Introduction
Cholesteatoma is a chronic middle ear disease, which
is pathologically displayed as a benign tumor with excessive
squamous epithelial cell proliferation. Nevertheless, it can
clinically manifest a malignant nature by destroying adjacent bony
structures and nerves, resulting in hearing loss, tinnitus,
dizziness, facial paralysis, brain abscess, meningitis, and
hydrocephalus (1). To date, the
only treatment for cholesteatoma is surgical resection as no drug
treatments are currently available, although recurrence with
complications after surgery is quite high (2).
Previous reports have indicated that the underlying
molecular mechanisms of cholesteatoma are regulated by growth
factors and inflammatory mediators (3–6). In
recent years, epigenetic regulation, such as dysregulation of
microRNAs (miRNAs), has been demonstrated to play a crucial role in
cholesteatoma formation (7–11). Most experiments have focused on
discovering the messenger RNA (mRNA) targets of miRNAs and
elucidating their regulatory mechanisms on the assumption that
following buffering of the expression of an miRNA, the expression
of targeted mRNAs is correspondingly disturbed (7,8).
However, a single miRNA can regulate the expression of hundreds of
mRNAs and each mRNA can be regulated by several miRNAs (12), all of which complicate the
functional research of the molecular mechanism of
cholesteatoma.
Recent years, in the research of therapeutic targets
of diseases, endogenous miRNA ‘sponges’, also termed competing
endogenous RNAs (ceRNAs), which contain tandem repeats of miRNA
recognition elements (MREs), have shed new light on the
investigation of the function of miRNAs (13,14).
ceRNAs can act as natural miRNA sponges and thus influence the
expression of miRNAs. All RNA transcripts that share common MREs
can function as ceRNAs, such as protein coding genes, pseudogenes,
long non-coding RNAs (lncRNAs) (13,15),
and recently discovered circular RNAs (circRNAs) (16).
In our previous study, we reported that lncRNAs had
ceRNA potential in cholesteatoma formation (15). Unlike lncRNAs, circRNAs are a novel
class of noncoding RNAs characterized by the unique structure
wherein the 3′ and 5′ ends are covalently joined in a closed loop
structure without polarity or poly (A) tail (17,18).
The special structure ensures the much higher stability of circRNAs
than linear transcripts, protecting circRNAs from exonucleolytic
decay (16,19,20).
Studies have shown that circRNAs can function as endogenous miRNA
sponges, which can efficiently soak up miRNAs and buffer their
activities, resulting in the upregulated expression of
miRNA-targeted genes (21). The
circRNA-mediated ceRNA network has been verified to play crucial
roles in many disease processes, such as bladder cancer (22), cardiovascular diseases (23), and Alzheimer's disease (24). Despite the marked regulatory
potential in disease, however, no reports have elucidated whether
circRNAs can play a role in cholesteatoma.
In our present study, we explored the differentially
expressed profile of circRNAs between cholesteatoma and matched
normal skin tissues by using microarray analysis for the first
time. The reliability of microarray expression data was confirmed
by quantitative RT-PCR. By constructing the
circRNA-lncRNA-miRNA-mRNA ceRNA network with bioinformatics
approaches, we explored the ceRNA potential of circRNAs in the
pathogenesis of cholesteatoma.
Materials and methods
Patients and specimens
All specimens were obtained from 3 female and 4 male
patients aged 18 to 32-years-old (mean age 26.3 years, 2 female and
2 male patients for microarray analysis and real-time qPCR
validation, 1 female and 2 male patients for real-time qPCR
validation), who underwent surgical procedures for unilateral
middle ear cholesteatoma between June 2016 and November 2016 at the
Department of Otorhinolaryngology, Peking Union Medical College
Hospital, Beijing, China. All patients in this study met the
following inclusion criteria: Patients presented with acquired
cholesteatoma, the resected mass was identified as cholesteatoma by
pathological examination, and no antitumor treatments were given
before surgery. Patients that had previous middle ear surgeries or
combined with other middle ear tumors were excluded. All specimens
were stored at −80°C after collection for subsequent RNA
extraction.
RNA extraction and quality
control
Total RNAs were extracted from cholesteatoma and
post-auricular skin tissues using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. The concentrations of the RNA samples were determined
by OD260 using a NanoDrop ND-1000 instrument (Thermo Fisher
Scientific, Inc.). The integrity of RNAs was assessed by
electrophoresis on a denaturing agarose gel and with an Agilent
2100 Bioanalyzer (Agilent Technologies, Inc.).
Microarray assay
Microarray analysis was used to detect
differentially expressed circRNAs between cholesteatoma and
post-auricular skin tissues. Sample labelling and array
hybridization were performed according to the manufacturer's
protocol (Arraystar Inc.). The sample preparation and microarray
hybridization were performed based on the Arraystar's standard
protocols including RNA purification, amplification, and
transcription into fluorescent cRNA. The labelled cRNAs were then
hybridized onto an assembled RNA expression microarray slide
(Arraystar). After washing, the arrays were scanned using the
Agilent G2505C Scanner. Agilent Feature Extraction software
(version 11.0.1.1) was used to analyze acquired array images.
Quantile normalization and subsequent data processing were
performed using the R software limma package (version 3.22.7)
(25) and GeneSpring GX v12.1
(Agilent Technologies). Differentially expressed circRNAs between
two samples were identified through fold change filtering.
Hierarchical clustering was performed to show the distinguishable
RNA expression patterns among the samples.
Real-time qPCR validation
Extracted RNA was reverse transcribed to synthesize
cDNA for RT-qPCR analysis. RNA (3 µg) was mixed with 1 µl Random N9
primers (0.5 µg/µl) (Invitrogen; Thermo Fisher Scientific, Inc.)
and 1.6 µl dNTP Mix (HyTest Ltd.), and then the mixture was put on
ice for 2 min followed by incubation at 65°C for 5 min. The reverse
transcription system was subsequently prepared, comprising the
above mixture, 0.2 µl SuperScript III RT (Invitrogen; Thermo Fisher
Scientific, Inc.), 4 µl 5X First-Strand Buffer (Invitrogen; Thermo
Fisher Scientific, Inc.), 1 µl 0.1 M DTT (Promega) and 0.3 µl RNase
inhibitor (Epicentre, Inc.). This reaction system underwent
successive incubation in water at a temperature of 37°C for 1 min,
50°C for 60 min and then 70°C for 15 min until reverse
transcription was completed. The selected circRNAs and primers for
RT-qPCR were designed using Primer 5.0 software (Primer-E Ltd., UK)
(Table I) and synthesized by
Generay Biotech. For all samples, β-actin was used as an internal
control. RT-qPCR was performed using the ViiA 7 Real-time PCR
System (Applied Biosystems; Thermo Fisher Scientific, Inc.) with a
SYBR expression assay system (Takara). The PCR reaction conditions
were as follows: An initial denaturation at 95°C for 10 min,
followed by 40× PCR cycles at 95°C for 10 sec and 60°C for 60 sec,
then annealing and extension at 95°C for 10 sec, 60°C for 60 sec,
and finally 95°C for 15 sec. Each sample was assayed in triplicate.
The 2−ΔΔCq method was used to determine the relative
expression level of each circRNA (26). We used an unpaired t-test to compare
the expression of circRNAs between cholesteatoma and normal skin
samples. A P-value <0.05 was considered to be statistically
significant.
 | Table I.Primers for RT-qPCR analysis. |
Table I.
