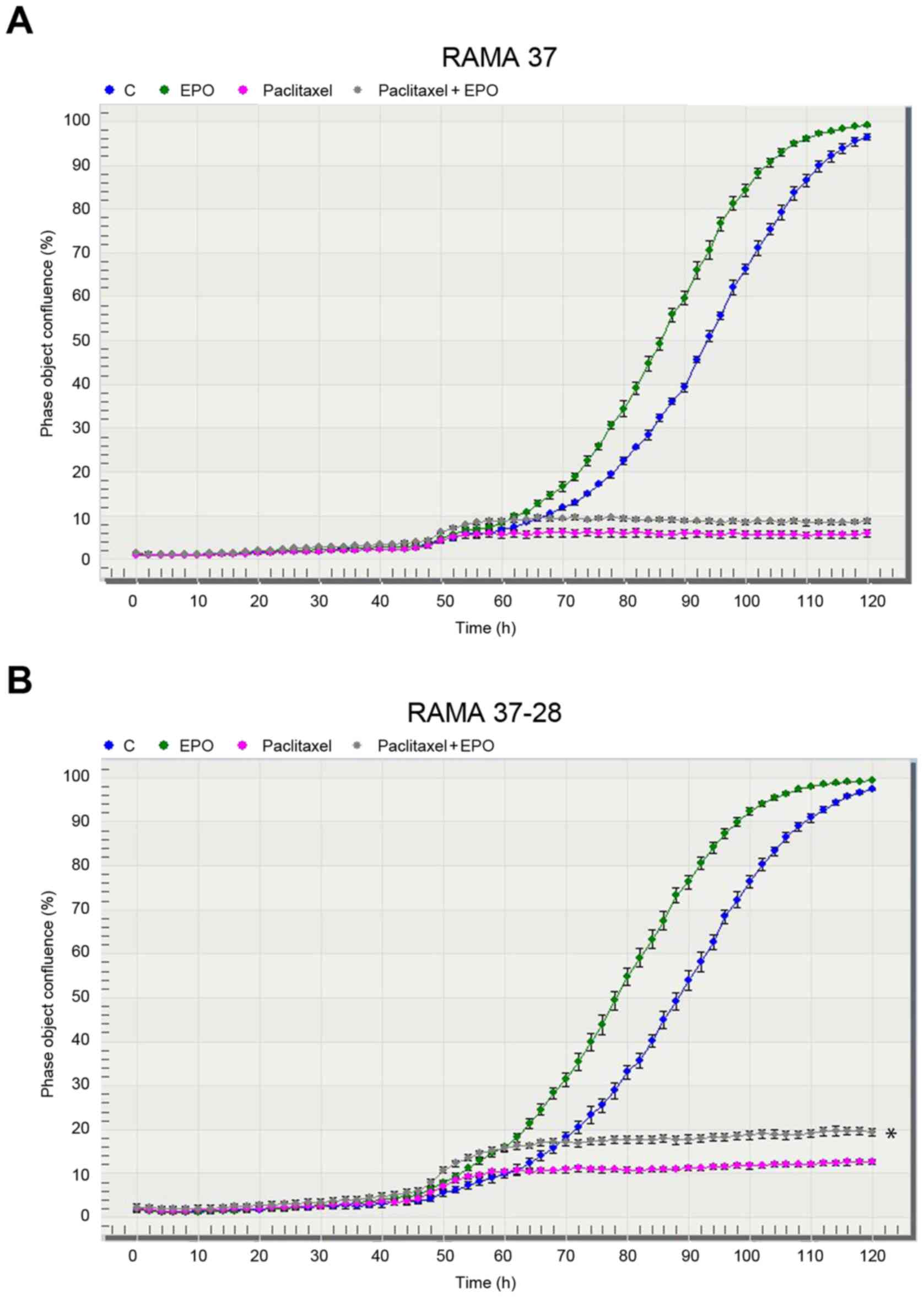
IncuCyte ZOOM system
The effects of EPO, paclitaxel, and their
combination on in vitro proliferation of the
EPOR-overexpressing RAMA 37-28 cells was also monitored by the
IncuCyte ZOOM system (Figs. 2B and
3B). We determined the stimulation of
RAMA 37-28 cells either after single EPO or after the combination
of EPO and paclitaxel. Our results, as demonstrated in Fig. 2A and B, showed that a more pronounced
stimulation and/or protection was observed in the RAMA 37-28 cells
with stably expressed human EPOR, although low stimulation was also
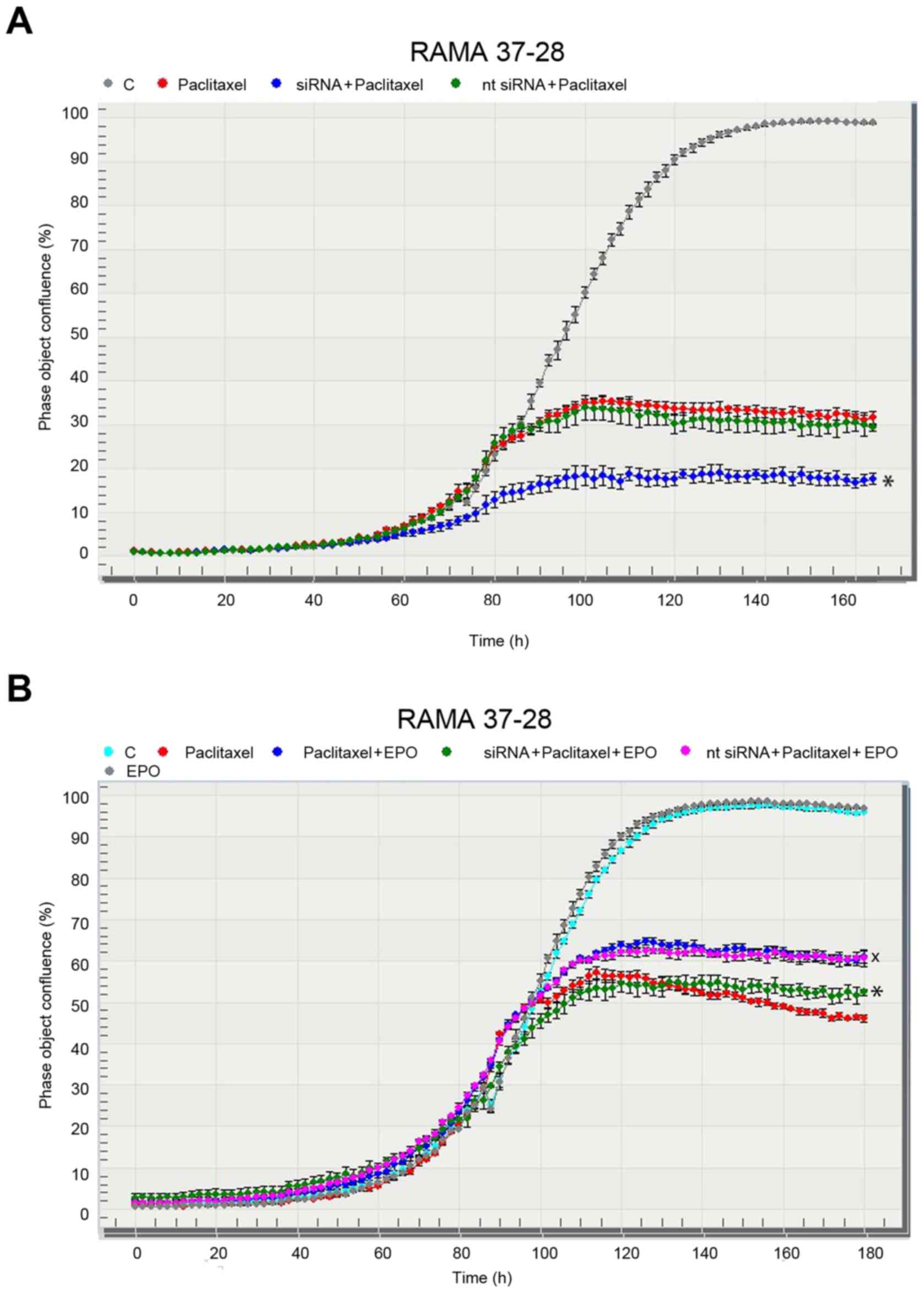
observed in the parental RAMA 37 cells. We chose a specific siRNA
against human EPOR to confirm the role of EPOR in the potential
resistance of RAMA 37-28 cells to paclitaxel. Indeed, the decrease
in RAMA 37-28 cell proliferation, as a result of EPOR silencing in
the group treated with siRNA and paclitaxel, confirmed the role of
EPOR in paclitaxel resistance (Fig.
3A). The RAMA 37-28 cells in the negative control group treated
with nt siRNA did not exhibit a difference in cell proliferation in
