Introduction
Tumor angiogenesis is a hallmark of liver cancer
that is necessary for tumor growth and progression (1–3).
Vascular endothelial growth factor (VEGF) is crucial for the
development of tumor angiogenesis and exerts its functions by
interacting with the tumor microenvironment (4). Drugs targeting the VEGF receptor
(VEGFR) for antitumor and anti-angiogenic purposes were considered
to be promising for clinical tumor therapy (4,5).
However, clinical data have indicated that traditional
anti-vascular treatment was limited and vasculogenic mimicry (VM)
formation may contribute to tumor development (3).
VM formed by malignant tumor cells serves a role in
tumor growth and metastasis, leading to poor patient prognosis
(6). The presence of VM may trigger
several cascades that increase the availability of oxygen and
nutrients from endothelial vessels to tumor tissues, thereby
promoting tumor progression (7).
The development of vasculogenic mimicry (VM) is similar to that of
endothelium-dependent vessels (EDVs) (6,8).
Vascular endothelial-cadherin (VE-cadherin), epithelial cell kinase
(EphA2), phosphoinositide 3-kinase-α (PI3K-α), matrix
metalloproteinases (MMPs), laminin 5 (Ln-5) γ2 chain,
hypoxia-inducible factor-1α (HIF-1α), focal adhesion kinase (FAK)
and p38 (9–13) are proteins and signaling pathways
that promote cell proliferation, migration, invasion and matrix
remodeling during VM formation (6).
For example, VE-cadherin induces EphA2 phosphorylation by
modulating the interaction of EphA2 with its membrane-bound ligand.
Additionally, PI3K activation via FAK and ERK1/2-MAPK further
mediates MMP14 and MMP2, resulting in VM formation (14–16).
In ovarian cancer, the expression of VE-cadherin, EphA2 and MMPs
was revealed to be upregulated by VEGF factor A, promoting matrix
plasticity and VM formation (17).
Supervillin (SVIL) is an actin and
membrane-associated protein that belongs to the largest sub-family
of the villin/gelsolin superfamily (18,19).
In tumor cells, SVIL comprises several isoforms, which have been
implicated at each step of tumor development, including cell
survival, migration and metastasis (20–22).
As previously reported, these proteins are involved in cell
spreading, lamellipodia extension, actin filament assembly and
focal adhesion maturation and/or disassembly (19,23–25).
Furthermore, SVIL promotes cancer cell survival by regulating p53
levels (24). A previous study
revealed that hypoxia induced an increased SVIL expression, leading
to cancer metastasis and poor survival in patients with liver
cancer (22). However, the
functional relationships between SVIL and tumor angiogenesis/VM
formation in liver cancer have not yet been fully elucidated.
Materials and methods
Cell culture, small interfering
(si)RNA transfection and VEGF treatment
Human umbilical vein endothelial cells (HUVECs) were
purchased from AllCells LLC (cat. no. H-001F-C) and cultured with
HUVEC medium (cat. no. H-004; AllCells LLC) in a 0.25%
gelatin-coated culture flask. HepG2, Bel7405 and MHCC-97H liver
cancer cell lines were cultured in DMEM medium (cat. no. C11995;
Gibco; Thermo Fisher Scientific, Inc.) and provided by Professor ZY
Tang (Liver Cancer Institute, Fudan University, Shanghai, China),
which were used in previous studies (22,26).
These cell lines were characterized by DNA fingerprinting and
isozyme detection. All cell lines used in the present study were
regularly authenticated via morphologic observation and tested for
the absence of mycoplasma contamination. Samples were last tested
for mycoplasma in March 2017.
Cells were transfected with SVIL Stealth siRNA: E4
double stranded (ds)RNA, E5 dsRNA, E11 dsRNA and negative control
dsRNA (all Invitrogen; Thermo Fisher Scientific, Inc.; all 40 nM)
using Lipofectamine® RNAi-MAX (Invitrogen; Thermo Fisher
Scientific, Inc.). The dsRNA targeting sequences were as follows:
E4 dsRNA, 5′-CUCACUUUGAAUGUAGAGAACCAUC-3′; E5 dsRNA,
5′-UUCUGCUGAAGUUAUAGGUUGGGUU-3′; E11 dsRNA,
5′-AGCAUAUUUAGAUUCCUUAUGGCUG-3′.
HepG2 cells were treated with or without 50 µg/l
recombinant human VEGF (Novus Biologicals, LLC) at the indicated
time-points.
The Cancer Genome Atlas (TCGA) data
analysis
The TCGA (https://tcga-data.nci.nih.gov/tcga/) cohort included
normal tissues (n=50) and tumor tissues (n=347). The relationship
between the expression of genes in liver cancer was analyzed using
this database.
Immunohistochemistry (IHC)
staining
A liver cancer tissue microarray consisting of 173
pathological samples was purchased from US Biomax, Inc. Liver
cancer tissue microarrays were immunohistochemically treated with
antibodies against SVIL (dilution 1:1,000; cat. no. S8695;
Sigma-Aldrich, Inc.) (24), cluster
of differentiation (CD) 31 (dilution 1:1,500; product no. 3528;
Cell Signaling Technology, Inc.), CD34 (dilution 1:50; product no.
3569; Cell Signaling Technology, Inc.), CD146 (dilution 1:500; cat.
no. GTX60775; GeneTex, Inc.) and CD248 (dilution 1:500; cat. no.
564993; BD Biosciences). Tissue was sectioned and incubated in
65–75°C for 90 min. Samples were then placed in xylene in 25°C for
10 min and re-treated with xylene in 25°C for a further 10 min. The
sections were sequentially placed in 100, 95 and 80% ethanol, after
which purified water was used for 5 min. Samples were placed into a
repair box with antigen repair solution (citrate buffer). A
pressure cooker was heated under 1,600 W to automatically deflate
for 2 min and samples were removed for 2 min for cooling. The
antigen retrieval solution was discarded and sections were rinsed
with PBS. The samples were permeabilized with 0.5% Triton X-100
(prepared in PBS) at room temperature for 20 min. Sections were
transferred to a wet box and freshly prepared 3% hydrogen peroxide
was added in 25°C for 10 min to remove endogenous peroxidase
blocking solution. Samples were subsequently incubated for 10 min
at room temperature and rinsed with PBS. During washing, slides
were immersed in PBS 3 times for 3 min each and blotted dry. Normal
goat serum (cat. no. AR0009; Boster Biological Technology co., Ltd)
was added dropwise for blocking at room temperature for 30 min.
Each slide was dropped with a sufficient quantity of diluted
primary antibodies into a wet box and incubated overnight at 4°C.
Rabbit/mouse secondary antibodies (MaxVision TM HRP-Polymer
anti-Mouse IHC Kit; cat. no. 5001; MXB Biotechnologies) were
subsequently added and incubated for 1 h at room temperature, after
which slides were washed with PBS. DAB was developed for 3 min in
25°C and the degree of staining was assessed under a light
microscope (magnification, ×100 and ×200). Samples were then rinsed
again with PBS or tap water for 1 min. The samples were then
counterstained with hematoxylin in 25°C for 3 min. Samples were
subsequently rinsed with tap water for 1 min. Subsequently,
dehydration, transparency, sealing, and microscopic examination
were performed. The KF-PRO Digital Slide Scanning System (Kongfong
Biotech International Co., Ltd.) was used to visualize the
resulting signal.
Matrigel tube formation assay
After incubation for 30 min at 37°C,
2×104 cells were added to a 96-well plate coated with 35
µl Matrigel at a concentration of 8.8 mg/ml. Following incubation
for 6 h (liver cancer cells) or at the indicated time-points (0, 2,
4, 6 and 8 h) (HUVEC cells) at 37°C, three non-overlapping light
microscopic images were randomly obtained at low-power
magnifications (magnification, ×100). Total tube length and the
number of branching points formed by endothelial or liver cancer
cells per field were measured using Angio Tool 64 0.6a software
(National Cancer Institute).
Western blotting
Cells were lysed in RIPA lysis buffer (cat. no.
