Introduction
Several integrated analyses of whole-genome and
whole-exome sequencing data and transcriptomics have been reported
for cohorts of prostate cancer (PCa) in Western countries. In these
cohorts, the incidence of androgen-inducible fusion oncogenes
generated by chromosomal alterations involving erythroblastosis
virus E26 transformation-specific related gene (ERG) was
reported to be >50% (1–4). In addition to ERG-associated
fusion events, variants of SPOP and MED12 and
deletions of chromosome 5q21/6q21 have been reported as common
genomic alterations (5,6). Recently, BRCA1, BRCA2 (5,7) and
HOXB13 (5,8) were identified as new therapeutic
targets or tumor markers, on which new molecular pathway analyses
are currently being conducted.
We previously reported that PCa harboring the
TMPRSS2-ERG fusion gene was less frequent in Japan compared
with Western countries (9). Another
group supported this finding in an independent Japanese cohort
together with a Chinese cohort, suggesting that this low frequency
is characteristic of PCa in Asians (10). Although the exact frequency of
SPOP variations has not yet been determined in Japanese
patients, TMPRSS2-ERG and SPOP variations occur in a
mutually exclusive manner in Western countries (6,11), and
it has been reported that there may be a clinical benefit in
classifying patients into TMPRSS2-ERG-positive and
SPOP-mutated groups (9).
This background suggests the potential benefits of also performing
thorough investigations of the genomic alterations in Japanese
patients, as well as in patients from Western countries.
It has been indicated that the profiling of genetic
alterations alone is insufficient to obtain a comprehensive
understanding of the tumorigenesis pathway; trans-omics studies are
required for this purpose. However, such studies are
resource-intensive due to the need for genomic, transcriptomic,
proteomic, epigenetic, or more omics analyses on the same tumor
specimen, followed by integration of the results and identification
of the biological pathways. In the present study, the gene network
model data of The Cancer Network Galaxy (TCNG; Human Genome Center,
University of Tokyo; http://tcng.hgc.jp/index.html) was used to deduce the
characteristics of PCa in the Japanese population, using the
limited gene variation data that were obtained in the present
study. TCNG is a database of gene networks estimated from
high-throughput biological data using a Bayesian network (12,13).
Some genetic variants disrupt the balance of the regulatory
relationships between genes, which may result in cancer; as such,
if this approach is extended to include a higher number of cases,
the above analyses may enable a comprehensive overview of PCa in
Japanese patients.
Materials and methods
PCa patients and tumor specimens
A total of 21 PCa patients who underwent radical
prostatectomy between 2011 and 2014 at the Department of Urology,
Yokohama City University Graduate School of Medicine, were included
in the present study. Several parts of each resected prostate that
had been indicated to contain cancer tissues by preoperative
examinations were embedded in OCT compound (Sakura Finetek Japan)
and immediately stored at −80°C. The patient clinical information
is summarized in Table I. All the
patients were Japanese, with a mean age of 67 years (range, 51–76
years), and serum prostate-specific antigen (PSA) values ranging
from 4.4 to 31.0 ng/ml (mean, 10.4±7.36 ng/ml). All tumors were
diagnosed as non-metastatic adenocarcinomas and assigned a Gleason
score of 6–9 at the Department of Pathology. When multiple Gleason
scores had been assigned to one patient, the highest score was
used, as shown in Table I.
 | Table I.Clinical information for 21 prostate
cancer patients. |
Table I.
Clinical information for 21 prostate
cancer patients.
| Case | Age (years) | pTNMa | Gleason
Scoreb | Histology | PSA (ng/ml) |
|---|
| 1 | 65 | pT2cN0M0 | 3+3=6 | Adenocarcinoma | 7.2 |
| 2 | 69 | pT3aN0M0 | 4+5=9 | Adenocarcinoma | 11.0 |
| 3 | 62 | pT2cN0M0 | 3+4=7 | Adenocarcinoma | 5.6 |
| 4 | 76 | pT2cN0M0 | 3+4=7 | Adenocarcinoma | 8.2 |
| 5 | 63 | pT3b, N0 | 4+3=7 | Adenocarcinoma | 14.0 |
| 6 | 56 | pT2cN0M0 | 3+4=7 | Adenocarcinoma | 4.4 |
| 7 | 61 | pT2cN0M0 | 4+3=7 | Adenocarcinoma | 5.3 |
| 8 | 71 | pT3aN0M0 | 3+4=7 | Adenocarcinoma | 15.2 |
| 9 | 76 | pT3aN0M0 | 3+4=7 | Adenocarcinoma | 15.6 |
| 10 | 59 | pT2cN0M0 | 4+4=8 | Adenocarcinoma | 11.4 |
| 11 | 75 | pT2cN0M0 | 4+5=9 | Adenocarcinoma | 5.6 |
| 12 | 65 | pT3aN0M0 | 3+4=7 | Adenocarcinoma | 5.4 |
| 13 | 71 | pT3aN0M0 | 4+5=9 | Adenocarcinoma | 31.0 |
| 14 | 71 | pT2cN0M0 | 4+4=8 | Ductal
adenocarcinoma | 7.6 |
| 15 | 71 | pT2cN0M0 | 3+4=7 | Adenocarcinoma | 8.9 |
| 16 | 75 | pT1cN0M0 | 4+4=8 | Adenocarcinoma | 5.1 |
| 17 | 67 | pT2cN0M0 | 3+5=8 | Adenocarcinoma | 29.1 |
| 18 | 73 | pT3aN0M0 | 3+5=8 | Adenocarcinoma | 7.4 |
| 19 | 67 | pT2cN0M0 | 3+4=7 | Adenocarcinoma | 7.2 |
| 20 | 51 | pT2cN0M0 | 3+4=7 | Adenocarcinoma | 4.8 |
| 21 | 61 | pT3aN0M0 | 4+3=7 | Adenocarcinoma | 8.2 |
Design of the original panels for
detection of genetic alterations
In order to profile the genetic alterations in
Japanese patients with PCa in an efficient as well as highly
sensitive manner, two original panels were prepared for the
targeted sequencing of genes that were reported to be altered in
previous whole-genome or whole-exome sequencing studies in Western
countries. The KCC71 panel was for DNA samples designed to detect
single-nucleotide variations (SNVs) and small insertions and
deletions (indels) in 71 PCa-related genes and driver genes
reported by whole-exome and whole-genome sequencing (4–11,14,15)
(Table SI). The PCaFusion panel
was for RNA samples designed to detect transcripts derived from 38
previously reported fusion transcripts, together with 8 control
transcripts (Table SI). In
addition, the PCaFusion panel was designed to detect fusion
transcripts with different exonic junctions from the same fusion
partners (1–5,14–18).
The multiplex-PCR primer sets for the KCC71 and PCaFusion panels
are provided in Tables SIIA and
SIIB.
