Introduction
Glucocorticoids (GCs) are essential circadian
steroid hormones that regulate perinatal development, memory,
immune system function, and metabolism, as well as process
emotional input (1) GCs are
frequently used to treat rheumatoid arthritis due to their potent
anti-inflammatory effect. The potent immunosuppressive effects of
GCs are mediated by a series of transcriptional events, primarily
by binding to the cytosolic glucocorticoid receptor (GR).
Translocated GR can directly bind to a canonical GC response
element or act indirectly by interaction with other transcription
factors (2). The generally accepted
view of anti-inflammatory actions on macrophages is through the
suppression of NF-κB activity, thus inhibiting the transcription of
proinflammatory genes (3,4). Accumulating evidence has demonstrated
that other than the ability to suppress macrophage production of
proinflammatory mediators, GCs can directly induce specific changes
in cell survival, proliferation, and phagocytosis to suppress
inflammation (5). Direct GC action
on macrophages was suggested to suppress immunocompetence and
promote antitumor gene transcription via GR activity. Synthetic
glucocorticoids, such as dexamethasone (Dex), display potent
inflammatory suppressive properties and are used to treat various
inflammatory and autoimmune conditions (2,6).
Macrophages are resident phagocytic cells found in
lymphoid and non-lymphoid tissues. Macrophages are critical
effectors involved in steady-state tissue homeostasis, via the
clearance of apoptotic cells, production of growth factors, and
regulation of inflammation and the innate immune response.
Macrophages are the first line of defense against microorganisms,
which is accomplished by phagocytosis and the production of
inflammatory cytokines. In addition, inflammatory chemoattractants,
induced by distant primary tumors, influence the attraction of
macrophages in secondary sites before metastasis. The presence of
macrophages within tumors indicates a poor prognosis, as they
enhance angiogenesis and metastases. However, the migration of
activated macrophages has not yet been elucidated.
Krüppel-like factor 9 (KLF9), also called
basic transcription element-binding protein-1 (Bteb1), is a
ubiquitously expressed member of the C2H2-type zinc finger family
(7). A recent study suggested that
KLF9 plays an important role in regulating animal development and
differentiation of various cell types (8). KLF9 can be induced by several
physiological or pathological stresses. Current experimental
evidence indicates that KLF9 plays a key hormone-dependent role in
liver gluconeogenesis (9). Notably,
a recent study indicated that KLF9 is induced by NF-E2-like basic
leucine zipper transcriptional activator (Nrf2), thereby promoting
cell oxidative stress (7).
Cyclooxygenase-2 (COX-2) levels are increased
in tumors by several proinflammatory cytokines, which are secreted
by macrophages, lymphocytes, and even epithelial cells (10,11).
Increased COX-2 levels have been reported in many malignant
gastrointestinal tumors, including cholangiocarcinoma. Inhibition
of COX-2 may suppress the development of epithelial cell
malignancies in the gastrointestinal tract (12–14).
In addition, COX-2 has been revealed to modulate the fate of colon
cancer cell lines by generating prostanoids (15–17).
These observations indicate that COX-2 expression and
prostanoid generation may play a fundamental role in the initiation
and promotion of cancers arising from inflamed tissues, such as
cholangiocarcinoma (18). However,
it is unknown whether COX-2 can modulate apoptosis of immune cells,
such as macrophages.
In the present study, the inhibitory effects and
underlying molecular mechanisms of GCs on the inflammatory response
were further investigated in lipopolysaccharide (LPS)-stimulated
macrophages. Results indicated that KLF9 significantly suppressed
LPS-induced intracellular reactive oxygen species (ROS) production
and COX-2, while promoting the production of inflammatory factors
in macrophages.
Materials and methods
Cell culture
RAW 264.7, murine macrophage cell line (TIB-71) and
HepG2, liver cancer cell line (HB-8065) were purchased from the
ATCC and were cultured in DMEM with 10% FBS purchased from Gibco;
Thermo Fisher Scientific, Inc. and 1% penicillin-streptomycin in a
humidified 5% CO2 incubator at 37°C. During maintenance,
the cells were sub-cultured every 3 or 4 days.
Real-time quantitative PCR
Total RNA was extracted from RAW 254.7 cells using
the TRIzol-based method (product no. 10296010; Invitrogen; Thermo
Fisher Scientific, Inc.) and reverse-transcribed using Super-Script
III reverse transcriptase (product no. 11752250; Invitrogen; Thermo
Fisher Scientific, Inc.). The realtime quantitative-PCR was
performed using the SYBR Green PCR Master mix (product no. A6001;
Promega Corp.). The real-time quantitative-PCR conditions were 95°C
for 30 sec, followed by 30 cycles at 95°C for 30 sec, 57°C for 1
min, and 72°C for 30 sec. All quantitative-PCR data were normalized
to the level GAPDH. Specific primers used are listed in Table I.
 | Table I.The sequences of the primers used for
RT-qPCR. |
Table I.
The sequences of the primers used for
RT-qPCR.
| Primer name | Sequence
(5′-3′) |
|---|
| KLF9 | F:
5′-CGAGCGGCTGCGACTACCTG-3′ |
|
| R:
5′-GGGCTGTGGGAAGGACTCGAC-3′ |
| COX-2 | F:
5′-CATCCCCTTCCTGCGAAGTT-3′ |
|
| R:
5′-GGCCCTGGTGTAGTAGGAGA-3′ |
| IL-6 | F:
5′-CCACGGCCTTCCCTACTTC-3′ |
|
| R:
5′-TTGGGAGTGGTATCCTCTGTGA-3′ |
| IL-1β | F:
5′-AGTTGACGGACCCCAAAAGAT-3′ |
|
| R:
5′-GGACAGCCCAGGTCAAAGG-3′ |
| TNF-α | F:
5′-CACCGTCAGCCGATTTGC-3′ |
|
| R:
5′-TTGACGGCAGAGAGGAGGTT-3′ |
| GAPDH | F:
5′-CAAGGCTGTGGGCAAGGT-3′ |
|
| R:
5′-GGAAGGCCATGCCAGTGA-3′ |
Chromatin immunoprecipitation (ChIP)
assay
The RAW 264.7 cells (2×107) were fixed
with 1% formaldehyde at room temperature for 30 min, then chromatin
was extracted and the cells were sonicated to shear the chromatin
and immunoprecipitated with 2–5 µg antibodies specific for GR
(product no. SAB4501309; Sigma-Aldrich; Merck KGaA), KLF9 (product
no. ab227920; Abcam) or non-specific IgG (product no. sc-2027;
Santa Cruz Biotechnology, Inc.). Then, the immunoprecipitants were
isolated using protein G agarose beads (product no. 15920010;
Invitrogen; Thermo Fisher Scientific, Inc.), followed by extensive
washing and elution with 2% SDS in 0.5 M NaHCO3. After reversing
the cross-links, the input DNA and immunoprecipitated DNA fragments
were quantified by qPCR in both the input and precipitated samples.
