Introduction
Gallbladder cancer is a very rare disease with an
incidence of 1.13/100,000 and a 5-year survival rate for all stages
at approximately 5%; it has highly variable characteristics and
presentations (1–3). Some research articles have reported that
gallbladder cancer is diagnosed late and that treatment is
ineffective. Resection is still the most effective and most likely
curative treatment for gallbladder cancer (4). However, numerous patients present late
in the course of the disease, when surgical intervention is no
longer beneficial (5). Therefore, the
prognosis of patients with gallbladder cancer remains poor
(1,2).
It is necessary to study the molecular mechanisms that effect the
progression of gallbladder cancer, as these may be potential
therapeutic targets for gallbladder cancer treatment.
To the best of our knowledge, receptor-interacting
serine/threonine-protein kinase 1 (RIP-1) is overexpressed in
gallbladder cancer tissues, while it has low expression in normal
tissues (6). A previous study by our
groups showed that increased RIP-1 expression is common in
gallbladder cancer tissues (6). It
was also found that RIP-1 expression was markedly correlated with
the clinical stage in patients with gallbladder cancer (6). There was evidence that a reduction of
RIP-1 expression in gallbladder cancer cells can exert inhibitory
effects on the ability of cancer cells to grow and invade in
vitro and in vivo (6).
Furthermore, the underlying mechanisms of RIP-1 affecting
gallbladder cancer cell biological behaviors were studied, and it
was found that RIP-1-nuclear factor κ-B (NF-κB) and activator
protein 1 (AP-1)-vascular endothelial growth factor-C (VEGF-C)
signaling pathways play an important role in RIP-1 promoting
gallbladder cancer cell growth and invasion (6). These are consistent with a previous
study, which demonstrated that RIP-1 is an independent prognostic
factor in glioblastomas (7).
Previous studies have found that RIP-1 plays a vital
role during cellular stress caused by different factors, such as
inflammation and DNA damage (8–12). These
responses activated RIP-1, with signals being transmitted when
activated RIP-1 activated the transcription factors NF-κB and AP-1
(8–12). These further triggered the expression
of downstream genes, and promoted cell survival and differentiation
(8–12). The activation of NF-κB in human cancer
is very common (13), and in a
previous study by our group, it was demonstrated that NF-κB
activation plays a vital role in gallbladder cancer progression
(14). Although a previous study by
our group revealed that RIP-1 could upregulate VEGF-C expression
though the RIP-1-NF-κB and AP-1 signaling pathways (6), the precise mediation mechanisms of this
upregulation are still unknown.
In our previous study, the authors found that
knockdown of RIP-1 expression suppressed the growth of subcutaneous
xenograft tumors of gallbladder cancer cells (6). In the animal model of gallbladder cancer
established by our group, liver, lung and lymph node metastasis,
and ascites had not developed (6).
These findings were consistent with a study by Horiuchi et
al (15), who suspected that the
subcutaneous xenograft gallbladder cancer model cannot provide a
suitable microenvironment for tumor cells. The findings of our
previous study therefore support the view that orthotopic xenograft
models of gallbladder cancer are better than the subcutaneous
xenograft gallbladder cancer model for studying the biological
characteristics of gallbladder cancer (16).
In the present study, the authors investigated the
specific mechanisms by which RIP-1 regulates the expression of
VEGF-C. This was performed to support the hypothesis that RIP-1
promotes growth and lymph node metastasis in gallbladder cancer.
Furthermore, the authors of the current study modified the
orthotopic xenograft model of gallbladder cancer to further study
RIP-1 and its influence on the biological behavior of gallbladder
cancer.
Materials and methods
Cell culture
The gallbladder cancer cell line GBC-SD and NOZ were
purchased from Shanghai Institutes for Biological Science
(Shanghai, China). SGC-996 was provided by the Tumor Cytology
Research Unit, Medical College, Tongji University (Shanghai,
China). NOZ cells were obtained from a 48-year-old female patient
with gallbladder cancer, which was established by Health Science
Research Resources Bank in Japan (17). All three cell lines were cultured in
Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10%
fetal bovine serum (FBS; both Life Technologies; Thermo Fisher
Scientific, Inc.). The three gallbladder cancer cell lines were
incubated at 37°C under 95% air and 5% CO2.
Plasmid constructs
Genomic DNA was extracted from NOZ cells using the
DNeasy Blood & Tissue kit (Qiagen, Inc.), which was used as a
template for polymerase chain reaction (PCR) amplification. The
plasmid pGL3B-2000 was constructed by ligation of the longest
PCR-produced VEGF-C promoter (nucleotides −2,000 to +1, relative to
the transcription start site). The longest fragment contained the
HindIII and XhoI restriction site, which was connected to the
pGL3-Basic vector (Promega Corporation). Different kinds of VEGF-C
promoter plasmids were constructed, including pGL3B-1500,
pGL3B-1000, pGL3B-487, pGL3B-332, pGL3B-190 and pGL3B-332NF-κBmut.
A total of 1 µg plasmids in each lane were resolved by
electrophoresis using 2% ethidium bromide-stained agarose gels.
Bands were visualized by densitometric scanning gels. Analyses were
performed using Quantity One software (version 4.6; Bio-Rad
Laboratories, Inc.). The plasmids were stored by the Fujian
Institute of Hepatobiliary, Union Hospital, Fujian Medical
University (Fuzhou, China). The primers for PCR amplification are
as follows: Forward, 5-TGACTCGAGCTGCCCCTGCGCCCGCCGCC-3,
reverse, 5-TGAAAGCTTCCTCCCCTTCCCCGAAGT-3 (Xhol and
HindIII restriction sites are shown in italics). pGL3B-332
was used as a template for the construction of AP-1-binding site
mutants. The authors constructed the mutant plasmids using overlap
PCR technology (18). The mutant
plasmids were named pGL3B-332AP-1 mut, and the NF-κB and AP-1
co-mutant plasmids were constructed and named pGL3B-332 mut. To
obtain the overexpression plasmids, pcDNA3.1-NF-κB(p65) and
pcDNA3.1-AP-1, PCR was carried out with a superscript library (Life
Technologies; Thermo Fisher Scientific, Inc.) using the template
human full-length cDNA sequence of NF-κB(p65)/AP-1. The
PCR-generated NF-κB(p65)/AP-1 gene was inserted into the
XhoI-HindIII sites of pcDNA3.1 (Promega Corporation).
All of the above constructs were confirmed by DNA sequencing, which
was performed by Bosune Biological Company. The primers for PCR
amplification are shown in Table
I.
 | Table I.List of oligonucleotides used in the
current study. |
Table I.
List of oligonucleotides used in the
current study.
| Oligonucleotides
names | Sequences
(5′-3′) |
|---|
|
|---|
| A, pGL3B-332
mutation |
|---|
| Mut AP-1 F |
CGCAGGCAGAGGGCGCGTTTTTCATGCCCTGCCCCTGCG |
| Mut AP-1 R |
CGCAGGGGCAGGGCATGAAAAACGCGCCCTCTGCCTGCG |
| Mut NF-κB F |
AGGCGAGGGAAACGAAGAGCTCCAGGGAGA |
| Mut NF-κB R |
TCTCCCTGGAGCTCTTCGTTTCCCTCGCCT |
| P1 co-primer F |
TACGGGAGGTACTTGGAGCGG |
| P1 co-primer R |
TTCCAGGAACCAGGGCGTATC |
|
| B,
pcDNA3.1-expression plasmids |
|
| AP-1 F |
GCCTCGAGGCCACCATGACTGCAAAGATGGAAACG |
| AP-1 R |
GCAAGCTTTCAAAATGTTTGCAACTGCTGCGTT |
| NF-κB F |
GCCTCGAGGCCACCATGGACGAACTGTTCCCCCT |
| NF-κB R |
GCAAGCTTTTAGGAGCTGATCTGACTCA |
| RIP-1 F |
GCCTCGAGGCCACCATGCAACCAGACATGTCCTTG |
| RIP-1 R |
GCAAGCTTGGGTTAGTTCTGGCTGACGTAAATC |
|
| C,
Electrophoretic mobility shift assay |
|
| Biotin-labeled
probe |
Biotin-GAGGGAAACGGGGAGCTCCAGGGAG |
| NF-κB cold
probe |
AGTTGAGGGGACTTTCCCAGGC |
| NF-κB mutation
probe |
AGTTGAGGAAACTTGCCCAGGC |
| Biotin-labeled
probe |
Biotin-AGAGGGCGCGTCAGTCATGCC |
| AP-1 cold
probe |
AGAGGGCGCGTCAGTCATGCC |
| AP-1 mutation
probe |
AGAGGGCGCGTTTTTCATGCC |
|
| D, Chromatin
immunoprecipitation assay |
|
| AP-1 F |
CGAGGGAGAGTGAGAGGGGAGGGCA |
| AP-1 R |
GCGGCGGGCGCAGGGGCAGGGCATG |
| NF-κB F |
GACAGGGGCGGGGAGGGAGA |
| NF-κB R |
CTCACTCTCCCTCGGAAGCCGTCTC |
Western blot analysis
These experiments and results analysis were carried
out as described previously (6,14). Cells
were lysed with Western IP cell lysis buffer (Beyotime Institute of
Biotechnology) containing PMSF (Amresco, LLC) on ice for 30 min.
Cell protein (30 µg/lane) was separated by SDS-PAGE on a 10% gel
and transferred onto a 0.45 µM PVDF membrane (GE Healthcare). The
membrane was blocked with 0.5% bovine serum album (Amresco, LLC) at
room temperature for 2 h. The following monoclonal primary
antibodies were used in the western blot analysis: Mouse anti-human
RIP-1 antibody (cat. no. ab72139; 1:1,000; Abcam), rabbit
anti-human AP-1 (cat. no. ab32137; 1:1,000; Abcam), mouse
anti-human NF-κB (p65; cat. no. 8242; 1:500; Cell Signaling) and
mouse anti-human β-actin (cat. no. sc-47778; 1:1,500; Santa Cruz
Biotechnology, Inc.) overnight at 4°C. The membranes were washed
three times with TBST (0.1% Tween-20) for 10 min each at room
temperature. The membranes were incubated with anti-rabbit
immunoglobulin (Ig)G (cat. no. sc-2357) and mouse IgGκ (cat. no.
sc-516102; both dilution 1:4,000; Santa Cruz Biotechnology, Inc.)
horseradish peroxidase-conjugated secondary antibodies at room
temperature for 1 h. Visualization of the immunoreactive proteins
was performed by chemiluminescence kit (BeyoECL Plus; Beyotime
Institute of Biotechnology). The intensities of band signals were
quantified using Quantity One densitometric software (version
4.6.3); the relative intensity of the target bands was normalized
to that of β-actin.