Sample preparation and target
sequencing with the original panels
To obtain DNA and RNA samples, a thin-sliced section
was prepared from each stored frozen specimen, embedded in OCT
compound and stained with hematoxylin and eosin (HE). Based on
information on the area of tumor tissues in the HE-stained section,
PCa tissues were obtained directly from the remaining OCT-embedded
specimen, from which DNA and RNA were extracted using ZR-Duet
DNA/RNA miniprep (Zymo Research), following the manufacturer's
protocol. DNA and RNA were quantified with Qubit 2 (Thermo Fisher
Scientific, Inc.). To assess the DNA and RNA quality, ratios of
optical densities, A260/A280 and A260/A230, were further evaluated
by NanoPhotometer (Implen). A total of 10 ng of genomic DNA or
total RNA was used to create panel libraries for each specimen.
Library amplification was performed using Ion Torrent AmpliSeq™
technology, along with sequencing with the Ion PGM next-generation
sequencer (Thermo Fisher Scientific, Inc.).
Variant call and validation
Torrent Suite v4.0.2 and Ion Reporter version 4.4
(Thermo Fisher Scientific, Inc.) softwares were used to process and
analyze the sequenced data from the Ion PGM. Quality control
reports were obtained from the Torrent Suite. To identify somatic
variants, the SNVs and indels with a coverage rate of ≥20 and with
coding amino acid sequence substitutions when compared with the
UCSC hg19 reference genome sequence were first selected. Next,
single-nucleotide polymorphisms (SNPs) were excluded by using the
sequences as queries against the data in the databases COSMIC
(http://cancer.sanger.ac.uk/cosmic),
dbSNPs (NCBI, NIH; http://www.ncbi.nlm.nih.gov/projects/SNP/), the 1000
Genomes Project (http://www.1000genomes.org), and other publicly
accessible databases. For SNVs for which it remained unclear
whether they were SNPs or somatic variants after database analysis,
Sanger sequencing on DNA from the non-neoplastic counterpart of
each specimen was performed to obtain definitive results. Finally,
sequence alterations with an allele frequency of >5% were
defined as somatic variants in the present study.
Fusion transcript detection
Torrent Suite v4.0.2 and Ion Reporter version 4.4
were used to process and analyze the sequenced data from the
PCaFusion panel. Quality Check reports were obtained from the
Torrent Suite server. The unclear fusion transcripts identified by
the PCaFusion panel were further verified by reverse-transcription
(RT)-PCR followed by Sanger sequencing of the products.
Results
Sequencing statistics
A summary of the sequencing statistics is presented
as a representative case of KCC71 panel analysis in Fig. S1A. The means of the obtained reads
and coverage were 3,902,663 and 1,850, respectively. The data on
average alignment ratios revealed that 97.6% of the total reads
were aligned properly to the hg19 human genome reference sequence.
A summary of the sequencing statistics as a representative case of
the PCaFusion panel analysis is shown in Fig. S1B. The mean number of obtained
reads was 539,860. The data on the average alignment ratios
revealed that 88.1% of the total reads were aligned properly to the
hg19 human genome reference sequence.
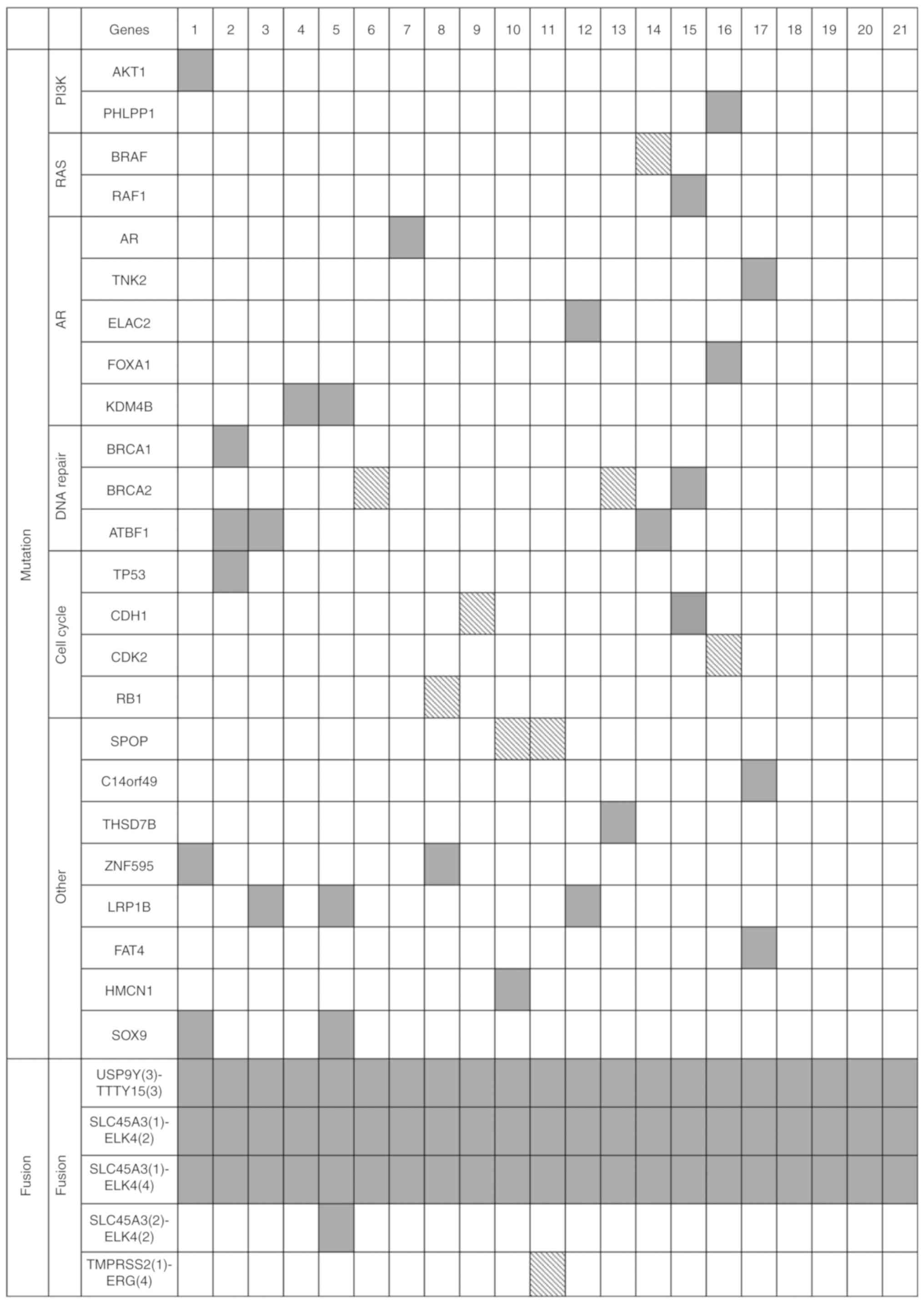
Gene variants by KCC71 panel
analysis
As indicated in Materials and methods, confirmed
somatic nucleotide sequence alterations of non-synonymous SNVs and
indels, with or without frameshifts, were considered as somatic
variants in the present study. Somatic variants were detected in 17
of 21 patients by the present panel analyses. No variants were
detected in 4 patients (cases 18–21). A total of 33 somatic
variants were identified in 24 of 71 PCa-related genes in the KCC71
panel. The results are summarized in Fig. 1 (detailed information is provided in
Table SIII).