P0013K; Beyotime Institute of Biotechnology) to obtain a protein
lysate. Then the protein quantification was determined by BCA. Each
8% SDS gel was infused with 40 µg of protein product. Total protein
was separated by SDS-PAGE and transferred to nitrocellulose
membranes. Samples were then blocked with 4% skim milk powder in
Tris-buffered saline with Tween-20 (TBST) for 1 h at room
temperature. Primary antibodies were diluted in 4% skim milk powder
with TBST and incubated overnight at 4°C. Membranes were probed
with targeted primary antibodies: SVIL (dilution 1:1,000; cat. no.
S8695; Sigma-Aldrich, Inc.), β-tubulin (dilution 1:5,000; cat. no.
EM0103; HuaBI), JNK (dilution 1:1,000; product no. 9252; Cell
Signaling Technology, Inc.), p-JNK (dilution 1:1,000; product no.
9255; Cell Signaling Technology, Inc.), p38 (dilution 1:1,000;
product no. 9212; Cell Signaling Technology, Inc.), p-p38 (dilution
1:1,000; product no. 9211; Cell Signaling Technology, Inc.),
VE-cadherin (dilution 1:1,000; product no. 2500; Cell Signaling
Technology, Inc.). After washing with TBST three times, the
membranes were incubated with horseradish peroxidase-conjugated
anti-rabbit IgG antibodies (cat. no. 7074s; Cell Signaling
Technology, Inc.) for 1 h at 25°C. Proteins were visualized using
an ECL luminescent solution (cat. no. 180-5001; Tanon Science and
Technology Co., Ltd.).
Cell viability and proliferation
assays
For cell viability, cells were seeded in 96-well
plates and treated with MTT (5 mg/ml) for 4 h at 37°C. DMSO (150
µl) was subsequently added and plates were measured at 450 nm (CMax
Plus; Molecular Devices).
For cell proliferation, cells were cultured in a
12-well plate at 37°C. Subsequently, 50 µM EdU (cat. no. C10310-3;
Guangzhou RiboBio Co., Ltd.) was added for 2 h, after which plates
were analyzed according to the manufacturer's protocol. For the
BrdU assay, 10–20,000 cells were added per well to a 12-well plate.
After cells reached a density of 30–40%, transient transfection was
performed and the culture medium was changed after 4–6 h. Following
48 h of culture, 10 µm BrdU was added to each well plate and
incubated for 4 h. Subsequently, the samples were fixed in 4%
paraformaldehyde precooled on ice for 20 min, after which slides
were washed three times with PBS. Samples were then further
incubated with PBS containing 1.5 mol HCl for 10 min at room
temperature and 0.2% Triton X-100 for 10 min. PBS was used to wash
samples in triplicate. BrdU antibodies were diluted to 1:1,000 and
300 µl was added to each well. After incubation at 4°C overnight,
samples were washed three times with PBS and cell nuclei were
stained with 0.5 µg/ml DAPI at 25°C for 2 min. Distilled water was
used to wash off the PBS, glycerin was used to seal the samples and
nail polish was sealed around the film.
Cell migration and spreading
assays
For cell migration, 2×105/100 µl cells
were placed in a Transwell chamber and cultured for 16 h at 37°C.
Samples were fixed with 4% paraformaldehyde at 4°C for 15 min,
stained with 0.1% crystal violet at 25°C for 10 min and counted
using a light microscope (magnification, ×100).
To determine the degree of cell spreading, samples
were seeded in 12-well plates coated with 10 µg/ml fibronectin and
observed at the indicated time-points. Briefly, 800 µl of diluted
Fibronectin (10 µg/ml) was added to each well of the 12-well plate,
and coated at room temperature for 4 h. The transfected cells were
digested and resuspended, the cell density was adjusted to
4–5×105 cells after counting the cells, and adding them
to the coated 12-well plate. After cells adhered to the wall, cells
were observed and images were captured every 30 min.
VEGF release assay
Secreted VEGF was quantified using a VEGF ELISA kit
(cat. no. DVE00; R&D Systems, Inc.). The absorbance of each
well was measured at a range of wavelengths based on the
manufacturer's protocol. A Microplate ELISA Analyzer (CMax Plus;
Molecular Devices, LLC) was used to obtain absorbance data.
Reverse transcription PCR
(RT-PCR)
HepG2 cells were transiently transfected with siRNA,
after which total RNA was extracted using TRIzol® (cat.
no. 15596026; Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was
subsequently obtained using an RT kit (cat. no. AH311-02; Beijing
Transgen Biotech Co., Ltd.) and mRNA levels of associated factors
were determined via RT-PCR with cDNA as a template. The reaction
was performed in 2X EasyTaq® PCR SuperMix (cat. no.
AS111-11; TransGen Biotech) and the thermocycling conditions were
followed according to the instructions. The primer sequences were
as follows: 5′ to 3′: Cyclooxygenase 2 (COX2) forward,
ATGATTGCCCGACTCCCTTG and reverse, CCCCACAGCAAACCGTAGAT; EphA2
forward, AGCATCAACCAGACAGAGCC and reverse, AGCATGCCCTTGTACACCTC;
N-cadherin forward, CCTGGATCGCGAGCAGATAG and reverse,
CCGTGGCTGTGTTTGAAAGG; VE-cadherin forward TCAAGCCCATGAAGCCTCTG and
reverse, CCGGTCAAACTGCCCATACT; MMP-2 forward, AACACCTTCTATGGCTGCCC
and reverse, GCCGTACTTGCCATCCTTCT; MMP-9 forward,
TCCTTATCGCCGACAAGTGG and reverse, AGCGGTCCTGGCAGAAATAG; MMP-12
forward, AACCAACGCTTGCCAAATCC and reverse, GGCCCGATTCCTTGGAAGTT;
MMP-14 forward, ACATCTTCCTGGTGGCTGTG and reverse,
GTACTCGCTATCCACTGCCC; MMP-25 forward, AAGCGAACCCTGACATGGAG and
reverse, CGCCTTCCCATAGAGTTGCT; Mig-7 forward, AGAGGAAAACTGAGGCTGCC
and reverse CCGAGTGACAATCTGGGCTT.
In vivo tumorigenicity assays
Healthy nude mice (20 male; age, 3–4 weeks old;
weight, 12–14 g) were purchased at the Institute of Model Animal
Research of Nanjing University and cultured in an SPF environment.
Cells (1×107/200 µl) were injected into the right side
of the back of nude mice. After one week, tumor size was measured
(~10 mm3), and in vivo SVIL siRNA (50 µl; 10
nmol) was injected directly into tumor tissue. Injections were
administered three times in the first week and then twice a week,
for 4 consecutive weeks. The weight of mice and tumor growth was
subsequently measured. RNA interference (RNAi) modified with 2′-OMe
(Guangzhou RiboBio Co., Ltd.) with the following targeting
sequences were utilized: Negative control,
5′-AAUUCUCCGAACGUGUCACGU-3′; E4 RNAi,
5′-CUCACUUUGAAUGUAGAGAACCAUC-3′; E5 RNAi,
5′-UUCUGCUGAAGUUAUAGGUUGGGUU-3′; E11 RNAi,
5′-AGCAUAUUUAGAUUCCUUAUGGCUG-3′. Animal experiments were performed
according to the guidelines of the Animal Use and Care Committees
at Hefei Institutes of Physical Science, CAS.
Statistical analysis
Data are presented as the mean ± standard deviation.
Continuous variables were analyzed using an unpaired Student's
t-test for comparisons between two groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
SVIL is upregulated in liver cancer
and is localized to tumor vessels
To determine the expression of SVIL in liver cancer
tissue, SVIL levels were analyzed in 50 normal liver tissues and
347 liver cancer tissues obtained from The Cancer Genome Atlas
(TCGA) database. The results revealed that SVIL was significantly
increased in liver cancer compared with normal liver tissue
(Fig. 1A). Immunohistochemical
staining of liver cancer further indicated that SVIL levels were
significantly increased in liver cancer, and positively associated
with liver cancer stage (Fig.
1B).