Transfection and dual-luciferase
reporter assay
NOZ cells were seeded at a density of
2×105 cells per well in 12-well plates. DNA transfection
was performed in by use of Lipofectamine 2000 (Life Technologies;
Thermo Fisher Scientific, Inc.), in accordance with the
manufacturer's protocol. The small interfering (si)RNAs for NF-κB
and AP-1 were 5'-CUCAAGAUCUGCCGAGUGA-3' and
5'-CCTCAGCAACTTCAACCC-3', respectively, and were synthesized by
Beyotime Institute of Biotechnology; 100 pmol of each siRNA were
transfected into the cells. At 48 h after transfection, the mRNA
and proteins were extracted. NOZ cells were also co-transfected
with the siRNAs and different VEGF-C promoter vectors, and cells
were lysed 48 h after transfection. A total of 20 µg of cell lysate
was used for the detection of intracellular luciferase activity,
following the manufacturer's protocol of
Dual-Luciferase® Reporter (DLR™) Assay system (Promega
Corporation). The Renilla luciferase expression vector
pRL-TK (Promega Corporation) was used for normalization and the
promoter-less vector pGL3-Basic served as the negative control.
Luminescence measurement was performed on a luminometer (Orion II
Microplate Luminometer; Berthold Detection Systems GmbH). Each
transfection was performed in duplicate and data were expressed as
the mean ± standard deviation of three separate experiments.
Identification of putative
transcription factor binding sites
A computer-based search for potential transcription
factor binding site motifs was carried out on TESS (http://www.cbil.upenn.edu/cgi-bin/tess/tess) (19), TFBIND (http://tfbind.hgc.jp/) (20) and TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCH.html)
(21).
Nuclear extraction and electrophoretic
mobility shift assay (EMSA)
Transfected cells (107) were transfected
and the Nuclear Extraction kit (Beyotime Institute of
Biotechnology) was used to extract nuclear proteins. The 5-biotin
end-labeled oligonucleotides were synthesized by Beyotime Institute
of Biotechnology and used as probes. Unlabeled oligonucleotide cold
probes or mutated cold probes with NF-κB and AP-1 mutation sites
were used as competitors. The oligonucleotide sequences are shown
in Table I. Nuclear protein was
subjected to hybridization to these oligonucleotide probes. Nuclear
proteins, including the interaction of NF-κB/AP-1 with the dsDNA
probe, were tested by use of the Chemiluminescent Nucleic Acid
Detection Module kit (cat no. 89880; Thermo Fisher Scientific,
Inc.), in accordance with the manufacturer's protocol.
Briefly, the binding buffer mixture was prepared of
1X binding buffer supplemented with 5% glycerol, 200 mM KCl and 100
mM MgCl2 (cat. no. 20148X; Thermo Fisher Scientific,
Inc.). A total of 7 µg of extracted nuclear proteins were incubated
with 1 µg of poly(deoxyinosinic-deoxycytidylic) and 0.02 pmol
labeled probe in a final volume of 10 µl for 20 min at room
temperature. The competition assay was performed using 0.5 pmol
biotin probes and 50 pmol cold/mut probes, as well as 50–100 or
50–200 fold molar excess of cold probes or cold mutated probes,
which added to other component for 5 min before adding
biotin-labeled probes. Then, proteins were subjected to
electrophoresis on a 6% non-denaturing pre-made polyacrylamide gel
(Invitrogen; Thermo Fisher Scientific, Inc.). The complexes were
transferred to a nylon membrane (GE Healthcare) and fixed for 3 min
using a UV crosslinker (UVP, LLC). The biotin end-labeled DNA was
detected by the addition of a streptavidin-horseradish peroxidase
conjugate and a chemiluminescent substrate from the
Chemiluminescent Nucleic Acid Detection Module kit intensities of
band signals were quantified using Quantity One densitometric
software (version 4.6.3). The experimental results are shown in
bars.
Chromatin immunoprecipitation
(ChIP)-PCR
Transfected NOZ cells (107) were cultured
and cross-linked using formaldehyde (final concentration, 1%) at
room temperature for 10 min. Following this, 125 mM glycine was
added for 5 min to stop cross-linking from occurring at room
temperature. Cells were rinsed twice with ice-cold PBS and then
collected by centrifuging at 3,500 × g for 5 min at 4°C. The cells
were resuspended in 0.75 ml FA lysis buffer (Abcam) and placed on
ice for 15 min. Cell lysates were sonicated for 20 min.
Immunoprecipitation was conducted with anti-NF-κB and anti-AP-1
antibodies (both 1:200) overnight at 4°C followed by 80 µl of
pre-blocked protein A/G beads overnight at 4°C. DNA was then
recovered by phenol/chloroform extraction and ethanol precipitation
in the presence of glycogen [glycogen: 10 µl (5 mg/ml); ethanol: 25
µl], and dissolved in 100 µl sterile distilled water. The target
DNA fractions were amplified by PCR [LA Taq® DNA
polymerase (Takara Bio, Inc.) 28 cycle of denaturation at 95°C for
30 sec, annealing at 56°C for 30 sec, and extension at 72°C for 30
sec followed by 10 min for final extension at 72°C], and the
primers used are as follows: NF-κB site in VEGF-C promoter forward,
5′-GACAGGGGCGGGGAGGGAGA-3′, and reverse,
5′-CTCACTCTCCCTCGGAAGCCGTCTC-3′; and AP-1 site in VEGF-C promoter
forward, 5′-CGAGGGAGAGTGAGAGGGGAGGGCA-3′, and reverse,
5′-GCGGCGGGCGCAGGGGCAGGGCATG-3′.
Establishment of the orthotopic
xenograft model by modified methods
A total of 15 male 4–6 week-old athymic BALB/c nu/nu
mice weighing 15–20 g were purchased from the Shanghai SLAC
Laboratory Animals Company, and were raised in the Experimental
Center of Fujian Medical University in a climate-controlled room at
a temperature of 20–26°C (termed barrier system), with a relative
humidity of 40–70% and light/dark cycle of 12/12 h and free access
to food and water. The mice were treated according to the
institutional guidelines, and the study was approved by the Ethics
Committee of the Medical Faculty of the Fujian Medical
University.
To build the orthotopic xenograft model of
gallbladder cancer in nude mice, untreated NOZ cells [NOZ-control
(con) group], NOZ cells were treated with a negative control (NC)
siRNA (NOZ-NC group), and NOZ cells were treated with a siRNA
against RIP-1 (NOZ-RIPsi group) cells were used (6). The NC sequence was
5′-TTCTCCGAACGTGTCACGT-3′ and the RIPsi sequence was
5′-GCACAAATACGAACTTCAA-3′. These sequences were obtained from
Genechem Co., Ltd. These were stored by the Fujian Institute of
Hepatobiliary, Union Hospital, Fujian Medical University. A total
of 2 µg of each siRNA were transfected into NOZ cells in DMEM with
10% FBS at ~90% confluency at 37°C under 95% air and 5%
CO2 for 72 h. The culture medium was replaced with DMEM
containing 2 µg/ml puromycin (Sigma-Aldrich; Merck KGaA). When the
stably transfected cells we obtained, the cells were continuously
maintained in 1 µg/ml of puromycin for later experiments, and
termed the NOZ-NC group and the NOZ-RIPsi group.
To establish the mouse model, differently treated
NOZ cells were harvested at a concentration of 107
cells/ml. These cells were placed on ice for the following
experiments. The nude mice were anesthetized a mixed anesthetic
(ketamine 80 mg/kg, xylazine 8 mg/kg). The mice did not consume
water or food for 4 h before anesthesia. After anesthesia,
conventional complexing iodine was used to disinfect the skin of
the mice. An abdominal midline incision was made (approximately 1.0
cm), after which the gallbladder of the nude mice was exposed. The
gallbladder was located in the middle of liver lobes. A total of 20
µl of suspended cells was mixed with 20 µl Matrigel.
The 40 µl mixture was then taken up by a modified
insulin syringe. The operation was performed on ice to keep the
Matrigel in a liquid state. The syringe was then used to puncture
through the gallbladder, the syringe was then withdrawn from the
puncture site into the gallbladder, leaving a hole on one side. The
gallbladder bile them leaked onto a swab. With the needle still
inside, 40 µl of the cell mixture was slowly injected into the
empty gallbladder. After 30 sec, when the cells suspension
solidified, the syringe was withdrawn from the gallbladder. The
gallbladder and liver were placed in the abdominal cavity, and then
the abdominal wall was sutured. There were five mice in each
experimental group. After regaining consciousness, the mice were
raised in a sterile environment. The physical condition of the mice
was monitored for 4 days after surgery, and then recorded once
every 2 days for several weeks. A total of 5 weeks later, the mice
were euthanized by means of cervical dislocation and the mice were
dissected in a sterile environment. Tumor samples were fixed in 10%
formaldehyde solution at room temperature for 24 h and some samples
were kept in liquid nitrogen.
Hematoxylin and eosin staining,
immunohistochemistry, and evaluation
The primary tumors and lymph node were fixed in 10%
neutral buffer formalin for 24 h at room temperature. These tissues
were then trimed and embedded in paraffin. Samples were cut in
4-µm-thick serial sections, and stained with hematoxylin for 3 min
and eosin for 10 sec both at room temperature. The samples were
then observed under an optical microscope (DM4000B; Leica) at a
magnification of ×400.
Immunohistochemistry and its evaluation were carried
out as previously described (6).
Briefly, the sections were blocked in 10% normal goat serum (Vector
Laboratories, Inc.) at room temperature for 15 min and then the
sections were incubated in 3% H2O2 at room
temperature for 35 min to inactivate endogenous peroxidase.
Thereafter, primary antibodies were incubated with 4-µm-thick
slices overnight in humidified boxed at 4°C, followed by incubation
with 60 µl ready-to-use biotin-conjugated secondary antibodies from
the UltraSensitive S-P kit (cat. no. 9710; Fuzhou Maixin Biotech,
Co., Ltd.) at room temperature for 1 h. Then sections were then
washed in TBST three times (5 min each) and incubated with
streptavidin-peroxidase from the UltraSensitive S-P kit in dark at
room temperature for 10 min. DAB (ZSJQB Co., Ltd.) for 4–6 min at
room temperature and hematoxylin for 3 min at room temperature. The
samples were then observed with a DM4000B microscope at a
magnification of ×400. The following primary antibodies were used:
Mouse monoclonal anti-human RIP-1 (1:500), rabbit polyclonal
anti-human VEGF-C (1:150; cat. no. ab9546; Abcam) and rabbit
polyclonal anti-mouse lymphatic vessel endothelial hyaluronic acid
receptor 1 (LYVE-1; cat. no. AF2125; 1:250; R&D Systems, Inc.).
