Introduction
Acute myeloid leukemia (AML) is a heterogeneous
disease defined by clonal expansion of immature myeloid cells that
infiltrate the bone marrow and other tissues (1). The therapeutic strategies AML have
remained largely unchanged over the past 30 years, and the disease
is currently curable in 35–40% of patients aged ≤60 years, and in
5–15% of those aged >60 years (2). The development of next-generation
sequencing (NGS) technologies have led to the identification of
eight functional categories of significantly mutated genes,
including tumor-suppressor genes, such as tumor protein p53
(TP53) (3). TP53
mutations occur in 8–14% of the cases (4) and they are associated with complex
karyotype AML (5), in particular
typical complex karyotype (6) and
chromothripsis (7,8), and confer a very poor prognosis
(4,9). Moreover, the p53-transcriptional
program is generally silenced in aneuploid AML (10), and TP53 mutations and
aneuploidy define a specific molecular subgroup in the recent AML
genomic classification and prognostic stratification (9).
TP53 is the most frequently mutated gene in
cancer (11,12) and is a critical regulator of several
genes involved in DNA repair [e.g., growth arrest and DNA damage
(GADD45)], cell cycle [e.g., cyclin-dependent kinase
inhibitor 1A (CDKN1A)], apoptosis [e.g., BCL2-associated X
protein, BCL2-binding component 3] and angiogenesis (13). Under normal conditions, p53 is
maintained at a low level and is mainly regulated by
E3-ubiquitin-ligase mouse double minute 2 (MDM2), which promotes
its proteasomal degradation (14).
Moreover, its rapid turnover is associated with correct folding by
chaperone proteins, such as heat shock protein 90 (Hsp90), which
has been proven to be fundamental for the stability and DNA-binding
capacity of the wild-type (wt) protein (15). Under stress conditions, p53 is
phosphorylated by various sensor kinases, promoting the
dissociation of the p53-MDM2 complex and p53 stabilization and
activation (16,17).
Kevetrin (thioureidobutyronitrile or 3-cyanopropyl
carbamimidothioate hydrochloride, C5H10ClN3S) is a small-molecule
compound that exhibits p53-dependent and -independent activity in
solid tumors, including lung, breast, colon and ovarian cancer cell
and xenograft models (18–20). In TP53-wt models, kevetrin
induces cell cycle arrest and apoptosis through the alteration of
the E3 ligase processivity of MDM2 and activation and stabilization
of p53, with increased expression of its targets, including protein
21 (p21) and p53-upregulated modulator of apoptosis (18,19). A
p53-independent upregulation of p21 expression was observed in
TP53-mutant ovarian cancer cell lines (20). Accordingly, in a phase I trial
evaluating the effect of kevetrin on solid cancers, 48% of patients
exhibited a ≥10% increase in p21 expression in the peripheral blood
7–24 h after the treatment initiation (NCT01664000). Moreover, it
was hypothesized that kevetrin induced downregulation of histone
deacetylase 6 (HDAC6), negatively affected the HDAC6-Hsp90
chaperone axis, resulting in degradation of p53 in the mutated
models (19). Kevetrin treatment
led to the forced expression of the pro-apoptotic protein BID and
decreased levels of the anti-apoptotic protein MCL1. It also had an
effect on the Rb-E2F tumor suppressor pathway by downregulating
E2F1 and its target genes in TP53-mutant and wt models
(19).
To the best of our knowledge, the present study is
the first to investigate the effects of kevetrin exposure on AML
cell lines and primary cells characterized by different TP53
mutational status.
Materials and methods
Cell lines and culture
Four AML cell lines, MOLM-13 (AML M5), KASUMI-1 (AML
M2), OCI-AML3 (AML M4) and NOMO-1 (AML M5) were obtained from the
American Type Culture Collection, and were mycoplasma-tested and
authenticated using the LGC Standards Cell Line Authentication
service. The cell lines were cultured at 37°C in a 5%
CO2 atmosphere at a density of 0.3×106
cells/ml in complete medium, in T75 flasks. MOLM-13 and KASUMI-1
cells were cultured in RPMI-1640 (Euroclone) supplemented with 20%
heat-inactivated FBS (GE Healthcare), 2 mM L-glutamine (GE
Healthcare), 100 U/ml penicillin, 100 µg/ml streptomycin (GE
Healthcare) and 0.2% Mycozap (Lonza, Inc.). OCI-AML3 cells were
cultured in α-MEM (Lonza, Inc.) with 20% FBS, 2 mM L-glutamine, 100
U/ml penicillin and 100 µg/ml streptomycin. NOMO-1 cells were grown
in RPMI-1640 with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin
and 100 µg/ml streptomycin.
Drug
Kevetrin powder was kindly provided by Innovation
Pharmaceuticals, dissolved in sterile water in a 3.4 mM stock
solution, stored at 4°C and used within 1 month. Cells were seeded
in 96-well or 6-well plates at 0.5×106/ml in 100 and
3,000 µl of medium, respectively, and treated with increasing drug
concentrations (85–340 µM), according to peak plasma concentrations
measured in the phase I clinical trial (NCT01664000). For pulsed
experiments, cells were exposed to the drug for 6 h and then washed
and replated in complete medium [wash-out (wo)]. After 66 h, cells
were reseeded in fresh medium containing the drug for 6 h, followed
by a 66-h wo. The pulsed treatment was repeated 2–3 times.
Primary cell cultures
Samples were collected at Istituto Scientifico
Romagnolo per lo Studio e la Cura dei Tumori (IRST) IRCCS from 4
AML patients at diagnosis (inclusion criteria: Age ≥18 years,
confirmed AML diagnosis, available clinical data for review and
obtained written informed consent) between December 2018 and
October 2019 (Table SI). Bone
marrow mononuclear cells (BMMCs) and peripheral blood mononuclear
cells (PBMCs) were collected by density gradient centrifugation
using Lymphosep (BioWest SAS), then lysed in RLT buffer (Qiagen,
Ltd.) supplemented with 1% β-mercaptoethanol, and/or cryopreserved
in 90% FBS and 10% DMSO (Sigma-Aldrich; Merck KGaA). After thawing,
BMMCs were primed for 24 h with a cytokine cocktail [20 ng/ml
Fms-related tyrosine kinase 3 ligand (FLT3-L), interleukin (IL)-3,
IL-6, stem cell factor and granulocyte colony-stimulating factor
(Miltenyi Biotec GmbH)] and live cells [collected using the Dead
Cell Removal Kit (Miltenyi Biotec GmbH)] were then treated with
increasing doses of kevetrin (85–340 µM) for 48 h.