ChIP primer sequences are listed in Table II.
| Table II.The sequences of the primers used for
ChIP-qPCR. |
Table II.
The sequences of the primers used for
ChIP-qPCR.
| Primer name | Sequence
(5′-3′) |
|---|
| KLF9 ChIP | F:
5′-AGAGGCGCGGCGCGGCAGG-3′ |
|
| R:
5′-GTCGGAGTCCCAGAGAAAC-3′ |
| COX-2 ChIP | F:
5′-CCACTACGTCACGTGGAGT-3′ |
|
| R:
5′-GCTGAGTTCCTTCGTGAGCA-3′ |
Western blot analysis
Whole-cell extracts were extracted from cultured RAW
264.7 cells in lysis buffer (20 mM Tris-Cl pH 7.5, 140 mM NaCl, 1
mM CaCl2 and MgCl2, 10 mM NaF, 1% NP-40, 10%
glycerol, 2 mM Na-Vanadate, and 1 mM PMSF) supplemented with
protease inhibitor cocktail (Roche Diagnostics). Homogenates were
sonicated and centrifuged at 4°C for 15 min, and the supernatants
were used for western blotting. Protein concentrations of the
samples in the experiments were determined by BCA (Bio-Rad protein
assay kit). Proteins (20–50 µg) were subjected to 10%
SDS-polyacrylamide gels. Proteins were separated by SDS-PAGE and
transferred to a PVDF membrane (EMD Millipore). After being blocked
with 5% non-fat milk, the membranes were incubated with the
antibodies overnight at 4°C. Immunoblotting was performed using the
following primary antibodies: KLF9 (1:1,000; product no. A7196;
ABclonal), COX-2 (1:1,000; product no. A1253; ABclonal), β-tubulin
(1:1,000; product no. AC010; ABclonal), caspase-9 (1:500; product
no. ab52298; Abcam), cleaved caspase-9 (1:500; product no. ab2324,
Abcam), caspase-3 (1:500; product no. ab13847; Abcam), cleaved
caspase-3 (1:500; product no. ab2302, Abcam), cytochrome c
(Cyt-c) (1:500; product no. A0225; ABclonal). Then the PVDF
membranes were incubated with the secondary antibodies for 1 h at
room temperature. HRP-conjugated secondary antibody (1:5,000;
product no. sc-2357 and sc-2005; Santa Cruz Biotechnology, Inc.)
was used according to the manufacturer. ECL reagent was used for
visualization (Genestar). Western blots were quantified by
densitometry using ImageJ (1.52q for Windows; National Institutes
of Health).
ELISA assays
RAW 264.7 cells (1×106 cells/well) were
transfected with Lenti-KLF9 for 48 h in 6-well plates, then
LPS (1 µg/ml) was added to the cultured wells for another 24 h. The
cultured supernatant was collected and the levels of PGE2, IL-1β,
IL-6, and TNF-α were assessed using ELISA assays. ELISA kits were
purchased from R&D Systems for IL-1β, IL-6 and TNF-α (product
no. A54609) and PGE2 (product no. A50432) was purchased from
EpiGentek.
Transient transfection and luciferase
reporter assays
RAW264.7 cells were transiently co-transfected with
KLF9-Luc or COX-2-Luc and the respective expressed plasmid
(GR or KLF9) using Lipofectamine 2000 (product no.
11668019; Invitrogen; Thermo Fisher Scientific, Inc.). After 48 h,
the cells were treated with Dex or LPS for another 3 h. Then,
reporter gene assays were performed using a luciferase assay system
(product no. E2810; Promega Corp.). The internal reference was
determined by measuring the TK (mammalian vector for weak
constitutive expression of humanized Renilla luciferase)
bioluminescence intensity.
TUNEL assay
A TUNEL assay was performed to detect the apoptotic
RAW 264.7 cells. The TUNEL assay was performed using the In
Situ Cell Death Detection kit (Roche Diagnostics). The images
for assessing the apoptotic cells were obtained by fluorescence
microscope (ZEISS AG). TUNEL-positive cells in different groups
were calculated by randomly selecting six different fields and at
least six data of each group were used for analysis. ImageJ was
used to count the percentage of apoptotic cells subjected for
statistical analysis. The nucleus was counterstained with DAPI
fluorescence dye (product no. c1005; Beyotime Institute of
Biotechnology) for total cell count.
Cell co-culture system
Transwell plates (product no. 3378; Corning, Inc.)
were used to construct a co-culture system. In brief, serum-free
single cell suspensions (3×104 cells/ml) were prepared.
The upper Transwell chamber was filled with 100 µl HepG2 cells,
while RAW 264.7 cells with RPMI-1640 medium (20% FBS) was added
into the lower chamber. Cells were cultured in a humidified 5%
CO2 incubator at 37°C. During maintenance, the cell
proliferation assay was performed once a day for 5 consecutive
days.
Cell proliferation assay
A Cell Counting Kit-8 (product no. ab228554; Abcam)
was used to assess cell proliferation. Cells were cultured at 37°C
in a 5 % CO2 incubator. Subsequently, 10 µl CCK-8
reagent was added into each well and the plate was incubated for
1–4 h. OD values at 450 nm were assessed for cell
proliferation.
Statistical analyses
The quantitative data are represented as the mean ±
SD of at least three independent experiments. Two-tailed Student's
t-test was used to compare the differences between 2 groups.
One-way ANOVA with Bonferroni correction was used for multiple
comparisons. Statistical significance was defined as P<0.05
(*P<0.05; **P<0.01; ***P<0.005; ****P<0.001, as
indicated in the figures and legends). Analysis was performed using
GraphPad Prism (GraphPad Software, Inc.).