The expression of RIP-1 and VEGF-C were evaluated using the
Image-Pro Plus 6.0 software (Media Cybernetics, Inc.). Three slices
were randomly selected for each group and five fields were selected
for each slice. The expression of RIP-1 and VEGF-C was
semi-quantitatively analyzed in fifteen fields using the following
formula: Mean optical density = integral optical density / the
positive area.
Microlymphatic vessel counting
The 4-µm-thick slices were observed at low
magnification (×100) using a DM4000B microscope to detect areas
with the most intense staining and apparent highest occurrence of
microvessels, thus named ‘the hotspot’ (22). Two physiologists independently
evaluated the slides for microvessels using a DM4000B microscope at
a magnification of ×400. Brown vessels without red blood cells in
their lumen were considered microlymphatic vessels. Vessels with a
single immunoreactive layer of endothelial cells or endothelial
cells were counted as a blood vessel (23). The density of positive cells in
microlymphatic vessels were counted with a DM4000B microscope at a
magnification of ×400.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 5 software (GraphPad Software, Inc.). Data were
analyzed by ANOVA with Tukey's post hoc test when more than two
groups were compared or Student's t-test when two groups were
compared. The data were expressed as the mean ± standard deviation.
A P-value of <0.05 was considered to indicate a statistically
significant difference.
Results
Region of −332 nt to +1 is crucial for
the functions of the VEGF-C promoter and for the effect of RIP-1 on
VEGF-C promoter activity
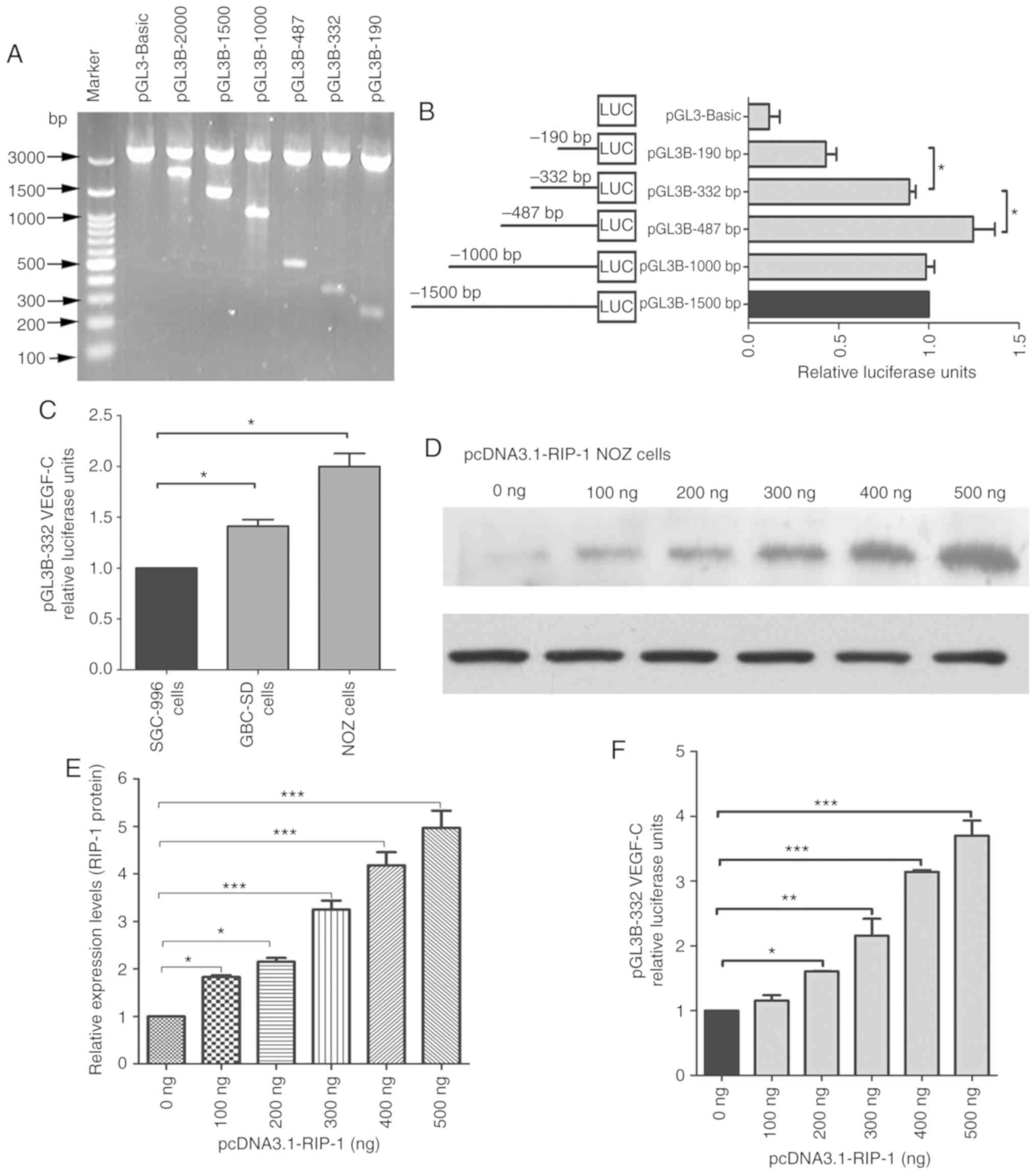
Gel electrophoresis was used to detect the
constructed plasmids, including various promoter deletions of
VEGF-C (Fig. 1A). These plasmids were
transfected transiently into NOZ cells. As shown in Fig. 1B, the cells revealed similar
luciferase activities for pGL3B-1500, pGL3B-1000 and pGL3B-332,
while luciferase activities were significantly increased for
pGL3B-487 and decreased for pGL3B-190 compared with pGL3B-332. This
indicates that the region of −332 to +1 nt is key to the activities
of the VEGF-C promoter. To investigate the luciferase activities of
the VEGF-C promoter in gallbladder cancer cell lines, SGC-996,
GBC-SD and NOZ cell lines were used to test pGL3B-332 luciferase
activity. As shown in Fig. 1C,
luciferase activity were significantly higher in NOZ cells and
GBC-SD cells compared with that in SGC996 cells. The luciferase
activity was highest in NOZ cells, moderate in GBC-SD and lowest in
SGC-996. The expression of the RIP-1 protein was assessed by
western blot analysis after transfecting different concentrations
of pcDNA3.1-RIP-1 plasmids in NOZ cells for 48 h. RIP-1 protein
expression in NOZ cells significantly increased in a dose-dependent
manner of pcDNA3.1-RIP-1 concentration compared with the untreated
cells (Fig. 1D and E). RIP-1
overexpression significantly increased the relative luciferase
activity of the pGL3B-332 plasmid compared with the untreated group
and this increase was dependent on pcDNA3.1-RIP-1 concentration
(Fig. 1F). These results suggested
that RIP-1 affects the luciferase activity of the VEGF-C
promoter.
Regulation of the VEGF-C promoter by
the transcription sites of NF-κB and AP-1
TESS and TFBIND were used to search for putative
transcription factor binding sites. Previously, our team found that
the NF-κB site on the VEGF-C promoter regulates its transcriptional
activity (24). That study focused on
the regulation of NF-κB by RIP-1. NF-κB and two AP-1 sites
(overlapped partially and very similiar in the DNA sequences) were
identified in the −332 nt to +1 region of the promoter (Fig. 2A). A mutated NF-κB site and two
mutated AP-1 site in pGL3B-332 were constructed, and the promoter
luciferase activity of the mutant plasmids was measured. As
revealed in Fig. 2B, both
pGL3B-332-NF-κB mut and pGL3B-332-AP-1 mut had significantly lower
activity than the non-mutated control plasmid, pGL3B-332. In
addition, the co-mutation of the NF-κB site and two AP-1 sites,
pGL3B-332 mut, had significantly lower luciferase activity than
that of pGL3B-332, pGL3B-332- NF-κB mut and pGL3B-332-AP-1 mut
plasmids. This suggests that the NF-κB site and two AP-1 sites may
work together to encourage the luciferase activity of the VEGF-C
promoter. Overexpression of RIP-1 significantly increased the
relative luciferase activity of the pGL3B-332 plasmid and the
luciferase activity also increased with increasing concentrations
of pcDNA3.1-RIP-1 (Fig. 2C and D).
However, the NF-κB mutation site, AP-1 mutation sites, and
co-mutation NF-κB and AP-1 sites markedly impaired the ability of
RIP-1 to increase the luciferase activity of the pGL3B-332
promoter. These results further indicate that RIP-1 regulates the
luciferase activity of the VEGF-C promoter through NF-κB and
AP-1.
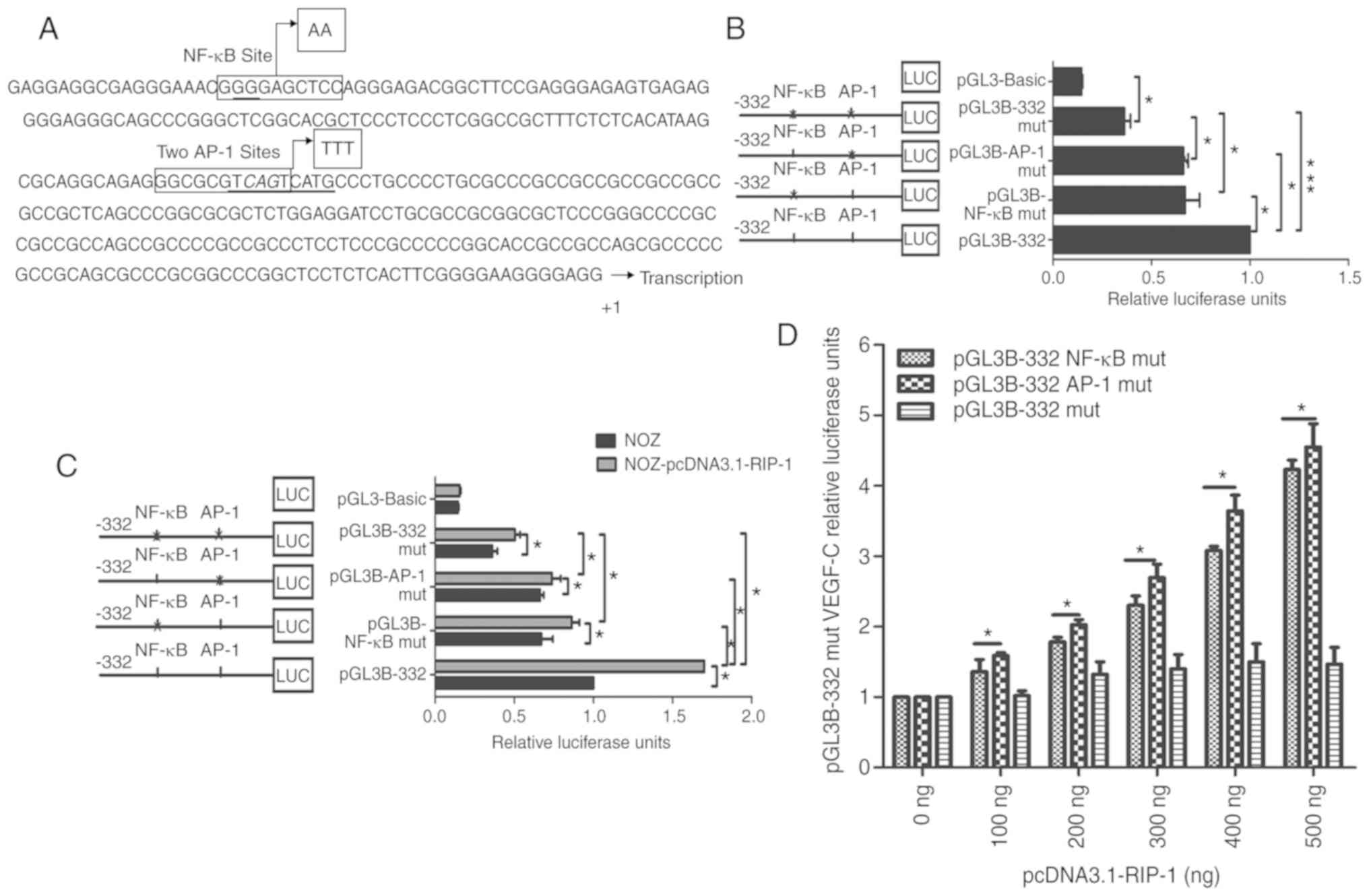 | Figure 2.NF-κB and AP-1 sites were crucial for
the pGL3B-332 relative luciferase activities. (A) Nucleotide
sequence of the −332 to +1 nt fragment of the VEGF-C promoter. The
promoter start site is G and G is set to +1. The NF-κB binding
sites and the AP-1 binds sites in the nucleotide region are framed.