Cell viability assay
Cell viability was determined using the CellTiter
96® AQueous One Solution Cell Proliferation Assay
(Promega Corporation), according to the manufacturer's
instructions. The optical density was determined after 3 h at a
wavelength of 490 nm by the Thermo Multiskan EX microplate reader
(Thermo Fisher Scientific, Inc.). Cell viability in primary samples
was evaluated by the trypan blue exclusion assay.
Annexin V staining
Phosphatidylserine externalization was evaluated
using the fluorescein isothiocyanate (FITC) Annexin V Apoptosis
Detection kit (eBioscence; Thermo Fisher Scientific, Inc.). After
treatment, cells were incubated with 25 µl/ml of Annexin V-FITC for
15 min at 37°C in a humidified atmosphere in the dark. Prior to
flow cytometric analysis, propidium iodide (PI) was added to a
final concentration of 5 µg/ml. Flow cytometric analysis was
performed using a FACSCanto flow cytometer (Becton, Dickinson and
Co.) equipped with 488 nm (blue) and 633 nm (red) lasers, and
10,000 events were recorded for each sample. Data acquisition and
analysis were performed using FACSDiva software v.6.1.3. (Becton,
Dickinson and Co.). In primary samples, Annexin V staining was
combined with surface markers using the following antibodies:
CD45-APC Vio770 (cat. no. 130-110-635), CD33-APC (cat. no.
130-111-020), CD14-PerCP Vio 700 (cat. no. 130-110-523), CD3-PE
(cat. no. 130-113-139) (all from Miltenyi Biotec GmbH, dilution
1:50) and CD19-PE Cy7 (cat. no. 302216, 1:10, BioLegend, Inc.).
Mitochondrial membrane potential (ΔΨm)
depolarization analysis
Mitochondrial membrane polarization was evaluated
using the Mito-PT JC-1 Assay Kit (ImmunoChemistry Technologies,
LLC). Following kevetrin exposure, cells were incubated according
to the manufacturer's instructions in JC-1 working solution for 15
min at 37°C and 5% CO2 in a humidified atmosphere in the
dark, suspended in 1X assay buffer and analyzed by flow cytometry.
A positive control treated with 50 µM of carbonyl cyanide
3-chlorophenylhydrazone was used for each experiment.
TUNEL assay
Fragmented DNA was detected by the TUNEL assay
(Roche Diagnostics GmbH). After each treatment, samples were fixed
in 1% formaldehyde on ice for 15 min, suspended in 70% ice-cold
ethanol and stored overnight at −20°C. Cells were incubated for 5
min at 4°C in PBS containing 0.1% Triton X-100 (Bio-Rad
Laboratories, Inc.). Thereafter, samples were suspended in 50 µl of
solution containing TdT and FITC-conjugated dUTP deoxynucleotides
1:1 (Roche Diagnostics GmbH) and incubated for 90 min at 37°C in a
humidified atmosphere in the dark. Samples were counterstained with
2.5 µg/ml of PI (MP Biomedicals, LLC) and 10 kU/ml of RNAse
(Sigma-Aldrich; Merck KGaA) for 30 min at 4°C in the dark, then
analyzed by flow cytometry. A positive control treated with 80
kU/ml of DNase (Sigma-Aldrich; Merck KGaA) was included for each
experiment.
Active caspase-3 assay
The percentage of active caspase-3 was measured
using the FITC Active Caspase-3 Apoptosis Kit (BD Biosciences).
After treatment, cells were collected, washed with ice-cold PBS 1X
and incubated for 20 min at 4°C in Cytofix/Cytoperm buffer (BD
Biosciences). The samples were then incubated with 20 µl of
anti-active caspase-3 antibody (cat. no. 550480, 1:5, BD
Biosciences) for 30 min at room temperature in the dark, washed in
Perm/Wash buffer and analyzed by flow cytometry. A positive control
treated with 5 µM camptothecin was used for each experiment.
Cell cycle analysis
After treatment, cells were washed in 1X PBS, fixed
in 70% ice-cold ethanol, stained with 10 µg/ml PI (MP Biomedicals),
10 kU/ml RNAse (Sigma-Aldrich; Merck KGaA) and 0.01% Nonidet™ P40
(NP40; Sigma-Aldrich; Merck KGaA) overnight at 4°C in the dark and
analyzed by flow cytometry. Data analysis was performed using
ModFit 4.1 (DNA Modelling System, Verity Software House, Inc.).
NGS and variant calling
DNA was extracted from primary mononuclear cells
(BMMCs or PBMCs) using the Maxwell® RSC Blood DNA Kit
(Promega Corporation) according to the manufacturer's instructions.
The mutational profile of patients was determined using SOPHiA
Myeloid Solution™ (SOPHiA GENETICS), a CE-IVD marked molecular
diagnostic application. Libraries were prepared according to the
manufacturer's instructions and quantified using the
Qubit® dsDNA High Sensitivity Assay on a
Qubit® Fluorometer (Thermo Fisher Scientific, Inc.). The
median average of amplicons was determined by capillary
electrophoresis using Agilent High Sensitivity DNA kit (Agilent
Technologies, Inc.). The pooled libraries were paired-end (2×301)
and sequenced with v3 chemistry on a MiSeq™ instrument (Illumina,
Inc.), as described in the manufacturer's protocol. FASTQ
sequencing files were uploaded onto the SOPHiA DDM®
platform (version 4), which uses patented advanced technologies for
variant calling and annotation. Human Genome Build 19 (Hg19) was
used as the reference for sequence alignment. A minimum coverage
depth of 1,000× was recommended. A filtering tool was set to
exclude known single-nucleotide polymorphisms, variants localized
in intronic and UTR regions and synonymous variants. Only exonic
and splice site variants with a variant allele frequency (VAF)
≥2.5% were evaluated.
Gene expression profiling
RNA was isolated using TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.) from cells untreated
or treated for 6 and 48 h with kevetrin at 340 µM. Labeled
single-stranded complementary DNA was prepared from 100 ng of RNA
and hybridized to Human Transcriptome Array 2.0 (Thermo Fisher
Scientific, Inc.), according to the manufacturer's recommendations.
Three independent replicates of each condition per cell line were
analyzed. Data quality control and normalization were carried out
by Expression Console software v1.4.1.46 (Thermo Fisher Scientific,
Inc.), while supervised analysis was performed with Transcriptome
Analysis Console software v.3.0 (Thermo Fisher Scientific, Inc.).