Results
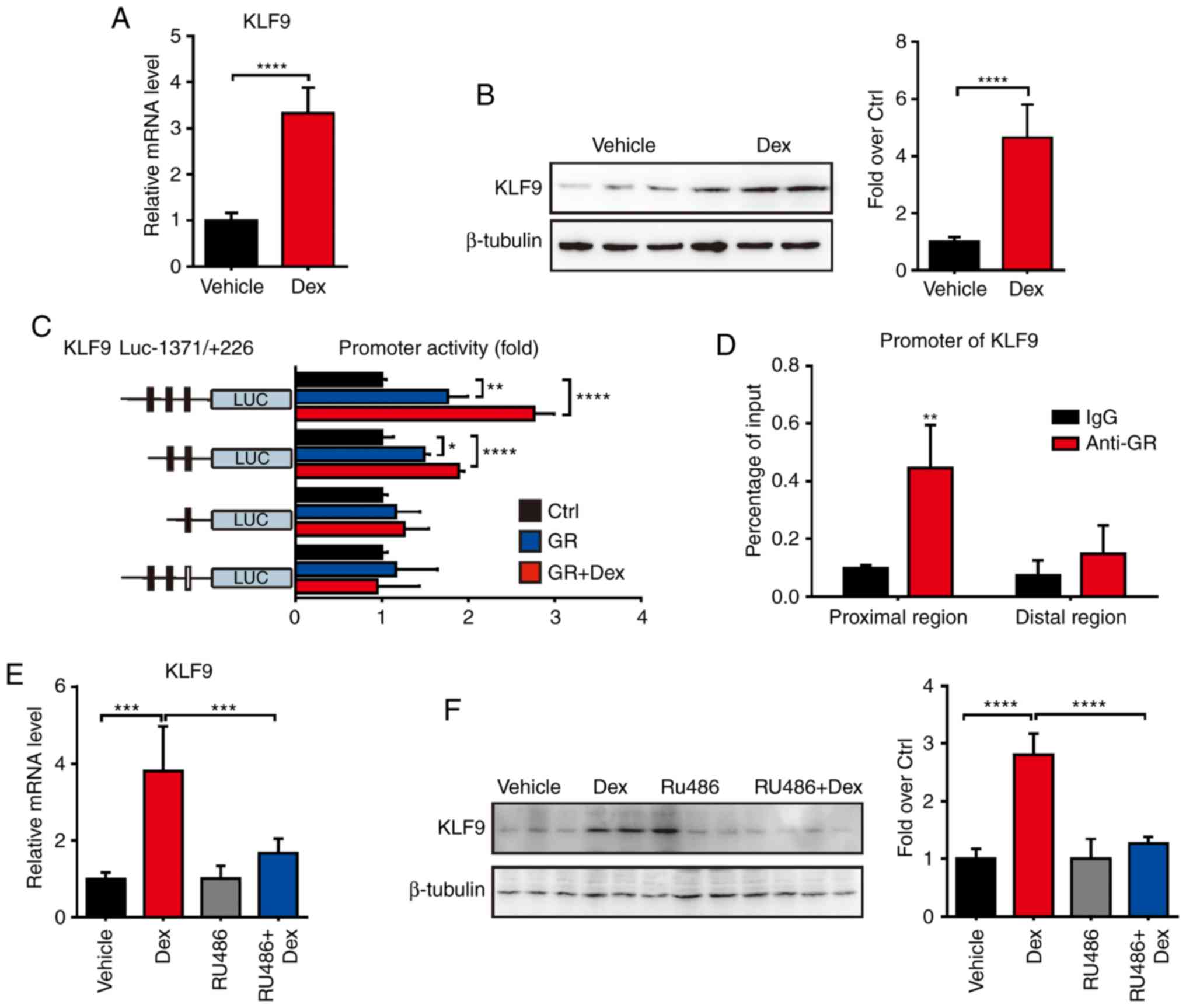
Dex induces KLF9 expression in
macrophage cells through GR
The molecular mechanism of GCs which promote
macrophages apoptosis and tumor growth remains largely unclear. In
order to investigate the regulated effect of GCs, the expression
level of KLF9, which plays a key role in
mitochondria-dependent cell death, was first examined in
macrophages after Dex treatment. The results revealed that Dex
significantly induced the expression of KLF9 in RAW 264.7
cells (Fig. 1A and B). As is
recognized, the transcriptional regulation of the function of GCs
is largely due to GR, an important hormone-sensitive transcription
factor of the nuclear receptor superfamily. To further investigate
the regulated function of Dex, promoter activity analysis of
KLF9 was performed. The −1371 to +226 region of KLF9
promoter which was fused to the luciferase reporter gene system
(KLF9-Luc) was constructed. Then, GR expression
plasmid or control plasmid was co-transfected with KLF9-Luc
into RAW 264.7 cells. The results revealed that GR caused
significant activation of the promoter with or without Dex
(Fig. 1C). Subsequently, a
potential GR response element site in the KLF9 promoter that
mediated the stimulatory effect of Dex/GR was revealed using the
promoter deletion and mutation assays. ChIP assays confirmed that
the GR was recruited to the KLF9 promoter region and
mediated gene expression after Dex treatment (Fig. 1D). In another experiment, it was
revealed that RU486, a GR antagonist, almost completely abolished
the Dex/GR-mediated increased KLF9 level in RAW 264.7 cells
(Fig. 1E and F). These results
indicated that KLF9 was a GC- regulated gene in macrophage
cells, and it was directly regulated by GR.
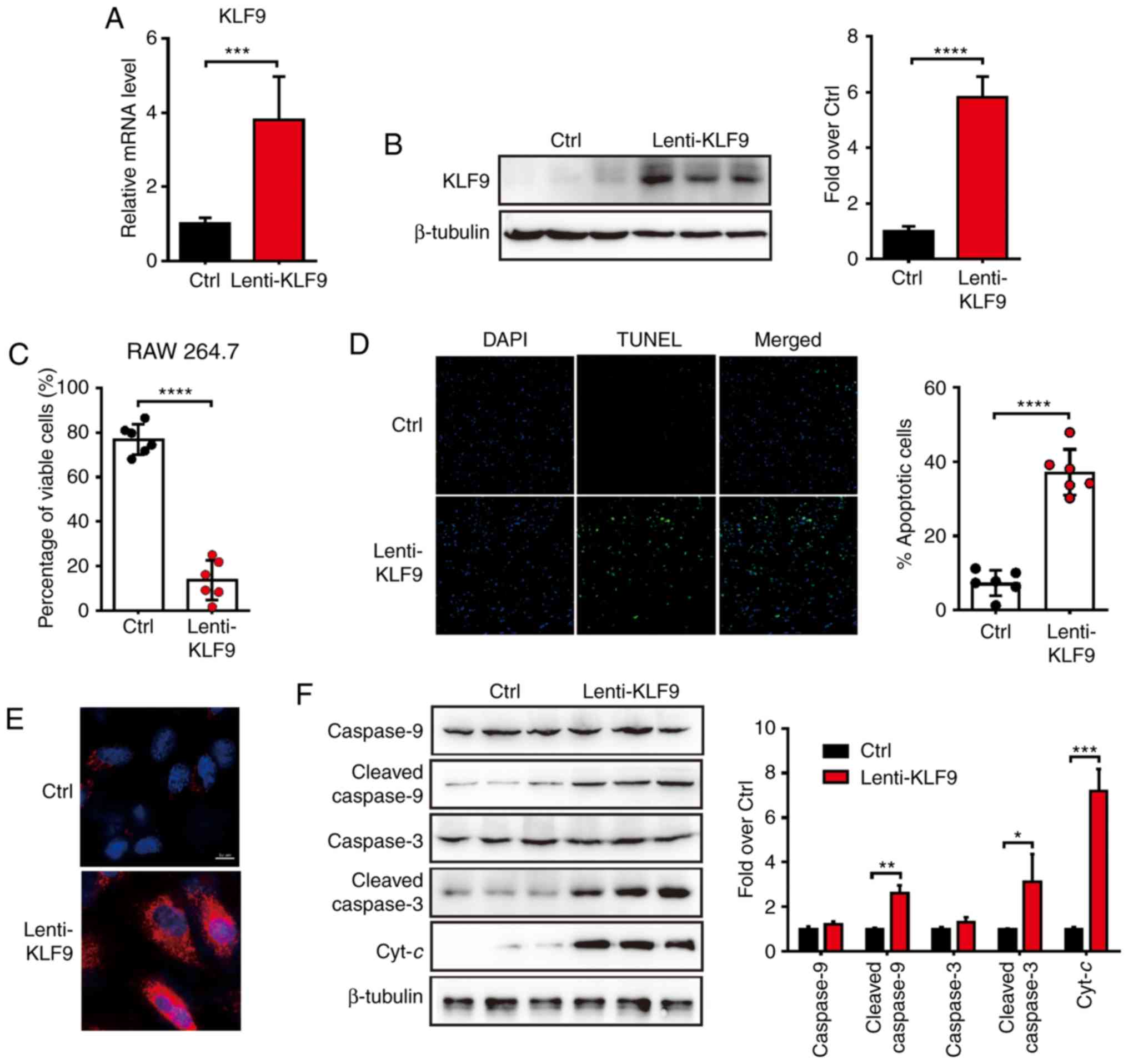
Overexpression of KLF9 induces
apoptosis in macrophage cells
GCs were previously demonstrated to activate the
intrinsic apoptotic pathways in a tumor model (4). in the present study, it was proposed
that KLF9, which was induced by Dex, would activate apoptosis in
macrophage cells. To investigate whether overexpression of