any way, and it showed the same trend in proliferation as the group
treated only with paclitaxel. Moreover, an EPO stimulating
(protecting) effect, when combined with paclitaxel as opposed to
paclitaxel alone, was also successfully minimized using siRNA
(Fig. 3B). The specificity and the
efficiency of the applied siRNA were confirmed by western blot
analysis using a specific anti-EPOR antibody A82. In contrast, the
same technique also demonstrated the unspecificity of the nt
siRNA.
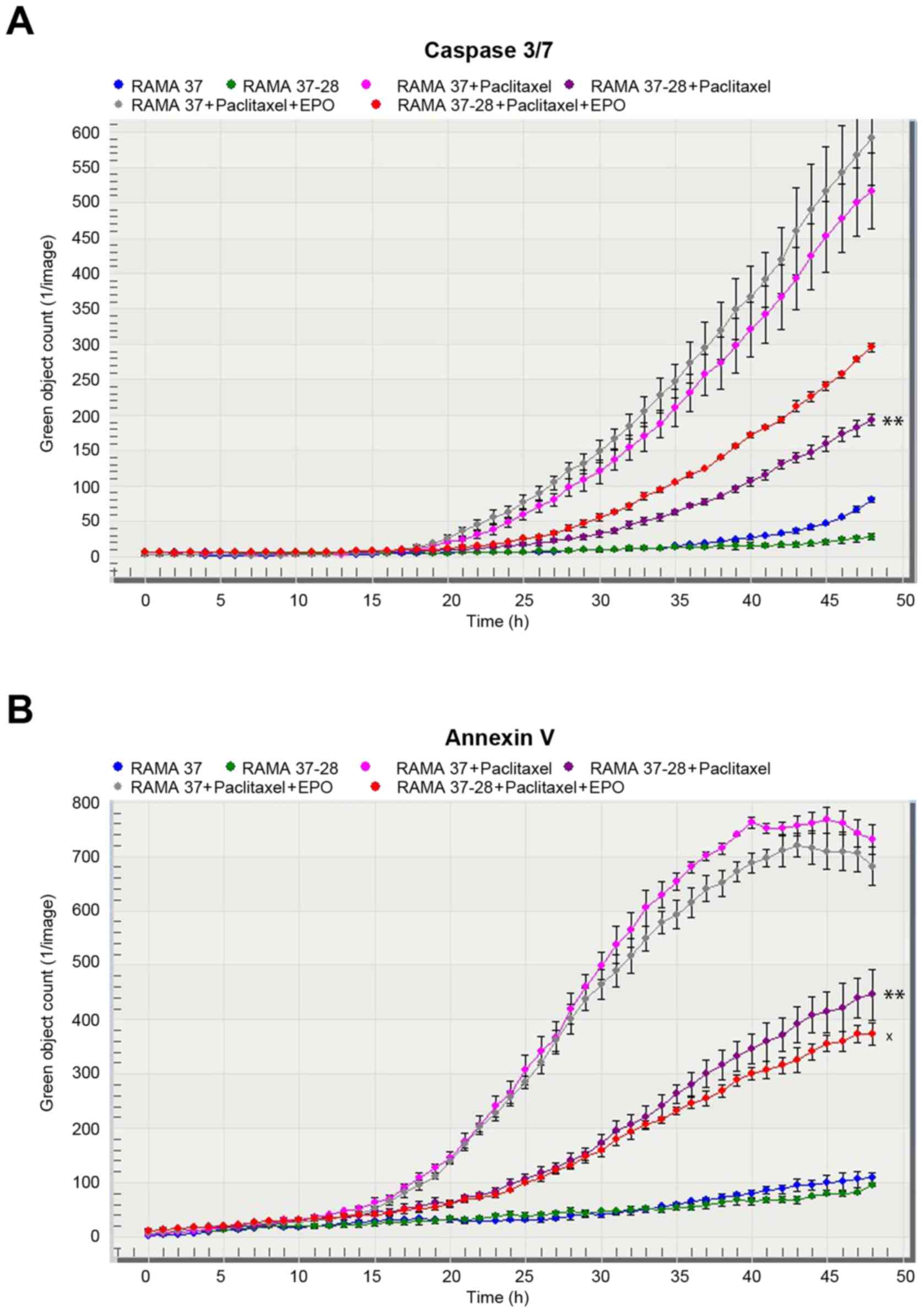
Analysis of apoptosis
The rate of apoptosis induced by paclitaxel and
represented by caspase 3/7 (Fig. 4A)
and Annexin V (Fig. 4B) activation
was negatively correlated with the EPOR expression in the RAMA
cells. Indeed, RAMA 37-28 cells showed a lower rate of apoptosis
compared to RAMA 37 cells after paclitaxel treatment. In this
regard, the addition of EPO to paclitaxel-treated cells weakened
the effect of paclitaxel in terms of decreased activation of
Annexin V reagent. On the contrary, such a weakening effect of EPO
was not observed in the case of caspase 3/7 activation. Similarly,
EPOR silencing did not significantly affect the activation of both