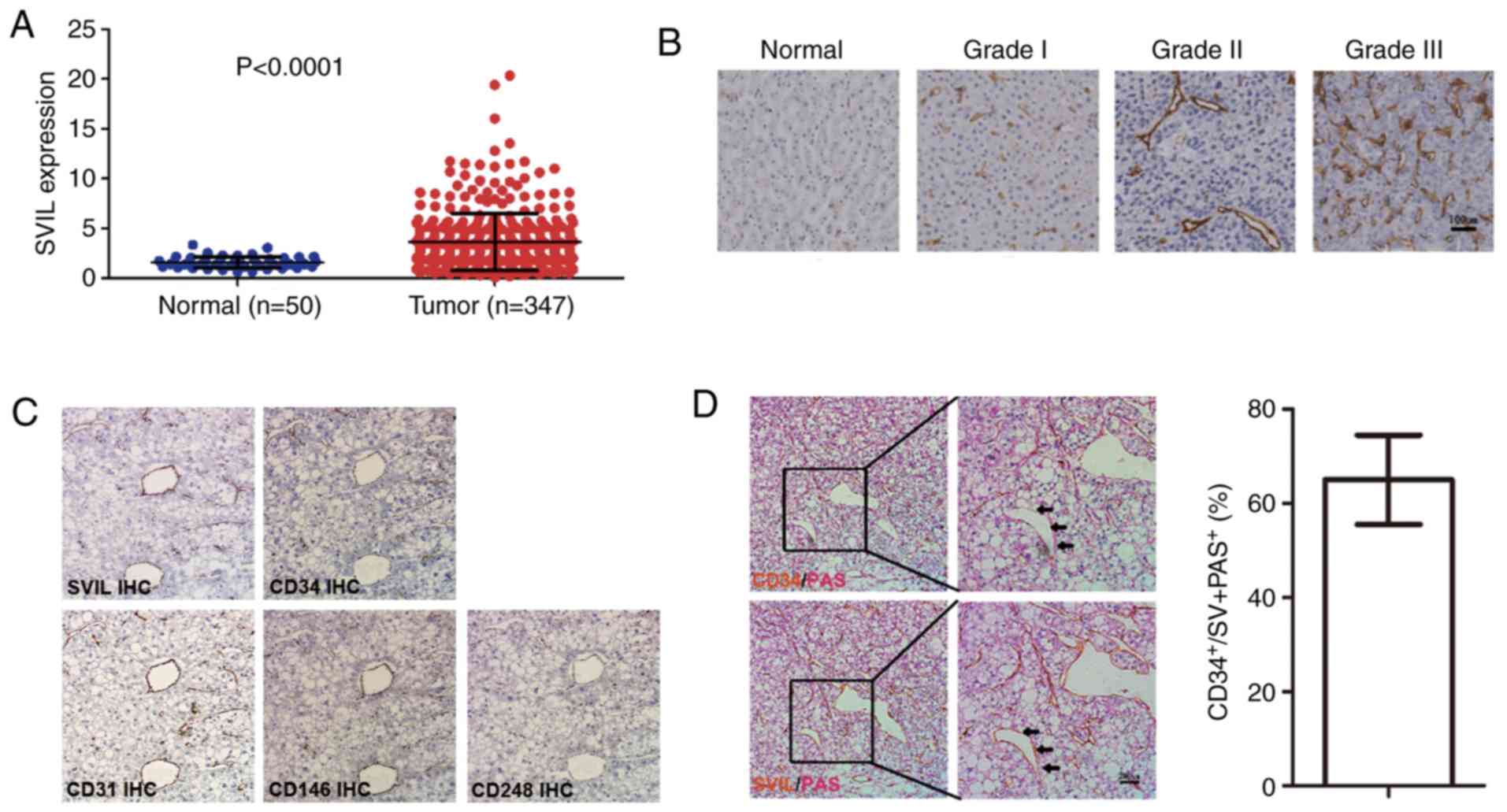 | Figure 1.SVIL is highly expressed in liver
cancer and localized to new tumor vessels. (A) Analysis of
microarray data from The Cancer Genome Atlas. (B)
Immunohistochemistry was performed to detect the expression of SVIL
in normal liver tissue, grade I, grade II and grade III liver
cancer tissue samples (DAB staining; magnification, ×100). (C)
Liver cancer tissue was serially sectioned and analyzed for SVIL,
CD31, CD34, CD146 and CD248 expression (DAB staining;
magnification, ×100). (D) CD34/PAS staining and SVIL/PAS staining
were performed on the same liver cancer tissue area (DAB staining;
magnification, ×100 and ×200). Statistics of the proportion of
CD34-labeled endothelial blood vessels in SVIL-labeled tumor blood
vessels. Data are presented as the mean ± standard deviation. SVIL,
supervillin; TCGA, The Cancer Genome Atlas; PAS, periodic
acid-Schiff. |
In addition to hepatoma cells, SVIL was expressed in
tumor vessels. Tumor vessels of liver cancer primarily include EDV
formed via endothelial cells and VM formed via tumor cells
(25,27). The expression of SVIL, CD31, CD34,
CD146 and CD248 were assessed in serial liver cancer tissue
sections via immunohistochemical staining. The results revealed
that in the liver cancer samples, certain SVIL-labeled cells were
co-located with CD31 and CD34 endothelial cells, exhibiting close
proximity to CD146- or CD248-positive pericytes (smooth muscle
cells for microvessels; Fig. 1C).
Additionally, the results demonstrated that certain SVIL-labeled
cells were present on neovascular-like structures formed by
non-endothelial cells, presenting as
CD34−/PAS+ and therefore indicating that SVIL
may be expressed in vascular mimetic structures (Fig. 1D). The VM structure accounted for
~40% after determining the number of EDVs labeled with CD34 and the
number of tumor blood vessels marked via SVIL (Fig. 1D).
Collectively, the results indicated that SVIL served
a role in liver cancer angiogenesis, particularly in EDV and VM
development.
SVIL-mediated biological function of
endothelial cell promotes endothelium-dependent vessel
development
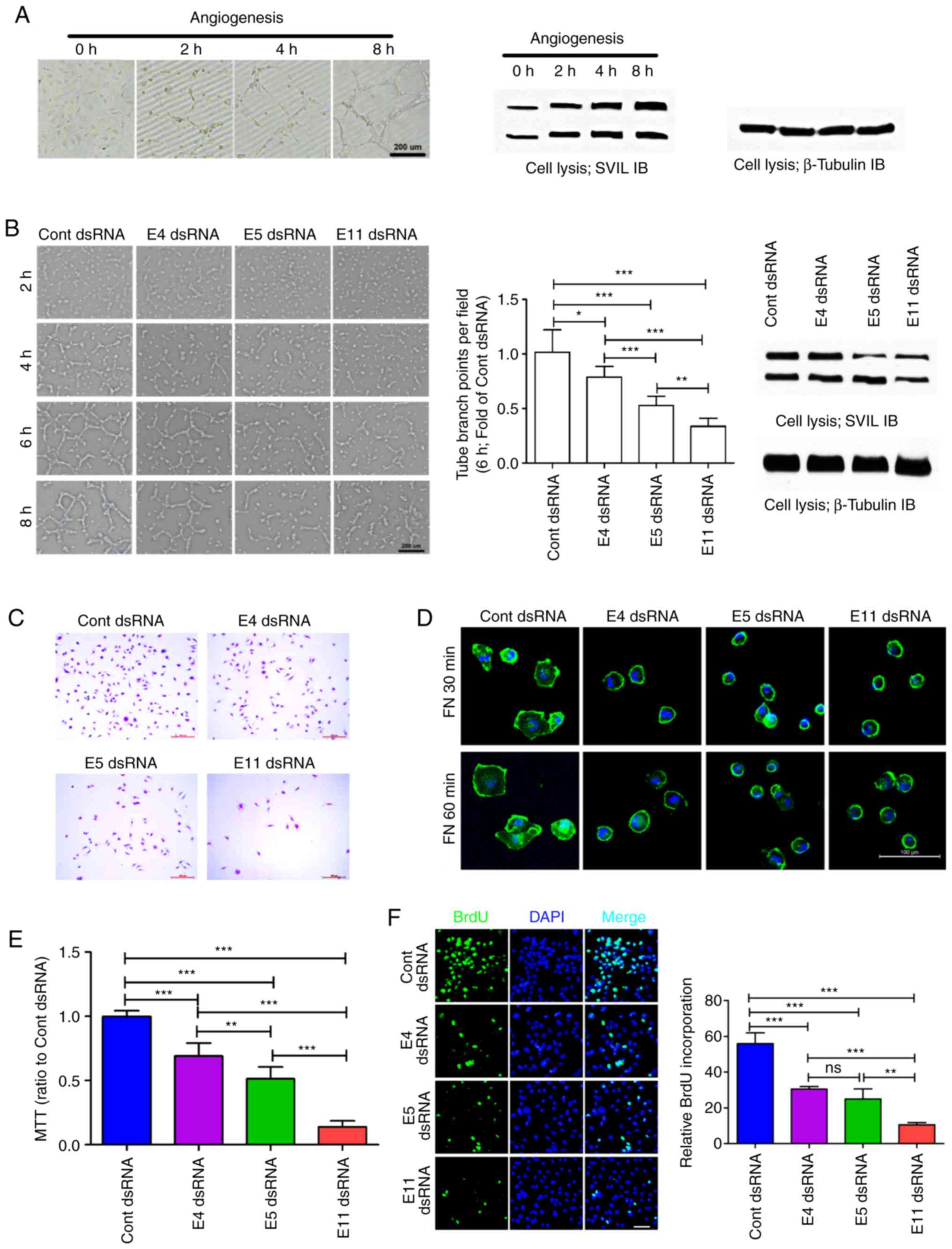
The expression of SVIL was upregulated during
angiogenesis in HUVEC cells (Fig.