KLF9 would induce the apoptosis of macrophages, the
KLF9 lentivirus-expressing system (Lenti-KLF9) was
generated and infected RAW 264.7 cells. The mRNA and protein level
of KLF9 were dramatically elevated after Lenti-KLF9
transfected (Fig. 2A and B). The
proportion of viable and apoptotic cells were analyzed by Trypan
blue exclusion experiment and TUNEL analysis 48 h after
Lenti-KLF9 transfection. The proportion of viable cells was
significantly decreased in the KLF9 overexpression group
compared to the control group (Fig.
2C). Consistent with the decreased number of viable cells, the
number of apoptotic cells was increased (Fig. 2D). It was therefore determined
whether the decrease in cell viability could be attributed to the
induction of apoptosis. Subsequently, the analysis of mitochondrial
generated ROS was performed by a MitoSOX staining experiment to
determine whether the mitochondrial-mediated apoptosis pathway was
involved. The results revealed that cells in the KLF9 group
exhibited significantly more positive ROS generation than the
control group (Fig. 2E). To further
examine the mitochondrial effect induced by KLF9, the proteins of
the mitochondrial apoptosis pathway in macrophage cells were next
assessed. Western blot analysis revealed that the apoptosis-related
protein levels of procaspase-9, cleaved caspase-9, procaspase-3,
cleaved caspase-3 and Cyt-c of the KLF9 group were increased
compared to the control group (Fig.
2F). Collectively, it was concluded that KLF9 may play an
important role in the macrophage cells apoptosis pathway by
promoting mitochondrial ROS generation and Cyt-c
release.
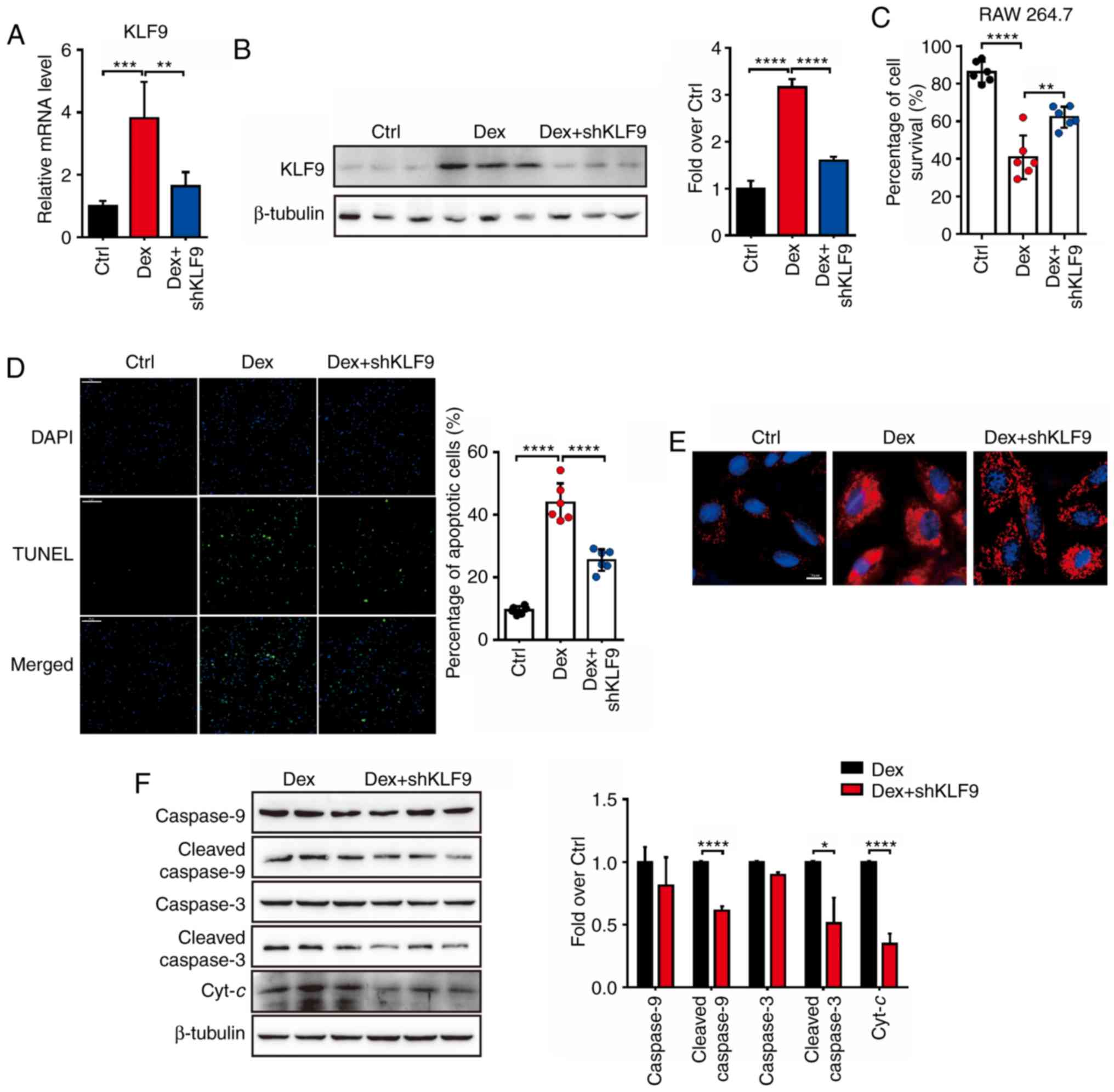
KLF9 knockdown protects against
dexamethasone-induced apoptosis in macrophage cells
It was previously revealed that KLF9 mediated the
signaling pathway of Dex-induced mitochondrial injury and
macrophage cells apoptosis. Therefore, it was hypothesized whether
decreased KLF9 expression level would inactivate the
intrinsic apoptotic pathways induced by Dex. The shKLF9
lentivirus-knockdown system (Lenti-shKLF9) was generated and