reagents (data not shown).
Activation of key proteins
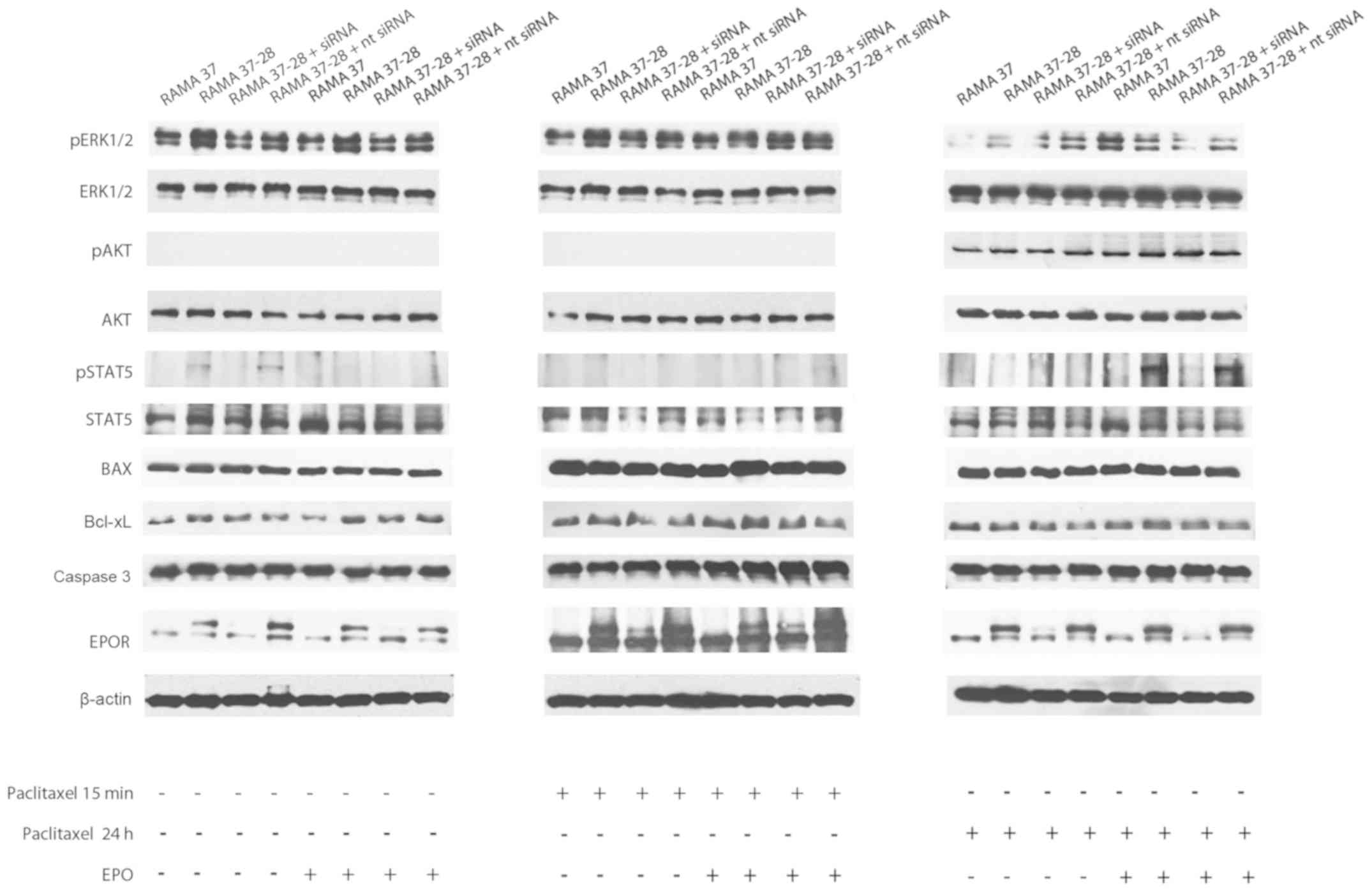
Subsequently, we focused on the key proteins
activated in response to paclitaxel and/or EPO (Fig. 5 and Table
I). Indeed, protein levels of phosphorylated ERK1/2 (pERK1/2)
were higher in RAMA 37-28 cells compared to RAMA 37 cells even
under control conditions. In this regard, the rate of pERK1/2 was
higher in RAMA 37-28 and RAMA 37-28 + nt siRNA and lower in the
group with siRNA. The addition of EPO had no further effect on
pERK1/2 levels. A very similar situation occurred 15 min after
paclitaxel administration when pERK1/2 was higher in RAMA 37-28
compared to RAMA 37 cells in both paclitaxel as well as paclitaxel
+ EPO conditions. Interestingly, a higher rate of pERK1/2 was still
observed in RAMA 37-28 and RAMA 37-28 + nt siRNA cells compared to
RAMA 37 and RAMA 37-28 cells with EPOR siRNA in the presence of
paclitaxel at 24 h. On the contrary, RAMA 37 cells showed a higher
pERK1/2 level compared to RAMA 37-28 cells 24 h after the
administration of the combination of paclitaxel and EPO (Fig. 5 and Table
I).