2A). To determine the potential role of SVIL in HUVEC
angiogenesis, Stealth RNAi™ dsRNAs were used to target sequences
within the SVIL coding exon 4 (E4 dsRNA), coding exon 5 (E5 dsRNA),
and coding exon 11 (E11 dsRNA). As described previously, each
Stealth siRNA that targeted the different axons of SVIL in HUVEC
cells reduced the level of each isoform by ≥75%. Furthermore,
transfection of SVIL-specific RNAi resulted in a 25% (E4 dsRNA),
40% (E5 dsRNA) and 55% (E11 dsRNA) reduction in tube formation
during EDV angiogenesis (Figs. 2B
and S1A).
The roles of SVIL in HUVEC migration, spread and
proliferation were assessed in the present study as these
biological functions are important to HUVEC angiogenesis (28). The results revealed that SVIL
knockdown inhibited HUVEC migration (Figs. 2C and S1C), spread (Figs. 2D and S1B), viability (Fig. 2E) and proliferation (Fig. 2F), to different degrees. Thus, the
results indicated that SVIL served an important role in HUVEC
angiogenesis, particularly regarding cell migration, spread and
proliferation.
SVIL-mediated biological function of
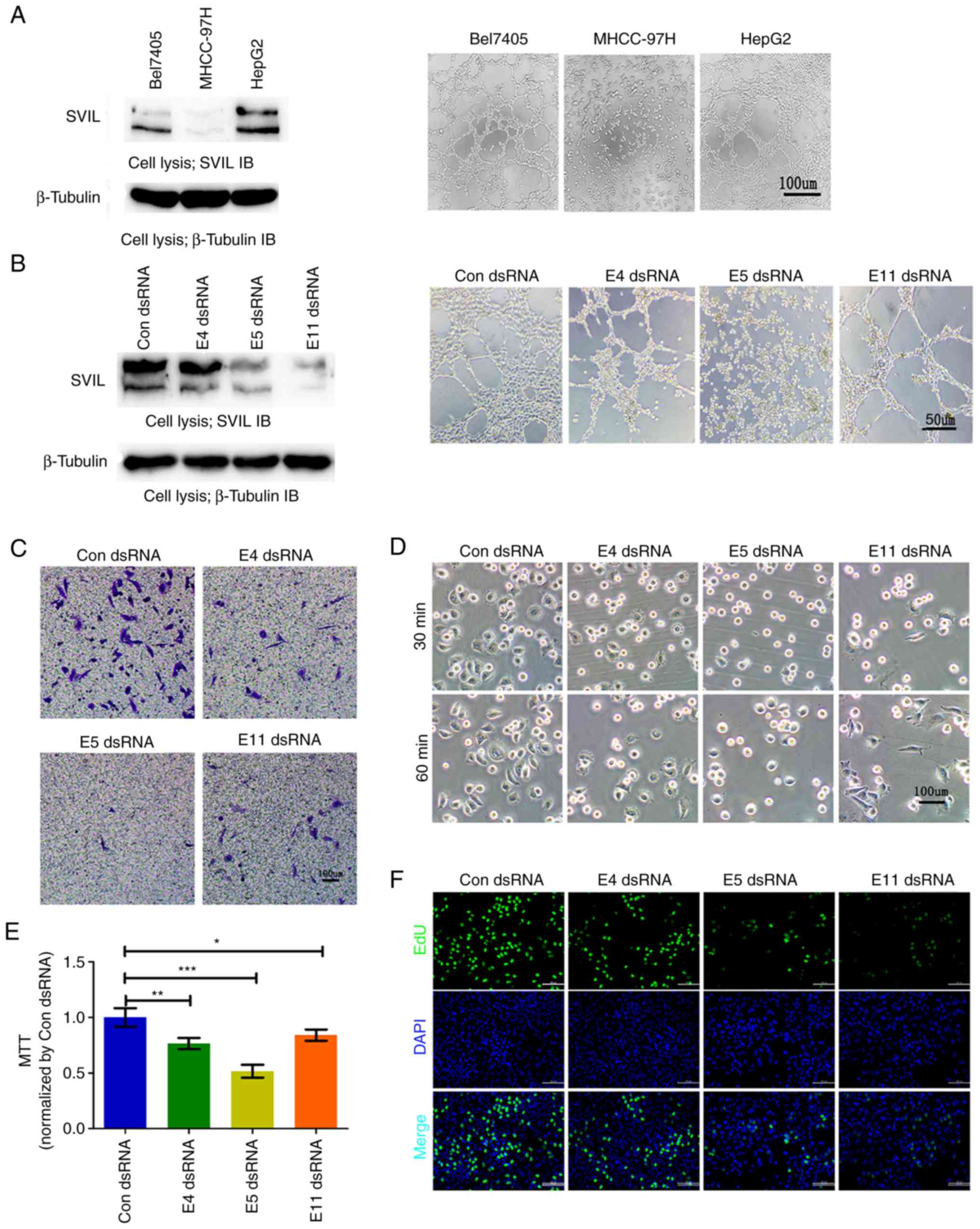
hepatoma carcinoma cells promotes the formation of VM
VM formation may be a primary factor for the failure
of traditional anti-vascular treatment (6). The present study therefore analyzed
the role of SVIL in the progression of VM development. The results
revealed that SVIL expression levels were positively associated
with VM formation in liver cancer. This may have been due to HepG2
cells expressing greater quantities of SVIL, gaining a stronger
ability to develop VM when compared with other liver cancer cells
(Fig. 3A). SVIL knockdown with
specific dsRNA resulted in a reduction in VM formation, to varying
degrees (Figs. 3B and S2A). Similar
to the results of HUVEC angiogenesis, SVIL knockdown in HepG2 cells
demonstrated decreased cell migration (Figs. 3C and S2B), spread (Figs. 3D and S2C), viability (Fig. 3E), and proliferation (Figs. 3F and S2D). The results indicated
that SVIL served an important role in VM formation, potentially by
regulating the survival, migration and proliferation of hepatoma
cells.