A total of 7 probable pathogenic variants in 5 genes
were identified in the present KCC71 panel analyses. Evident driver
gene variants in the literature were found in BRAF (p.K601E)
(22) and SPOP (p.F102V and
p.F133L) (5,6). Although not well characterized as
driver genes in the literature, somatic variants with a high
pathogenic score predicted by FATMM (23–25)
were also identified in CDH1 (p.E880K) and RB1
(p.R621S). Two variants found in BRCA2, namely p.I1859fs and
p.R2318ter, which may result in premature termination and
truncation of the BRCA2 polypeptide, were considered as pathogenic,
although the identical alterations did not appear in COSMIC.
The remaining 26 variants were considered as
variants of uncertain/unknown significance (VUSs), including
AR (p.K610E), CDH1 (p.G62V), FOXA1
(p.R265-K267 del) and TP53 (p.V31I). These variants were
found in the COSMIC v82 database with labels of ‘n/a’ or ‘neutral’
based on FATHMM score. The CDH1 (p.G62V) variant is not
registered in COSMIC; however, the identical mutation was reported
as a germline mutation detected in families with hereditary diffuse
gastric cancer (26). This
non-synonymous mutation was in a region encoding a pro-domain and
is generally considered to be non-pathogenic (23–25).
The remaining 21 somatic mutations did not appear in COSMIC.
Fusion transcripts by the PCaFusion
panel analysis
The existence of gene fusion transcripts was
analyzed to identify the presence of fusion genes with the original
PCaFusion panel. All 8 non-fusion transcripts evaluated as positive
controls were detected in all specimens. In the present study,
fusion transcripts were designated as follows: The 5′ gene symbol
(number of the exon located at the fusion site)-the 3′ partner gene
symbol (exon number). For SLC45A3-ELK4 fusion transcripts,
SLC45A3(1)-ELK4(2) and SLC45A3(1)-ELK4(4) were
detected in all cases. By contrast, SLC45A3(2)-ELK4(2) was
identified in only 1 case (case 5). USP9Y-TTTY15 fusion
transcripts were detected in all examined cases. Among the
TMPRSS2-ERG fusion gene transcripts,
TMPRSS2(1)-ERG(4) was identified in only one case (case 11).
No other fusion transcripts were identified in the PCaFusion panel
analysis. The results are summarized in Fig. 1.
Comparative analysis between the
variants detected by the panels and the variants registered in
cBioPortal
A comparative analysis of the results of the KCC71
panel with TCGA and other big data registered in cBioPortal
(http://www.cbioportal.org) was
performed. Briefly, the frequency of somatic variants in the
aforementioned public databases were examined, including the
databases of TCGA, Broad Institute, Freed Hutchinson Cancer
Research Center, and Memorial Sloan Kettering Cancer Center
(hereafter referred to as ‘cBioPortal databases’) for the 71 genes
in the KCC71 panel, and this was compared with the frequency
obtained in the present study. The total frequency of somatic
variants in the 71 genes, calculated as the total variant number
identified per examined case, was higher compared with that in the
cBioPortal databases (present study, 33 different variants in 21
cases; summary of cBioPortal databases, 849 variants in 1,656
cases) (Fig. S2, and Tables SIV and SV). This difference was particularly
notable for the frequencies of variants in ATBF1, BRCA2 and
LRP1B, which were all ≥10% compared with those in the
cBioPortal databases. By contrast, the frequency of variants of
TP53 was low (1 in 21 cases, 4.8%; Fig. 1). To compare those mutation
frequencies in terms of pathways, the ratio between the number of
patients with and without mutations in the genes in a particular
pathway was calculated. Then, the ratios from our database and TCGA
databases were compared using Fisher's exact test. We observed that
those ratios in genes belonging to the androgen receptor (AR;
-log10 Fisher's P-value=2.67) and DNA Repair (-log10 Fisher's
P-value=4.40) signaling pathways were particularly high compared
with those in TCGA database (Tables
II, SIV and SV).
| Table II.Comparison of the frequency of
somatic variants identified between the KCC71 analysis and TCGA
database evaluated by signaling pathway. |
Table II.
Comparison of the frequency of
somatic variants identified between the KCC71 analysis and TCGA
database evaluated by signaling pathway.
| Signaling
pathway | KCC (%) | TCGA (%) | P-value (Fisher's
log10) |
|---|
| PI3K | 9.5 | 9.4 | 0.22 |