Functional annotation clustering was performed using David
Bioinformatics Resources 6.8 (National Institute of Allergy and
Infectious Diseases, National Institutes of Health) (21). Gene set enrichment analysis (GSEA)
was performed with GSEA software v.3.0 (Broad Institute) (22,23).
Western blot analysis
After a 48-h treatment, total protein extracts were
prepared in ice-cold lysis buffer (0.5% NP40, 250 mM NaCl, 50 mM
HEPES, 5 mM EDTA and 0.5 mM EGTA) containing phosphatase inhibitor
cocktail 2 (Sigma-Aldrich; Merck KGaA), protease inhibitor
(Clontech Laboratories, Inc.), and DTT (Invitrogen; Thermo Fisher
Scientific, Inc.). Before use, extract concentrations were
normalized using a BCA protein assay kit (Bio-Rad Laboratories,
Inc.). Proteins (50–100 µg) were separated by SDS-PAGE using 4–20%
polyacrylamide precast gel (Bio-Rad Laboratories, Inc.) and were
transferred to PVDF membranes using a TransBlot Turbo system
(Bio-Rad Laboratories, Inc.). Blocking and antibody incubations
were performed in Tris-buffered saline with 0.1% Tween-20 (TBST)
plus 5% dry milk or in TBST plus 5% BSA (Sigma-Aldrich; Merck
KGaA). The following antibodies were used: Anti-phosphorylated-p53
(Ser15) (cat. no. 9286, clone 16G8, 1:1,000), anti-p53 (cat. no.
2527, clone 7F5, 1:1,000) and anti-p21 (cat. no. 2946, clone DCS60,
1:2,000), all from Cell Signaling Technologies, Inc. Detection was
performed using horseradish peroxidase-conjugated anti-rabbit (cat.
no. NA934, 1:5,000) and anti-mouse (cat. no. NA931, 1:10,000)
secondary antibodies (GE Healthcare) with WesternBright Sirius
(Advansta, Inc.) or SuperSignal West Femto (Thermo Fisher
Scientific, Inc.) substrates. Membranes were imaged on a ChemiDoc
MP system (Bio-Rad Laboratories, Inc.). Membranes were stripped and
reprobed with antibody against β-actin (cat. no. ab49900, clone
AC-15, 1:25,000, Abcam) as normalizer. QuantityOne 4.6.8 software
(Bio-Rad Laboratories, Inc.) was used for analysis.
Immunofluorescence
Cells were harvested after a 48-h treatment, washed
in 1X PBS and fixed in 10% non-buffered formalin in a 37°C water
bath for 1 h. Samples were then washed in 1X PBS and resuspended in
1 ml of 70% EtOH. Slides were prepared by cytospin centrifugation
at 133 × g for 5 min at room temperature and 5 min incubation at
room temperature in ethanol solutions with increasing concentration
(50–70–100%). Blocking was performed with 1% BSA and 0.3% Triton
X-100 in 1X PBS for 1 h. Slides were incubated overnight at 4°C
with anti-p53 antibody (clone 7F5, 1:1,600 Cell Signaling
Technologies, Inc.), washed and stained with goat anti-rabbit Alexa
Fluor 594 secondary antibody (1:1,000, Invitrogen; Thermo Fisher
Scientific, Inc.) for 1 h at room temperature. The samples were