The nucleotides in boxes next to arrows are the mutated
oligonucleotides. AP-1 sites include two overlapping AP-1 binding
sites (one site in a box, the other site underlined). (B)
Luciferase activity analysis of VEGF-C promoter following the
co-transfection of pRL-TK plasmids and mutated plasmids into NOZ
cells with. pGL3-Basic vector served as the negative control. (C)
pcDNA3.1-RIP-1 plasmid was co-transfected into NOZ cells with
pGL3B-NF-κB mut, pGL3B-AP-1 mut or pGL3B-332 mut plasmids. (D) NOZ
cells were treated with different concentrations of pcDNA3.1-RIP-1,
and then the cells were co-transfected with the pRL-TK plasmid and
the pGL3B-NF-κB mut, pGL3B-AP-1 mut or pGL3B-332 mut plasmid. The
relative luciferase units were tested after 48 h. *P<0.05,
***P<0.001 as indicated. Each transfection was carried out in
duplicate and the data were expression as the mean ± standard
deviation of three experiments. pGL3B-NF-κB mut, NF-κB mutant
plasmid; pGL3B-AP-1 mut, AP-1 mutant plasmid; pGL3B-332 mut, NF-κB
and AP-1 mutant plasmid; mut, mutant; NF-κB, nuclear factor κ-B;
AP-1, activator protein 1; LUC, luciferase; RIP-1,
receptor-interacting serine/threonine-protein kinase 1; VEGF-C,
vascular endothelial growth factor-C. |
The transcription factors NF-κB and
AP-1 regulate the luciferase activity of the VEGF-C promoter
To investigate the effect of the transcription
factors NF-κB and AP-1 on the VEGF-C promoter, NF-κB and AP-1
eukaryotic expression vectors, pcDNA3.1-NF-κB and pcDNA3.1-AP-1
plasmids, were constructed. The knockdown interference RNA
sequences, NF-κB-siRNA and AP-1 siRNA, were also synthesized. After
the pcDNA3.1-NF-κB and pcDNA3.1-AP-1 plasmids were transfected into
NOZ cells for 48 h, western blotting was used to test the protein
expression of NF-κB and AP-1 in NOZ cells. As shown in Fig. 3A-D, NF-κB and AP-1 protein expression
significantly increased in the pcDNA3.1-NF-κB and pcDNA3.1-AP-1
groups compared with the control group (pcDNA-3.1). It was also
found the luciferase activity of pGL3B-332 was significantly
increased when co-transfected with pcDNA3.1-NF-κB or pcDNA3.1-AP-1
compared with the control group (Fig.
3E).
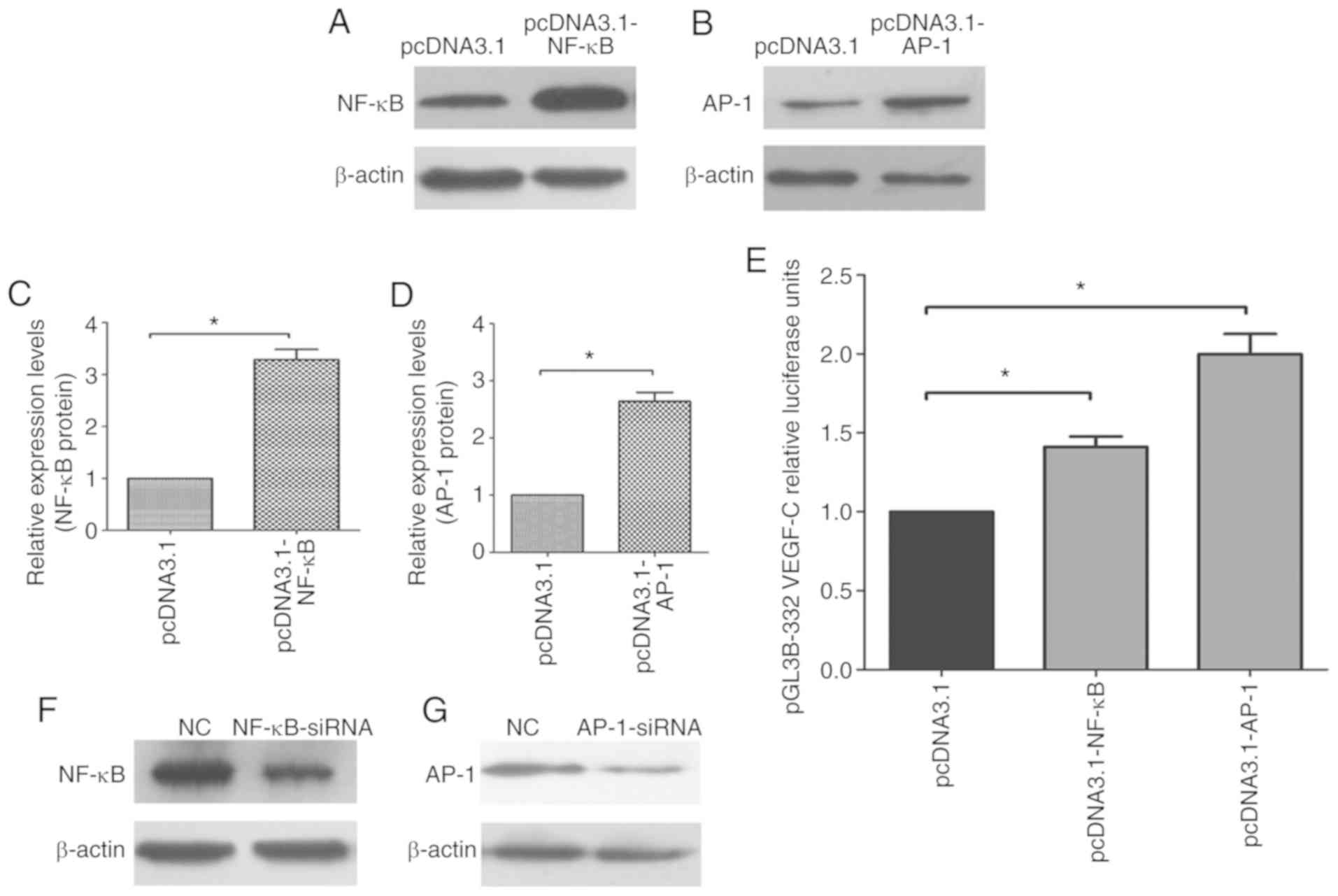 | Figure 3.NF-κB and AP-1 are key transcription
factors for the VEGF-C promoter. Western blot analysis of (A) NF-κB
and (B) AP-1 in NOZ cells transfected with pcDNA3.1-NF-κB,
pcDNA3.1-AP-1 or an empty pcDNA3.1 vector. Quantification of
relative (C) NF-κB and (D) AP-1 protein expression levels in the
NOZ cells. *P<0.05 vs. empty pcDNA3.1. (E) Luciferase activity
analysis of VEGF-C promoter following the co-transfection of the
pGL3B-332 plasmid and different pcDNA3.1 plasmids. pRL-TK plasmids
were used for normalization. *P<0.05 as indicated. (F) NF-κB and
(G) AP-1 in NOZ cells transfected with NF-κB-siRNA, AP-1-siRNA or
an NC siRNA. Quantification of relative (H) NF-κB and (I) AP-1
protein expression levels in the NOZ cells. *P<0.05 vs. NC. (J)
Luciferase activity analysis of VEGF-C promoter following the
co-transfection of the pGL3B-332 plasmid and different siRNAs.
*P<0.05 as indicated. (K) VEGF-C promoter luciferase activity
following the co-transfection of NOZ cells with the pGL3B-332 or
pGL3B-332 NF-κB mut plasmid and different concentrations of
pcDNA3.1-NF-κB. (L) VEGF-C promoter luciferase activity following
the co-transfection of NOZ cells with the pGL3B-332 or pGL3B-332
AP-1 mut plasmid and different concentrations of pcDNA3.1-AP-1. The
experiments were duplicated three times. *P<0.05 as indicated.
pcDNA3.1, overexpressing plasmid; pGL3B-NF-κB mut, NF-κB mutant
plasmid; pGL3B-AP-1 mut, AP-1 mutant plasmid; pGL3B-332 mut, NF-κB
and AP-1 mutant plasmid; mut, mutant; NF-κB, nuclear factor κ-B;
AP-1, activator protein 1; VEGF-C, vascular endothelial growth
factor-C; NC, negative control siRNA. |
Furthermore, NF-κB-siRNA and AP-1-siRNA were
transfected into NOZ cells to knockdown the endogenous NF-κB and
AP-1 proteins. After 48 h, the proteins were harvested, and western
blotting was performed to detect the protein expression of NF-κB
and AP-1. The protein expression of NF-κB and AP-1 was
significantly downregulated in NOZ cells compared with the control
group (Fig. 3F-I). NF-κB-siRNA and
AP-1-siRNA were then co-transfected into NOZ cells with pGL3B-332.
The luciferase activity of pGL3B-332 was significantly decreased
with NF-κB-siRNA and AP-1-siRNA compared with the control group
(Fig. 3J).