Primers for RT-qPCR analysis.
| circRNAs | Primer
sequences |
|---|
|
hsa_circRNA_006562 | F:
5′-ACGAGAAGACCCGCAAGATTAC-3′ |
|
| R:
5′-GCGTTCAGACCTAAGGCTCATC-3′ |
|
hsa_circRNA_084725 | F:
5′-GTAACACTCAGGTCCGTAGAAGA-3′ |
|
| R:
5′-CAGACTGGCTCATACTCGTGT-3′ |
|
hsa_circRNA_101458 | F:
5′-TTTAGACCGTCTGGCTACACC-3′ |
|
| R:
5′-CGTTCTGGGTTGATTCTGTTC-3′ |
|
hsa_circRNA_101965 | F:
5′-CATCCGATCCAGGTGTTTTAC-3′ |
|
| R:
5′-TCAGAAACTTGATCCTGGTGTCT-3′ |
|
hsa_circRNA_102747 | F:
5′-TGTGCTTTCTGGAGGGTCTACT-3′ |
|
| R:
5′-TGCCTCATCACCAACCATAAG-3′ |
|
hsa_circRNA_103276 | F:
5′-TGTTTTCACCAGTCACATCTCTT-3′ |
|
| R:
5′-CCCAGCCCTCAGTTGTATTC-3′ |
| β-actin | F:
5′-GTGGCCGAGGACTTTGATTG-3′ |
|
| R:
5′-CCTGTAACAACGCATCTCATATT-3′ |
GO and KEGG pathway analyses
Gene ontology (GO) provides a ‘framework for the
model of biology’ (http://www.geneontology.org). The ontology describes
the genes, gene product functions, and their inter-relationships.
It classifies functions into three aspects, biological process
(BP), cellular component (CC) and molecular function (MF). Fisher's
exact test was used to elucidate the overlap between the gene list
and the GO annotation list. The -log10 (P-value) was applied to
denote the significance of the GO term enrichment in the analyzed
genes. A lower P-value indicated a more significant GO term
(recommended P-value <0.05). Pathway analysis was performed to
predict molecular interactions and reaction networks by mapping
genes to Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/). The -log10 (P-value) was
used to denote the significance of the pathway correlations,
wherein a lower P-value indicated a more significant correlation
(recommended P-value <0.05).
ceRNA network analysis
Significantly differentially expressed circRNAs were
subjected to ceRNA network analysis. The potential miRNA
recognition elements (MREs) were predicted based on the sequences
of circRNAs and mRNAs. miRNA binding seed sequence sites were
predicted using miRanda (27)
(http://www.microrna.org/microrna/)
and TargetScan (28) (http://www.targetscan.org/). Then miRNAs were
optimized and selected by parameter settings of Context ≤-0.10 and
Context+ ≤-0.10. ceRNAs were filtered based on matching
abilities.
Statistical analysis
In the microarray data analysis, when comparing two
groups of profile differences, the statistical significance of the
difference was estimated using an unpaired t-test. SPSS 20.0
software (IBM, Inc.) was used for statistical analysis. In the
RT-qPCR validation, the data are expressed as the means ± SE using
GraphPad Prism 6.05 software (GraphPad Software, Inc.). Unpaired
t-tests were used to compare the expression of circRNAs between two
groups. P<0.05 was considered to indicare a statistically
significant difference.
Results
Significant differential expression
profiles of circRNAs are found in cholesteatoma compared to matched
normal skin tissues
In the present study, a total of 13,247 circRNAs
were detected by microarray analysis. By comparing cholesteatoma
and normal skin groups, circRNA expression patterns between the
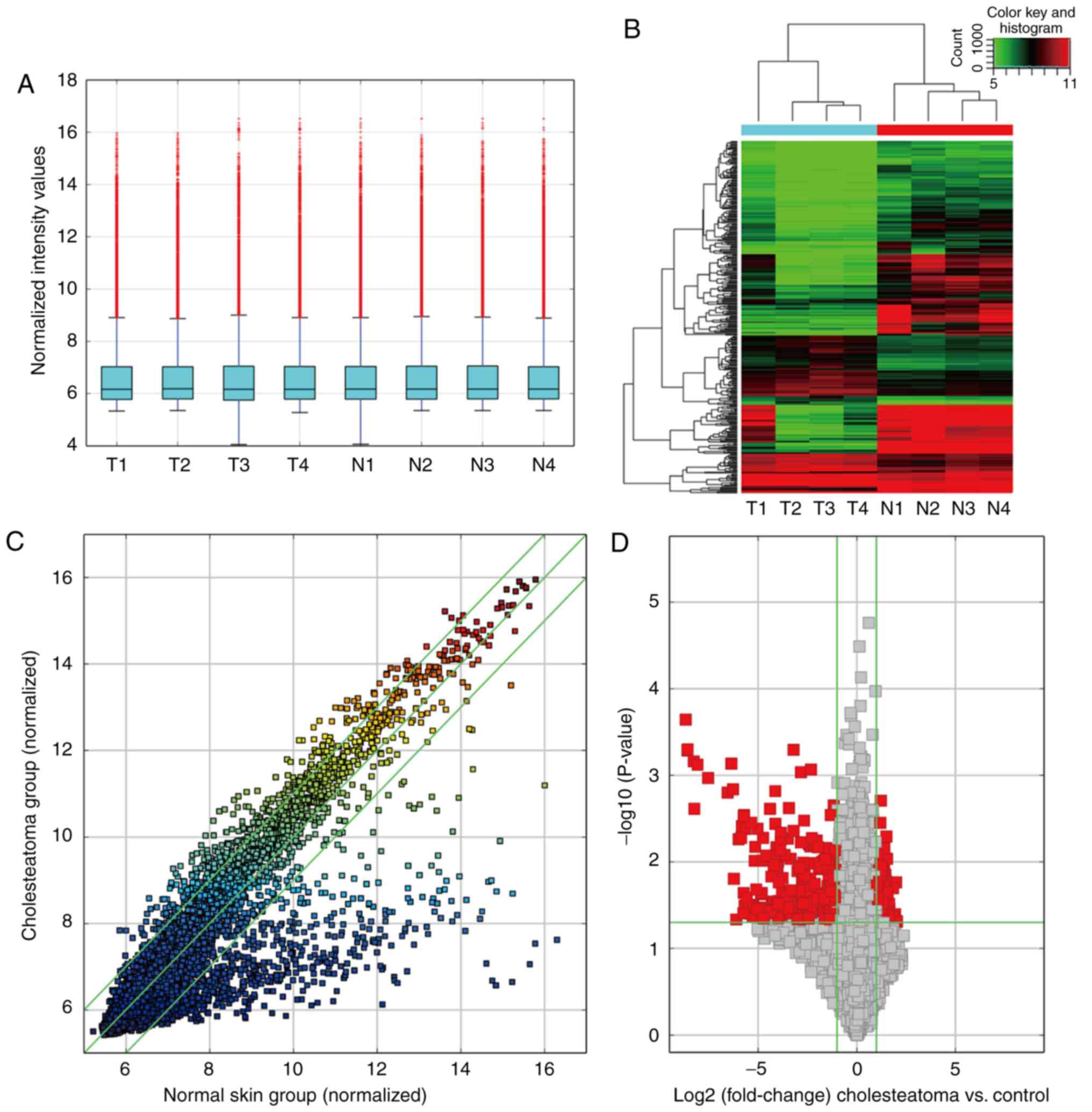
groups were elucidated as being quite different. A box plot was
used to provide a convenient manner in which to visualize and
compare the distributions of expression values for the two groups
(a total of 8 samples) after normalization (Fig. 1A). Hierarchical clustering showed
distinguishable circRNA expression profiling between the two groups
(Fig. 1B). The Scatter-Plot
provided a visualization method for reproducibility distinguishing
the circRNA expression between the two compared groups, indicating
that circRNA expression profiles in cholesteatoma differed markedly
from those of normal skin tissues (Fig.