it was revealed that the mRNA and protein level of KLF9 was
suppressed by Lenti-shKLF9 with Dex treatment (Fig. 3A and B). Initially, as assessed by
the Trypan blue, it was observed that RAW 264.7 cells which were
treated with Dex and transfected with Lenti-shKLF9 exhibited
a significant increased survival rate compared to Dex treatment
alone (Fig. 3C). In addition, TUNEL
assay revealed that the number of apoptotic cells in the Dex
treatment group was higher than in the Dex+shKLF9 treatment
group, which was exposed to Dex and transfected with Lenti-shKLF9
for 72 h (Fig. 3D). These results
indicated that suppressed KLF9 level could markedly inhibit the
apoptosis of RAW 264.7 cells induced by Dex. The analysis of
MitoSOX-positive staining revealed that RAW 264.7 cells in the Dex
+ shKLF9 group exhibited significant less ROS generation
than the Dex group (Fig. 3E). The
apoptosis-related protein levels were next investigated and it was
revealed that procaspase-9, cleaved caspase-9, procaspase-3,
cleaved caspase-3 and Cyt-c protein levels of the Dex +
shKLF9 group were decreased compared to the Dex group
(Fig. 3F). Collectively, it was
concluded that Dex could influence the viability of macrophage
cells by the KLF9-mediated mitochondrial apoptosis
pathway.
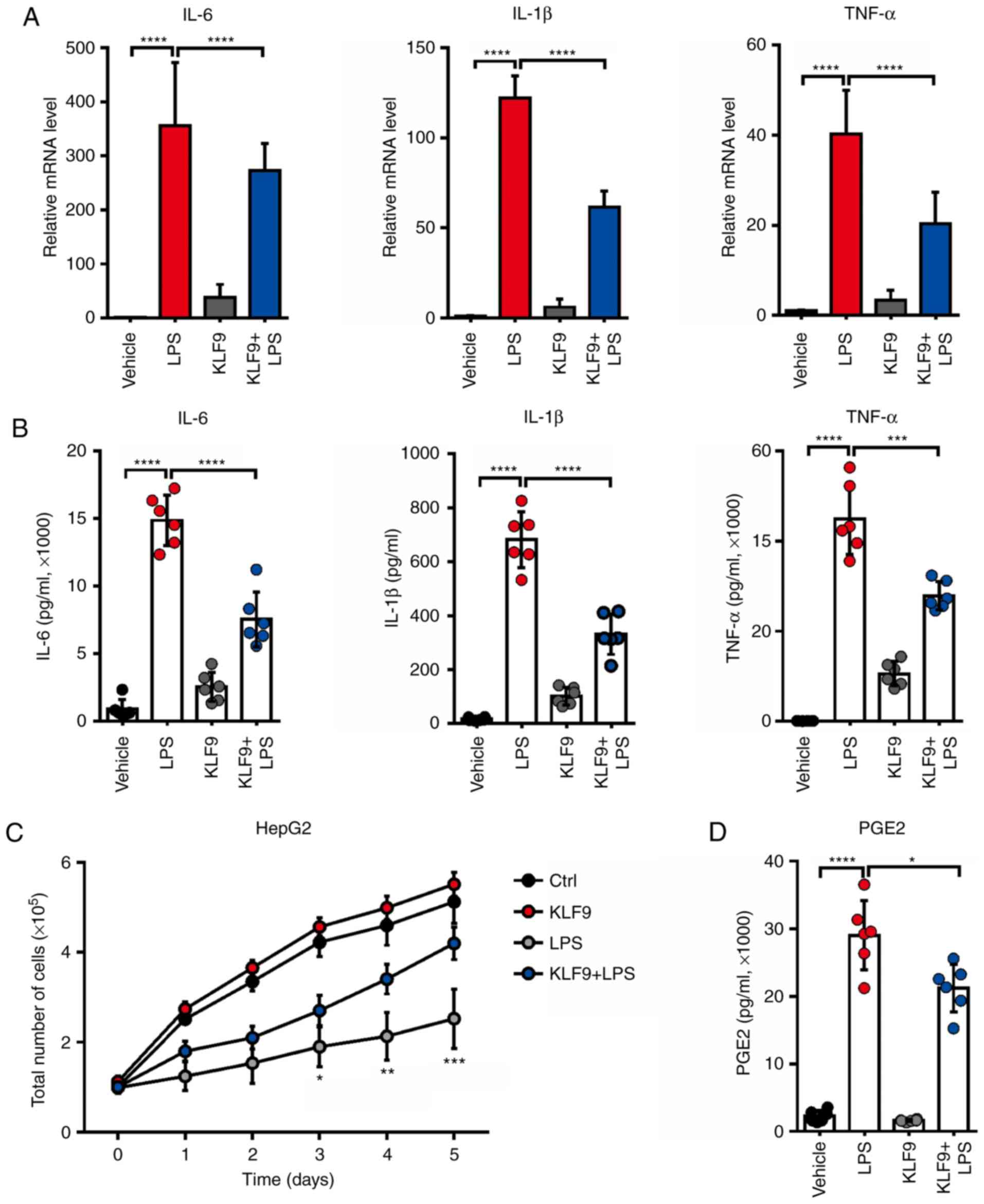
KLF9 overexpression reduces the
LPS-induced inflammatory cytokine release in RAW 264.7 cells
It was previously determined that KLF9 mediated
macrophage apoptosis which was induced by Dex. Furthermore, it was
then determined whether KLF9 influenced the function of
macrophages. It is recognized that LPS can stimulate inflammatory
cytokine production. Thus, the expressed and secreted level of
inflammatory cytokines including IL-1β, IL-6, and TNF-α, which are
potent antitumor factors released from macrophage cells, were
assessed. The results revealed that LPS significantly increased the
mRNA levels of these inflammatory cytokines, and KLF9 could
decrease such variations (Fig. 4A).
In addition, KLF9 also reduced LPS-induced secretion of these
inflammatory cytokines (Fig. 4B).