| RAS | 9.5 | 3.6 | 0.72 |
| AR | 28.6 | 6.2 | 2.67 |
| DNA Repair | 28.6 | 2.6 | 4.40 |
| Cell cycle | 23.8 | 17.8 | 0.49 |
| other | 52.4 | 33.2 | 1.20 |
Pathways of PCa predicted by TCNG
network analysis
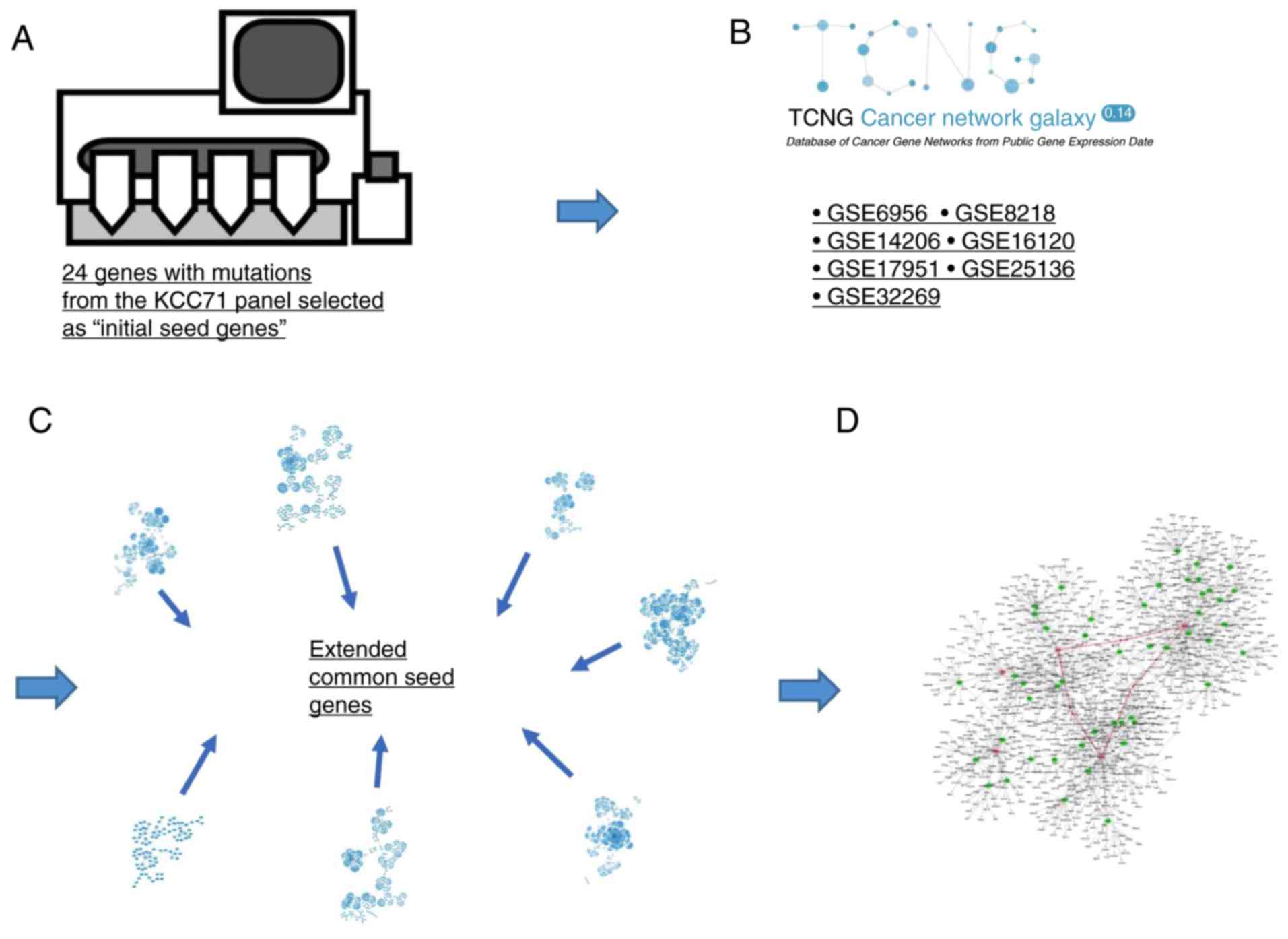
Our network analysis consisted of four steps as
explained below (Fig. 2A-D). In the
first step, genes with mutations from the KCC71 panel analysis were
selected as ‘initial seed genes’ (Fig.
2A). In the second step, the 7 gene networks of PCa in TCNG
were selected as graphical representations of the regulatory
relationships between genes. The Gene Expression Omnibus (GEO) ID
and information from each selected gene network for PCa are shown
in Fig. 2B and Table SVI. In the third step, the ‘initial
seed genes’ were mapped on the 7 PCa gene networks, and a
subnetwork around the ‘initial seed genes’ was extracted from each
of the gene networks; each subnetwork consisted of the ‘initial
seed genes’ and downstream genes within the two passes around the
initial seed genes (Fig. 2C). Genes
with one path from ‘initial seed genes’ are referred to as ‘child
genes’ and genes with a path from the child genes are referred to
as ‘grandchild genes’. The obtained subnetworks show the regulation
around the seed genes. Next, we attempted to identify ‘extended
common seed genes’ that are shared among ≥6 subnetworks. In the
last step, a putative ‘core network’ of PCa was estimated by
integrating subnetworks around ‘the extended common seed genes,’
referred to as ‘extended subnetworks’ (Fig. 2D). The above network operations were
conducted by using the functions of igraph, a package of R version
3.5.0. The subnetworks (relationships) of gene regulation were
demonstrated by graphical visualization using Cytoscape software
version 3.5.1 (19), as shown in
Fig. S3, and the core network was
presented using igraph.
Two publicly available tools, Database for
Annotation, Visualization, and Integrated Discovery (DAVID;
Laboratory of Human Retrovirology and Immunoinformatics, http://david.ncifcrf.gov/home.jsp) (20) and Reduce + Visualize Gene Ontology
(REVIGO; Redjer Boskivic Institute; http://revigo.irb.hr) (21), were then used to investigate the
biological functions associated with the gene groups involved in
the core network. DAVID v6.8, which mainly provides typical batch
annotation and Gene Ontology (GO) term enrichment analysis, was
used to highlight the most relevant GO terms associated with a
given gene list, in order to elucidate the biological meaning
behind a large list of genes. Enrichment analysis was performed
using DAVID for the core network genes listed in Table SVII, and a functional annotation
chart including a list of GO IDs and P-values for the enrichment
tests was obtained. The functional annotation chart report of DAVID
shows categories, enriched terms associated with a gene list of
interest, related term search, genes involved in the term, and
percentages or modified Fisher's exact P-values. REVIGO was used to
summarize the results obtained from DAVID (21). REVIGO provides a functional
interpretation of genes defined by GO with statistical methods. A
list of GO IDs and P-values was entered from the functional
annotation chart report of DAVID. The REVIGO GO tree map shows a
two-level hierarchy of GO terms.
The workflow for the reconstruction of the core
network of PCa was schematically summarized with TCNG (Fig. 2A-D). The 24 genes with somatic
variations from the KCC71 panel analysis (Fig. 1) were selected as ‘initial seed
genes.’ Although only 5 well-characterized pathogenic driver gene
mutations were identified in the present analysis and the remaining
19 genes were considered as VUSs, all ‘initial seed genes’ were
reported to be involved in PCa in the literature and databases
(Materials and methods, Sample preparation and target sequencing
with the original panels). Subnetworks were extracted from 7 public
PCa gene networks in TCNG (Table
SVI), which included initial seed genes (parent nodes), and
parent-child and child-grandchild genes in the network (Fig. S3). ‘Extended common seed genes’,
TNK2, SOX9, CDH1, FOXA1 and TP53, that were commonly
included in the subnetworks, were extracted. To identify the core
network of PCa s examined, extended subnetworks around the
‘extended common seed genes’ were further extracted from each of
the original 7 PCa networks, integrated, and the core network was
finally reconstructed.