1C). A volcano plot was utilized to display the differentially
expressed circRNAs with statistical significance between the
cholesteatoma and normal skin groups (Fig. 1D). Therefore, by setting a threshold
of fold change >2.0 and P<0.05, a total of 355 significantly
differentially expressed circRNAs were discriminated in
cholesteatoma compared with normal skin tissues. Among these, 101
circRNAs were identified to be upregulated, whereas 254 were
downregulated (fold change >2.0, P<0.05). The microarray
profile and RNAseq data sets have been deposited into Gene
Expression Omnibus (GEO) with accession number GSE102715
(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102715).
Category characteristics of
dysregulated circRNAs and their chromosomal distributions
In the present study, all the significantly
differentially expressed circRNAs (fold change >2.0, P<0.05)
were classified into five categories: Exonic (76%), antisense (3%),
intronic (12%), sense-overlapping (8%), and intergenic (1%). Among
the upregulated circRNAs, 79 were exonic, 3 antisense, 12 intronic,
and 7 sense-overlapping. Among the downregulated circRNAs, 189 were
exonic, 9 antisense, 30 intronic, 22 sense-overlapping, and 4
intergenic. These results clearly demonstrated that exonic circRNAs
account for the majority of all significantly differentially
expressed circRNAs.
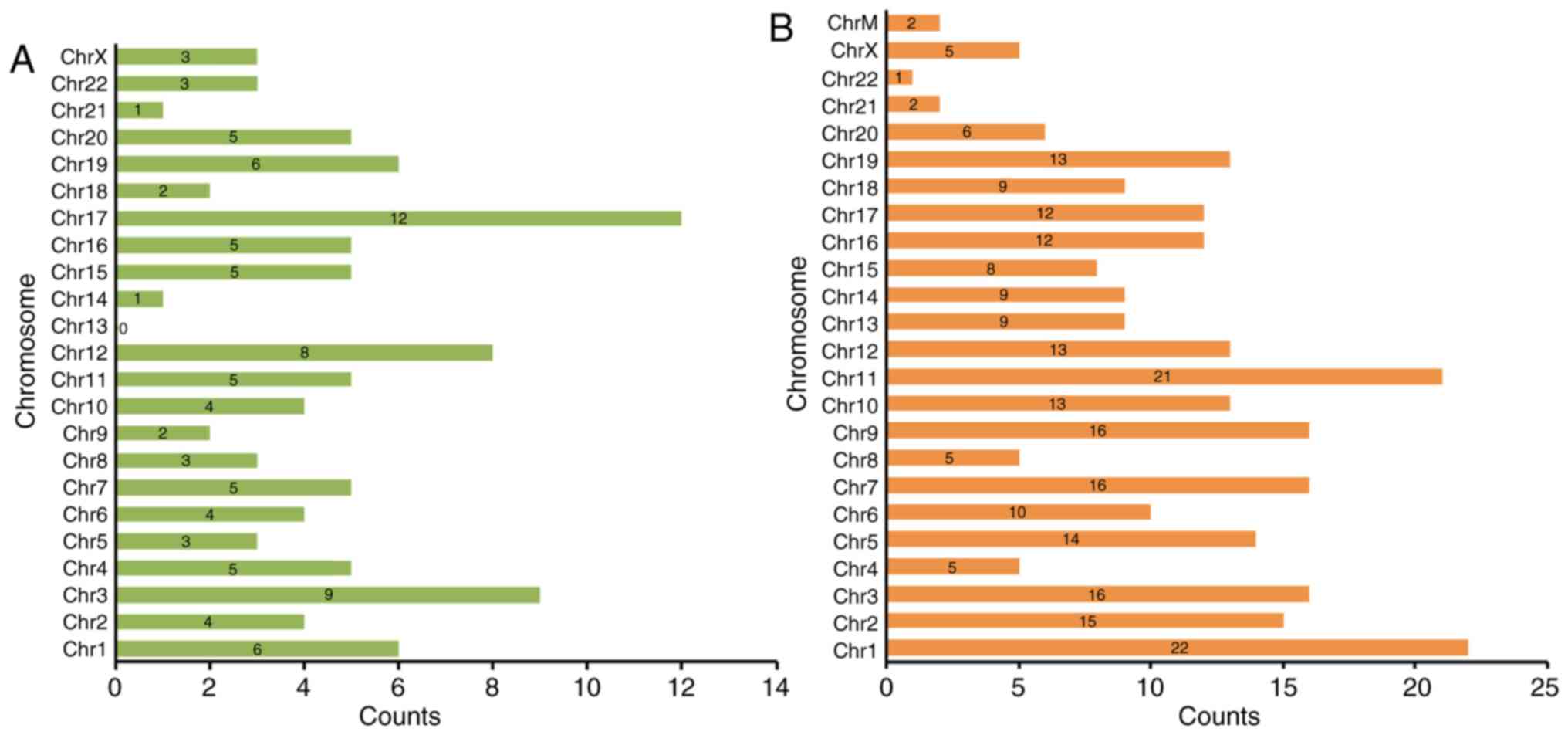
Furthermore, we also analyzed the chromosome
distributions of all significantly differentially expressed
circRNAs (fold change >2.0, P<0.05) (Fig. 2). The dysregulated circRNAs
originated from almost all human genomes, including chromosomes and
the mitochondrial genome (chrM). For the upregulated circRNAs, 12
were located on chromosome 17 (chr17) and 9 on chr3 (Fig. 2A). Among the downregulated circRNAs,
22 were located on chr1 and 21 on chr11 (Fig. 2B).
Microarray expression results are
validated as highly reliable by quantitative reverse
transcription-polymerase chain reaction (RT-qPCR)
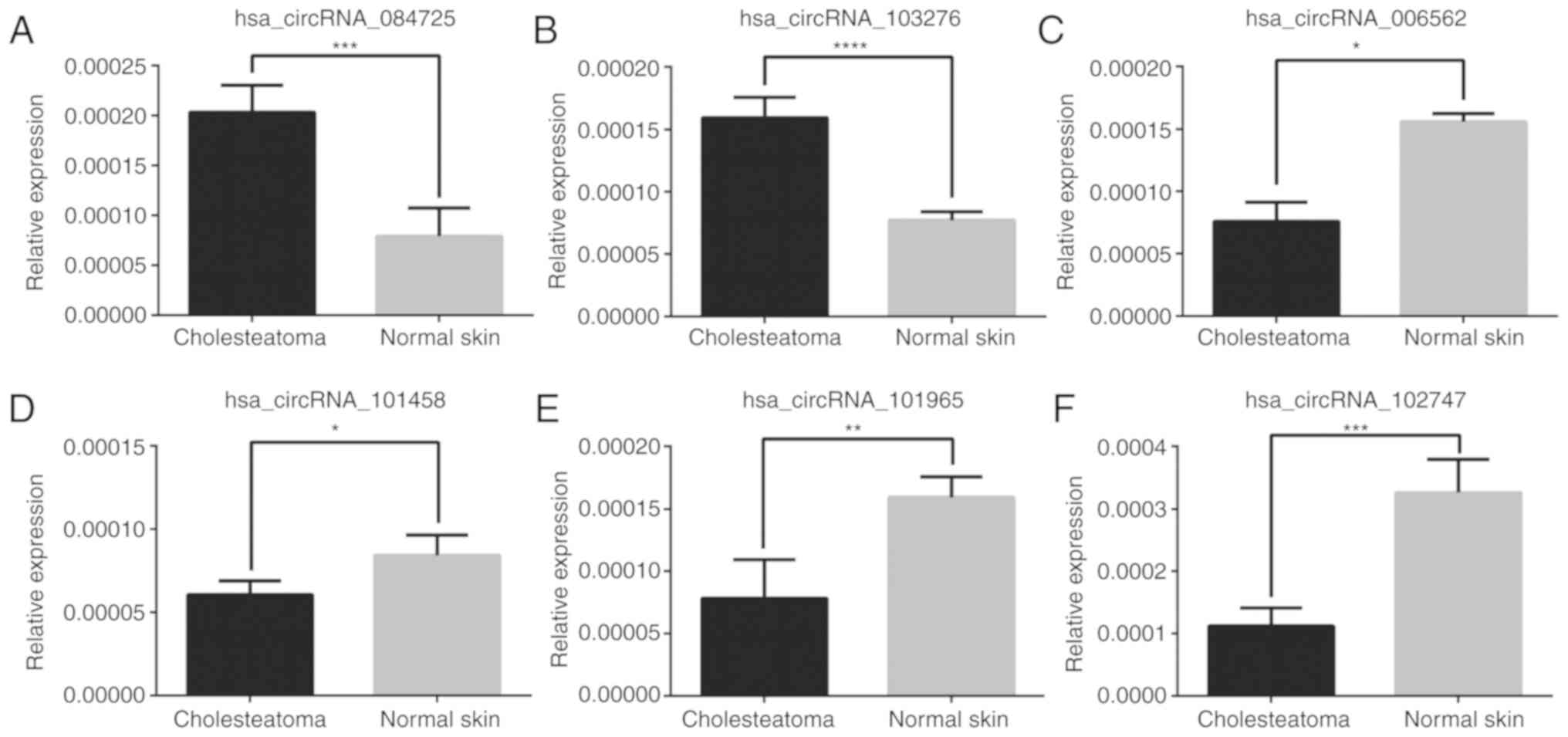
To validate our microarray data, we randomly
selected 6 circRNAs (fold change >2 and P<0.05 from among 355
significantly dysregulated circRNAs for further validation in 7
pairs of cholesteatoma and matched skin tissues (4 pairs of
original tissues and 3 another pairs of cholesteatoma and normal
skin tissues) using RT-qPCR. As depicted in Fig. 3, the relative expression levels of
validated circRNAs were consistent with those in the microarray
data, which indicated our microarray analysis results as being
highly reliable.