 | Table I.Effect of paclitaxel and/or EPO
treatment on RAMA 37 and RAMA 37-28 cell signaling: Effect of EPOR
siRNA. |
Table I.
Effect of paclitaxel and/or EPO
treatment on RAMA 37 and RAMA 37-28 cell signaling: Effect of EPOR
siRNA.
| A, Effect of EPO
treatment |
|---|
|
|---|
|
| Ratio according to
control | Ratio according to
EPO treatment |
|---|
|
|
|
|
|---|
| Protein target | RAMA 37 | RAMA 37-28 | RAMA 37-28 +
siRNA | RAMA 37-28 + nt
siRNA | RAMA 37 | RAMA 37-28 | RAMA 37-28 +
siRNA | RAMA 37-28 + nt
siRNA |
|---|
| pERK1/2 | 1.00±0.12 |
1.93±0.22a | 0.93±0.08 | 1.46±0.14 | 0.75±0.09 | 1.21±0.24 | 0.82±0.11 | 1.35±0.18 |
| pAKT | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| pSTAT5 | 1.00±0.08 | 1.28±0.13 | 1.01±0.20 | 1.36±0.21 | 1.17±0.35 | 1.23±0.22 | 0.84±0.18 | 1.18±0.27 |
| Bax | 1.00±0.16 | 1.05±0.08 | 1.25±0.26 | 1.22±0.32 | 1.10±0.31 | 1.10±0.27 | 1.19±0.23 | 1.20±0.19 |
| Bcl-xL | 1.00±0.06 |
1.72±0.09a | 1.22±0.23 |
1.42±0.08a | 0.26±0.02 |
2.03±0.31b | 1.37±0.17 |
2.21±0.27b |
| Caspase 3 | 1.00±0.11 | 1.12±0.28 | 1.08±0.17 | 1.10±0.10 | 1.02±0.33 | 0.86±0.09 | 0.98±0.21 | 0.98±0.15 |
| EPOR | 1.00±0.10 |
4.36±0.22c | 1.08±0.12 |
10.98±0.63c | 1.17±0.29 |
5.91±0.49c | 1.43±0.37 |
5.42±0.51c |
|
| B, Effect of
paclitaxel (15 min) or paclitaxel (15 min) combined with
EPO |
|
|
| Ratio according
to paclitaxel treatment (15 min) | Ratio according
to paclitaxel treatment (15 min) + EPO |
|
|
|
|
| Protein
target | RAMA 37 | RAMA
37-28 | RAMA 37-28 +
siRNA | RAMA 37-28 + nt
siRNA | RAMA 37 | RAMA
37-28 | RAMA 37-28 +
siRNA | RAMA 37-28 + nt
siRNA |
|
| pERK1/2 | 1.00±0.15 |
1.68±0.26a | 1.40±0.46 |
2.02±0.21a | 1.15±0.08 |
1.78±0.18a | 1.39±0.32 |
1.73±0.23a |
| pAKT | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| pSTAT5 | 1.00±0.13 | 0.83±0.27 | 0.72±0.08 | 0.71±0.18 | 0.85±0.26 | 1.39±0.09 | 1.17±0.33 |
1.67±0.16a |
| Bax | 1.00±0.30 | 0.86±0.18 | 0.83±0.17 | 0.91±0.31 | 0.77±0.22 | 1.12±0.25 | 0.87±0.23 | 0.90±0.09 |
| Bcl-xL | 1.00±0.08 |
1.44±0.09a | 0.80±0.16 | 0.89±0.16 | 1.36±0.24 |
1.86±0.11a | 0.97±0.14 | 0.52±0.03 |
| Caspase 3 | 1.00±0.11 | 0.82±0.05 | 1.11±0.21 | 1.14±0.12 | 1.31±0.23 | 1.36±0.41 | 1.41±0.40 | 1.42±0.25 |
| EPOR | 1.00±0.09 |
1.98±0.24b | 1.30±0.32 |
2.37±0.17b | 1.15±0.20 |
2.09±0.19b | 1.44±0.18 |
2.65±0.30b |
|
| C, Effect of
paclitaxel (24 h) or paclitaxel (24 h) combined with EPO |
|
|
| Ratio according
to paclitaxel treatment (24 h) | Ratio according
to paclitaxel treatment (24 h) + EPO |
|
|
|
|
| Protein
target | RAMA 37 | RAMA
37-28 | RAMA 37-28 +
siRNA | RAMA 37-28 + nt
siRNA | RAMA 37 | RAMA
37-28 | RAMA 37-28 +
siRNA | RAMA 37-28 + nt
siRNA |
|
| pERK1/2 | 1.00±0.15 |
3.73±0.47b | 1.73±0.58 |
8.00±0.82c |
10.73±0.93c |
5.93±0.72b |
2.89±0.63a |
4.81±0.77b |
| pAKT | 1.00±0.07 | 1.47±0.23 | 1.20±0.36 |
1.55±0.24a | 1.74±0.59 |
1.92±0.22a |
1.87±0.31a |
1.77±0.24a |
| pSTAT5 | 1.00±0.11 | 0.32±0.03 | 0.71±0.06 | 0.76±0.08 | 0.65±0.07 |
1.81±0.25a | 1.46±0.47 |
2.47±0.31b |
| Bax | 1.00±0.09 | 0.93±0.10 | 0.81±0.13 | 0.89±0.20 | 0.92±0.11 | 0.98±0.23 | 0.91±0.19 | 0.92±0.21 |
| Bcl-xL | 1.00±0.08 | 0.66±0.07 | 0.52±0.06 | 0.28±0.02 | 0.57±0.03 | 0.78±0.08 | 0.43±0.06 | 0.32±0.03 |
| Caspase 3 | 1.00±0.10 | 0.84±0.09 | 0.76±0.08 | 0.90±0.11 | 0.93±0.09 | 0.96±0.12 | 1.00±0.20 | 0.85±0.11 |
| EPOR | 1.00±0.07 |
2.34±0.22b | 0.57±0.19 |
2.22±0.19b | 0.56±0.08 |
2.78±0.32b | 0.15±0.02 |
2.32±0.28b |
No phosphorylation of AKT (pAKT) was observed in the
control and EPO conditions in both RAMA 37-28 as well as RAMA 37
cells. On the other hand, paclitaxel activated AKT signal
transduction only at 24 h, not at the 15-min time point. Notably,
paclitaxel did not induce significant pAKT changes in
EPOR-overexpressing cells and/or experimental groups: RAMA 37-28