As shown in Fig. 3K,
the change in pGL3B-332 promoter luciferase activity was
pcDNA3.1-NF-κB plasmid concentration-dependent as 200–500 ng
pcDNA3.1-NF-κB plasmid significantly increased luciferase activity
compared with the control group. However, the pGL3B-332-NF-κB mut
plasmid's promoter luciferase activity did not significantly
increase. In addition, pGL3B-332 and pGL3B-332-AP-1 mut plasmids
were co-transfected with pcDNA3.1-AP-1 into the NOZ cells to
analyze the change in luciferase activity. The activity of the
pGL3B-332 promoter was enhanced in a pcDNA3.1-AP-1
concentration-dependent manner compared with the control group.
However, the pGL3B-332-AP-1 mut promoter's luciferase activity was
not markedly enhanced (Fig. 3L).
These results show that the transcription factors NF-κB and AP-1
regulate the luciferase activity of the VEGF-C promoter.
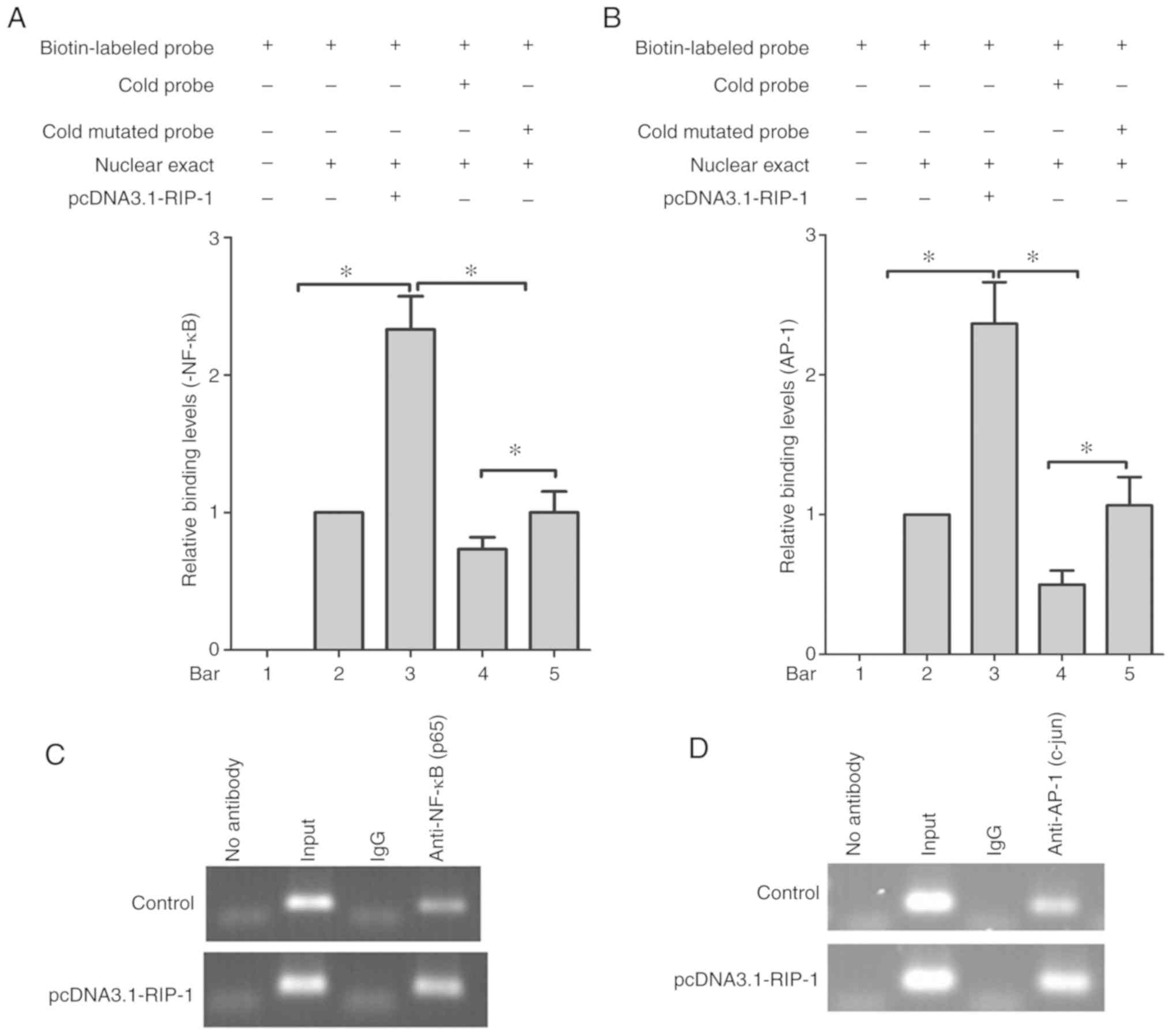
RIP-1 promotes the binding of NF-κB
and AP-1 to the VEGF-C promoter
The effects of NF-κB and AP-1 on the VEGF-C promoter
were further confirmed by EMSA of nuclear extracts from NOZ cells.
The results revealed that the nuclear extract could change the
position of the biotin-labeled NF-κB and AP-1 probe in
electrophoretic mobility, due to DNA and protein complex formation
(bar 1 and 2; Fig. 4A and B). The
RIP-1 overexpression plasmid increased the combined rates of the
nuclear extract, and the NF-κB and AP-1 binding sites, which were
included in the nucleotide sequences of the biotin-labeled probe
(bar 3; Fig. 4A and B). In addition,
a competition assay showed that pre-incubation with a 100-fold
molar excess of cold probe diminished the intensity of the bands,
but the cold mutated probes did not (bars 4 and 5; Fig. 4A and B).
To investigate whether NF-κB and AP-1 are associated
with the VEGF-C promoter in vivo, ChIP experiments using
either NF-κB or AP-1 antibodies and PCR amplification were
performed. As shown in Fig. 4C and D,
112 and 120 bp DNA fragments covering NF-κB and AP-1 sites in the
VEGF-C promoter were amplified, and chromatin was
immunoprecipitated with anti-NF-κB or anti-AP-1 antibodies. In
addition, RIP-1-promoted NF-κB and AP-1 bound to the VEGF-C
promoter. The same bands were obtained with DNA input, and the
normal IgG and no antibody controls did not give rise to the