Gene ontology and pathway analyses
suggest that circRNAs may regulate multiple biological functions in
cholesteatoma pathogenesis
CircRNAs are generated from the splicing of
protein-coding genes and can regulate the functions of their parent
genes. Therefore, to preliminarily understand the functions of
circRNAs in cholesteatoma, we conducted gene ontology (GO) and
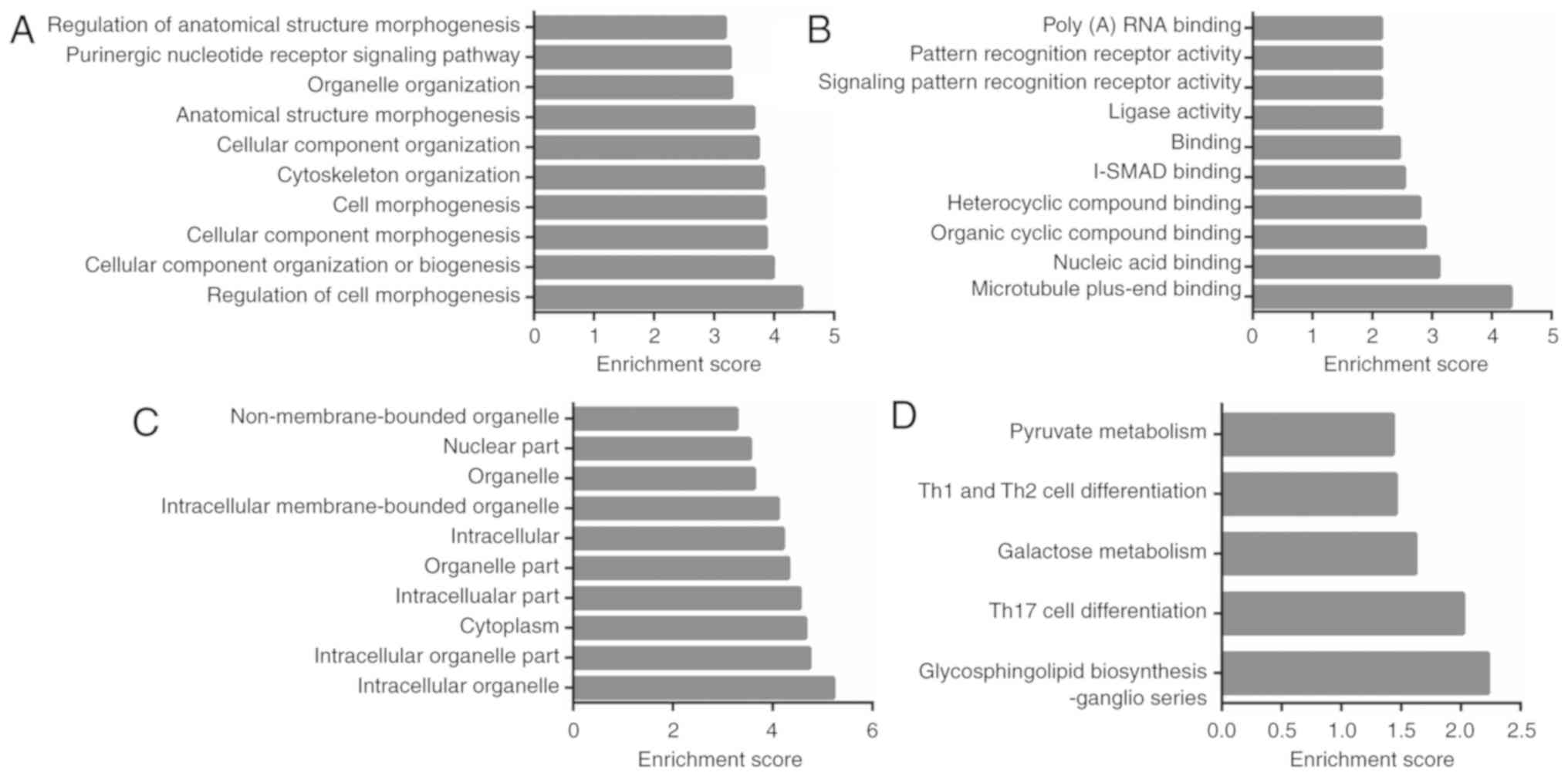
pathway analyses of parent genes of the dysregulated circRNAs. We
examined three aspects, biological process (BP), cellular component
(CC), and molecular function (MF) in the GO analysis. Generally,
212 BP, 64 CC, and 30 MF GO terms were found to be significantly
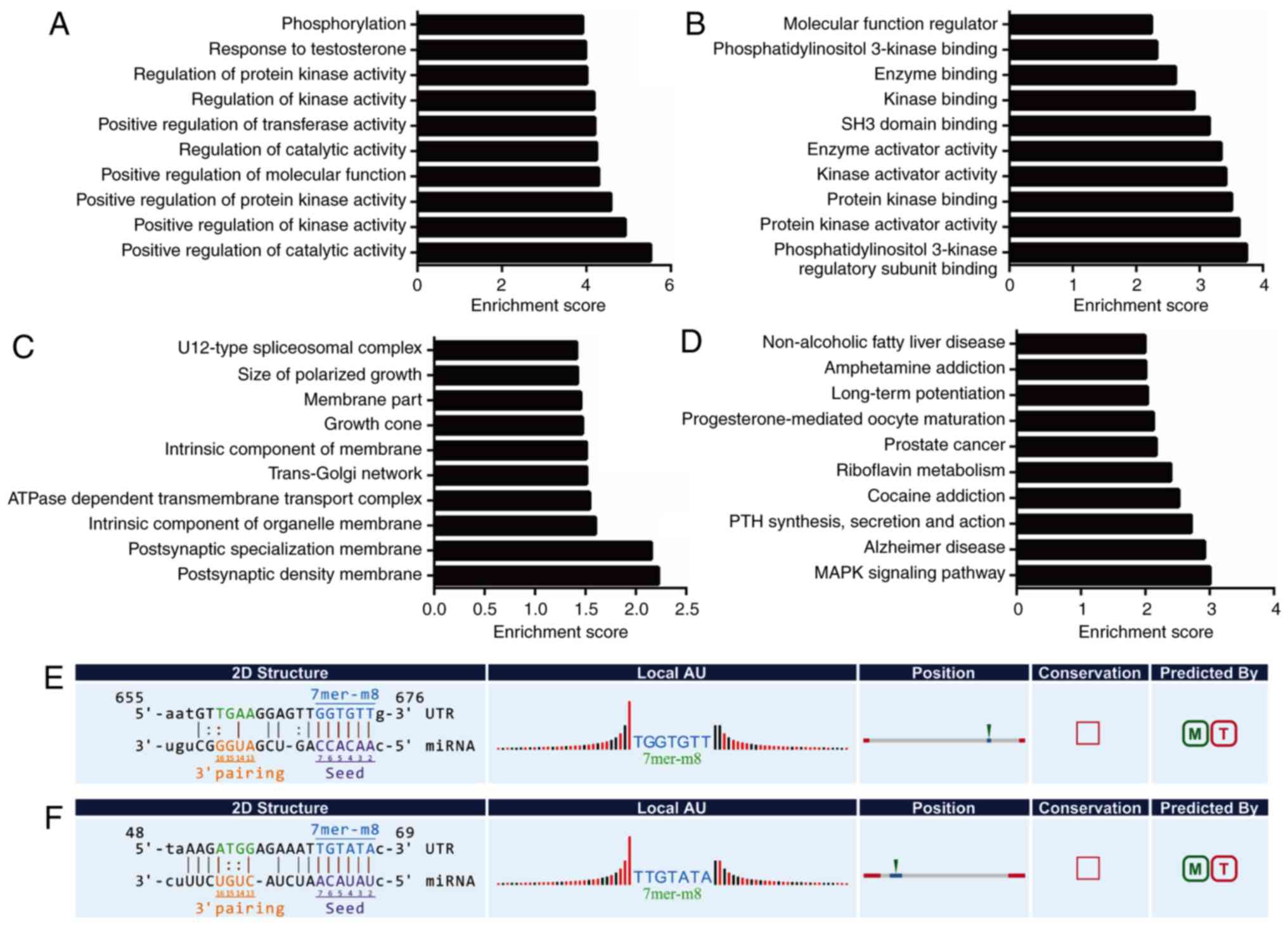
enriched (P<0.05) and the top-10 enriched GO terms of BP, CC, MF
are listed (Fig. 4A-C). The
majority of biological functions in the GO analysis consisted of
cell morphogenesis, cell cycle, cell communication, stimulus
response, and metabolic processes. By mapping the pathway analysis
of parent genes to the Kyoto Encyclopaedia of Genes and Genomes
(KEGG), 5 significantly enriched pathways were found in
cholesteatoma (P<0.05, Fig. 4D).
These 5 pathways, glycosphingolipid biosynthesis, Th17 cell
differentiation, galactose metabolism, Th1 and Th2 cell
differentiation, and pyruvate metabolism, all correlated with cell
growth, cell proliferation, cell migration, cell survival, and
inflammation (29–33).
ceRNA network analysis-elucidated
circRNAs have ceRNA potential in cholesteatoma
According to the ceRNA hypothesis, all RNA
transcripts communicate with each other by harboring multiple miRNA
binding sites (i.e. MREs). In this model, MREs act as an ‘RNA
language’ during the cross-talk of non-coding and coding RNAs
(13). In our previous study, we
confirmed that lncRNAs had ceRNA potential in cholesteatoma
formation (15) (the microarray
profile and RNAseq data sets have been deposited into GEO with
accession number GSE102673 http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102673).
Therefore, to explore the regulatory potential of circRNAs in the
pathogenesis of cholesteatoma, we selected 2 significant
differentially expressed circRNAs (hsa-circRNA-102747,