washed three times in 1X PBS and mounted using ProLong Antifade
DAPI (Invitrogen; Thermo Fisher Scientific, Inc.). Cells were
imaged with a N-SIM E laser confocal microscope (Nikon Corporation)
at a magnification of ×60 and analyzed with NIS Elements software
5.11 (Nikon Corporation) and ImageJ software 1.52a (National
Institutes of Health).
Statistical analysis
Statistical analysis was carried out with GraphPad
Prism 8.0.1 software (GraphPad Software, Inc.). Comparisons between
two groups were performed using Student's t-test, whereas multiple
comparisons were performed using one-way analysis of variance with
Dunnett's post hoc test. Values represent the mean ± standard
deviation of three independent experiments. P<0.05 was
considered to indicate statistically significant differences.
Results
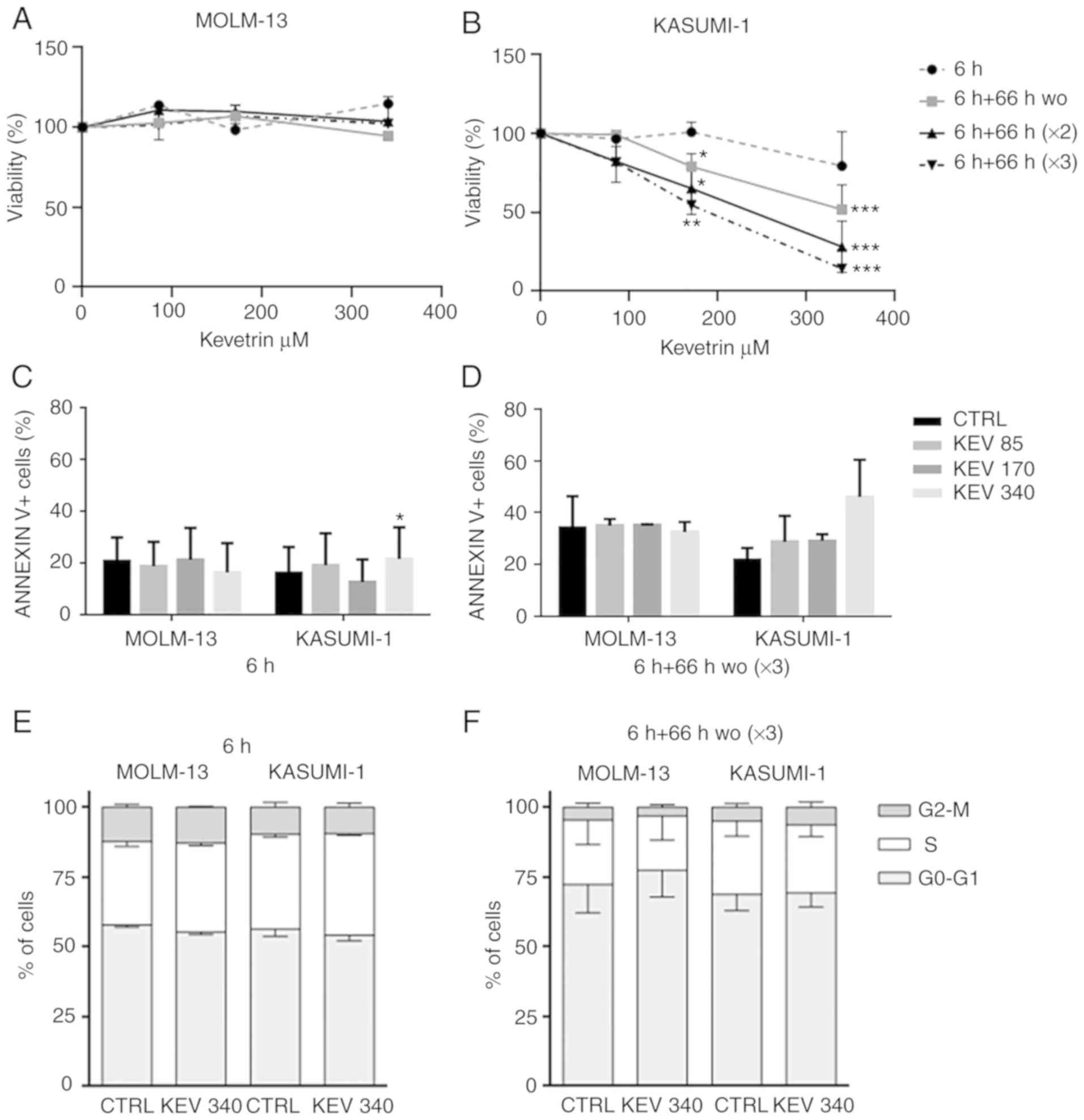
Kevetrin-pulsed treatment decreases
the viability of only the KASUMI-1 cell line
The response of MOLM-13 (TP53-wt) and
KASUMI-1 [mutated (homozygous single-nucleotide variant, p.R248Q)]
cells to kevetrin exposure was analyzed, initially using a pulsed
treatment to simulate the dosing schedules used in clinical
practice. Cell viability was evaluated after exposure to kevetrin
doses of 85, 170 and 340 µM for 6 h, or for 6 h followed by a 66-h
wo. The latter schedule was repeated consecutively two and three
times, respectively. No effect on the viability of MOLM-13 cells
was observed (Fig. 1A), whereas a
dose- and time-dependent decrease was evident in the KASUMI-1 cell
line (Fig. 1B). The role of
kevetrin treatment in apoptosis induction was then evaluated by
examining phosphatidylserine externalization following exposure to
the highest concentration in KASUMI-1 cells, detecting a trend
towards increased percentage of Annexin V+ cells
(Fig. 1C and D and Fig. S1A and B). Kevetrin treatment did
not induce cell cycle alterations (Fig.
1E and F and Fig. S1C and
D).
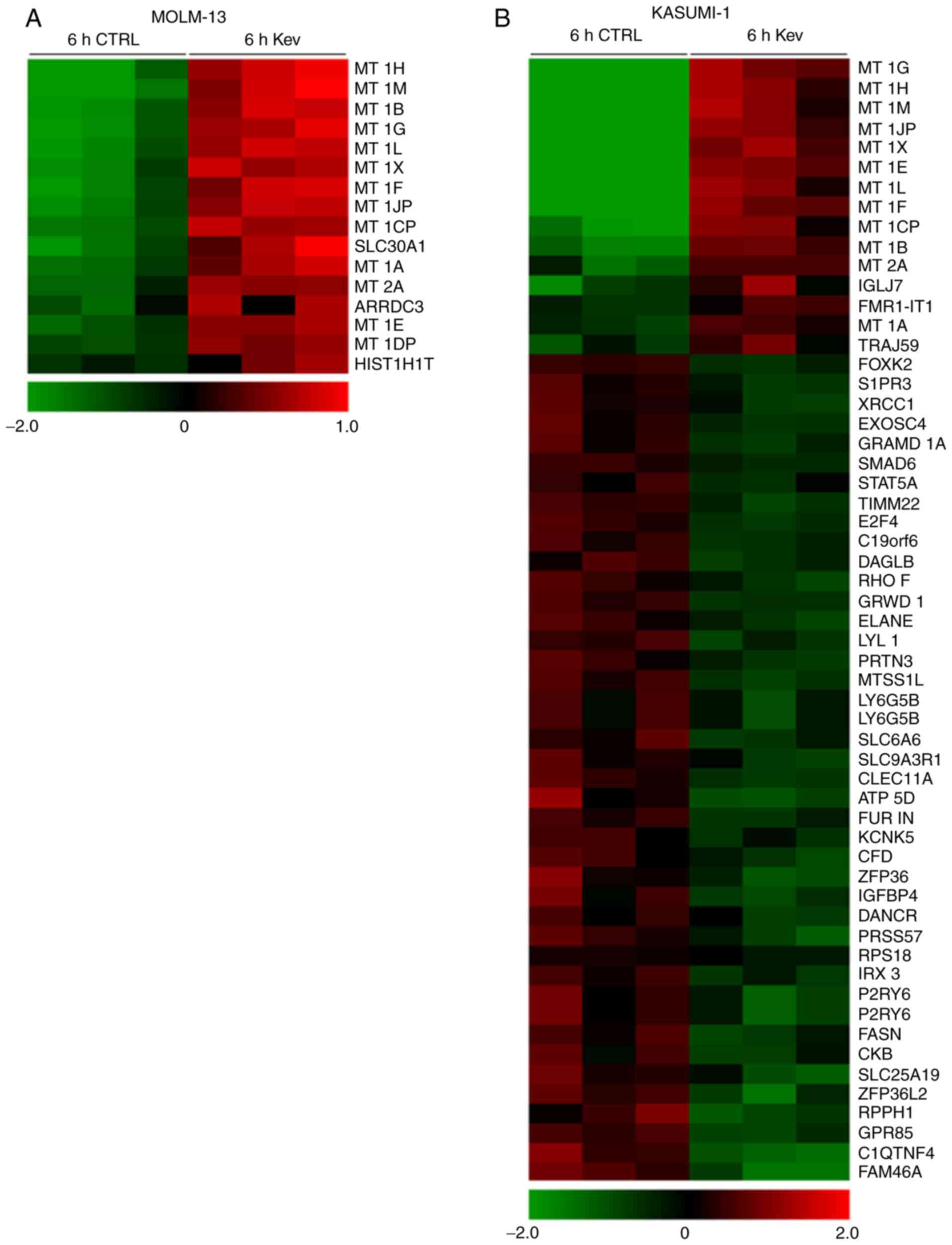
Short-term kevetrin treatment induces
metallothionein (MT) expression in AML cells
Gene expression profile analysis of MOLM-13 and
KASUMI-1 cells was performed following treatment with kevetrin or
the vehicle for 6 h to identify the transcriptional program
regulated by short-term kevetrin exposure. This strategy enabled us
to select early kevetrin-responsive genes. The overall
transcriptional program was barely affected and we observed a
differential expression of a restricted subset of genes (Fig. 2A and B), including the regulator of
WNT/β-catenin signaling forkhead box K2 and the transcription
factor signal transducer and activator of transcription 5A
(STAT5A), which were downregulated 2-fold in KASUMI-1 cells
after treatment (Fig. 2B).