These results indicated that KLF9 could destroy the function of
macrophages by reducing the inflammatory cytokine production and
secretion. Subsequently, it was assessed whether KLF9
overexpression in macrophages had an effect in HepG2 liver cancer
cell proliferation using an in vitro co-culture system. The
results demonstrated that the supernatant of macrophages stimulated
with LPS significantly suppressed HepG2 liver cancer cell
proliferation, however, this effect was alleviated following
transfection with KLF9 (Fig.
4C). Then, it was assessed whether KLF9 could regulate the
level of PEG2, which plays a key role in mediating the
microenvironment of liver cancer. Notably, it was revealed that
KLF9 significantly suppressed LPS-induced PGE2 production in
macrophage cells compared to the LPS group and there was a slight
difference between the control and KLF9 group without LPS treatment
(Fig. 4D).
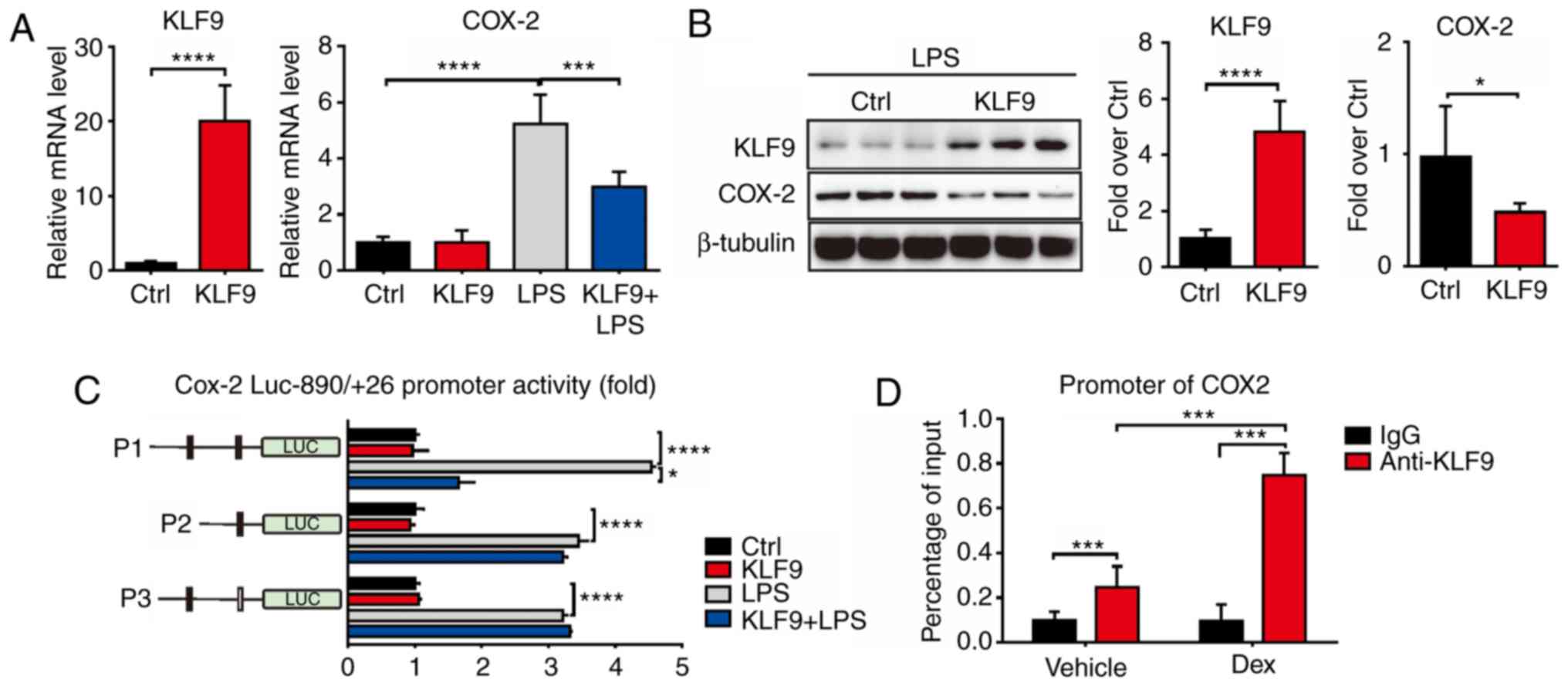
KLF9 suppresses COX-2 levels
The response to LPS of KLF9-suppressed RAW 264.7
cells largely resided in the domain of transcriptional regulation.
Thus, it was determined whether KLF9 directly regulated the
expression of COX-2. The mRNA and protein levels of
COX-2 were decreased by KLF9 compared to the control group
as determined using the Real-time PCR and western blot assays in
RAW 264.7 cells (Fig. 5A and B). To
further investigate the regulated mechanism, promoter activity
experiments were conducted to examine whether KLF9 directly bound
to the DNA element of COX-2. The promoter region of
COX-2 was fused to a luciferase reporter gene and was
co-transfected with KLF9 expression plasmid into RAW264.7
cells. Notably, KLF9 significantly inhibited COX-2 promoter
activity in RAW264.7 cells under LPS-inducing conditions. To
determine the region responsible for KLF9 suppression, a series of
deletions of COX-2 promoter was constructed. As revealed,
KLF9 significantly suppressed the P1 promoter activity, whereas the
suppression was almost completely abolished with the transfection
of P2 and mutant promoters under LPS-inducing conditions (Fig. 5C). ChIP assays using RAW264.7 cell
extracts indicated that KLF9 proteins were recruited to the
COX-2 promoter, and this regulation was enhanced during Dex
treatment (Fig. 5D). These data
revealed the functional involvement of KLF9 in the regulation of
COX-2 gene transcription.
Discussion
GCs have been used for the early treatment of
rheumatoid arthritis, and have since become the most common therapy
for inflammatory disorders (4). GCs
remain very effective anti-inflammatory and immunosuppressive
agents used in the treatment of numerous autoimmune diseases
(1). However, recent research has
suggested that chronic use of GCs is associated with tumorigenesis.
Although various modes of action are still debated, it is broadly
accepted that GCs mediate their anti-inflammatory and
immunosuppressive effects by affecting macrophage viability and
antitumor functionality (4).
Several studies have demonstrated that tumor-derived KLF9 may act
as a tumor suppressor by promoting apoptosis (8,19,20).
Decreased KLF9 suppresses oxidative stress and apoptosis,
subsequently promoting cancer progression (7). In fact, KLF9 overexpression in
tumor cells can induce apoptosis via excessive oxidative stress
production. However, whether macrophage-derived KLF9 exerts the
same effect is yet to be elucidated. Previous research has reported
that Dex promotes macrophage apoptosis and decreases Cyt-c,
caspase-3, and caspase-9 expression (21). Research has demonstrated that Dex
promoted macrophage apoptosis partly by affecting the mitochondrial
apoptosis pathway. The present findings indicated that Dex induced
GR recruitment to the KLF9 promoter, consequently increasing
the levels of KLF9, and increasing mitochondrial ROS production,
leading to mitochondrial-dependent apoptosis of macrophages.