SVIL knockdown inhibits the formation
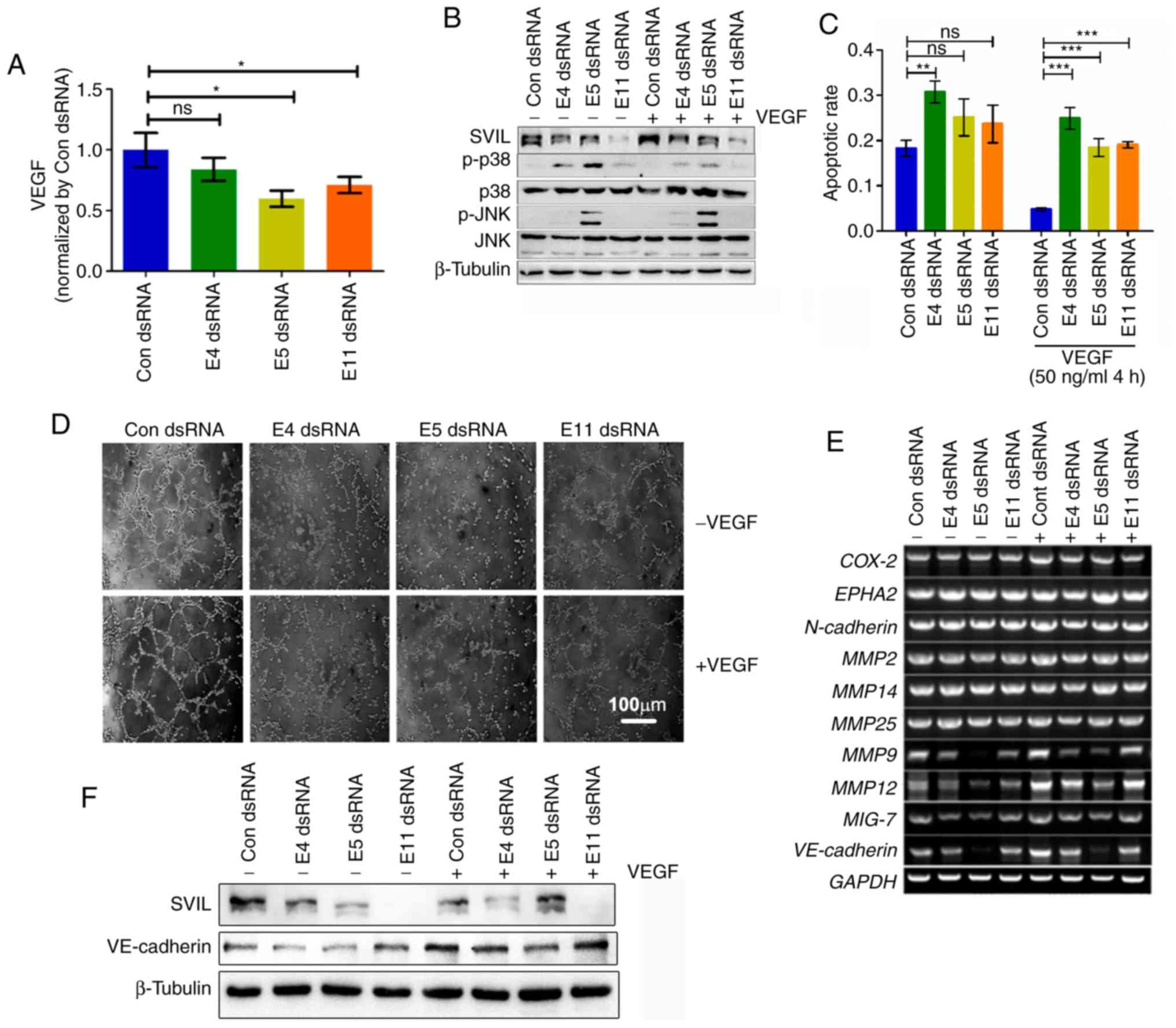
of vasculogenic mimicry via the VEGF-p38 axis
Increasing evidence has indicated that VEGF is
crucial for the development of VM and can be secreted from tumor
cells (17,29). In the present study, it was
demonstrated that SVIL knockdown significantly suppressed VEGF
secretion in the culture media of HepG2 cells, indicating that
there was an interaction between VM and tumor cells which may lead
to tumor progression (Fig. 4A).
The MAPK signaling pathway has been demonstrated to
mediate cell survival and migration, and serve an important role in
VM formation (11,30,31).
It was demonstrated in a previous study that SVIL was associated
with MAPK activation (22,32,33).
As hypothesized, SVIL knockdown increased p38 activation and the
phosphorylation level of JNK compared with the control group
(Fig. 4B). Furthermore, cellular
apoptosis was increased (Figs. 4C
and S3A) and cell population was decreased (Fig. S3B). The results indicated that SVIL
may regulate tumor cell survival and p38 activation to ensure VM
development.
As VEGF secretion and p38 signaling are
indispensable for VM formation (6,11,34),
the present study investigated potential crosstalk between the VEGF
and p38 signaling pathways. The results revealed that VEGF
abolished the SVIL knockdown-induced reduction of tube formation in
VM (Figs. 4D and S4) and
downregulated p38 phosphorylation levels. However, the
phosphorylation level of JNK did not change, indicating that p38
phosphorylation may be downstream of SVIL-mediated VEGF secretion
(Fig. 4B).
As previously reported, VE-cadherin, N-cadherin,
COX-2, EphA2, MMPs and Mig-7 are involved in VM formation (6,35,36).
In the present study, SVIL downregulation induced alterations of
the VM formation transcriptional network, particularly via
VE-cadherin, MMP9, MMP12 and Mig-7. With the addition of VEGF, the
expression levels of these transcriptional factors were rescued by
SVIL knockdown (Fig. 4E and F).
Collectively, the results suggested that SVIL promoted VM
development by activating the VEGF-p38 axis and inducing
VM-associated transcriptional factors.
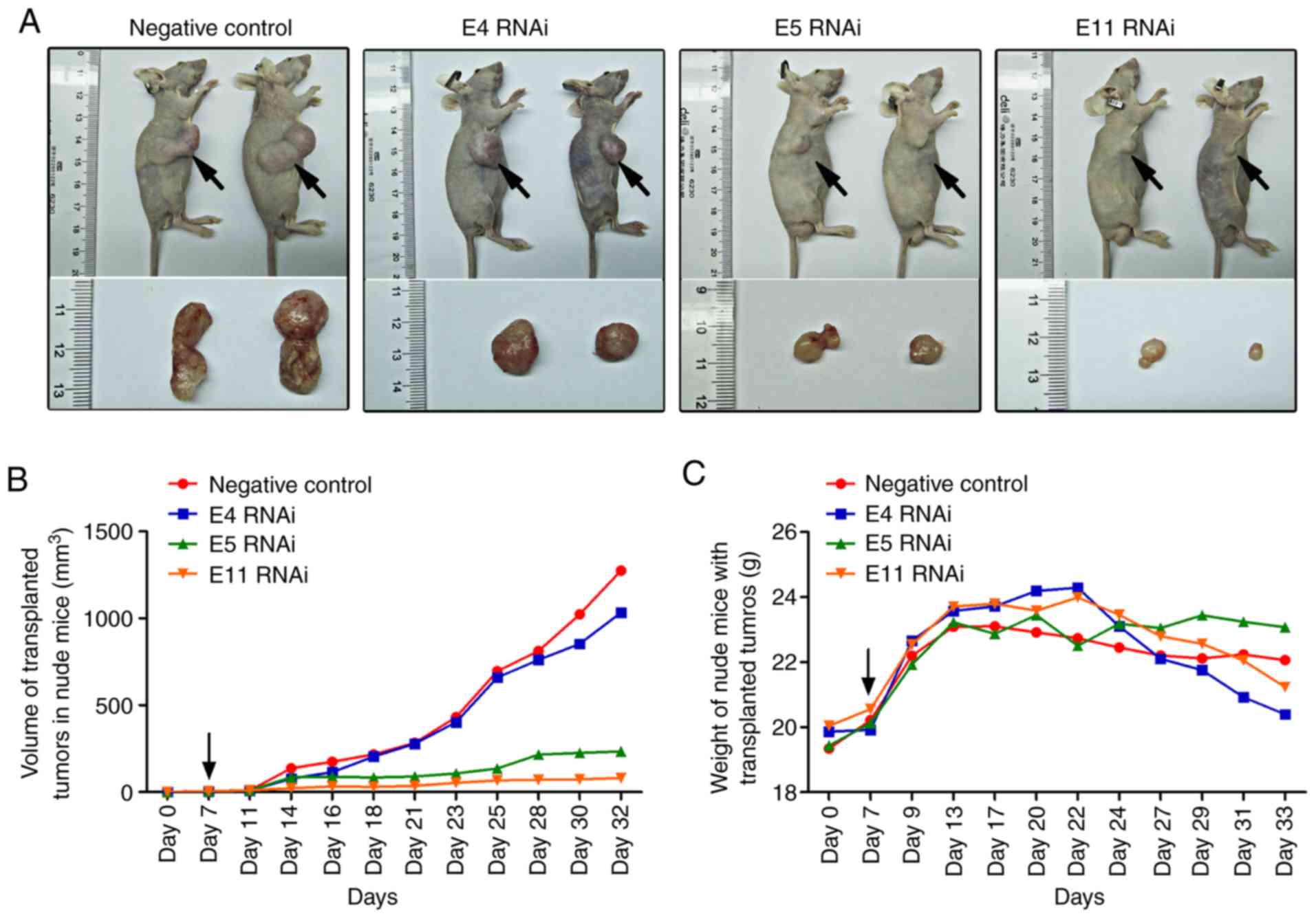
SVIL knockdown suppresses liver tumor
growth in vivo
As tumor angiogenesis promotes the proliferation,
growth, invasion and metastasis of hepatoma cells (1,2,37), the
present study investigated whether SVIL knockdown inhibited tumor
growth in vivo. Hepatoma cells were injected into the right
side of the back of each nude mouse. After a week, in vivo
SVIL siRNA was injected directly into the tumor tissue. The results
revealed that injection of in vivo SVIL siRNA significantly
inhibited tumor growth, particularly E5 RNAi and E11 RNAi (Fig. 5A and B). However, mouse weight did
not differ compared with the negative control RNAi-treated mice
(Fig. 5C).