Moreover, MT 1 and 2, which are involved in the scavenging of
oxygen-free radicals, were upregulated by kevetrin exposure in both
cell lines (Fig. 2A and B). In
addition, KASUMI-1 cells exhibited a significant downregulation of
genes in relation to p53 activity, including E2F transcription
factor 4 (E2F4), glutamate-rich WD repeat containing 1
(GRWD1), solute carrier family 6 member and elastase,
neutrophil expressed (ELANE) (Fig. 2B).
Continuous kevetrin treatment induces
apoptosis in AML cell lines
The effects of prolonged kevetrin exposure were
investigated by evaluating cell viability after treatment for 24,
48 and 72 h (Fig. S2) using the
same concentration range tested in the pulsed experiments. In
addition to being tested on MOLM-13 and KASUMI-1 cells, continuous
kevetrin treatment was also tested on TP53-wt OCI-AML3 cells
and TP53-mutant (heterozygous frameshift deletion, p.C242fs)
NOMO-1 models. Cell viability was barely altered after 24 h of
treatment (Fig. 3A). At 48 h, a
significant decrease in cell viability was observed in MOLM-13
cells at the highest concentration and in OCI-AML3 cells at 170 and
340 µM (Fig. 3B). A dose-dependent
inhibition was also detected in KASUMI-1 and NOMO-1 cells (Fig. 3B). To better define the mechanism of
action of kevetrin, apoptosis and cell cycle progression were
analyzed. In KASUMI-1 cells, a significant apoptosis induction was
observed after 24 h of treatment at the highest concentration
(Fig. 3C and Fig. S3). After a 48-h exposure to the
highest kevetrin concentration (340 µM), a significant increase was
observed in Annexin V+ cells among MOLM-13 (54.95±5.63%
in kevetrin-treated cells vs. 12.53±6.15% in the control) and
NOMO-1 (60.93±2.63% in kevetrin-treated cells vs. 22.90±4.63% in
the control) cells, and a mild but significant effect in OCI-AML3
cells (10.03±3.79% in kevetrin-treated cells vs. 2.60±0.70% in the
control; Fig. 3D and Fig. S4). KASUMI-1 cells exhibited a
dose-dependent response at 48 h, with 79.70±4.57% of apoptotic
cells at 340 µM (compared with 13.18±0.80% in the control; Fig. 3D and Fig. S4). Apoptotic data in MOLM-13 and
KASUMI-1 cells were confirmed in terms of mitochondrial
depolarization, DNA fragmentation and caspase-3 activation
(Fig. S5A-F). No cell cycle
alterations were observed in MOLM-13 and KASUMI-1 cells, while
NOMO-1 and OCI-AML3 cells displayed an accumulation of cells in the
G0/G1 phase and a decrease of S phase cells after 24 and 48 h of
kevetrin treatment at any concentration (Fig. 3E and F and Fig. S6A-D).
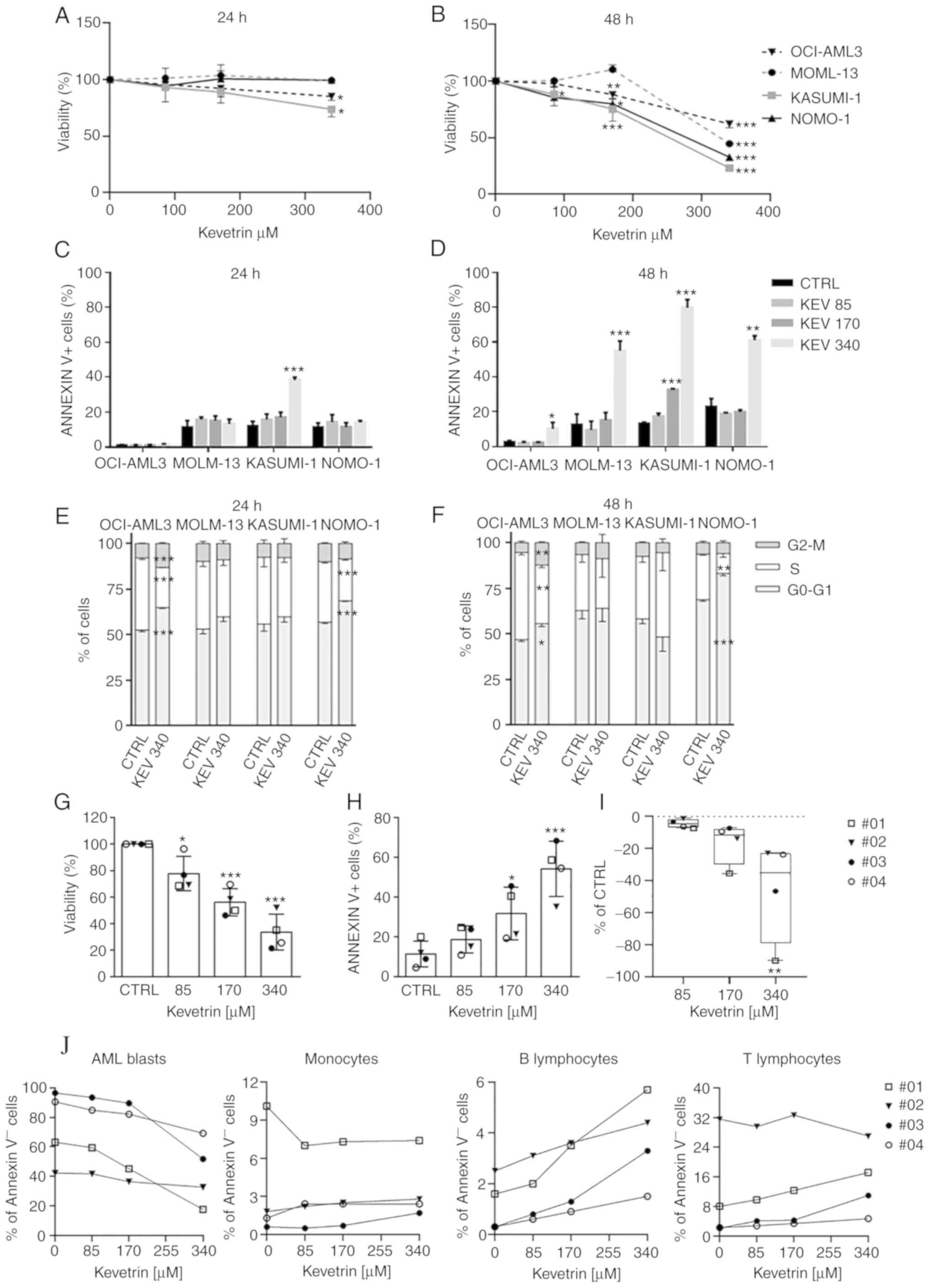 | Figure 3.Effect of continuous kevetrin
treatment on viability and apoptosis induction in AML cell lines
and primary samples. (A and B) Viability of TP53-wt OCI-AML3
and MOLM-13 and TP53-mut KASUMI-1 and NOMO-1 cell lines
treated with different concentrations of kevetrin (85, 170 and 340
µM) for 24 and 48 h. (C and D) Quantification of Annexin
V+ cells in AML cell lines treated with kevetrin at
different concentrations (85, 170 and 340 µM) for 24 and 48 h. (E
and F) Cell cycle analysis of cell lines treated with 340 µM
kevetrin for 24 and 48 h. Values represent the mean ± standard