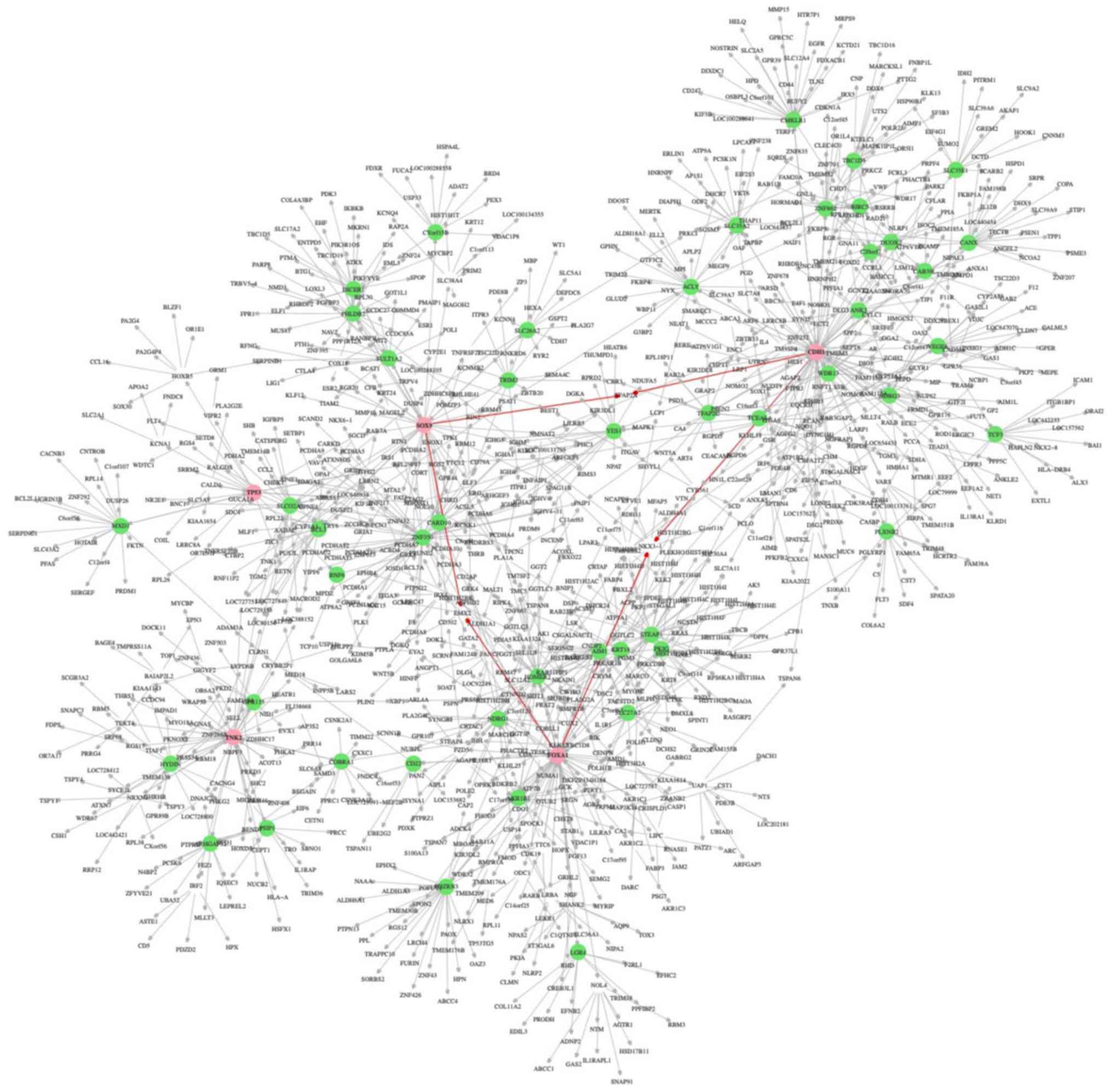
The core network around the ‘extended common seed
genes’ was further analyzed. The 3 extended common seed genes,
SOX9, CDH1 and FOXA1, were connected via edges with
each other through the genes EMX2, NKX3-1 and TFAP2A,
and formed a closed loop (Fig. 3,
red arrows). EMX, NKX3-1 and TFAP2A were the only
genes in the network located between the extended common genes. The
top 15 genes with high connectivity (hub genes), are listed in
Table SVIII. All 5 extended common
genes are listed in Table SIV, but
none of the initial seed genes appears in it. Only AR and
SPOP as the initial seed genes appear in the final network,
with few edges.
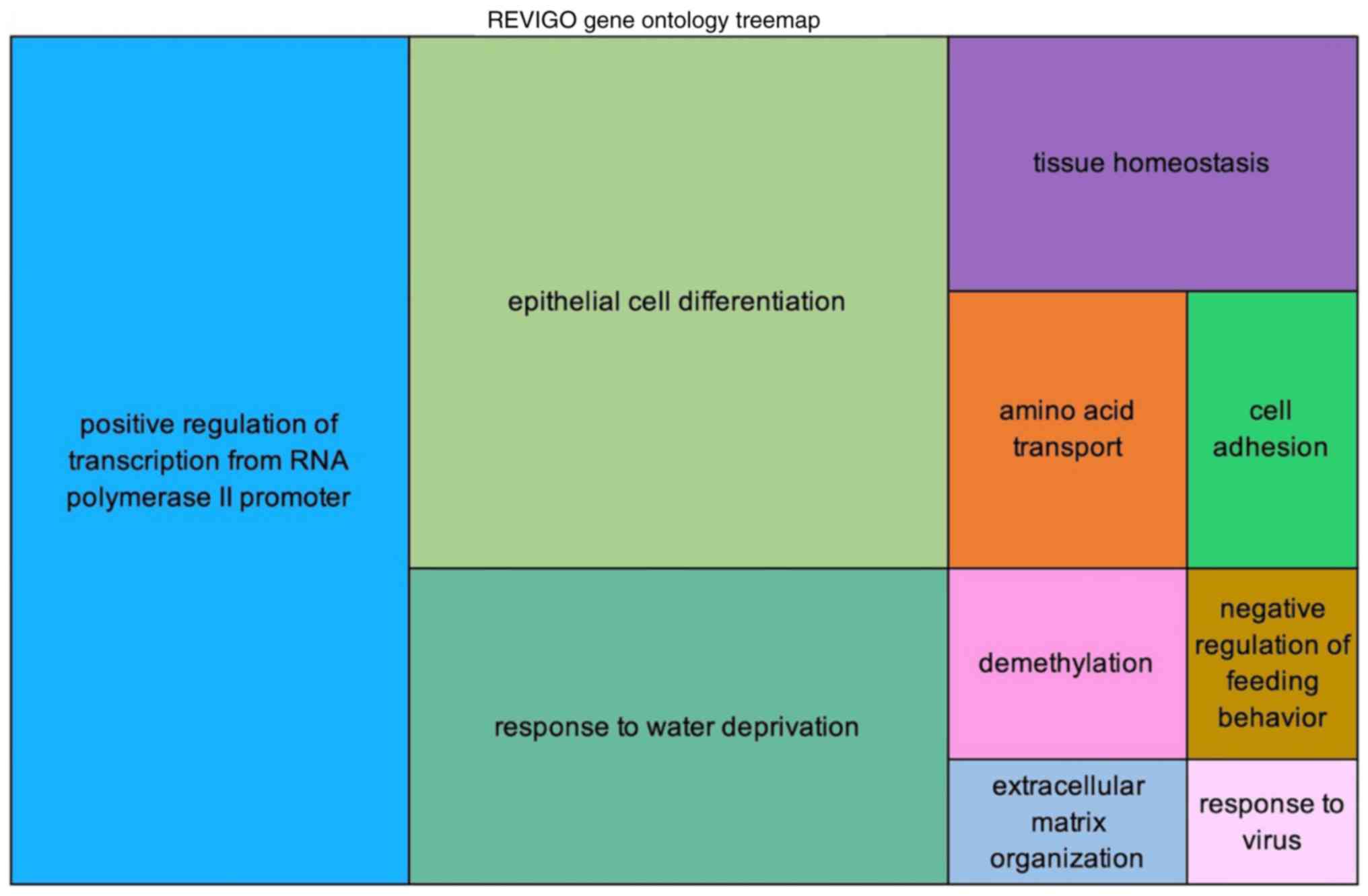
The enrichment analysis using DAVID and REVIGO found
50 GO terms with adjusted P-values (Table SIX). REVIGO generates tree maps of
the GO terms, as shown in Fig. 4
and Fig. S4. The GO terms are
joined into ‘superclusters’ of loosely related terms and depicted
with different colors. The most significant GO term found was
‘positive regulation of transcription from RNA polymerase II
promoter’, followed by ‘epithelial cell differentiation’, ‘response
to water deprivation’, ‘tissue homeostasis’, and ‘amino acid
transport’.