and RAMA 37-28 + nt siRNA compared to RAMA 37 and RAMA 37-28 +
siRNA. In addition, a 24-h treatment with paclitaxel + EPO
potentiated the pAKT signalization nonspecifically in each
experimental group compared to single paclitaxel therapy (Fig. 5 and Table
I).
Similar to pERK1/2, the phosphorylation of STAT5
(pSTAT5) in RAMA 37-28 cells occurred without EPO stimulation and
disappeared after the silencing of EPOR using siRNA in these cells.
No additional pSTAT5 was found in RAMA 37-28 cells either after
their incubation with EPO or after paclitaxel treatment, regardless
of incubation time. On the contrary, significant pSTAT5 was
monitored in RAMA 37-28 cells treated with the combination of
paclitaxel and EPO, but only at the 24-h time point. The
significance of EPOR in pSTAT5 of RAMA 37-28 cells is evident from
their comparison with RAMA 37 cells and also from the silencing of
EPOR using specific siRNA. Indeed, RAMA 37 cells did not manifest
STAT5 signalization under any tested conditions (Fig. 5 and Table
I).
The roles of pro-apoptotic protein Bax and
anti-apoptotic protein Bcl-xL were also considered. In this regard,
RAMA 37-28 (including experimental groups with siRNA and nt siRNA)
and RAMA 37 cells did not show any difference in the Bax protein
level under all conditions tested. Neither paclitaxel (in any time
of treatment) nor EPO addition altered the level of Bax protein
(Fig. 5 and Table I). On the other hand, the level of
Bcl-xL was higher in the control RAMA 37-28 cells when compared to
the level in the RAMA 37 cells, and silencing of EPOR did not lower
the level of the Bcl-xL protein. Moreover, the level of Bcl-xL in
the RAMA 37-28 cells was even more pronounced under EPO treatment
without paclitaxel treatment. On the contrary, although the Bcl-xL
level remained elevated in the RAMA 37-28 cells after paclitaxel
and paclitaxel + EPO treatments at 15 min, it decreased under
control conditions in each group at 24 h. In the case of caspase 3,
no significant differences were observed in the level of this
protein between experimental groups (Fig.
5 and Table I).
With the aim to show stable overexpression of EPOR
in RAMA 37-28 cells and to confirm the results of EPOR silencing
using siRNA, we decided to demonstrate the EPOR level in all
experimental groups. In this regard, both control groups (without
paclitaxel) and groups with paclitaxel (15 min or 24 h) showed an
equal EPOR level in each experimental group. Furthermore, EPO did
not have any effect on the level of EPOR in our experimental group
(Fig. 5 and Table I).
Discussion
Both the adverse outcomes of various clinical trials
(1,2)
of cancer patients undergoing recombinant EPO support therapy as
well as the confirmation of the presence of EPOR on the cancer cell
surface are attracting great research interest. The evidence of the
effects of recombinant EPO in regards to tumor progression in
cancer patients, however, is unclear in several clinical trials.
For example, in the case of Breast Cancer-Anemia and the Value of
Erythropoietin (BRAVE) study there was no significant difference in
overal survival (15). Furthermore,
recent clinical studies failed to confirm the negative effect of
recombinant EPO (16–18). To answer the question of whether the
presence of EPOR can somehow affect the proliferation and apoptosis
of cancer cells, we chose breast cancer cells and monitored their
response to paclitaxel chemotherapy. Although paclitaxel is one of
the most promising anti-cancer agents in clinical use, the
development of paclitaxel resistance in cancer cells decreases the
effectiveness of this drug. Poor response of oncological patients
to paclitaxel raises the need to identify new markers for
paclitaxel susceptibility in cancer cells.