Discussion
Liver cancer is a solid malignant tumor with a dense
vascular network, a high level of metastasis, high recurrence and
poor prognosis (38). It has been
revealed that tumor angiogenesis serves an important role in the
proliferation, growth, invasion and metastasis of hepatoma cells.
Recent studies have focused on the inhibition of tumor angiogenesis
and the development of small-molecule targeted drugs to inhibit
angiogenesis (39–41). There are two major forms of tumor
angiogenesis during tumorigenesis: EDV and VM. EDV is composed of
endothelial cells, while VM is constructed via in situ
cancer cells. SVIL is an actin-associated protein that is involved
in various cell movements, such as adhesion and migration (41). A previous study revealed that SVIL
promoted the malignant progression of liver cancer (22). Therefore, the present study
investigated whether SVIL was associated with EDV and VM. The
results indicated that SVIL promoted EDV development and induced VM
formation. Furthermore, decreased SVIL ultimately inhibited VM
formation by suppressing VEGF secretion to activate p38-induced
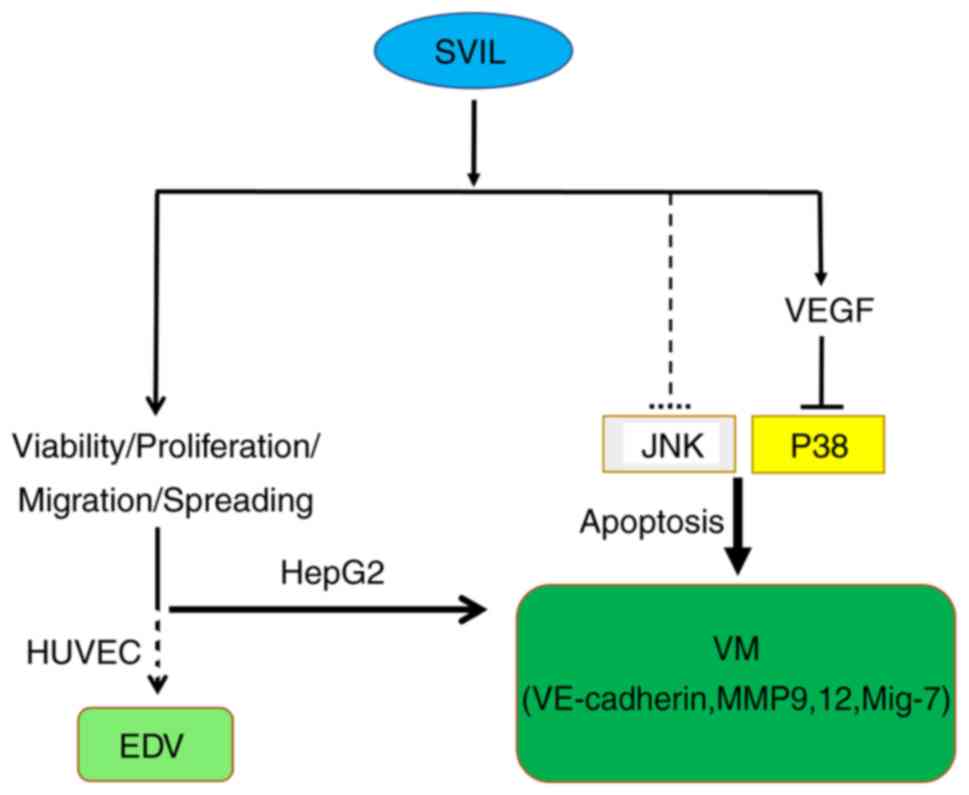
tumor cell apoptosis (Fig. 6).
The results of the present study revealed that SVIL
was highly expressed in liver cancer compared with normal liver
tissues and localized to tumor neovascular sites. Additionally, VM
accounted for ~40% of the total number of new blood vessels,
indicating the significance of VM in tumor formation. Further
studies revealed that SVIL expression was closely associated with
the migration, spread, viability and proliferation of endothelial
cells, as well as tumor cells in liver cancer. Changes in
biological function affected the formation of new blood vessels.
SVIL was revealed to serve an important role in each step of tumor
development, including cell survival, migration and metastasis.
SVIL was revealed to also regulate tumor cell survival by
inhibiting the ubiquitin-specific-processing protease 7-dependent
deubiquitination of p53 (24). SVIL
has also been revealed to serve an important role in cell migration
and invasion (21). In podosomes
and invadopodia, SVIL was revealed to recombine the cortical actin
cytoskeleton, promote the formation of podosomes and invadopodia,
and promote ECM degradation, thereby inducing tumor cell motility
(42,43).
The MAPK is involved in the neovascularization
process, and the ERK/JNK/p38-MAPK pathways
separately/co-executively perform positive or negative functions
(44). In a previous study, it was
determined that SVIL promoted invasion, metastasis, and
epithelial-mesenchymal transition (EMT) processes by activating the
RhoA/ROCK-ERK/p38 signaling pathway during hypoxia (22). However, under normoxic conditions,
reduced SVIL may activate the p38 signaling pathway and cell
apoptosis, which may be accompanied by JNK activation. It has been
reported that the p38 inhibitor, SB202190, induces angiogenesis by
reducing apoptosis, thereby increasing DNA synthesis and cell
proliferation, and enhancing cell differentiation, which involves
fibroblast growth factor (FGF)-2 (44). In the present study, SVIL knockdown
significantly suppressed VEGF secretion in HepG2 cell culture
media. VEGF treatment also inhibited p38, but not the JNK, pathway
and partially rescued VM formation, suggesting that SVIL regulated
VM formation via the VEGF-p38 axis. VEGF was secreted from tumor
cells, which may, in turn, promote tumor metastasis and
angiogenesis. This may partly explain the role of SVIL in
endothelial cell angiogenesis.
VM formation is a complex process that involves a
variety of pathways and signaling molecules, including factors
associated with tumor cell invasion, migration, apoptosis and
matrix remodeling, such as VE-cadherin, N-cadherin, EphA2, MMPs and
COX-2 (6). The results of the
present study revealed that changes in VE-cadherin, MMP9, MMP12 and
Mig-7 levels were positively associated with SVIL and recovered
following VEGF treatment. Transfection of E5 dsRNA demonstrated
more significant alterations in VE-cadherin, MMP9, MMP12 and Mig-7
levels when compared with E4 and E11 dsRNA. This may be dependent
on the ratio of the three isoforms in tumor cells and the
differences in dsRNA levels in target sites. Among the
aforementioned molecules, VE-cadherin is an important adhesion
protein. VE-cadherin regulates EphA2, which is an important factor
in VM formation (45,46). The p38/MAPK signal that occurs
during VE-cadherin regulation often regulates cell membrane
permeability and cytoskeletal remodeling (47). p38 inhibition, but not ERK-MAPK, was
revealed to significantly reduce the loss of membrane-associated
VE-cadherin (48,49). MMPs are vital for the degradation
and integration of the matrix. Twist1 was revealed to promote MMP2
and MMP9 activation, thereby inducing liver cancer invasion during
the EMT process (50). In addition,
epigallocatechin-3-gallate inhibited pancreatic tumor cell growth,
invasion, angiogenesis and metastasis. This involved promoting p38
and JNK activities and significantly reducing MMP9 and MMP12
(51). Mig-7 is overexpressed in
highly invasive melanoma and invasive melanoma conversely, and it
increases the Ln-5γ2 chain domain III fragment, thereby promoting
tumor cell migration, migration and VM formation (52). Collectively, the results of the
present study indicated that SVIL promoted VM development by
activating the VEGF/p38 axis and inducing VM-associated
transcriptional factors.