Discussion
The present study attempted to elucidate the
molecular pathways involved in PCa in Japanese patients by starting
with a limited number of cases and with a new bioinformatics
analysis using TCNG. Starting the analysis with 24 genes harboring
mutations as initial seed genes, we reached a core network
involving 3 genes that were not included among the initial seed
genes, but 2 of those had been well-characterized in relation to
PCa. This may demonstrate the validity of this analytical approach,
but further estimation with a larger numbers of cases is
required.
In the present study, 21 surgically removed PCa
specimens without any neoadjuvant treatments were analyzed using
our original DNA and RNA panels for PCa profiling. Both panels
functioned appropriately, as revealed by sequencing statistics and
the results obtained with the positive control set for the RNA
panel. The well-characterized pathogenic TMPRSS2-ERG fusion
gene transcript was identified in only 1 case (1/21, 4.8%) in the
PCaFusion panel analysis, which was an unexpectedly low frequency
when compared with that in previous reports, even in Japanese or
Chinese cohorts in which the rate was significantly lower compared
with that in cohorts from Western countries (9,27). By
contrast, two other transcription-mediated chimeric RNAs,
SLC45A3-ELK4 and USP9Y-TTTY15 fusion transcripts,
were detected in all examined cases. Although enriched in cancer
tissues, the pathogenicity of these fusion RNAs remains unclear,
and they were found to be expressed in both cancerous and adjacent
non-cancerous prostatic tissues. Highly sensitive methods, such as
RT-PCR, have demonstrated these RNAs in almost all examined
specimens (28,29). As our PCaFusion panel analysis is a
PCR-mediated amplicon sequencing technology, the obtained results
were compatible with those in previous reports. A similar
transcription-mediated chimeric RNA, SDK1-AMACR, was not
found in the present study, although Chinese cohorts identified the
fusion transcript in 23–24% of examined cases (26,29).
Despite their similar origins in East Asia, Chinese and Japanese
PCa patients appear to differ in their genetic or epigenetic
background.
The KCC71 panel analysis identified 33 different
genetic variants associated with PCa in the Japanese. Two cases
contained SPOP mutations (2/21, 9.5%) in the hotspots in the
MATH domain. SPOP, encoding the E3 ubiquitin ligase, is the
most frequently mutated gene, with mutations found in 6–15% of PCa
cases across multiple cohorts, in a manner mutually exclusive with
the presence of the fusion gene TMPRSS2-ERG. SPOP
mutation is known to be associated with certain clinicopathological
characteristics, such as serum PSA level, pathological parameters
and patient prognosis (6,11). The frequency of SPOP mutation
in the present study was comparable with that in previous reports,
which may indicate the absence of major bias in our cohort;
however, the reason for the low frequency of TMPRSS2-ERG is
unclear. BRCA2 truncating inactivating mutations were also
identified in 2 cases (9.5%). BRCA2 mutation is rare, with a
frequency of ~2% in early-onset PCa (30); it has also been shown to be
associated with a higher Gleason score and poor prognosis (31,32).
Regarding our BRCA2-mutated cases, one had a Gleason score
of 9 and the other had a score of 7. The evaluation of BRCA2
mutation with biopsy or surgical specimens may also be useful for
selecting the treatment modality for Japanese patients.
Other pathogenic variants were found in CDH1,
BRAF and RB1, with 1 mutation per gene. The sample size
was small and precise comparison of the mutation frequency of each
gene with that in previous cohorts was not the principal objective
of this research. However, the overall frequency of mutated genes
and somatic variations in the 71 selected genes was higher compared
with that calculated from public big data, such as TCGA. This may
be a characteristic of PCa in Japanese patients, but further
investigation in large cohorts is required.
In recent years, efforts have intensified to obtain
novel meaningful insights into biological pathways involved in
cancer by utilizing big data in the life sciences. We herein
attempted to develop a new approach to extracting a common core
network related to PCa in the Japanese population by integrating
information on mutated genes identified in KCC71 panel analysis and
multiple gene networks of PCa in TCNG. Subsequently, we developed a
new way of exploring cancer-related gene interactions. TCNG is a
database of cancer gene regulatory networks estimated from publicly
available cancer gene expression data, in the GEO database, by
using Bayesian network models (12,13).
In addition, by combining data and examining common genes that
constitute the core network, it is possible to characterize the
interactions among genes that may cause cancer. The core network
may be considered as the center of the pathogenic pathway.
The central genes of the network estimated here were
identified as the ‘extended common seed genes’ of PCa. All 5
identified common genes were initial seed genes and have been well
characterized as being associated with cancer, including PCa.
Surprisingly, only 3 genes were revealed to connect common genes to
each other, namely TFAP2A (between CDH1 and
SOX9), EMX2 (between SOX9 and FOXA1),
and NKX3-1 (between FOXA1 and CDH1). Although
none of these 3 genes was involved with the initial seed genes,
NKX3-1 is a well-known prostate-specific tumor suppressor
(33) and has been implicated in
prostatic epithelial cell differentiation (34) and the maintenance of luminal stem
cells (35). TFAP2A, also
referred to as AP-2 or AP2TF/TFAP2, is a
transcription factor and its tumor suppressor properties were also
reported in cancers including PCa (36–38).
As neither NKX3-1 nor TFAP2A were involved with the
initial seed genes, which were the starting point of the present
analysis, this may support the reliability of this analysis. By
contrast, although EMX2, a homeobox-containing transcription
factor, was characterized as a tumor suppressor gene in cancers
such as colorectal cancer (39),
malignant pleural mesothelioma or lung cancer (40,41),
to the best of our knowledge no report on PCa has yet been
published. Research on EMX2 may elucidate the
biological/pathological characteristics of PCa in Japanese
patients.