Whereas some studies have demonstrated the presence
of EPOR in cancer cells (19), other
studies have failed to demonstrate high expression of EPOR in
cancer cells and EPO-mediated cell stimulation (20,21). The
discrepancy in data in terms of the level of EPOR in cancer cells
appears to reflect differences in the specificity of used anti-EPOR
antibodies or methodological approaches. The problem of
unspecificity of anti-EPOR antibodies comes from the possible
detection of proteins with improper molecular weight or non-EPOR
molecules with the same molecular weight as EPOR (22,23).
The probability of the stimulation of cancer cell
growth by EPOR was tested at different levels of EPOR expression in
rat mammary adenocarcinoma cells RAMA. Indeed, since RAMA 37 and
RAMA 37-28 cells differ in the level of EPOR expression but not in
the expression of estrogen receptor α, β and G-protein coupled ER,
they represent a suitable model for evaluating the effects of
EPO/EPOR on cell physiology (13).
The present study confirmed our previous results
(13) and demonstrated a difference
in the proliferation of EPOR-overexpressing RAMA 37-28 cells
compared to low EPOR-expressing RAMA 37 cells. Even in the absence
of EPO, RAMA 37-28 cells proliferated slower compared to RAMA 37
cells. Because the slower proliferation of RAMA 37-28 cells would
seem to imply greater resistance to paclitaxel, siRNA was used
against EPOR to confirm the role of EPOR in the response of RAMA
37-28 cells to paclitaxel. Indeed, a decrease in the proliferation
and increase in the rate of apoptosis after EPOR silencing was
observed in the study of Cao et al (24) in the case of glioma stem cells. Paragh
et al (25) showed the
presence of phosphorylated EPOR signaling components in A2780 human
ovarian adenocarcinoma cells, even when the cells were not exposed
to exogenous EPO. In this regard, EPOR knockdown in breast cancer
cell lines reduces pAKT levels, which suggests its involvement in
transmission of signals, including phosphorylation and activation
of AKT (12). Moreover, Ueda et
al (26) demonstrated the role of
JAK2 point mutation in EPOR activation and myeloproliferative
neoplasms. Indeed, the EPO-independent EPOR activation needs to be
elucidated in more detail. We confirmed the proposed cell
stimulation by single EPO and/or by its combination with paclitaxel
using IncuCyte monitoring, with pronounced proliferation observed
in EPOR-overexpressing RAMA 37-28 cells. A slightly weaker but
nevertheless stimulating effect of EPO in control and paclitaxel
conditions was found also in RAMA 37 cells where EPOR had an almost
undetectable level. This finding could be explained by the
existence of receptors other than EPOR, e.g., β common receptor
and/or ephrin type-B receptor 4 (4)
through which EPO may affect cancer cells. On the contrary, this
study clearly demonstrated a decrease in RAMA 37-28 cell
proliferation after EPOR silencing in both paclitaxel as well as
paclitaxel + EPO groups compared to nt siRNA.
Our findings are in line with an in vivo
study by Todaro et al (27),
which showed an increased progression of metastases in the case of
combined therapy of paclitaxel + EPO compared to single paclitaxel
in human breast cancer stem-like cell (BCSC)-derived
orthotopic/metastatic xenografts. Furthermore, our results are
consistent with other in vivo studies of mouse models, where
the silencing of EPOR expression using short hairpin RNAs prevented
the progression of melanoma (28) or
prostate cancer (29). Moreover, EPO
protected BCSCs from chemotherapy and enhanced metastatic tumor
progression through early activation of AKT and ERK signalization
and later through an increase in Bcl-xL protein level. Similarly,
early after stimulation of RAMA 37-28 cells by EPO, Shi et
al (30) observed a significant
activation of PI3K/AKT, RAS/ERK, and JAK2/STAT5 pathways, while in
RAMA 37 cells, they observed no activation of the JAK2/STAT5
pathway and only minor activation of PI3K/AKT and RAS/ERK pathways.
On the contrary, Swift et al (7) showed the inability of EPO to induce
intracellular signaling in NCI-H661 cells.
In our in vitro model, EPOR-overexpressing
RAMA 37-28 cells, as compared to parental RAMA 37 cells, revealed
higher phosphorylation of ERK1/2 in control conditions and higher
AKT signal transduction later after paclitaxel addition. Indeed,
AKT/ERK signal transductions were shown to be active in
paclitaxel-resistant gastric cancer cell lines, and it seems that
AKT/ERK activation might have led to the development of paclitaxel
and/or multidrug resistance of cancer cells (31). Furthermore, the silencing of EPOR gene
expression confirmed the role of the ERK1/2 pathway in our
EPOR-induced paclitaxel protection. On the other hand, increased
Bcl-xL level in RAMA 37-28 cells did not change after EPOR
silencing, and its level was even more pronounced under EPO
treatment, regardless of the presence or absence of paclitaxel.
Whereas in BCSC model Bcl-xL level was induced by EPO (27), in hepatocellular carcinoma cells
SNU-398 (32) was caused by
paclitaxel treatment, and in our RAMA 37-28 cells Bcl-xL increase
resulted from the simple EPOR overexpression. Consistent with these
findings, we also observed a decreased rate of caspase 3/7
activation and Annexin V in cells with EPOR overexpression compared
to the rate of apoptosis induced by paclitaxel in RAMA 37-28 vs.
RAMA 37 cells. On the contrary, the antiapoptotic effect of EPO in
RAMA 37-28 cells was only demonstrated by Annexin V, not by caspase
3/7 activity. The lower sensitivity of IncuCyte monitoring of
caspase 3/7 activity compared to enzymatic ELISA or flow cytometric
analysis could explain this discrepancy. Moreover, neither Annexin
V nor caspase 3/7 demonstrated any significant antiapoptotic effect
of EPO in RAMA 37 cells at the endpoints of the monitoring.