In summary, the results elucidated the important
role of SVIL in the progression of malignant liver cancer and tumor
angiogenesis, both in EDV and VM. Particularly, decreased SVIL
levels inhibited VM formation by reducing VEGF secretion to
activate the p38 pathway and regulate VE-cadherin/MMP9/12/Mig-7
transcription. As a result, SVIL may be considered as a potential
tumor vascular biomarker and a promising therapeutic target for
patients with liver cancer.
Supplementary Material
Supporting Data
Acknowledgments
We would like to thank the members of the technical
assistance team at the Center of Medical Physics and Technology,
Hefei Institutes of Physical Science.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant. nos. 31571433,
81773131, and 81872066), the Anhui Provincial Natural Science
Foundation (grant. nos. 1608085MH180 and 1808085QH272), the
Innovative program of the Development Foundation of Hefei Center
for Physical Science and Technology (grant. no. 2017FXCX008), and
the CASHIPS Director's Fund (grant. no. YZJJ201704).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZF, CZ, and ZW conceived and designed the article.
ZW, CZ and ZZ, jointly completed cytology experiments and
histological examination of liver specimens. The manuscript was
written by CZ and critically reviewed by all participating authors.
The data collection and statistical analysis were performed by ZZ,
LH, and HW. All authors have read and approved the final manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Study protocols were approved by the Institutional
Review Board of the Cancer Hospital of Hefei Institutes of Physical
Science, Chinese Academy of Sciences (CAS). Animal experiments were
performed according to the guidelines of the Animal Use and Care
Committees at Hefei Institutes of Physical Science, CAS.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests related to this work.
Glossary
Abbreviations
Abbreviations:
|
COX-2
|
cyclooxygenase-2
|
|
EDV
|
endothelium- dependent vessel
|
|
EMT
|
epithelial-mesenchymal transition
|
|
EphA2
|
epithelial cell kinase
|
|
FAK
|
focal adhesion kinase
|
|
FGF
|
fibroblast growth factor
|
|
HIF-1α
|
hypoxia-inducible factor-1α
|
|
Mig-7
|
migration-inducing protein 7
|
|
MMPs
|
matrix metalloproteinases
|
|
PI3K-α
|
phosphoinositide 3-kinase alpha
|
|
SVIL
|
supervillin
|
|
TCGA
|
The Cancer Genome Atlas
|
|
VE-cadherin
|
vascular endothelial-cadherin
|
|
VEGF
|
vascular endothelium growth factor
|
|
VM
|
vasculogenic mimicry
|
References
|
1
|
Zhu AX, Duda DG, Sahani DV and Jain RK:
HCC and angiogenesis: Possible targets and future directions. Nat
Rev Clin Oncol. 8:292–301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fernandez M, Semela D, Bruix J, Colle I,
Pinzani M and Bosch J: Angiogenesis in liver disease. J Hepatol.
50:604–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang Z, Dabrosin C, Xin Y, Fuster MM,
Arreola A, Rathmell WK, Generali D, Nagaraju GP, El-Rayes B,
Ribatti D, et al: Broad targeting of angiogenesis for cancer
prevention and therapy. Semin Cancer Biol. 35 (Suppl):S224–S243.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hicklin DJ and Ellis LM: Role of the
vascular endothelial growth factor pathway in tumor growth and
angiogenesis. J Clin Oncol. 23:1011–1027. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Viallard C and Larrivée B: Tumor
angiogenesis and vascular normalization: Alternative therapeutic
targets. Angiogenesis. 20:409–426. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Qiao L, Liang N, Zhang J, Xie J, Liu F, Xu
D, Yu X and Tian Y: Advanced research on vasculogenic mimicry in
cancer. J Cell Mol Med. 19:315–326. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Döme B, Hendrix MJ, Paku S, Tóvári J and
Tímár J: Alternative vascularization mechanisms in cancer:
Pathology and therapeutic implications. Am J Pathol. 170:1–15.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang W, Lin P, Sun BC, Cai WJ, Han CR, Li
L, Lu HH and Zhang JM: Role of vasculogenic mimicry and
endothelium-dependent vessel in metastasis of laryngeal cancer.
Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 47:400–405. 2012.(In
Chinese). PubMed/NCBI
|
|
9
|
Elpek GÖ: Angiogenesis and liver fibrosis.
World J Hepatol. 7:377–391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Delgado-Bellido D, Serrano-Saenz S,
Fernández-Cortés M and Oliver FJ: Vasculogenic mimicry signaling
revisited: Focus on non-vascular VE-cadherin. Mol Cancer.
16:652017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ling G, Ji Q, Ye W, Ma D and Wang Y:
Epithelial-mesenchymal transition regulated by p38/MAPK signaling
pathways participates in vasculogenic mimicry formation in SHG44
cells transfected with TGF-β cDNA loaded lentivirus in vitro
and in vivo. Int J Oncol. 49:2387–2398. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang H, Lin H, Pan J, Mo C, Zhang F, Huang
B, Wang Z, Chen X, Zhuang J, Wang D and Qiu S: Vasculogenic mimicry
in prostate cancer: The roles of EphA2 and PI3K. J Cancer.
7:1114–1124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Trapani F, Metcalf RL, Polanski R, Fusi A,
Hodgkinson C, Nonaka D, Hendrix MJ, Morrrow C, Blackhall F, Simpson
KL and Dive C: 264 Vasculogenic mimicries in small cell lung
cancer. European J Cancer. 50 (Suppl 6):S882014. View Article : Google Scholar
|
|
14
|
Brantley DM, Cheng N, Thompson EJ, Lin Q,
Brekken RA, Thorpe PE, Muraoka RS, Cerretti DP, Pozzi A, Jackson D,
et al: Soluble Eph A receptors inhibit tumor angiogenesis and
progression in vivo. Oncogene. 21:7011–7026. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hess AR and Hendrix MJ: Focal adhesion
kinase signaling and the aggressive melanoma phenotype. Cell Cycle.
5:478–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheng N, Brantley DM, Liu H, Lin Q,
Enriquez M, Gale N, Yancopoulos G, Cerretti DP, Daniel TO and Chen
J: Blockade of EphA receptor tyrosine kinase activation inhibits
vascular endothelial cell growth factor-induced angiogenesis. Mol
Cancer Res. 1:2–11. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang JY, Sun T, Zhao XL, Zhang SW, Zhang
DF, Gu Q, Wang XH, Zhao N, Qie S and Sun BC: Functional
significance of VEGF-a in human ovarian carcinoma: Role in
vasculogenic mimicry. Cancer Biol Ther. 7:758–766. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Y, Takizawa N, Crowley JL, Oh SW,
Gatto CL, Kambara T, Sato O, Li XD, Ikebe M and Luna EJ: F-actin
and myosin II binding domains in supervillin. J Biol Chem.
278:46094–46106. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pestonjamasp KN, Pope RK, Wulfkuhle JD and
Luna EJ: Supervillin (p205): A novel membrane-associated, F-actin-
binding protein in the villin/gelsolin superfamily. J Cell Biol.
139:1255–1269. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oh SW, Pope RK, Smith KP, Crowley JL, Nebl
T, Lawrence JB and Luna EJ: Archvillin, a muscle-specific isoform
of supervillin, is an early expressed component of the costameric
membrane skeleton. J Cell Sci. 116:2261–2275. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen X, Yang H, Zhang S, Wang Z, Ye F,
Liang C, Wang H and Fang Z: A novel splice variant of supervillin,
SV5, promotes carcinoma cell proliferation and cell migration.