deviation of three biological replicates. (G) Cell viability and
(H) percentage of Annexin V+ cells in bone marrow
mononuclear cells from AML patients exposed to increasing
concentrations of kevetrin. (I) Percentage of AML blasts relative
to control. Values represent the mean ± standard deviation. Symbols
indicate samples from AML patients (no. 01: TP53-mut AML;
nos. 02, 03 and 04: TP53-wt AML). (J) Percentage of Annexin
V− cells in AML blasts, monocytes and lymphocytes.
Blasts were defined as CD33+CD14− cells in
the CD45/side scatter ‘blast gate’, monocytes as
CD33+CD14+, B lymphocytes as CD19+
and T lymphocytes as CD3+. *P<0.05, **P<0.01,
***P<0.001. AML, acute myeloid leukemia; KEV, kevetrin; CTRL,
control; wt, wild-type. |
Kevetrin induces apoptosis in primary
AML blast cells
BMMCs from 4 patients were treated with increasing
kevetrin concentrations to evaluate its effect on primary AML
cells. The mutational status of patients was assessed by targeted
NGS (Table SII). One of the cases
carried a TP53 mutation [no. 01: NM_000546, c.764T>A,
p.(Ile255Asn), VAF 92.3%]. A dose-dependent decrease in cell
viability (85 µM: 77.8±12.9%, 170 µM: 56.1±10.3% and 340 µM:
33.6.1±13.5%; Fig. 3G) and a
significant increase in the proportion of apoptotic cells (170 µM:
31.8±13.3% and 340 µM: 54.3±13.9% vs. control: 11.4±6.5%; Fig. 3H and Fig. S6E) were observed. Kevetrin
cytotoxicity was then assessed in BM cell populations and a
preferential activity against AML blast cells was noted, indicating
a significant dose-dependent decrease in the percentage of alive
cells with respect to control, with the TP53-mutant sample
exhibiting high sensitivity (Fig. 3I
and J). Conversely, the percentage of AnnexinV−
monocytes and B and T lymphocytes was only marginally affected by
kevetrin treatment (Fig. 3J),
suggesting a selective cytotoxic activity of this drug.
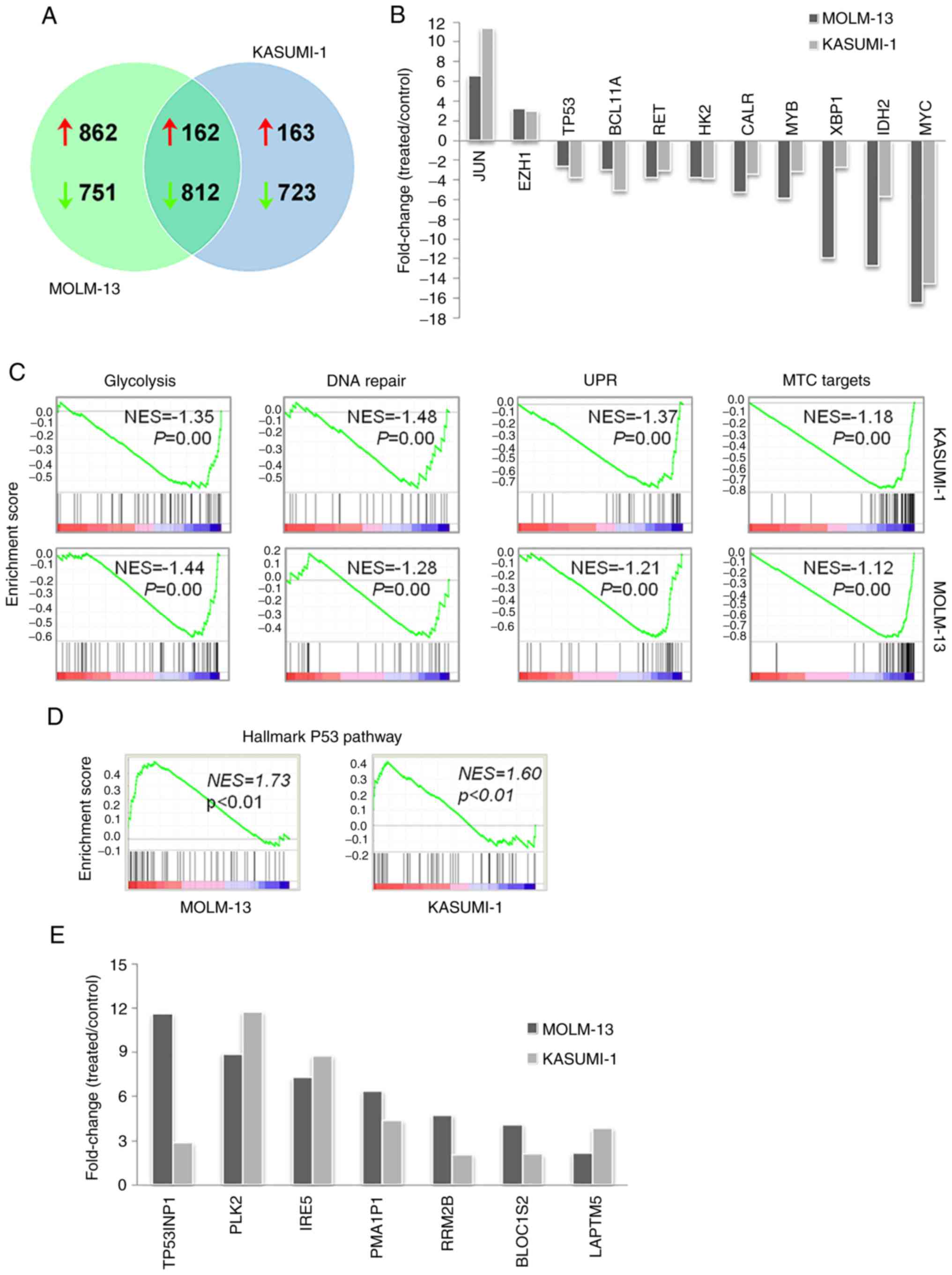
Prolonged kevetrin treatment results
in the alteration of a shared core transcriptional program in AML
cell lines
To better understand the molecular and biological
effects of kevetrin exposure, gene expression profiling of MOLM-13
and KASUMI-1 cells after a 48-h treatment was performed. A total of
1,024 upregulated and 1,563 downregulated genes were identified in
MOLM-13 cells, whereas KASUMI-1 cells exhibited increased
expression of 325 genes and decreased levels of 1,535 genes. Of
note, although a number of genes were uniquely altered, there was
also a high degree of overlap in the transcriptional changes
between MOLM-13 and KASUMI-1 cells, with a core transcriptional
program of 162 upregulated and 812 downregulated genes in both cell
lines (Fig. 4A). Enrichment
analysis of the core transcripts revealed that upregulated genes
were mainly involved in transcription, nucleosome assembly and
telomere organization (HIST4H4, HIST1H3H, HIST2H4B and
HIST1H4H), apoptosis (CASP10 and CASP8),
autophagy (GABARAPL1, ATG14 and WIPI1), nuclear
factor-κB pathway regulation (TRIM38, RIPK1, NFKBIA, IL1B,
HSPA1A, HSPA1B, S100A4, CASP10, PLK2, CASP8, TLR6, PTGS2 and
IL1B) and mitogen-activated protein kinase activity
(DUSP1, JUN, MAP3K8, DUSP10, IL1B, HSPA1A, HSPA1B, GADD45B
and DUSP22) (Table SIII).