Solar et al (33) were the first to demonstrate in
vitro development of a paclitaxel resistance phenotype in human
ovarian carcinoma A2780 cells as a result of EPO treatment. Indeed,
the results of our present study confirmed the stimulation of EPO
in mammary adenocarcinoma cells and the association between EPOR
and increased resistance of these cells to paclitaxel. The
hypothesized direct effect of EPO on cancer cells highlights the
importance of further studies on the EPO/EPOR interactions in
cancer cells and the possible modulation of their sensitivity not
only to paclitaxel, but also to other chemotherapeutics.
In conclusion, the higher paclitaxel resistance and
lower apoptosis rate of EPOR-overexpressing rat mammary
adenocarcinoma RAMA 37-28 cells indicate a strong association
between EPO/EPOR and tumor progression. The silencing of EPOR
expression under the presence of paclitaxel therapy led to a
decrease in RAMA 37-28 cell proliferation and thus confirmed the
role of EPOR in the sensitivity of these cells to this therapy.
Interestingly, compared to RAMA 37 cells, RAMA 37-28 cells also
showed a lower rate of apoptosis induced by paclitaxel and
monitored by caspase 3/7 activation and Annexin V. Moreover,
enhanced activation of signaling pathways mediated by pERK1/2 in
RAMA 37-28 cells was demonstrated to be essential for paclitaxel
resistance.
Acknowledgements
Cell lines RAMA 37 and RAMA 37-28 were kindly
donated by Dr Mohamed El-Tanani (Centre for Cancer Research and
Cell Biology, Queen's University, Belfast, Northern Ireland, UK).
Anti-EPOR primary antibody (A82) was provided by Amgen Company
(Thousand Oaks, CA, USA).
Funding
This study was supported by the Scientific Grant
Agency of the Ministry of Education of the Slovak Republic (grant
nos. VEGA1/0394/15 and VEGA1/0536/19) and the Internal Scientific
Grant System of the Faculty of Science of the Pavol Jozef Šafárik
University in Košice (grant no. VVGS-PF-2016-72617). This
publication is also the result of the project implementation: ‘The
University Medical Science and Technology Park in Košice (MediPark,
Košice-Phase II.)’ (code ITMS2014+313011D103), supported by the
Operational Programme Research and Innovation, funded by the
European Regional Development Fund.
Availability of data and materials
The datasets used during the present study which are
not provided in the manuscript are available from the corresponding
author upon reasonable request.
Authors' contributions
Conception and design of the study was carried out
by EZ and PS. Acquisition of the data was conducte by EZ, LI, PK
and BF. Analysis and interpretation of data was performed by EZ, PS
and LI. Writing, review and revision of the manuscript was carried
out by EZ, PS, LI, BF and PK. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The autors declare that they have no competing
interests.
References
|
1
|
Leyland-Jones B, Semiglazov V, Pawlicki M,
Pienkowski T, Tjulandin S, Manikhas G, Makhson A, Roth A, Dodwell
D, Baselga J, et al: Maintaining normal hemoglobin levels with
epoetin alfa in mainly nonanemic patients with metastatic breast
cancer receiving first-line chemotherapy: A survival study. J Clin
Oncol. 23:5960–5972. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Henke M, Laszig R, Rube C, Schäfer U,
Haase KD, Schilcher B, Mose S, Beer KT, Burger U, Dougherty C and
Frommhold H: Erythropoietin to treat head and neck cancer patients
with anaemia undergoing radiotherapy: Randomised, double-blind,
placebo-controlled trial. Lancet. 362:1255–1260. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Watowich SS: The erythropoietin receptor:
Molecular structure and hematopoietic signaling pathways. J
Investig Med. 59:1067–1072. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Debeljak N, Solar P and Sytkowski AJ:
Erythropoietin and cancer: The unintended consequences of anemia
correction. Front Immunol. 5:5632014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arcasoy MO, Jiang X and Haroon ZA:
Expression of erythropoietin receptor splice variants in human
cancer. Biochem Biophys Res Commun. 307:999–1007. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Osterborg A, Aapro M, Cornes P, Haselbeck
A, Hayward CR and Jelkmann W: Preclinical studies of erythropoietin
receptor expression in tumour cells: Impact on clinical use of
erythropoietic proteins to correct cancer-related anaemia. Eur J
Cancer. 43:510–519. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Swift S, Ellison AR, Kassner P, McCaffery
I, Rossi J, Sinclair AM, Begley CG and Elliott S: Absence of
functional EpoR expression in human tumor cell lines. Blood.
115:4254–4263. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marupudi NI, Han JE, Li KW, Renard VM,
Tyler BM and Brem H: Paclitaxel: A review of adverse toxicities and
novel delivery strategies. Expert Opin Drug Saf. 6:609–621. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Orr JW Jr: What constitutes the ‘optimal’
treatment environment of women with gynecologic cancer? Gynecol
Oncol. 89:1–3. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Larsson AM, Jirstrom K, Fredlund E,
Nilsson S, Rydén L, Landberg G and Påhlman S: Erythropoietin
receptor expression and correlation to tamoxifen response and
prognosis in breast cancer. Clin Cancer Res. 15:5552–5559. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Volgger B, Kurz K, Zoschg K, Theurl I,
Ciresa-König A, Marth C and Weiss G: Importance of erythropoetin
receptor expression in tumour tissue for the clinical course of
breast cancer. Anticancer Res. 30:3721–3726. 2010.PubMed/NCBI
|
|
12
|
Reinbothe S, Larsson AM, Vaapil M, Wigerup
C, Sun J, Jögi A, Neumann D, Rönnstrand L and Påhlman S:
EPO-independent functional EPO receptor in breast cancer enhances
estrogen receptor activity and promotes cell proliferation. Biochem
Biophys Res Commun. 445:163–169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ilkovičová L, Trošt N, Szentpéteriová E,
Solár P, Komel R and Debeljak N: Overexpression of the
erythropoietin receptor in RAMA 37 breast cancer cells alters cell
growth and sensitivity to tamoxifen. Int J Oncol. 51:737–746. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Niyazi M, Niyazi I and Belka C: Counting
colonies of clonogenic assays by using densitometric software.