Therefore, a molecular mechanism indicating KLF9 as an
immunosuppressor and an important regulator of GCs-induced
macrophage apoptosis is proposed. In the present study, it was
demonstrated that decreased KLF9 levels could alleviate Dex-induced
macrophage apoptosis. Altered levels of caspase-3, caspase-9, and
Cyt-c following overexpression of KLF9 in accordance
with exposure to Dex were also revealed. In addition, it has been
reported that KLF9 causes intracellular ROS accumulation by
suppressing transcription of the thioredoxin reductase 2 gene,
which plays a pivotal role in defense against oxidative damage
(7,19). In the present study, it was revealed
that KLF9 could promote the accumulation of intracellular ROS, thus
inducing macrophage apoptosis. The present data indicated a
hormone-responsive model of KLF9-dependent apoptosis regulation
through the mitochondrial pathway.
The increased understanding regarding GCs regulation
of tumor progression has drawn attention to the therapeutic
potential of modulating apoptosis and the antitumor functions of
macrophages. Suppression of inflammatory factors benefits tumor
growth via KLF9 overexpression in macrophages. The
contribution of tumor-derived COX-2 to tumorigenesis has been
examined in numerous studies (14,22–24).
COX-2 is associated with a poor prognosis across a range of human
cancers (13,25). In the present study, it was revealed
that KLF9 suppressed macrophage-derived COX-2 levels. COX-2 and
numerous inflammatory factors are induced during stimulation of
macrophages by LPS (24). This
suggests that elevated levels of macrophage-derived COX-2 is
strongly related to antitumor effects (14). While this implies that the
microenvironment may change, which would support tumor growth due
to prostaglandin production by COX-2 enzymatic activity, other
mechanisms are possible. Decreased levels of COX-2 are relative to
the abnormal immune function of macrophages (17,18).
In the present study, it was revealed that KLF9, a direct target of
GCs, could reduce LPS-induced COX-2 levels, indicating that KLF9 is
a potent inhibitor of immune function in macrophages.
Constitutive downregulation of KLF9 has been
revealed in many types of cancer. However, it was revealed that
KLF9 induction by GCs in macrophages, originally administered for
anti-inflammatory purposes, conferred the risk of tumorigenesis.
Furthermore, the molecular mechanism of the aforementioned effect
revealed that KLF9 is an important responder to small molecule
hormones and modulates oxidative injury and cell death. GR acts as
an upstream regulator to stimulate expression of KLF9. It
was also revealed that KLF9 directly bound to the COX-2 promoter,
and moderately suppressed the COX-2 transcriptional activity in a
luciferase reporter assay, indicating potential contribution to
COX-2 inactivation.
Collectively, these observations indicate that KLF9
mediates macrophage apoptosis by directly activating the
mitochondrial pathway. Most studies regarding KLF9 in cancer have
focused on global inhibition of KLF9, especially in the tumor cells
themselves, as a therapeutic target. Despite consensus that
interruption of KLF9 function in cancer cells reduces tumorigenesis
in in vivo models, an established macrophage hazard
associated with selectively suppressed KLF9 levels severely limits
their clinical use (26,27). This hazard arises, since in addition
to the desired inhibition of KLF9 in the injured tissue, unwanted
collateral augmentation of macrophage KLF9 levels reduces the
inflammatory response (11). By
avoiding increased levels of macrophage KLF9, specific small
molecule drugs targeting KLF9 may reduce the antitumor effect,
while providing the desired antitumor outcome. To the best of our
knowledge, the present finding are the first to specifically
investigate KLF9 levels in macrophages in relation to
tumorigenesis.
In summary, it is proposed that two functional
changes of GCs inducing KLF9 upregulation in macrophages may
contribute to this antitumor shift. Firstly, KLF9 induced
macrophage apoptosis through the mitochondrial-dependent pathway.
Increased KLF9 levels disrupted the mitochondrial membrane
potential and increased ROS, subsequently promoting the release of
Cyt-c, which is pivotal for the mitochondrial-dependent
apoptosis pathway. Conversely, KLF9 suppression restored the
membrane potential during macrophage exposure to Dex. KLF9
knockdown reversed Dex-induced cleaved caspase-3 level in
macrophages. These results indicated that KLF9 participated in
Dex-induced macrophage apoptosis through the
mitochondrial-dependent apoptosis pathway. Furthermore, it was
revealed that specific inhibition of COX-2 by KLF9 in macrophages
was administered for immunosuppression purposes incidentally
increasing the likelihood of cancer development. Since increased
KLF9 levels promote oxidative stress, downregulation may promote
the immune function of macrophages. Thus, it is important to
evaluate the therapeutic value of KLF9 in macrophages. Moreover, as
an important target of GCs, dual targeting of KLF9 could be
promising in both oxidative injury and cancer treatment.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
JL analyzed and interpreted the data and wrote the
manuscript. FA performed most of the experiments. GZ, WL and BL
assisted in the completion of the experiments. All authors read and
approved the final manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cari L, De Rosa F, Nocentini G and
Riccardi C: Context-dependent effect of glucocorticoids on the
proliferation, differentiation, and apoptosis of regulatory T
cells: A review of the empirical evidence and clinical
applications. Int J Mol Sci. 20(pii): E11422019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mylka V, Deckers J, Ratman D, De Cauwer L,
Thommis J, De Rycke R, Impens F, Libert C, Tavernier J, Vanden
Berghe W, et al: The autophagy receptor SQSTM1/p62 mediates
anti-inflammatory actions of the selective NR3C1/glucocorticoid
receptor modulator compound A (CpdA) in macrophages. Autophagy.
14:2049–2064. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Patil RH, Naveen Kumar M, Kiran Kumar KM,
Nagesh R, Kavya K, Babu RL, Ramesh GT and Chidananda Sharma S:
Dexamethasone inhibits inflammatory response via down regulation of
AP-1 transcription factor in human lung epithelial cells. Gene.
645:85–94. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Achuthan A, Aslam ASM, Nguyen Q, Lam PY,
Fleetwood AJ, Frye AT, Louis C, Lee MC, Smith JE, Cook AD, et al:
Glucocorticoids promote apoptosis of proinflammatory monocytes by
inhibiting ERK activity. Cell Death Dis. 9:2672018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li B, Wang Y, Yin L, Huang G, Xu Y, Su J,
Ma L and Lu J: Glucocorticoids promote the development of
azoxymethane and dextran sulfate sodium-induced colorectal
carcinoma in mice. BMC Cancer. 19:942019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang C, Nanni L, Novakovic B,
Megchelenbrink W, Kuznetsova T, Stunnenberg HG, Ceri S and Logie C:
Extensive epigenomic integration of the glucocorticoid response in
primary human monocytes and in vitro derived macrophages. Sci Rep.