In comparison with the generally Caucasian cohorts
in TCGA, the involvement of the AR pathway and the DNA repair
pathway were identified as characteristics associated with PCa in
the Japanese population. Although the AR pathway is clearly
significant worldwide, the potential involvement of the DNA repair
pathway in this disease was identified due to the two pathogenic
mutations that were identified in BRCA2. Momozawa et
al (43) reported the results
of germline mutation analysis of 7,636 Japanese PCa patients, and
found that the BRCA2, but not BRCA1, germline
pathogenic variant was significantly associated with PCa in
Japanese patients. This contradicts the Philadelphia Prostate
Cancer Consensus 2017 (42), based
generally on data from Western countries, which asserted that there
is high-grade evidence on the association of both BRCA1 and
BRCA2 with PCa (43). It is
possible that the disturbance of DNA repair, partly through the
inactivation of BRCA2, but not BRCA1, is involved in
PCa in Japanese patients. BRCA1 and BRCA2 are
currently considered as homologous recombination-related genes, and
associated differences in pathological phenotypes or the clinical
significance of their mutations, such as sensitivity to
poly(ADP-ribose) polymerase inhibitors, have not yet been well
addressed (44). BRCA2 may
warrant further research as a gene potentially associated with PCa
in the Japanese.
We herein analyzed the associations among limited
numbers of genes with somatic variations based on a Bayesian
network model. Using a statistical approach, it appeared possible
to predict the association among not only directly interacting
genes, but also ones that act indirectly. The analysis using a
statistical model may be effective when, for example, used to
predict drug targets, as it can predict signaling pathways even
from genes that are not directly associated with each other. In the
analyses, genes that do not harbor well-characterized pathogenic
mutations served as initial seed genes. Mutations with uncertain
significance in these genes should be further functionally
characterized in future research. In addition, although PCas are
known to be clonally heterogeneous tumors, the heterogeneity was
not considered in the present study. Additional larger studies
considering this heterogeneity are required to obtain an overall
understanding of PCa in the Japanese population.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank the members of the
Kanagawa Cancer Center Research Institute, Health Intelligence
Center, Human Genomic Center, the Institute of Medical Science,
University of Tokyo. Supercomputing resources were provided by
Human Genome Center, University of Tokyo.
Funding
The present study was supported by Grants-in-Aid for
Scientific Research (KAKENHI, nos. 17K1168, 16K19099 and
26860253).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article. Sequence data are
available upon request to the corresponding author; the request
must include a description of the research proposal.
Authors' contributions
RK, YM, RY designed the study and wrote the
manuscript; RK performed research and analyzed the data with the
assistance of ES; TK, YM, IA and HU prepared the clinical samples
and analyzed patient information; YT, SI, RY, AN, GT and ES
prepared the data from TCNG database and supervised the analyses.
SI, RY and SM supervised the research.
Ethics approval and consent to
participate
The ethical committees for investigations with
human materials at Yokohama City University Graduate School of
Medicine and Kanagawa Cancer Center approved the study. Informed
consent was obtained from all the participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tomlins SA, Rhodes DR, Perner S,
Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J,
Kuefer R, et al: Recurrent fusion of TMPRSS2 and ETS transcription
factor genes in prostate cancer. Science. 310:644–648. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Perner S, Demichelis F, Beroukhim R,
Schmidt FH, Mosquera JM, Setlur S, Tchinda J, Tomlins SA, Hofer MD,
Pienta KG, et al: TMPRSS2:ERG fusion-associated deletions provide
insight into the heterogeneity of prostate cancer. Cancer Res.
66:8337–8341. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Helgeson BE, Tomlins SA, Shah N, Laxman B,
Cao Q, Prensner JR, Cao X, Singla N, Montie JE, Varambally S, et
al: Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions
in prostate cancer. Cancer Res. 68:73–80. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumar-Sinha C, Tomlins SA and Chinnaiyan
AM: Recurrent gene fusions in prostate cancer. Nat Rev Cancer.
8:497–511. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Attard G, Parker C, Eeles RA, Schröder F,
Tomlins SA, Tannock I, DrakeZZC G and de Bono JS: Prostate cancer.
Lancet. 387:70–72. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barbieri CE, Baca SC, Lawrence MS,
Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van
Allen E, Stransky N, et al: Exome sequencing identifies recurrent
SPOP, FOXA1 and MED12 mutations in prostate cancer.
Nature Genet. 44:685–689. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cavanagh H and Rogers KM: The role of
BRCA1 and BRCA2 mutations in prostate, pancreatic and
stomach cancers. Hered Cancer Clin Pract. 1(13): 162015. View Article : Google Scholar
|
|
8
|
Witte JS, Mefford J, Plummer SJ, Liu J,
Cheng I, Klein EA, Rybicki BA and Casey G: HOXB13 mutation
and prostate cancer: Studies of siblings and aggressive disease.
Cancer Epidemiol Biomarkers Prev. 22:675–680. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miyagi Y, Sasaki T, Fujinami K, Sano J,
Senga Y, Miura T, Kameda Y, Sakuma Y, Nakamura Y, Harada M and
Tsuchiya E: ETS family-associated gene fusions in Japanese
prostate cancer: analysis of 194 radical prostatectomy samples. Mod
Pathol. 23:1492–1498. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haffner MC, Mosbruger T, Esopi DM, Fedor
H, Heaphy CM, Walker DA, Adejola N, Gürel M, Hicks J, Meeker AK, et
al: Tracking the clonal origin of lethal prostate cancer. J Clin
Invest. 123:4918–4922. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shoag J, Liu D, Blattner M, Sboner A, Park
K, Deonarine L, Robinson BD, Mosquera JM, Chen Y, Rubin MA and
Barbieri CE: SPOP mutation drives prostate neoplasia without
stabilizing oncogenic transcription factor ERG. J Clin Invest.
128:381–386. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tamada Y, Imoto S, Araki H, Nagasaki M,
Print C, Charnock-Jones DS and Miyano S: Estimating genome-wide
gene networks using nonparametric Bayesian network models on
massively parallel computers. IEEE/ACM Trans Comput Biol Bioinform.
8:683–697. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Imoto S, Goto T and Miyano S: Estimation
of genetic networks and functional structures between genes by
using Bayesian network and nonparametric regression. Pac Symp
Biocomput. 7:175–186. 2002.
|
|
14
|
Huang J, Wang JK and Sun Y: Molecular
pathology of prostate cancer revealed by next-generation
sequencing: Opportunities for genome-based personalized therapy.
Curr Opin Urol. 23:189–193. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Beltran H, Yelensky R, Frampton GM, Park
K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus
DM, et al: Targeted next-generation sequencing of advanced prostate
cancer identifies potential therapeutic targets and disease
heterogeneity. Eur Urol. 63:920–926. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu Y, Ren S, Jing T, Cai X, Liu Y, Wang
F, Zhang W, Shi X, Chen R, Shen J, et al: Clinical utility of a
novel urine-based gene fusion TTTY15-USP9Y in predicting prostate
biopsy outcome. Urol Oncol. 33:384.e9–20. 2015. View Article : Google Scholar
|
|
17
|
St John J, Powell K, Conley-Lacomb MK and
Chinni SR: TMPRSS2-ERG fusion gene expression in prostate tumor
cells and its clinical and biological significance in prostate
cancer progression. J Cancer Sci Ther. 4:94–101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rubin MA, Maher CA and Chinnaiyan AM:
Common gene rearrangements in prostate cancer. J Clin Oncol.