Radiat Oncol. 2:42007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aapro M, Leonard RC, Barnadas A, Marangolo
M, Untch M, Malamos N, Mayordomo J, Reichert D, Pedrini JL, Ukarma
L, et al: Effect of once-weekly epoetin beta on survival in
patients with metastatic breast cancer receiving anthracycline-
and/or taxane-based chemotherapy: Results of the breast
cancer-anemia and the value of erythropoietin (BRAVE) study. J Clin
Oncol. 26:592–598. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aapro M, Gascon P, Patel K, Rodgers GM,
Fung S, Arantes LH Jr and Wish J: Erythropoiesis-stimulating agents
in the management of anemia in chronic kidney disease or cancer: A
historical perspective. Front Pharmacol. 9:14982019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Leyland-Jones B, Bondarenko I, Nemsadze G,
Smirnov V, Litvin I, Kokhreidze I, Abshilava L, Janjalia M, Li R,
Lakshmaiah KC, et al: A randomized, open-label, multicenter, phase
III study of epoetin alfa versus best standard of care in anemic
patients with metastatic breast cancer receiving standard
chemotherapy. J Clin Oncol. 34:1197–1207. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nagarkar R, Gascón P, Šmakal M, Syrigos K,
Barrios C, Cárdenas Sánchez J, Zhang L, Tomita D, Park J and De
Oliveira Brandao C: A double-blind, randomized, placebo-controlled
phase 3 noninferiority study of darbepoetin alfa for anemia in
advanced NSCLC. MA02.05. J Thorac Oncol. 13 (Suppl):S3592018.
View Article : Google Scholar
|
|
19
|
Miller CP, Lowe KA, Valliant-Saunders K,
Kaiser JF, Mattern D, Urban N, Henke M and Blau CA: Evaluating
erythropoietin-associated tumor progression using archival tissues
from a phase III clinical trial. Stem Cells. 27:2353–2361. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hadland BK and Longmore GD:
Erythroid-stimulating agents in cancer therapy: Potential dangers
and biologic mechanisms. J Clin Oncol. 27:4217–4226. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jelkmann W, Bohlius J, Hallek M and
Sytkowski AJ: The erythropoietin receptor in normal and cancer
tissues. Crit Rev Oncol Hematol. 67:39–61. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Elliott S, Busse L, Bass MB, Lu H, Sarosi
I, Sinclair AM, Spahr C, Um M, Van G and Begley CG: Anti-Epo
receptor antibodies do not predict Epo receptor expression. Blood.
107:1892–1895. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Elliott S, Swift S, Busse L, Scully S, Van
G, Rossi J and Johnson C: Epo receptors are not detectable in
primary human tumor tissue samples. PLoS One. 8:e680832013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cao Y, Lathia JD, Eyler CE, Wu Q, Li Z,
Wang H, McLendon RE, Hjelmeland AB and Rich JN: Erythropoietin
receptor signaling through STAT3 is required for glioma stem cell
maintenance. Genes Cancer. 1:50–61. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Paragh G, Kumar SM, Rakosy Z, Choi SC, Xu
X and Acs G: RNA interference-mediated inhibition of erythropoietin
receptor expression suppresses tumor growth and invasiveness in
A2780 human ovarian carcinoma cells. Am J Pathol. 174:1504–1514.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ueda F, Tago K, Tamura H and
Funakoshi-Tago M: Three tyrosine residues in the erythropoietin
receptor are essential for janus kinase 2 V617F mutant-induced
tumorigenesis. J Biol Chem. 292:1826–1846. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Todaro M, Turdo A, Bartucci M, Iovino F,
Dattilo R, Biffoni M, Stassi G, Federici G, De Maria R and Zeuner
A: Erythropoietin activates cell survival pathways in breast cancer
stem-like cells to protect them from chemotherapy. Cancer Res.
73:6393–6400. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kumar SM, Zhang G, Bastian BC, Arcasoy MO,
Karande P, Pushparajan A, Acs G and Xu X: Erythropoietin receptor
contributes to melanoma cell survival in vivo. Oncogene.
31:1649–1660. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jeong JY, Hoxhaj G, Socha AL, Sytkowski AJ
and Feldman L: An erythropoietin autocrine/paracrine axis modulates
the growth and survival of human prostate cancer cells. Mol Cancer
Res. 7:1150–1157. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shi Z, Hodges VM, Dunlop EA, Percy MJ,
Maxwell AP, El-Tanani M and Lappin TR: Erythropoietin-induced
activation of the JAK2/STAT5, PI3K/Akt, and Ras/ERK pathways
promotes malignant cell behavior in a modified breast cancer cell
line. Mol Cancer Res. 8:615–626. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu G, Qin XQ, Guo JJ, Li TY and Chen JH:
AKT/ERK activation is associated with gastric cancer cell
resistance to paclitaxel. Int J Clin Exp Pathol. 7:1449–1458.
2014.PubMed/NCBI
|
|
32
|
Chun E and Lee KY: Bcl-2 and Bcl-xL are
important for the induction of paclitaxel resistance in human
hepatocellular carcinoma cells. Biochem Biophys Res Commun.
315:771–779. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Solar P, Feldman L, Jeong JY, Busingye JR
and Sytkowski AJ: Erythropoietin treatment of human ovarian cancer
cells results in enhanced signaling and a paclitaxel-resistant
phenotype. Int J Cancer. 122:281–288. 2008. View Article : Google Scholar : PubMed/NCBI
|