hsa-circRNA-101458, fold change >2.0, P<0.05, Table II), which shared common MREs with
each other, to generate a circRNA-lncRNA-miRNA-mRNA ceRNA network
(Fig. S1).
| Table II.Significantly differentially
expressed circRNAs for ceRNA network construction (fold change
>2.0, P<0.05). |
Table II.
Significantly differentially
expressed circRNAs for ceRNA network construction (fold change
>2.0, P<0.05).
| circRNA | Gene symbol | Fold change | P-value | Regulation |
|---|
|
hsa_circRNA_102747 | ACTR2 |
2.32100 | 0.0190592 | Downregulated |
|
hsa_circRNA_101458 | HERC2P3 | 27.86245 | 0.0380877 | Downregulated |
The network was shown to be composed of 2 circRNA
nodes, 31 lncRNA nodes, 48 miRNA nodes and 248 mRNA nodes. GO and
pathway analyses were also conducted for the ceRNA network. The
results displayed there were a total of 520 significantly enriched
GO terms (P<0.05) in the network, including 461 BP, 17 CC, 42 MF
terms and the top-10 enriched GO terms of BP, CC, MF are listed
(Fig. 5A-C). In the GO analysis, we
identified that almost all the GO processes were closely related
with various metabolic processes. In the KEGG pathway analysis, 24
enriched pathways were discovered, of which the MAPK signaling
pathway (hsa04010) has previously drawn the attention of
researchers owing to its function in cancer (34) (Fig.
5D).
In the network, we noted that circRNA-102747
interacted with miR-21-3p (Figs. 5E
and 6), a microRNA belonging to the
miR-21 family, which has been confirmed to promote the formation
and invasion of cholesteatoma (7,8).
Furthermore, circRNA-101458 was found to interact with miR
let-7a-3p (Figs. 5F and 7), a microRNA belonging to the miR let-7a
family, the upregulation of which has been considered to have an
antiproliferative effect and contribute to the benign nature of
cholesteatoma (11,35). The complementarity between the 2
candidate circRNAs and the 2 miRNAs was perfect according to
7mer-m8 matching types (Fig. 5E and
F). In addition, in the circRNA-102747-mediated ceRNA network,
circRNA-102747 and lncRNA-uc001kfc.1 both interacted with miR-21-3p
(Fig. 6); of which
lncRNA-uc001kfc.1 was considered to play a key role in
cholesteatoma pathogenesis in our previous research study (15).