9:27722019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bagati A, Moparthy S, Fink EE,
Bianchi-Smiraglia A, Yun DH, Kolesnikova M, Udartseva OO, Wolff DW,
Roll MV, Lipchick BC, et al: KLF9-dependent ROS regulate melanoma
progression in stage-specific manner. Oncogene. 38:3585–3597. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhong Z, Zhou F, Wang D, Wu M, Zhou W, Zou
Y, Li J, Wu L and Yin X: Expression of KLF9 in pancreatic cancer
and its effects on the invasion, migration, apoptosis, cell cycle
distribution, and proliferation of pancreatic cancer cell lines.
Oncol Rep. 40:3852–3860. 2018.PubMed/NCBI
|
|
9
|
Fink EE, Moparthy S, Bagati A,
Bianchi-Smiraglia A, Lipchick BC, Wolff DW, Roll MV, Wang J, Liu S,
Bakin AV, et al: XBP1-KLF9 axis acts as a molecular rheostat to
control the transition from adaptive to cytotoxic unfolded protein
response. Cell Rep. 25:212–223 e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Glinghammar B, Skogsberg J, Hamsten A and
Ehrenborg E: PPARdelta activation induces COX-2 gene expression and
cell proliferation in human hepatocellular carcinoma cells. Biochem
Biophys Res Commun. 308:361–368. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barnard ME, Hecht JL, Rice MS, Gupta M,
Harris HR, Eliassen AH, Rosner BA, Terry KL and Tworoger SS:
Anti-inflammatory drug use and ovarian cancer risk by COX1/COX2
expression and infiltration of tumor-associated macrophages. Cancer
Epidemiol Biomarkers Prev. 27:1509–1517. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lanocha-Arendarczyk N, Baranowska-Bosiacka
I, Kot K, Gutowska I, Kolasa-Wołosiuk A, Chlubek D and
Kosik-Bogacka D: Expression and activity of COX-1 and COX-2 in
Acanthamoeba sp.-Infected lungs according to the host
immunological status. Int J Mol Sci. 19(pii): E1212018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kern MA, Haugg AM, Koch AF, Schilling T,
Breuhahn K, Walczak H, Fleischer B, Trautwein C, Michalski C,
Schulze-Bergkamen H, et al: Cyclooxygenase-2 inhibition induces
apoptosis signaling via death receptors and mitochondria in
hepatocellular carcinoma. Cancer Res. 66:7059–7066. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen EP, Markosyan N, Connolly E, Lawson
JA, Li X, Grant GR, Grosser T, FitzGerald GA and Smyth EM: Myeloid
Cell COX-2 deletion reduces mammary tumor growth through enhanced
cytotoxic T-lymphocyte function. Carcinogenesis. 35:1788–1797.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen EP and Smyth EM: COX-2 and
PGE2-dependent immunomodulation in breast cancer. Prostaglandins
Other Lipid Mediat. 96:14–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Carey LC, Valego NK, Chen K and Rose JC:
Thyroid hormone regulates renocortical COX-2 and PGE2 expression in
the late gestation fetal sheep. Reprod Sci. 15:598–603. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Y, Zhou Y, Chen S, Hu Y, Zhu Z and
Wang Y, Du N, Song T, Yang Y, Guo A and Wang Y: Macrophage
migration inhibitory factor facilitates prostaglandin E2 production
of astrocytes to tune inflammatory milieu following spinal cord
injury. J Neuroinflammation. 16:852019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hwang JH, Ma JN, Park JH, Jung HW and Park
YK: Anti-inflammatory and antioxidant effects of MOK, a polyherbal
extract, on lipopolysaccharidestimulated RAW 264.7 macrophages. Int
J Mol Med. 43:26–36. 2019.PubMed/NCBI
|
|
19
|
Yang D, Lv Z, Zhang H, Liu B, Jiang H, Tan
X, Lu J, Baiyun R and Zhang Z: Activation of the Nrf2 signaling
pathway involving KLF9 plays a critical role in allicin resisting
against arsenic trioxide-induced hepatotoxicity in rats. Biol Trace
Elem Res. 176:192–200. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bagheri-Yarmand R, Sinha KM, Li L, Lu Y,
Cote GJ, Sherman SI and Gagel RF: Combinations of tyrosine kinase
inhibitor and ERAD inhibitor promote oxidative stress-induced
apoptosis through ATF4 and KLF9 in medullary thyroid cancer. Mol
Cancer Res. 17:751–760. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang Y, Wei H, Song T, Cai A, Zhou Y and
Peng J, Jiang S and Peng J: E4BP4 mediates glucocorticoid-regulated
adipogenesis through COX2. Mol Cell Endocrinol. 450:43–53. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Majumder M, Xin X, Liu L, Tutunea-Fatan E,
Rodriguez-Torres M, Vincent K, Postovit LM, Hess D and Lala PK:
COX-2 induces breast cancer stem cells via EP4/PI3K/AKT/NOTCH/WNT
axis. Stem Cells. 34:2290–2305. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin HY, Davis PJ, Tang HY, Mousa SA,
Luidens MK, Hercbergs AH and Davis FB: The pro-apoptotic action of
stilbene-induced COX-2 in cancer cells: Convergence with the
anti-apoptotic effect of thyroid hormone. Cell Cycle. 8:1877–1882.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Y, Zou L, Li T, Lai D, Wu Y and Qin S:
Mogroside V inhibits LPS-induced COX-2 expression/ROS production
and overexpression of HO-1 by blocking phosphorylation of AKT1 in
RAW264.7 cells. Acta Biochim Biophys Sin (Shanghai). 51:365–374.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chang J, Xue M, Yang S, Yao B, Zhang B,
Chen X, Pozzi A and Zhang MZ: Inhibition of 11β-Hydroxysteroid
dehydrogenase Type II suppresses lung carcinogenesis by blocking
tumor COX-2 expression as well as the ERK and mTOR signaling
pathways. PLoS One. 10:e01270302015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park J, Ha SH, Abekura F, Lim H, Magae J,
Ha KT, Chung TW, Chang YC, Lee YC, Chung E, et al:
4-O-Carboxymethylascochlorin inhibits expression levels of on
inflammation-related cytokines and matrix metalloproteinase-9
through NF-κB/MAPK/TLR4 signaling pathway in LPS-activated RAW264.7
cells. Front Pharmacol. 10:3042019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Greene ER, Huang S, Serhan CN and
Panigrahy D: Regulation of inflammation in cancer by eicosanoids.
Prostaglandins Other Lipid Mediat. 96:27–36. 2011. View Article : Google Scholar : PubMed/NCBI
|