Downregulated genes were mostly involved in cell cycle (e.g.,
CDK4 and CDC7), DNA repair (e.g., EXO1 and
FANCI), biosynthetic processes (e.g., PPAT involved
in purine nucleotide biosynthesis and FASN in fatty acid
synthesis), bioenergetics [e.g., glycolytic enzyme hexokinase 2
(HK2) and mitochondrial electron transport member
COX5A), translation (e.g., RPL34 and RPSA),
telomere maintenance (e.g., DKC1) and splicing (e.g.,
HNRNPR and PTBP1) (Table
SIV). The common core transcriptional program included critical
regulators of myelopoiesis and leukemogenesis, such as MYC,
MYB and BCL11A, leukemia-related genes involved in
unfolded protein response (UPR; XBP1 and CALR),
signaling (RET), glycolysis (HK2) and DNA methylation
[isocitrate dehydrogenase 2 (IDH2)], all of which were
downregulated by kevetrin treatment (Fig. 4B). Conversely, increased levels of
EZH1 and JUN were detected after exposure to
kevetrin. Notably, GSEA of microarray data revealed that
kevetrin-treated MOLM-13 and KASUMI-1 cells shared the
downregulation of glycolysis, DNA repair, UPR and MYC target gene
sets (Fig. 4C), in line with the
results obtained by pathway analysis.
Kevetrin affects the expression of p53
and its related proteins
The effects of kevetrin treatment on the p53 pathway
were then analyzed. High kevetrin doses induced a mild reduction in
p53 mRNA levels. However, treated cells were enriched with a
transcriptional gene signature involved in the p53 pathway
(Fig. 4D). A total of 24 bona
fide p53 targets (24)
exhibited increased expression in MOLM-13 cells, including p21
(CDKN1A), MDM2 and NOXA (PMAIP1) (Table I). The latter gene was also
upregulated in KASUMI-1 cells, along with 8 additional p53 targets
(Table I). Among these, BLOC1S2,
PLK2, RRM2B, TP53INP1, IER5 and LAPTM5 also exhibited
increased expression in MOLM-13 cells following kevetrin treatment
(Fig. 4E). To better understand the
dynamics of p53 expression and its related proteins under kevetrin
treatment, western blot and immunofluorescence analyses were
performed after a 48-h exposure to increasing kevetrin doses.
Western blot analysis did not reveal relevant alterations in the
level of total p53 or its active form (phosphorylated on Serine
15), with the exception of KASUMI-1 cells that exhibited reduced
p53 expression at the highest kevetrin dose. This may be associated
with the high numbers of apoptotic cells lost during the washing
steps required for pellet preparation. Immunofluorescence analysis
at the highest kevetrin dose revealed an increased p53 expression
compared with control cells, which resulted from a fraction of
intact cells displaying nuclear p53 staining and apoptotic cells
(identified by nuclear fragmentation) with very high p53 levels
(Fig. 5B-D). This phenomenon was
observed across all cell lines, particularly TP53-mutant
ones (Fig. 5C and D). MOLM-13 cells
generally displayed lower p53 expression levels across all
conditions, thus hampering the detection of its active form by
western blotting. Its target protein, p21, exhibited a
dose-dependent upregulation following kevetrin treatment in
TP53-wt models, in line with transcriptomic data obtained on
MOLM-13 cells (Fig. 5A and Table I).