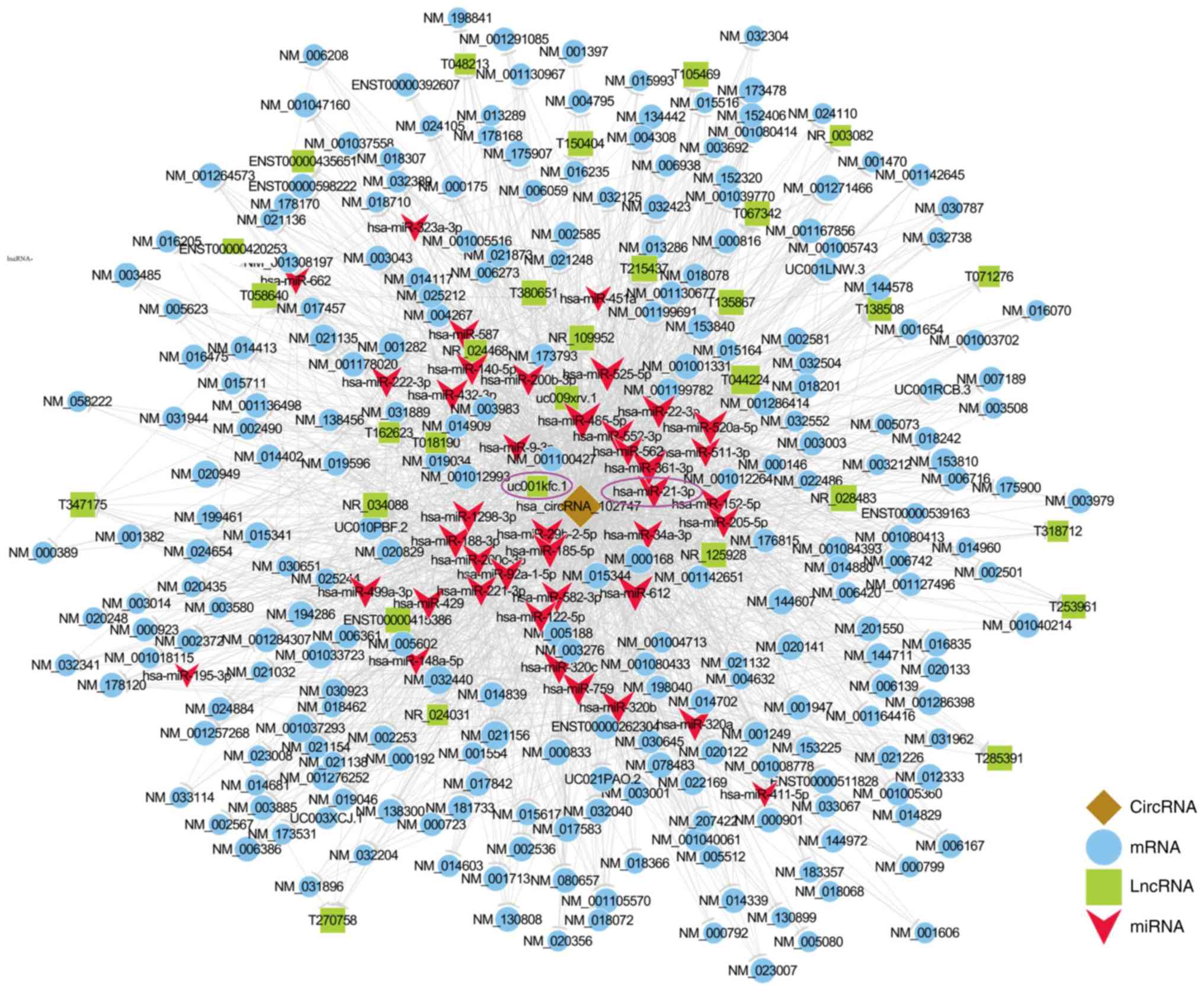 | Figure 6.circRNA-102747-mediated
circRNA-lncRNA-miRNA-mRNA ceRNA network. The network was based on
circRNA-lncRNA-miRNA-mRNA interactions. In the network,
hsa-miR-21-3p, which shares microRNA response element with both
circRNA-102747 and lncRNA-uc001kfc.1, was verified to play a vital
role in cholesteatoma. Both hsa-miR-21-3p and lncRNA-uc001kfc.1 are
circled in purple. Blue, mRNA; green, lncRNA; red, miRNA. circRNA,
circulating RNA; lncRNA, long non-coding RNA; miRNA, microRNA;
mRNA, messenger RNA; ceRNA, competing endogenous RNA. |
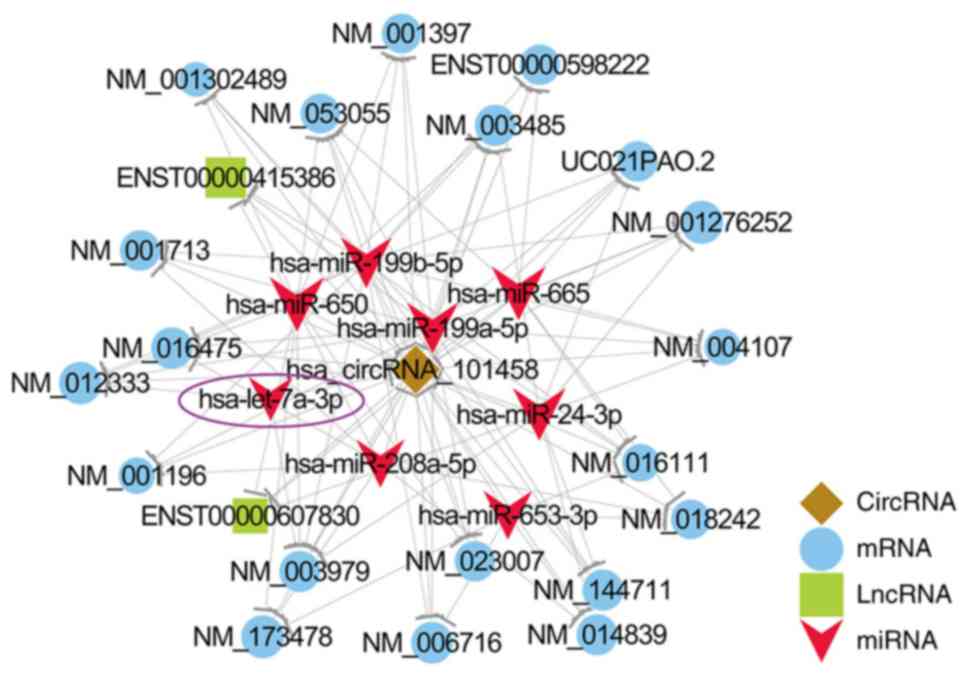 | Figure 7.circRNA-101458-mediated
circRNA-lncRNA-miRNA-mRNA ceRNA network. In the network, miR
let-7a-3p, which shares microRNA response element with both
circRNA-101458, was verified to play a vital role in cholesteatoma.
miR let-7a-3p is circled in purple. Blue, mRNA; green, lncRNA; red,
miRNA. circRNA, circulating RNA; lncRNA, long non-coding RNA;
miRNA, microRNA; mRNA, messenger RNA; ceRNA, competing endogenous
RNA. |
Discussion
In the present study, by microarray analysis, we
discovered that circulating RNA (circRNA) expression profiles in
cholesteatoma were significantly dysregulated compared with those
in normal skin tissues with 101 circRNAs upregulated and 254
downregulated, indicating that circRNAs may play crucial roles in
cholesteatoma. In the category analysis of differentially expressed
circRNAs, exonic circRNAs accounted for the largest type by 76%.
This is likely functionally relevant, because exonic circRNAs are
derived from exonic regions within coding genes directly through
back splicing by covalently linking the 3′ end of an exon with the
5′ end of either the same exon or the upstream exon. This signature
structure protects exonic circRNAs from ‘exon skipping’ and renders
them more inclined to regulate the linear coding RNAs from which
they are generated (36–39). In the chromosome distribution
analysis, we found that differentially expressed circRNAs were more
inclined to be located on chr17, chr1, and chr11. Furthermore,
Ecsedi et al demonstrated that chromosomal imbalances play
an important role in cell proliferation activation and bone
invasion (40); thus, our finding
may partially support this report. In addition, the RT-qPCR results
were verified to coincide with the microarray data, which indicated
that the microarray analysis was highly reliable. Together, these
circRNA profile analyses suggest that circRNAs have regulatory
function potential in the epigenetic regulatory mechanism of
cholesteatoma formation.
To preliminarily understand the functions of
circRNAs in cholesteatoma, we performed functional analysis on the
parental genes of the dysregulated circRNAs in cholesteatoma. This
revealed that the majority of related biological processes involved
cell morphogenesis, cell cycle, cell communication, stimulus
response, and metabolic processes. Pathway analysis elucidated that
‘glycosphingolipid biosynthesis’, ‘Th17 cell differentiation’,
‘galactose metabolism’, ‘Th1 and Th2 cell differentiation’, and
‘pyruvate metabolism’ were all enriched, which correlate with cell
growth, cell proliferation, cell migration, cell survival, and
inflammation associations (29–33).