immunoprecipitation of DNA fragments. This was found by PCR
amplification using two assays.
In conclusion, these results suggest that RIP-1
stimulates the transcription factors NF-κB and AP-1 to bind
directly to their corresponding consensus binding sites in the
VEGF-C promoter region.
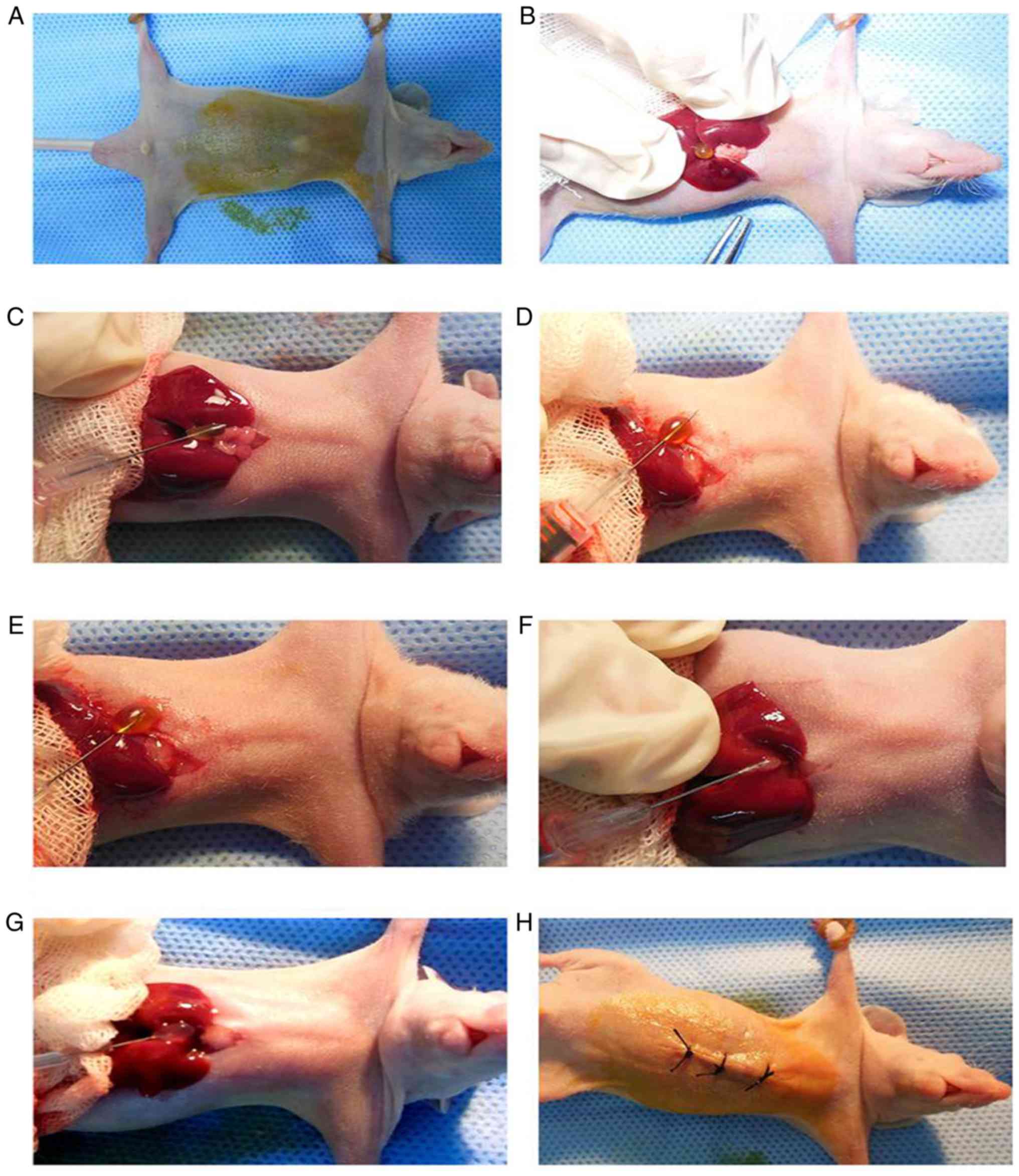
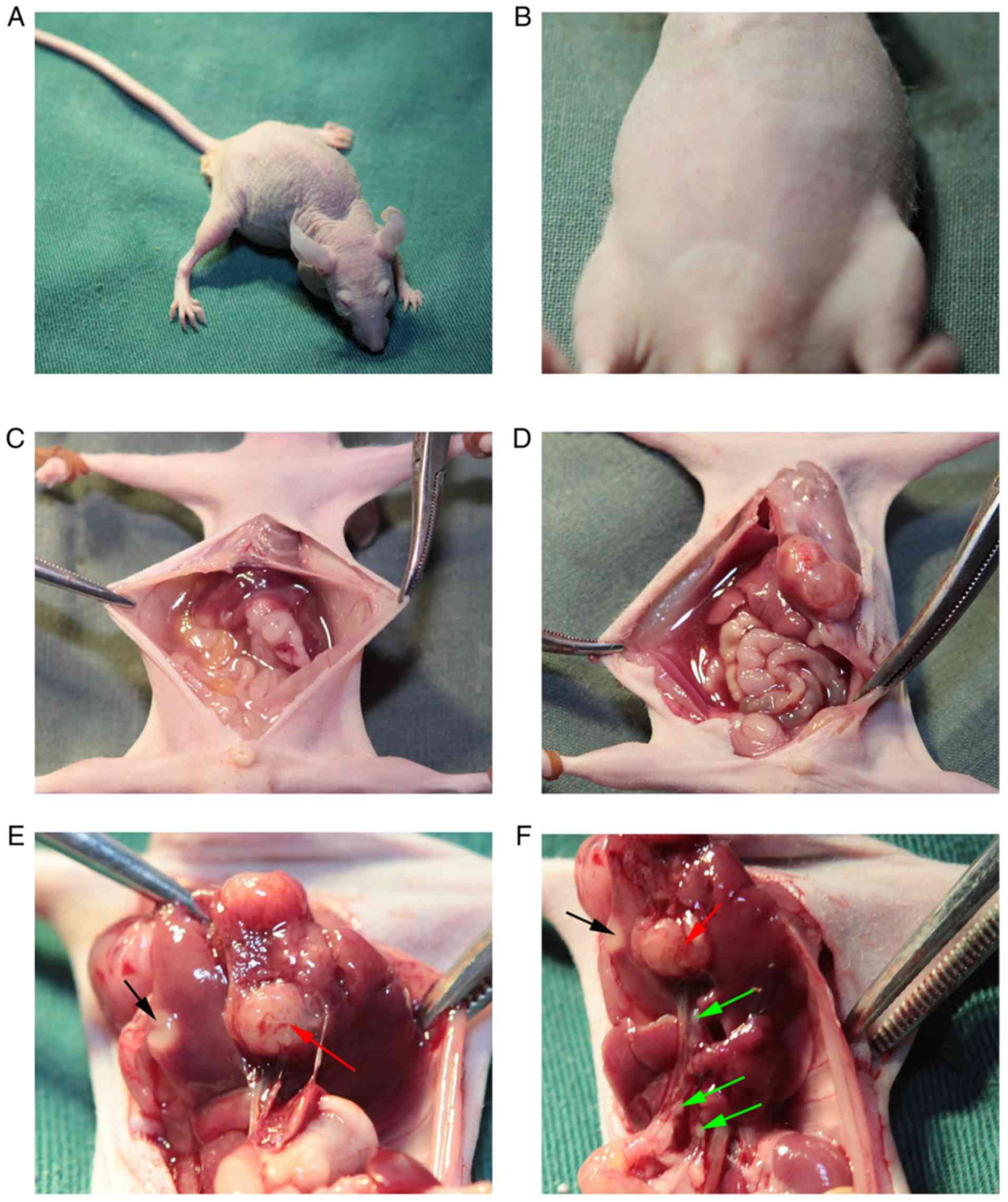
Establishment of the orthotopic
xenograft nude mouse model of gallbladder cancer
In our previous experiments, the authors established
the mouse model of subcutaneous tumors, which cannot illustrate the
biological function of the RIP-1 gene in lymph node metastasis
(6). In the current study, the
authors therefore tried to establish the mouse model of orthotopic
gallbladder cancer tumors using modified methods (Fig. 5). The mouse model was successfully
established by using NOZ cells and found that there were notable
swollen lymph nodes on the hepatoduodenal ligament. These were
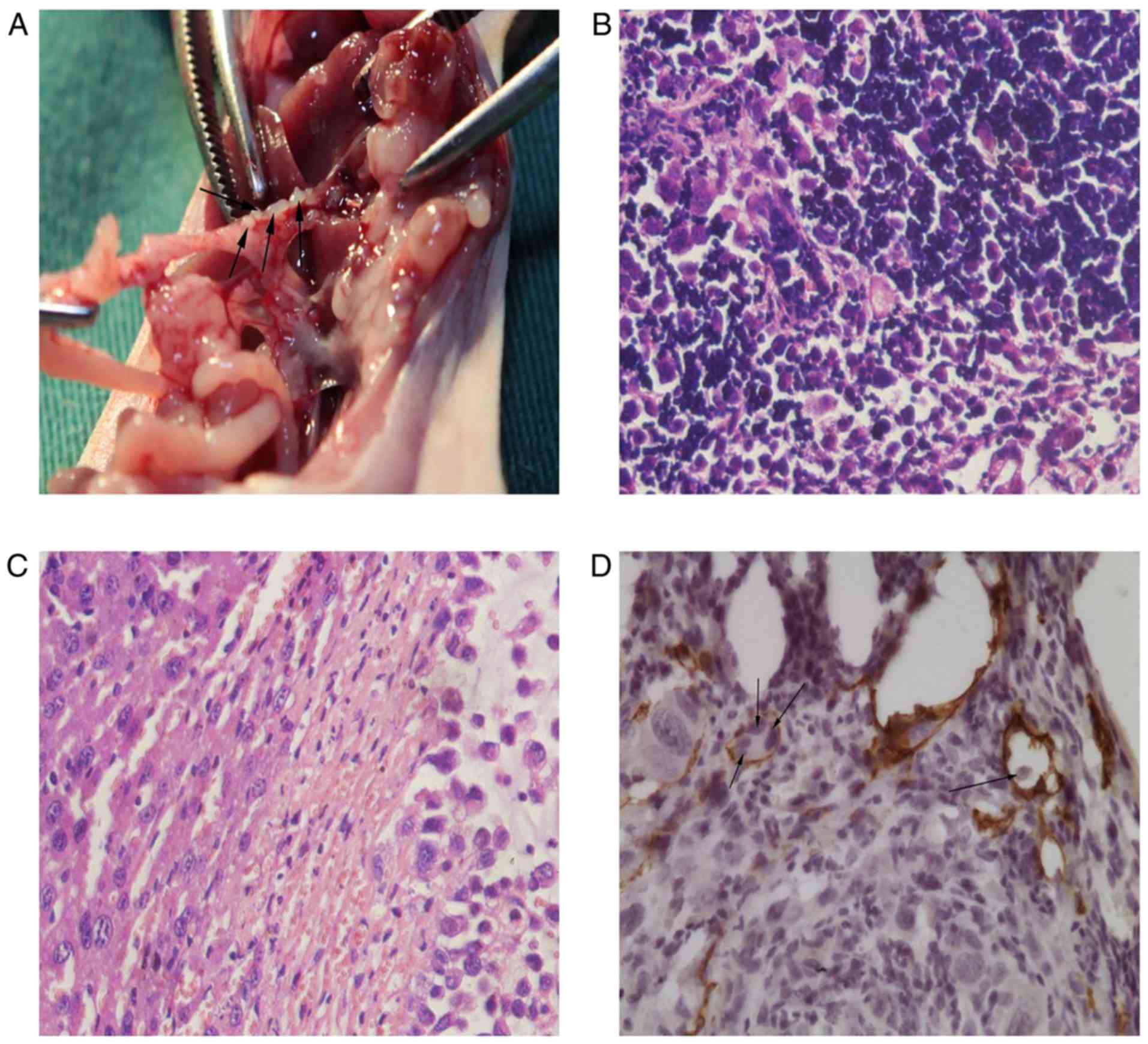
detected as mostly positive by pathology (Figs. 6F, 7A
and 7B). As shown in Fig. 7B, the lymph nodes were occupied by a
large amount of tumor cells. NOZ-con, NOZ-NC and NOZ-RIPsi were
orthotopically xenografted into the gallbladder of nude mice.
Post-inoculation, these mice were raised in a barrier system for 5
weeks. Some mice showed dyscrasia, and large tumors could be seen
and touched in the abdomens of the mice (Fig. 6A and B). When mice were sacrificed,
five of the four mice in the NOZ-con and NOZ-NC group tumors had
developed ascites, whereas none of the NOZ-RIPsi group mice had
ascites at the time of sacrifice (Fig.
6C and Table II). Fig. 6D showed the hemorrhagic ascites of the
mice at the time of autopsy. Hepatic metastasis or invasion were
macroscopically found in the livers of five mice in the NOZ-con
group and four in the NOZ-NC group, but only one mouse had liver
metastasis in the NOZ-RIPsi group (Figs.
6E and 7C, and Table II). Five mice in the NOZ-con group,
four mice in the NOZ-NC group and no mice in the NOZ-RIPsi group
had lymph node metastasis (Figs. 6F
and 7A, and Table II). No lung metastasis was detected
in all three groups (Table II).
| Table II.RIP-1 silencing inhibits the
progression of orthotopic xenograft nude mice models. |
Table II.
RIP-1 silencing inhibits the
progression of orthotopic xenograft nude mice models.