Biochem Biophys Res Commun. 482:43–49. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen X, Zhang S, Wang Z, Wang F, Cao X, Wu
Q, Zhao C, Ma H, Ye F, Wang H and Fang Z: Supervillin promotes
epithelial- mesenchymal transition and metastasis of hepatocellular
carcinoma in hypoxia via activation of the RhoA/ROCK-ERK/p38
pathway. J Exp Clin Cancer Res. 37:1282018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takizawa N, Ikebe R, Ikebe M and Luna EJ:
Supervillin slows cell spreading by facilitating myosin II
activation at the cell periphery. J Cell Sci. 120:3792–3803. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fang Z and Luna EJ: Supervillin-mediated
suppression of p53 protein enhances cell survival. J Biol Chem.
288:7918–7929. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jue C, Zhifeng W, Zhisheng Z, Lin C, Yayun
Q, Feng J, Hao G, Shintaro I, Hisamitsu T, Shiyu G and Yanqing L:
Vasculogenic mimicry in hepatocellular carcinoma contributes to
portal vein invasion. Oncotarget. 7:77987–77997. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ye QH, Zhu WW, Zhang JB, Qin Y, Lu M, Lin
GL, Guo L, Zhang B, Lin ZH, Roessler S, et al: GOLM1 modulates
EGFR/RTK cell-surface recycling to drive hepatocellular carcinoma
metastasis. Cancer Cell. 30:444–458. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gorrin-Rivas MJ, Arii S, Mori A, Takeda Y,
Mizumoto M, Furutani M and Imamura M: Implications of human
macrophage metalloelastase and vascular endothelial growth factor
gene expression in angiogenesis of hepatocellular carcinoma. Ann
Surg. 231:67–73. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lamouille S, Mallet C, Feige JJ and Bailly
S: Activin receptor-like kinase 1 is implicated in the maturation
phase of angiogenesis. Blood. 100:4495–4501. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu HB, Yang S, Weng HY, Chen Q, Zhao XL,
Fu WJ, Niu Q, Ping YF, Wang JM, Zhang X, et al: Autophagy-induced
KDR/VEGFR-2 activation promotes the formation of vasculogenic
mimicry by glioma stem cells. Autophagy. 13:1528–1542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Azad T, Janse van Rensburg HJ, Lightbody
ED, Neveu B, Champagne A, Ghaffari A, Kay VR, Hao Y, Shen H, Yeung
B, et al: A LATS biosensor screen identifies VEGFR as a regulator
of the Hippo pathway in angiogenesis. Nat Commun. 9:10612018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Paulis YWJ, Dinnes D, Soetekouw PMMB,
Nelson PJ, Burdach S, Loewe RP, Tjan-Heijnen VCG, von Luettichau I
and Griffioen AW: Imatinib reduces the vasculogenic potential of
plastic tumor cells. Current Angiogenesis. 1:64–71. 2012.
View Article : Google Scholar
|
|
32
|
Fang Z, Takizawa N, Wilson KA, Smith TC,
Delprato A, Davidson MW, Lambright DG and Luna EJ: The
membrane-associated protein, supervillin, accelerates
F-actin-dependent rapid integrin recycling and cell motility.
Traffic. 11:782–799. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu HP, Yu MC, Jiang MH, Chen JX, Yan DP,
Liu F and Ge BX: Association of supervillin with KIR2DL1 regulates
the inhibitory signaling of natural killer cells. Cell Signal.
23:487–496. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bai XL, Zhang Q, Ye LY, Liang F, Sun X,
Chen Y, Hu QD, Fu QH, Su W, Chen Z, et al: Myocyte enhancer factor
2C regulation of hepatocellular carcinoma via vascular endothelial
growth factor and Wnt/β-catenin signaling. Oncogene. 34:4089–4097.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ren K, Zhang J, Gu X, Wu S, Shi X, Ni Y,
Chen Y, Lu J, Gao Z, Wang C and Yao N: Migration-inducing gene-7
independently predicts poor prognosis of human osteosarcoma and is
associated with vasculogenic mimicry. Exp Cell Res. 369:80–89.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Folberg R, Hendrix MJ and Maniotis AJ:
Vasculogenic mimicry and tumor angiogenesis. Am J Pathol.
156:361–381. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang C, Xu Y, Cheng F, Hu Y, Yang S, Rao J
and Wang X: MiR-1301 inhibits hepatocellular carcinoma cell
migration, invasion, and angiogenesis by decreasing Wnt/β-catenin
signaling through targeting BCL9. Cell Death Dis. 8:e29992017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Colombo M: Liver cancer. Lancet.
3:551995.
|
|
39
|
Mashreghi M, Azarpara H, Bazaz MR, Jafari
A, Masoudifar A, Mirzaei H and Jaafari MR: Angiogenesis biomarkers
and their targeting ligands as potential targets for tumor
angiogenesis. J Cell Physiol. 233:2949–2965. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Niccoli Asabella A, Di Palo A, Altini C,
Ferrari C and Rubini G: Multimodality imaging in tumor
angiogenesis: Present status and perspectives. Int J Mol Sci.
18:E18642017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Casazza A, Fu X, Johansson I, Capparuccia
L, Andersson F, Giustacchini A, Squadrito ML, Venneri MA, Mazzone
M, Larsson E, et al: Systemic and targeted delivery of semaphorin
3A inhibits tumor angiogenesis and progression in mouse tumor
models. Arterioscler Thromb Vasc Biol. 31:741–749. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Crowley JL, Smith TC, Fang Z, Takizawa N
and Luna EJ: Supervillin reorganizes the actin cytoskeleton and
increases invadopodial efficiency. Mol Biol Cell. 20:948–962. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gimona M, Buccione R, Courtneidge SA and
Linder S: Assembly and biological role of podosomes and
invadopodia. Curr Opin Cell Biol. 20:235–241. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Matsumoto T, Turesson I, Book M, Gerwins P
and Claesson-Welsh L: p38 MAP kinase negatively regulates
endothelial cell survival, proliferation, and differentiation in
FGF-2-stimulated angiogenesis. J Cell Biol. 156:149–160. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kirschmann DA, Seftor EA, Hardy KM, Seftor
RE and Hendrix MJ: Molecular pathways: Vasculogenic mimicry in
tumor cells: Diagnostic and therapeutic implications. Clin Cancer
Res. 18:2726–2732. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hess AR, Seftor EA, Gruman LM, Kinch MS,
Seftor RE and Hendrix MJ: VE-cadherin regulates EphA2 in aggressive
melanoma cells through a novel signaling pathway: Implications for
vasculogenic mimicry. Cancer Biol Ther. 5:228–233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tremblay PL, Auger FA and Huot J:
Regulation of transendothelial migration of colon cancer cells by
E-selectin-mediated activation of p38 and ERK MAP kinases.
Oncogene. 25:6563–6573. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nwariaku FE, Chang J, Zhu X, Liu Z, Duffy
SL, Halaihel NH, Terada L and Turnage RH: The role of p38 map
kinase in tumor necrosis factor-induced redistribution of vascular
endothelial cadherin and increased endothelial permeability. Shock.
18:82–85. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kimura T, Mogi C, Sato K, Tomura H, Ohta
H, Im DS, Kuwabara A, Kurose H, Murakami M and Okajima F:
p2y5/LPA(6) attenuates LPA(1)-mediated VE-cadherin translocation
and cell-cell dissociation through G(12/13) protein-Src-Rap1.
Cardiovasc Res. 92:149–158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhao XL, Sun T, Che N, Sun D, Zhao N, Dong
XY, Gu Q, Yao Z and Sun BC: Promotion of hepatocellular carcinoma
metastasis through matrix metalloproteinase activation by
epithelial-mesenchymal transition regulator Twist1. J Cell Mol Med.
15:691–700. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shankar S, Ganapathy S, Hingorani SR and
Srivastava RK: EGCG inhibits growth, invasion, angiogenesis, and
metastasis of pancreatic cancer. Front Biosci. 13:440–452. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Petty AP, Garman KL, Winn VD, Spidel CM
and Lindsey JS: Overexpression of carcinoma and embryonic
cytotrophoblast cell-specific Mig-7 induces invasion and
vessel-like structure formation. Am J Pathol. 170:1763–1780. 2007.
View Article : Google Scholar : PubMed/NCBI
|