Considering that cholesteatoma is a disease caused by the
hyper-proliferation of keratinocytes, in the present study, the
functional analyses of the parental genes of dysregulated circRNAs
together implied that circRNAs may contribute to cholesteatoma
formation.
According to the competing endogenous RNA (ceRNA)
hypothesis, multiple miRNA binding sites (MREs) can act as an ‘RNA
language’ during the cross-talk of non-coding and coding RNAs
(13). To verify our hypothesis and
explore whether circRNAs could function as ceRNAs in the
pathogenesis of cholesteatoma, we selected 2 significantly
differentially expressed circRNAs (circRNA-102747, circRNA-101458,
fold change >2.0, P<0.05), which shared common MREs with each
other, to generate a circRNA-lncRNA-miRNA-mRNA ceRNA network
(Fig. S1). Functional analysis of
the ceRNA network revealed that multiple enriched GO processes in
the network were associated with various metabolic processes, such
as protein kinase activity, phosphatidylinositol 3-kinase (PI3K)
binding. PI3Ks are enzymes that catalyze the phosphorylation of
phosphatidylinositol (PtdIns). The PI3K pathway plays a key role in
the regulation of cell survival and proliferation (41). A previous study reported that
activation of the PI3K/Akt (Akt, i.e. serine kinase PKB, a
downstream effector of PI3K) signaling pathway protects epithelial
keratinocytes of cholesteatoma against programmed cell death
(42). Moreover, increased PI3K/Akt
signaling pathway activation has been proven to be related to
cholesteatoma epithelial hyper-proliferation (43,44).
In the KEGG pathway analysis of the ceRNA network, the most
enriched pathway was found to be the MAPK (mitogen-activated
protein kinase) signaling pathway, activation of which has
previously been proven to play an important role in the terminal
differentiation in cholesteatoma epithelium (42).
In the network, we noted that circRNA-102747 and
lncRNA-uc001kfc.1 both interacted with miR-21-3p, and
lncRNA-uc001kfc.1 was downregulated in cholesteatoma and was
considered to function as an ‘endogenous sponge’ for miR-21-3p in
cholesteatoma pathogenesis in our previous research (15). In the present study, compared with
normal skin tissues, circRNA-102747 was also confirmed to have low
expression in cholesteatoma by both microarray analysis and
RT-qPCR. The 2D structure of circRNA-102747 and miR-21-3p showed
that the complementarity between them was perfect with the miRNA
seed sequence AACACC. Furthermore, our previous study indicated
that the binding site of miR-21-3p on lncRNA-uc001kfc.1 was also
perfectly matching with the same miRNA seed sequence AACACC
(15). The miRNA seed sequence,
which is the nucleotides 2–7 of the 5′ region of the miRNA and
considered as the most conserved portion of the miRNAs, is
particularly important for miRNA recognition (12,45).
The perfectly matching types with the same miRNA seed sequence
further confirmed that both circRNA-102747 and lncRNA-uc001kfc.1
could share the same MRE with miR-21-3p. Therefore, we presume that
by using the MRE as ‘RNA language’ (13), circRNA-102747, together with
lncRNA-uc001kfc.1, functions as an ‘endogenous sponge’ for
miR-21-3p. In previous studies, miR-21 has been proven to be
upregulated in cholesteatoma (7).
Overexpression of miR-21 was found to promote cell proliferation
and invasion of keratinocytes, which contributed to the malignant
nature of cholesteatoma (7,11). In the crosstalk of
circRNA-102747/lncRNA-uc001kfc.1/miR-21-3p/targeted mRNAs, when
circRNA-102747 and lncRNA-uc001kfc.1 are downregulated in
cholesteatoma, hsa-miR-21-3p becomes transcriptionally active and
thus regulates targeted genes and results in the
hyper-proliferation of keratinocytes.
In the circRNA-101458-mediated ceRNA network, we
found that circRNA-101458 interacted with miR let-7a-3p, a microRNA
belonging to the miR let-7a family. The upregulation of miR let-7
can suppress cancer development by targeting specific oncogenes and
is considered to have an antiproliferative effect (35,46).
As is known, cholesteatoma is manifested by certain malignant
characteristics, such as excessive squamous epithelial cell
proliferation. However, cholesteatoma pathologically displays
benign tumors without abnormal keratinocyte mitosis. In previous
research, miR let-7a has been confirmed to be upregulated in
cholesteatomas and contribute to the benign nature of cholesteatoma
(11). In the present study,
circRNA-101458 was found to be significantly downregulated in
cholesteatoma. Herein we presume that circRNA-101458 may play a key
role in cholesteatoma pathogenesis and function as an ‘endogenous
sponge’ for let-7a-3p; in the crosstalk of circRNA-101458/miR
let-7a-3p/targeted mRNAs, when circRNA-101458 is downregulated in
cholesteatoma, miR let-7a-3p becomes transcriptionally active and
thus regulating targeted genes resulting in the anti-proliferation
of keratinocytes in cholesteatoma. Therefore, we formed the
hypothesis that in cholesteatoma, the
circRNA-102747/lncRNA-uc001kfc.1/miR-21-3p/targeted mRNAs may act
as the regulator of malignant characteristics, whereas
circRNA-101458/miR let-7a-3p/targeted mRNAs function as a benign
regulator. This suggests that circRNA-102747 and circRNA-101458
have ceRNA potential in the pathogenesis of cholesteatoma and can
be potentially therapeutic targets for the drug therapy of
cholesteatoma. In future studies, we will focus on the functional
studies and elucidate the ceRNA-mediated effects of circRNAs in the
pathogenesis of cholesteatoma.
Although we put forward the assumption that the
circRNA-miRNA-targeted mRNAs axis may be involved in the mechanism
of cholesteatoma pathogenesis, some limitations must be considered
in the present study. Firstly, the sample size in the study was
relatively small and the scale of circRNA profile might be
decreased with larger number of subjects. However, as there are few
similar studies and it is the first study of circRNAs in
cholesteatoma, it adds important knowledge to the current field.
Secondly, we only studied the circRNA profiles and their ceRNA
potential in the pathogenesis of cholesteatoma, while the
functional analysis is imperfect. Functional research, such as
selectively upregulating or downregulating the expression of
certain circRNAs in vitro or in vivo, are needed to
validate the exact role of circRNAs in cholesteatoma pathogenesis
in future studies.
In conclusion, our study elucidates the profiles of
circRNAs in cholesteatoma for the first time by using microarray
analysis. Significantly differentially expressed circRNAs were
found in cholesteatoma compared with the normal skin group and
their functions were predicted through GO and pathway analyses, the
results of which together indicate that circRNAs may contribute to
the pathogenesis of cholesteatoma. By constructing circRNA-mediated
ceRNA networks, we found that circRNAs may function as ceRNAs that
could sequester targeted miRNAs and influence the associated gene
functions. circRNAs may thus constitute promising therapeutic
agents for cholesteatoma. In future studies, we will focus on the
functional analysis of circRNAs to explore the precise molecular
mechanisms of cholesteatoma.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article or are available from
the corresponding author on reasonable request. The accession
number for circRNA-seq data in this study is GSE102715 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102715).
Authors' contributions
Conception and design of the study was carried out
by HY and JG. JG, QT, RX, XZ, SW, YZ and WL performed the
experiments. Acquisition of the data was carried out by JG and HY.
Analysis and interpretation of the data were the responsibility of
JG and HY. Reagents/materials/analysis tools were procured by JG,
SW, ZG and HY. Drafting of the article and revising it critically
for important intellectual content was carried out by JG and HY.
Final approval of the version was submitted by HY. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
All tissue donors participating in this study
provided written informed consent. Ethical approval for our study
was obtained from the Ethics Committee of Peking Union Medical
College Hospital (no. S-K292).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
financial interests.