| Condition | NOZ-RIPsi
group | NOZ-NC group | NOZ-con group |
|---|
| Tumors
formation | 4/5 (80) | 4/5 (80) | 5/5 (100) |
| Liver
metastasis | 1/5 (20) | 4/5
(80)a | 5/5
(100)a |
| Ascites | 0/5 (0) | 4/5
(80)a | 5/5
(100)a |
| Lymph node
metastasis | 0/5 (0) | 4/5
(80)a | 5/5
(100)a |
| Lung
metastasis | 0/5 (0) | 0/5 (0) | 0/5 (0) |
RIP-1 inhibition downregulates the
expression of VEGF-C in orthotopic xenograft gallbladder cancer in
nude mouse models
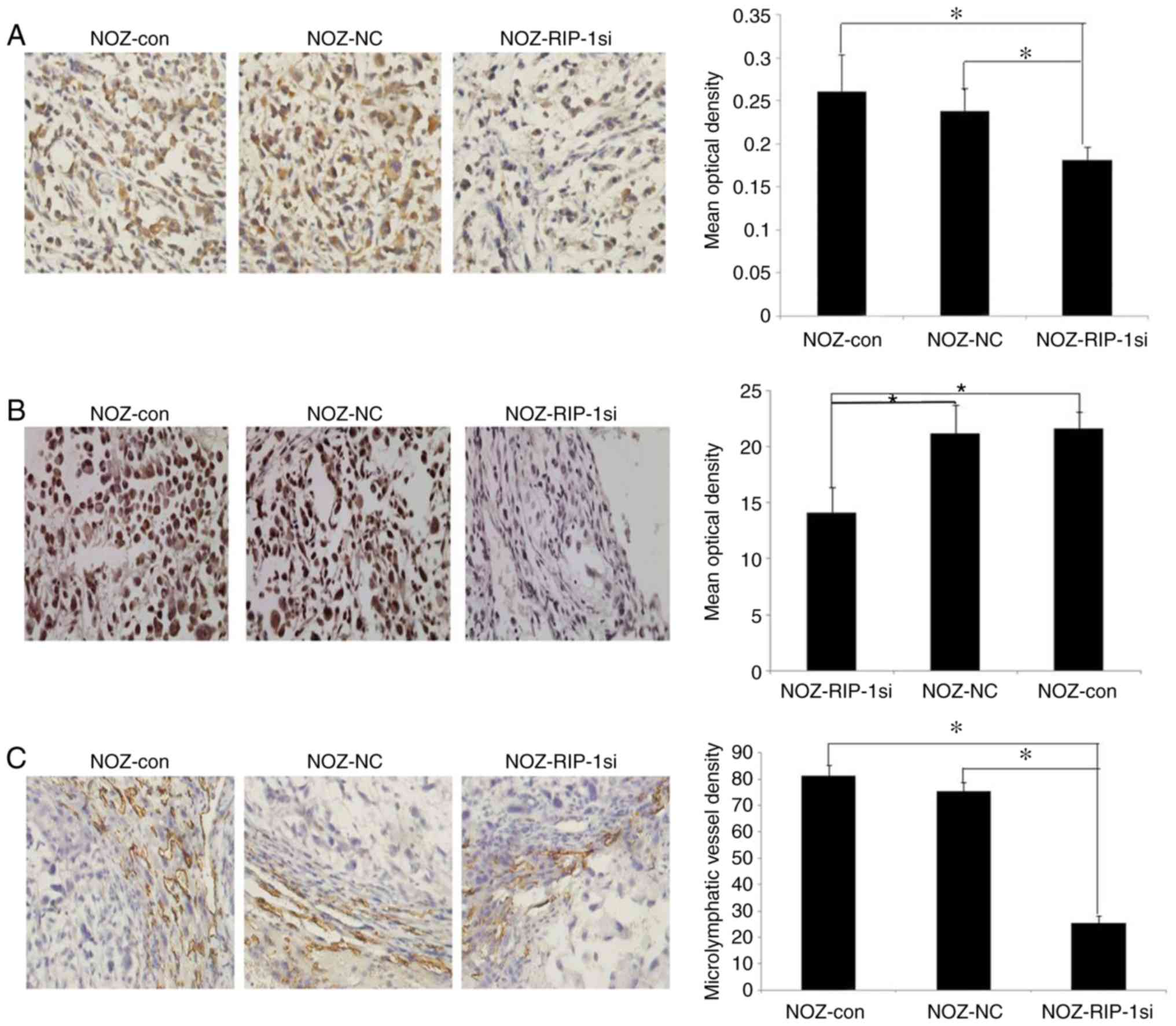
The expression of RIP-1 in all three groups of
orthotopic xenograft gallbladder tumors was analyzed. Compared with
the NOZ-con group (0.261±0.043) and the NOZ-NC group (0.238±0.027),
the expression level of RIP-1 was significantly decreased in the
NOZ-RIPsi group (0.182±0.015; Fig.
8A). Consistent with the expression of RIP-1, the VEGF-C
expression level was significantly lower in the NOZ-RIPsi group
(14.12±2.26) than in the NOZ-con (21.61±1.52) and NOZ-NC groups
(21.18±2.57; Fig. 8B).
RIP-1 inhibition downregulates the
microlymphatic vessel density (MLVD) in the xenograft tumor of
gallbladder cancer
The microlymphatic vessels of xenograft tumors of
gallbladder cancer were marked by LYVE-1 antibodies using the
immunohistochemical method. Of note, some tumor cells could be
found in the middle of brown-stained endothelial cell clusters
(Fig. 7D). The MLVD of orthotopic
xenograft tumors in the NOZ-RIPsi group (25.24±2.87) was
significantly lower than in the NOZ-con (75.52±3.17) and NOZ-NC
groups (81.10±4.13; Fig. 8C).
Discussion
Previous research has shown that RIP-1 was convened
quickly with FADD and TRAF2 when TNF-α binds to tumor necrosis
factor receptor superfamily member 1A (TNFR1), a TNF-α receptor,
and is a key molecule of the TNF-α-TNFR1 signal pathway (25). Some studies have demonstrated that
RIP-1 plays a vital role in this pathway and is essential in
promoting the survival of inflammatory and immune cells (26–28).
In our previous study, the authors confirmed that
RIP-1 was highly expressed in gallbladder cancer tissues, and RIP-1
expression is significantly correlated with the clinical stage and
lymph node metastasis in patients with gallbladder carcinoma
(6). This revealed that RIP-1 could
promote tumor growth and invasion (6). Experiments on the underlying mechanism
of tumor growth and invasion by RIP-1 showed that RIP-1 silencing
could decrease the expression of VEGF-C in NOZ cells (6). These were consistent with previous
studies (6,29,30). Han
et al (29) and Azijli et
al (30) have performed research
on leukemia and lung cancer cell lines, respectively. They found
that RIP-1 could promote the survival, proliferation, and migration
of leukemia and lung cancer.
VEGF-C is part of the VEGF family, which is
correlated with lymph node metastasis in many cancers, such as
gastric cancer (31) and lung cancer
(32). Since the lymphatic
endothelial cell-specific antibodies (LYVE-1 and D2-40) were found,
more studies have suspected that VEGF-C could induce
microlymphangiogenesis and lymph metastasis (24,33). In
addition, VEGF-C plays a very important role in tumor proliferation
and invasion (34,35). Lymph metastasis is the most important
means of gallbladder cancer diffusion (36). Previous research by our group has
reported that VEGF-C promotes growth and invasion in gallbladder
cancer (37).
In mammals, tens of thousands of non protein-coding
and protein-coding genes are regulated in specific manners
(38). These genes are primarily
regulated in combination via the interaction of a specific set of
transcription factors with their cognate cis-regulatory elements
(38). Thus, in the present study,
the authors investigated the specific mechanisms by which RIP-1
regulates VEGF-C expression.
The authors constructed the various fragments of the
VEGF-C promoter for a more precise location, and found that −332 to
−190 nt contained the largest relative luciferase activity within
the VEGF-C promoter. This suggested that this is the core region of
the VEGF-C promoter. Using bioinformatics software, two overlapping
AP-1 sites were found (GGCGCGTCAGT and CGTCAGTCATG at −207 to −192
bp overlapped to form GGCGCGTCAGTCATG) in the area of
−332 to −190 nt, which could regulate the activity of the VEGF-C
promoter. The NF-κB (GGGGAGCTCC) and AP-1 sites were confirmed and
may be crucial for VEGF-C promoter activity. The authors also
demonstrated that the transcription factors NF-κB and AP-1 could
bind to the −332 to −190 nt region of the VEGF-C promoter using
in vitro EMSA and ChIP assays. However, the −1,000 to −487
nt region of the VEGF-C promoter had noticeably increased relative
luciferase activity, which was even higher than the full length of
the VEGF-C promoter. This could potentially be due to some
inhibiting factors binding to the −1,000 to −487 nt region.
The pGL3B-332 plasmid was then chosen to conduct
further research. Different concentrations of pcDNA3.1-RIP-1 were
transfected into NOZ cells, and it was found that the relative
luciferase activity of pGL3B-332 increased with increasing
pcDNA3.1-RIP-1 plasmid concentration. When transfecting pGL3B-332,
pGL3B-NF-κB mut (NF-κB site mutation), pGL3B-AP-1 mut (AP-1 site
mutation), and pGL3B-332 mut (NF-κB and AP-1 sites co-mutation)
into NOZ cells, NF-κB and AP-1 site mutation markedly decreased the
relative luciferase activity of the pGL3B-332 VEGF-C promoter.
These results also showed that NF-κB and AP-1 sites were crucial
for VEGF-C promoter activity. Further, when endogenic NF-κB and
AP-1 were overexpressed or silenced in NOZ cells, the relative
luciferase activity of the pGL3B-332 promoter fragment increased
with the increasing amounts of NF-κB and AP-1 protein expression.
Together, the authors confirmed that RIP-1 could activate the
transcription factors NF-κB and AP-1 to combine with the −332 to
−190 nt region of the VEGF-C promoter, hence enhancing the
promoter's activity.
In a previous study by our group, a subcutaneous
xenograft nude mouse model of gallbladder cancer was built and
silencing the expression of RIP-1 by a siRNA was observed to
inhibit the growth of subcutaneous xenograft tumors (6). However, liver, lung, and lymph node
metastases were not observed, and ascites had not developed
(6). A study demonstrated that
carcinoma in the human gallbladder showed significant invasive
growth, irregular shape and unclear boundaries (15). However, the subcutaneous xenograft
model cannot simulate the microenvironment of gallbladder cancer
growth (15). Therefore, a previous
study by our group demonstrated that orthotopic xenograft models of
gallbladder cancer are better than subcutaneous xenograft
gallbladder cancer models for studying the biological
characteristics of gallbladder cancer (16).
In a previous orthotopic gallbladder cancer nude
mouse models by our group, there were many faults, such as the
anesthesia method and the method for emptying bile (16). In the present study, the anesthesia
method was modified, using a mixed anesthetic instead of
chloraldurate. The gallbladder of a nude mouse is very small and
sometimes finding it can be difficult. In a previous by our group,
the bile was removed using a syringe needle that punctured the
gallbladder. Then the needle re-entered the gallbladder, which was
very difficult. The described procedure could result in
unsuccessful model establishment. Therefore the procedure was
modified in the current study. The needle was used to puncture
through the gallbladder and then withdrawn back in to the
gallbladder, leaving a hole on one side. After the bile was
swabbed, the cells were injected into the empty gallbladder. Thus
the needle was not removed from the gallbladder until the end of
the procedure. The modified method increased the success rate of
the establishment of the orthotopic nude mouse model of gallbladder
cancer.
In the animal model of gallbladder cancer, the
authors observed that interference of RIP-1 by an siRNA suppressed
the progression of the orthotopic gallbladder cancer xenograft
tumor, consistent with a previous study by our group (6). It was also found that RIP-1 knockdown
significantly reduced lymphangiogenesis in the mouse model of
gallbladder cancer. Immunohistochemical analysis found that the
expression level of RIP-1 was notably decreased in the NOZ-RIPsi
group. The RIP-1 and VEGF-C expression level was lower in the
NOZ-RIPsi group, which was consistent with a previous in
vitro study (6). These results
are also consistent with studies on the functions of RIP-1 in other
cancers (7,29,30),
suggesting that RIP-1 played a vital role in promoting the tumor
progression. The mouse model established in the current study
conformed to the natural metastasis of gallbladder cancer in
vivo, in accordance with the theory proposed by Fidler
(39). In the orthotopic
transplantation model, the tumor could display its natural
biological characteristics (16). The
authors of the current study successfully established the
orthotopic mouse model of gallbladder cancer tumors, in addition to
the lymph node metastasis mouse model. The results suggest that
RIP-1 downregulation can inhibit gallbladder cancer growth and
lymphatic metastasis in gallbladder cancer.