29:3659–3668. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Gnome Res. 13:2498–2504. 2003. View Article : Google Scholar
|
|
20
|
Jiao X, Sherman BT, Huang da W, Stephens
R, Baseler MW, Lane HC and Lempicki RA: DAVID-WS: A stateful web
service to facilitate gene/protein list analysis. Bioinformatics.
28:1805–1806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Supek F, Bošnjak M, Škunca N and Šmuc T:
REVIGO summarizes and visualizes long lists of Gene Ontology terms.
PLoS One. 6:e218002011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Menzies AM and Long GV: Long, dabrafenib
and trametinib, alone and in combination for BRAF-mutant metastatic
melanoma. Clin Cancer Res. 20:2035–2043. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shihab HA, Gough J, Cooper DN, Stenson PD,
Barker GL, Edwards KJ, Day IN and Gaunt TR: Predicting the
functional, molecular and phenotypic consequences of amino acid
substitutions using hidden Markov models. Hum Mutat. 34:57–65.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shihab HA, Gough J, Cooper DN, Day INM and
Gaunt TR: Predicting the functional consequences of
cancer-associated amino acid substitutions. Bioinformatics.
29:1504–1510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shihab HA, Gough J, Mort M, Cooper DN, Day
IN and Gaunt TR: Ranking non-synonymous single nucleotide
polymorphisms based on disease concepts. Hum Genom. 8:112014.
View Article : Google Scholar
|
|
26
|
Simões-Correia J, Figueiredo J, Lopes R,
Stricher F, Oliveira C, Serrano L and Seruca R: E-Cadherin
destabilization accounts for the pathogenicity of missense
mutations in hereditary diffuse gastric cancer. PLoS One.
7:e337832012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ren S, Peng Z, Mao JH, Yu Y, Yin C, Gao X,
Cui Z, Zhang J, Yi K, Xu W, et al: RNA-seq analysis of prostate
cancer in the Chinese population identifies recurrent gene fusions,
cancer-associated long noncoding RNAs and aberrant alternative
splicings. Cell Res. 22:806–821. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kumar-Sinha C, Kalyana-Sundaram S and
Chinnaiyan AM: SLC45A3-ELK4 chimera in prostate cancer: Spotlight
on cis-splicing. Cancer Discov. 2:582–585. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Y, Mao XY, Liu X, Song RR, Berney D,
Lu YJ and Ren G: High frequency of the SDK1:AMACR fusion transcript
in Chinese prostate cancer. Int J Clin Exp Med. 8:15127–15136.
2015.PubMed/NCBI
|
|
30
|
Edwards SM, Kote-Jarai Z, Meitz J, Hamoudi
R, Hope Q, Osin P, Jackson R, Southgate C, Singh R, Falconer A, et
al: Two percent of men with early-onset prostate cancer harbor
germline mutations in the BRCA2 gene. Am J Hum Genet. 72:1–12.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mitra A, Fisher C, Foster CS, Jameson C,
Barbachanno Y, Bartlett J, Bancroft E, Doherty R, Kote-Jarai Z,
Peock S, Easton D; IMPACTEMBRACE, ; et al: Prostate cancer in male
BRCA1 and BRCA2 mutation carriers has a more aggressive phenotype.
Br J Cancer. 98:502–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cui M, Gao XS, Gu X, Guo W, Li X, Ma M,
Qin S, Qi X, Xie M, Peng C and Bai Y: BRCA2 mutations should be
screened early and routinely as markers of poor prognosis: Evidence
from 8,988 patients with prostate cancer. Oncotarget.
8:40222–40232. 2017.PubMed/NCBI
|
|
33
|
Abate-Shen C, Shen MM and Gelmann E:
Integrating differentiation and cancer: The Nkx3.1 homeobox
gene in prostate organogenesis and carcinogenesis. Differentiation.
76:717–727. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dutta A, Magnen C, Mitrofanova A, Ouyang
X, Califano A and Abate-Shen C: Identification of an NKX3.1-G9a-UTY
transcriptional regulatory network that controls prostate
differentiation. Science. 352:1576–1580. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Talos F, Mitrofanova A, Bergren SK,
Califano A and Shen MM: A computational systems approach identifies
synergistic specification genes that facilitate lineage conversion
to prostate tissue. Nat Commun. 8:146622017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ruiz M, Troncoso P, Bruns C and Bar-Eli M:
Activator protein 2alpha transcription factor expression is
associated with luminal differentiation and is lost in prostate
cancer. Clin Cancer Res. 7:4086–4095. 2001.PubMed/NCBI
|
|
37
|
Ruiz M, Pettaway C, Song R, Stoeltzing O,
Ellis L and Bar-Eli M: Activator protein 2alpha inhibits
tumorigenicity and represses vascular endothelial growth factor
transcription in prostate cancer cells. Cancer Res. 64:631–638.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jonckheere N, Fauquette V, Stechly L,
Saint-Laurent N, Aubert S, Susini C, Huet G, Porchet N, Van
Seuningen I and Pigny P: Tumour growth and resistance to
gemcitabine of pancreatic cancer cells are decreased by AP-2alpha
overexpression. Br J Cancer. 101:637–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aykut B, Ochs M, Radhakrishnan P, Brill A,
Höcker H, Schwarz S, Weissinger D, Kehm R, Kulu Y, Ulrich A and
Schneider M: EMX2 gene expression predicts liver metastasis
and survival in colorectal cancer. BMC Cancer. 17:5552017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Giroux Leprieur E, Hirata T, Mo M, Chen Z,
Okamoto J, Clement G, Li H, Wislez M, Jablons DM and He B: The
homeobox gene EMX2 is a prognostic and predictive marker in
malignant pleural mesothelioma. Lung Cancer. 85:465–471. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Okamoto J, Hirata T, Chen Z, Zhou HM,
Mikami I, Li H, Yagui-Beltran A, Johansson M, Coussens LM, Clement
G, et al: EMX2 is epigenetically silenced and suppresses growth in
human lung cancer. Oncogene. 29:5969–5975. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Giri VN, Knudsen KE, Kelly WK, Abida W,
Andriole GL, Bangma CH, Bekelman JE, Benson MC, Blanco A, Burnett
A, et al: Roel of genetic testing for inherited prostate cancer
risk: Philadelphia prostate cancer consensus conference2017. J Clin
Oncol. 36:414–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Momozawa Y, Iwasaki Y, Hirata M, Liu X,
Kamatani Y, Takahashi A, Sugano K, Yoshida T, Murakami Y, Matuda K,
et al: Germline pathogenic variants in 7636 japanese patients with
prostate cancer and 12366 controls. J Natl Cancer Inst. Jun
19–2019.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Venkitaraman AR: Cancer susceptibility and
the functions of BRCA1 and BRCA2. Cell. 108:171–182.
2002. View Article : Google Scholar : PubMed/NCBI
|