References
|
1
|
Louw L: Acquired cholesteatoma
pathogenesis: Stepwise explanations. J Laryngol Otol. 124:587–593.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Britze A, Moller ML and Ovesen T:
Incidence, 10-year recidivism rate and prognostic factors for
cholesteatoma. J Laryngol Otol. 131:319–328. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ergun S, Zheng X and Carlsoo B: Expression
of transforming growth factor-alpha and epidermal growth factor
receptor in middle ear cholesteatoma. Am J Otol. 17:393–396.
1996.PubMed/NCBI
|
|
4
|
Olszewska E, Wagner M, Bernal-Sprekelsen
M, Ebmeyer J, Dazert S, Hildmann H and Sudhoff H: Etiopathogenesis
of cholesteatoma. Eur Arch Otorhinolaryngol. 261:6–24. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kojima H, Shiwa M, Kamide Y and Moriyama
H: Expression and localization of mRNA for epidermal growth factor
and epidermal growth factor receptor in human cholesteatoma. Acta
Otolaryngol. 114:423–429. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kupper TS: The activated keratinocyte: A
model for inducible cytokine production by non-bone marrow-derived
cells in cutaneous inflammatory and immune responses. J Invest
Dermatol. 94 (6 Suppl):146s–150s. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Friedland DR, Eernisse R, Erbe C, Gupta N
and Cioffi JA: Cholesteatoma growth and proliferation:
Posttranscriptional regulation by microRNA-21. Otol Neurotol.
30:998–1005. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen X, Li X and Qin Z: MicroRNA-21
promotes the proliferation and invasion of cholesteatoma
keratinocytes. Acta Otolaryngol. 136:1261–1266. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang W, Chen X and Qin Z: MicroRNA let-7a
suppresses the growth and invasion of cholesteatoma keratinocytes.
Mol Med Rep. 11:2097–2103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li N and Qin ZB: Inflammation-induced
miR-802 promotes cell proliferation in cholesteatoma. Biotechnol
Lett. 36:1753–1759. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen X and Qin Z: Post-transcriptional
regulation by microrna-21 and let-7a microRNA in paediatric
cholesteatoma. J Int Med Res. 39:2110–2118. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bak RO and Mikkelsen JG: miRNA sponges:
Soaking up miRNAs for regulation of gene expression. Wiley
Interdiscip Rev RNA. 5:317–333. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao J, Tang Q, Zhu X, Wang S, Zhang Y, Liu
W, Gao Z and Yang H: Long noncoding RNAs show differential
expression profiles and display ceRNA potential in cholesteatoma
pathogenesis. Oncol Rep. 39:2091–2100. 2018.PubMed/NCBI
|
|
16
|
Jeck WR, Sorrentino JA, Wang K, Slevin MK,
Burd CE, Liu J, Marzluff WF and Sharpless NE: Circular RNAs are
abundant, conserved, and associated with ALU repeats. RNA.
19:141–157. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Memczak S, Jens M, Elefsinioti A, Torti F,
Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer
M, Loewer A, et al: Circular RNAs are a large class of animal RNAs
with regulatory potency. Nature. 495:333–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Salzman J, Gawad C, Wang PL, Lacayo N and
Brown PO: Circular RNAs are the predominant transcript isoform from
hundreds of human genes in diverse cell types. PLoS One.
7:e307332012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Starke S, Jost I, Rossbach O, Schneider T,
Schreiner S, Hung LH and Bindereif A: Exon circularization requires
canonical splice signals. Cell Rep. 10:103–111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo JU, Agarwal V, Guo H and Bartel DP:
Expanded identification and characterization of mammalian circular
RNAs. Genome Biol. 15:4092014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang M, Zhong Z, Lv M, Shu J, Tian Q and
Chen J: Comprehensive analysis of differentially expressed profiles
of lncRNAs and circRNAs with associated co-expression and ceRNA
networks in bladder carcinoma. Oncotarget. 7:47186–47200.
2016.PubMed/NCBI
|
|
23
|
Fan X, Weng X, Zhao Y, Chen W, Gan T and
Xu D: Circular RNAs in Cardiovascular Disease: An Overview. Biomed
Res Int. 2017:51357812017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang S, Zhu D, Li H, Li H, Feng C and
Zhang W: Characterization of circRNA-Associated-ceRNA networks in a
senescence-accelerated mouse prone 8 brain. Mol Ther. 25:2053–2061.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pasquinelli AE: MicroRNAs and their
targets: Recognition, regulation and an emerging reciprocal
relationship. Nat Rev Genet. 13:271–282. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Enright AJ, John B, Gaul U, Tuschl T,
Sander C and Marks DS: MicroRNA targets in Drosophila. Genome Biol.
5:R12003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
D'Angelo G, Capasso S, Sticco L and Russo
D: Glycosphingolipids: Synthesis and functions. FEBS J.
280:6338–6353. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Patel DD and Kuchroo VK: Th17 Cell Pathway
in Human Immunity: Lessons from genetics and therapeutic
interventions. Immunity. 43:1040–1051. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gray LR, Tompkins SC and Taylor EB:
Regulation of pyruvate metabolism and human disease. Cell Mol Life
Sci. 71:2577–2604. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Coelho AI, Berry GT and Rubio-Gozalbo ME:
Galactose metabolism and health. Curr Opin Clin Nutr Metab Care.
18:422–427. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kidd P: Th1/Th2 balance: The hypothesis,
its limitations, and implications for health and disease. Altern
Med Rev. 8:223–246. 2003.PubMed/NCBI
|
|
34
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Johnson CD, Esquela-Kerscher A, Stefani G,
Byrom M, Kelnar K, Ovcharenko D, Wilson M, Wang X, Shelton J,
Shingara J, et al: The let-7 microRNA represses cell proliferation
pathways in human cells. Cancer Res. 67:7713–7722. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jeck WR and Sharpless NE: Detecting and
characterizing circular RNAs. Nat Biotechnol. 32:453–461. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ashwal-Fluss R, Meyer M, Pamudurti NR,
Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N and
Kadener S: circRNA biogenesis competes with pre-mRNA splicing. Mol
Cell. 56:55–66. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lasda E and Parker R: Circular RNAs:
Diversity of form and function. RNA. 20:1829–1842. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ebbesen KK, Hansen TB and Kjems J:
Insights into circular RNA biology. RNA Bio. 14:1035–1045. 2017.
View Article : Google Scholar
|
|
40
|
Ecsedi S, Rakosy Z, Vizkeleti L, Juhász A,
Sziklai I, Adány R and Balázs M: Chromosomal imbalances are
associated with increased proliferation and might contribute to
bone destruction in cholesteatoma. Otolaryngol Head Neck Surg.
139:635–640. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huisman MA, De Heer E and Grote JJ:
Survival signaling and terminal differentiation in cholesteatoma
epithelium. Acta Otolaryngol. 127:424–429. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yune TY and Byun JY: Expression of PTEN
and phosphorylated Akt in human cholesteatoma epithelium. Acta
Otolaryngol. 129:501–506. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hawkins PT, Anderson KE, Davidson K and
Stephens LR: Signalling through Class I PI3Ks in mammalian cells.
Biochem Soc Trans. 34:647–662. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lim LP, Lau NC, Weinstein EG, Abdelhakim
A, Yekta S, Rhoades MW, Burge CB and Bartel DP: The microRNAs of
Caenorhabditis elegans. Genes Dev. 17:991–1008. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Park SM, Shell S, Radjabi AR, Schickel R,
Feig C, Boyerinas B, Dinulescu DM, Lengyel E and Peter ME: Let-7
prevents early cancer progression by suppressing expression of the
embryonic gene HMGA2. Cell Cycle. 6:2585–2590. 2007. View Article : Google Scholar : PubMed/NCBI
|