In conclusion, the current study researched the
mechanisms of RIP regulating VEGF-C, and further demonstrated that
RIP-1 promotes lymphatic metastasis in gallbladder cancer. The −332
to −190 nt region was the critical area for VEGF-C promoter
activity. Two overlapping AP-1 sites and one NF-κB site in the −332
to −190 nt region were also located, which could mediate the
activity of the VEGF-C promoter. In addition, it was confirmed that
RIP-1 could activate the transcription factors NF-κB and AP-1 to
bind to the −332 to −190 nt area, enhancing the activity of the
VEGF-C promoter. The authors of the current study successfully
established an orthotopic mouse model of gallbladder cancer tumors
by modified methods and established the lymph node metastasis mouse
model. The model revealed that downregulation of RIP-1 can decrease
lymphangiogenesis and lymphatic metastasis markedly, which could be
a potential therapeutic target for gallbladder cancer.
Acknowledgements
Not applicable.
Funding
The current study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81672468
and 81272373) and the National Clinical Key Specialty Construction
Project (General Surgery) of China (grant no. 2012).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC, FS and GZ conceived and designed the study. GZ,
QD and XC performed the experiments. NT and XW interpreted the
results. GZ and XC wrote the paper, but all authors contributed to
writing. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
the Medical Faculty of the Fujian Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no potential
conflict of interest.
References
|
1
|
Bartlett DL, Fong Y, Fortner JG, Brennan
MF and Blumgart LH: Long-term results after resection for
gallbladder cancer. Implications for staging and management. Ann
Surg. 224:639–646. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cubertafond P, Gainant A and Cucchiaro G:
Surgical treatment of 724 carcinomas of the gallbladder. Results of
the French surgical association survey. Ann Surg. 219:275–280.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Randi G, Malvezzi M, Levi F, Ferlay J,
Negri E, Franceschi S and La Vecchia C: Epidemiology of biliary
tract cancers: An update. Ann Oncol. 20:146–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shukla VK, Chauhan VS, Mishra RN and Basu
S: Lifestyle, reproductive factors and risk of gallbladder cancer.
Singapore Med J. 49:912–915. 2008.PubMed/NCBI
|
|
5
|
Miller G and Jarnagin WR: Gallbladder
carcinoma. Eur J Surg Oncol. 34:306–312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu G, Chen X, Wang X, Li X, Du Q, Hong H,
Tang N, She F and Chen Y: Expression of the RIP-1 gene and its role
in growth and invasion of human gallbladder carcinoma. Cell Physiol
Biochem. 34:1152–1165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park S, Hatanpaa KJ, Xie Y, Mickey BE,
Madden CJ, Raisanen JM, Ramnarain DB, Xiao G, Saha D, Boothman DA,
et al: The receptor interacting protein 1 inhibits p53 induction
through NF-kappaB activation and confers a worse prognosis in
glioblastoma. Cancer Res. 69:2809–2816. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hsu H, Huang J, Shu HB, Baichwal V and
Goeddel DV: TNF-dependent recruitment of the protein kinase RIP to
the TNF receptor-1 signaling complex. Immunity. 4:387–396. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kelliher MA, Grimm S, Ishida Y, Kuo F,
Stanger BZ and Leder P: The death domain kinase RIP mediates the
TNF-induced NF-kappaB signal. Immunity. 8:297–303. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hur GM, Lewis J, Yang Q, Lin Y, Nakano H,
Nedospasov S and Liu ZG: The death domain kinase RIP has an
essential role in DNA damage-induced NF-kappa B activation. Genes
Dev. 17:873–882. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Devin A, Lin Y and Liu ZG: The role of the
death-domain kinase RIP in tumour-necrosis-factor-induced
activation of mitogen-activated protein kinases. EMBO Rep.
4:623–627. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu ZG, Hsu H, Goeddel DV and Karin M:
Dissection of TNF receptor 1 effector functions: JNK activation is
not linked to apoptosis while NF-kappaB activation prevents cell
death. Cell. 87:565–576. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pacifico F and Leonardi A: NF-kappaB in
solid tumors. Biochem Pharmacol. 72:1142–1152. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu G, Du Q, Wang X, Tang N, She F and
Chen Y: TNF-α promotes gallbladder cancer cell growth and invasion
through autocrine mechanisms. Int J Mol Med. 33:1431–1440. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Horiuchi H, Kawamata H, Fujimori T and
Kuroda Y: A MEK inhibitor (U0126) prolongs survival in nude mice
bearing human gallbladder cancer cells with K-ras mutation:
Analysis in a novel orthotopic inoculation model. Int J Oncol.
23:957–963. 2003.PubMed/NCBI
|
|
16
|
Du Q, Jiang L, Wang XQ, Pan W, She FF and
Chen YL: Establishment of and comparison between orthotopic
xenograft and subcutaneous xenograft models of gallbladder
carcinoma. Asian Pac J Cancer Prev. 15:3747–3752. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Homma S, Hasumura S, Nagamori S and Kameda
H: Establishment and characterization of a human gall bladder
carcinoma cell line NOZ. Hum Cell. 1:95–97. 1988.PubMed/NCBI
|
|
18
|
Heckman KL and Pease LR: Gene splicing and
mutagenesis by PCR-driven overlap extension. Nat Protoc. 2:924–932.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rettino A, Rafanelli F, Genovese G,
Goracci M, Cifarelli RA, Cittadini A and Sgambato A: Identification
of Sp1 and GC-boxes as transcriptional regulators of mouse Dag1
gene promoter. Am J Physiol Cell Physiol. 297:C1113–C1123. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu YL, Peng XE, Wang D, Chen WN and Lin X:
Human liver fatty acid binding protein (hFABP1) gene is regulated
by liver-enriched transcription factors HNF3β and C/EBPα.
Biochimie. 94:384–392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ceelie H, Spaargaren-Van Riel CC, De Jong
M, Bertina RM and Vos HL: Functional characterization of
transcription factor binding sites for HNF1-alpha, HNF3-beta
(FOXA2), HNF4-alpha, Sp1 and Sp3 in the human prothrombin gene
enhancer. J Thromb Haemost. 1:1688–1698. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weidner N, Semple JP, Welch WR and Folkman
J: Tumor angiogenesis and metastasis-correlation in invasive breast
carcinoma. N Engl J Med. 324:1–8. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vermeulen PB, Gasparini G, Fox SB, Toi M,
Martin L, McCulloch P, Pezzella F, Viale G, Weidner N, Harris AL
and Dirix LY: Quantification of angiogenesis in solid human
tumours: An international consensus on the methodology and criteria
of evaluation. Eur J Cancer. 32A:2474–2484. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Du Q, Jiang L, Wang X, Wang M, She F and
Chen Y: Tumor necrosis factor-α promotes the lymphangiogenesis of
gallbladder carcinoma through nuclear factor-κB-mediated
upregulation of vascular endothelial growth factor-C. Cancer Sci.
105:1261–1271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
O'Donnell MA and Ting AT: RIP1 comes back
to life as a cell death regulator in TNFR1 signaling. FEBS J.
278:877–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hitomi J, Christofferson DE, Ng A, Yao J,
Degterev A, Xavier RJ and Yuan J: Identification of a molecular
signaling network that regulates a cellular necrotic cell death
pathway. Cell. 135:1311–1323. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ting AT, Pimentel-Muiños FX and Seed B:
RIP mediates tumor necrosis factor receptor 1 activation of
NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J.
15:6189–6196. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Devin A, Lin Y, Yamaoka S, Li Z, Karin M
and Liu Z: The alpha and beta subunits of IkappaB kinase (IKK)
mediate TRAF2-dependent IKK recruitment to tumor necrosis factor
(TNF) receptor 1 in response to TNF. Mol Cell Biol. 21:3986–3994.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Han W, Xie J, Fang Y, Wang Z and Pan H:
Nec-1 enhances shikonin-induced apoptosis in leukemia cells by
inhibition of RIP-1 and ERK1/2. Int J Mol Sci. 13:7212–7225. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Azijli K, Yuvaraj S, Peppelenbosch MP,
Würdinger T, Dekker H, Joore J, van Dijk E, Quax WJ, Peters GJ, de
Jong S and Kruyt FA: Kinome profiling of non-canonical TRAIL
signaling reveals RIP1-Src-STAT3-dependent invasion in resistant
non-small cell lung cancer cells. J Cell Sci. 125:4651–4661. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jüttner S, Wissmann C, Jöns T, Vieth M,
Hertel J, Gretschel S, Schlag PM, Kemmner W and Höcker M: Vascular
endothelial growth factor-D and its receptor VEGFR-3: Two novel
independent prognostic markers in gastric adenocarcinoma. J Clin
Oncol. 24:228–240. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Arinaga M, Noguchi T, Takeno S, Chujo M,
Miura T and Uchida Y: Clinical significance of vascular endothelial
growth factor C and vascular endothelial growth factor receptor 3
in patients with nonsmall cell lung carcinoma. Cancer. 97:457–464.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Achen MG, McColl BK and Stacker SA: Focus
on lymphangiogenesis in tumor metastasis. Cancer Cell. 7:121–127.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Timoshenko AV, Rastogi S and Lala PK:
Migration-promoting role of VEGF-C and VEGF-C binding receptors in
human breast cancer cells. Br J Cancer. 97:1090–1098. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Masood R, Kundra A, Zhu S, Xia G, Scalia
P, Smith DL and Gill PS: Malignant mesothelioma growth inhibition
by agents that target the VEGF and VEGF-C autocrine loops. Int J
Cancer. 104:603–610. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shimada H, Endo I, Togo S, Nakano A, Izumi
T and Nakagawara G: The role of lymph node dissection in the
treatment of gallbladder carcinoma. Cancer. 79:892–899. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen Y, Jiang L, She F, Tang N, Wang X, Li
X, Han S and Zhu J: Vascular endothelial growth factor-C promotes
the growth and invasion of gallbladder cancer via an autocrine
mechanism. Mol Cell Biochem. 345:77–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ernst P and Smale ST: Combinatorial
regulation of transcription. I: General aspects of transcriptional
control. Immunity. 2:311–319. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fidler IJ: Critical factors in the biology
of human cancer metastasis: Twenty-eighth G.H.A. Clowes memorial
award lecture. Cancer Res. 50:6130–6138. 1990.PubMed/NCBI
|