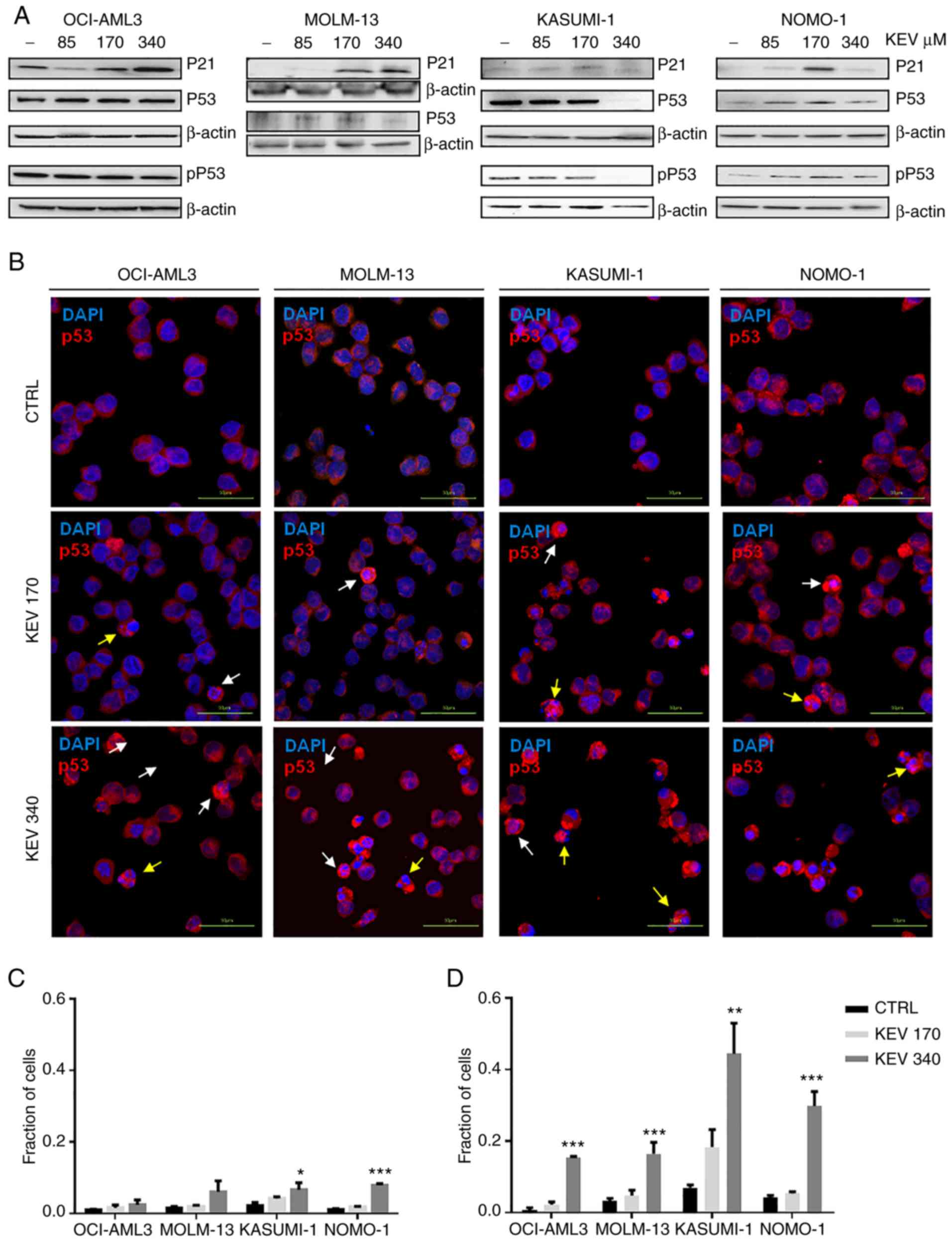 | Figure 5.Effect of kevetrin on the expression
of p53 in AML cell lines. (A) Representative western blots of
pSer15 p53 (pP53), P53 and P21 expression levels in MOLM-13,
KASUMI-1, OCI-AML3 and NOMO-1 cell lines treated with kevetrin at
85, 170 and 340 µM for 48 h. β-actin was used as loading control.
The figures show one representative of three independent
experiments. (B) Immunofluorescence analysis of p53 in AML cell
lines treated with 170 and 340 µM kevetrin for 48 h. The nuclei
were stained with DAPI. White and yellow arrows indicate intact
cells with nuclear p53 and nuclear-fragmented cells expressing high
p53, respectively. (C) Fraction of intact cells with nuclear p53
localization or (D) p53-high apoptotic cells in kevetrin-treated
cells. Values represent the mean ± standard deviation of 3
biological replicates (*P<0.05, **P<0.01, ***P<0.001).
AML, acute myeloid leukemia; KEV, kevetrin; CTRL, control. |
 | Table I.p53 targets modulated by kevetrin
treatment. |
Table I.
p53 targets modulated by kevetrin
treatment.
| Genes | MOLM-13 cells fold
change (treated/control) | KASUMI-1 cells fold
change (treated/control) |
|---|
| CDKN1A | 2.82 |
|
| MDM2 | 11.00 |
|
| BTG2 | 5.67 |
|
|
TNFRSF10B | 2.38 |
|
| ZMAT3 | 5.20 |
|
| PPM1D | 3.41 |
|
| FAS | 2.48 |
|
| NINJ1 | 2.48 |
|
| SERTAD1 | 3.66 |
|
| SESN1 | 10.67 |
|
| SLC30A1 | 2.28 |
|
| ARHGEF3 | 4.11 |
|
| FUCA1 | 2.33 |
|
| PRDM1 | 11.78 |
|
| RNF19B | 2.86 |
|
|
TNFRSF10D | 2.52 |
|
| ASCC3 | 2.21 |
|
| BLOC1S2 | 4.09 | 2.06 |
| PLK2 | 8.92 | 11.76 |
| RRM2B | 4.73 | 2.04 |
|
TP53INP1 | 11.64 | 2.85 |
| IER5 | 7.28 | 8.78 |
| LAPTM5 | 2.11 | 3.83 |
| PMAIP1 | 6.37 | 4.35 |
| ANKRA2 |
| 2.24 |
| CD82 |
| 2.74 |
Discussion
Tumor suppressor p53 is a fundamental regulator of
several responses to stress signals, such as DNA damage and
hypoxia, also modulating various genes that play a key role in cell
cycle arrest and DNA repair (13).
Recently, p53 was found to regulate metabolism, stem cell
maintenance, invasion and communication within the tumor
microenvironment (16), suggesting
a more comprehensive role in orchestrating cellular responses.
TP53 is one of the most frequently mutated genes in human
tumors (12) and mutant p53 exerts
a negative effect on the wt protein or acquires gain-of-function
properties (25,26). The aim of the present study was to
evaluate the cellular and molecular effects of kevetrin on AML
models with different TP53 mutational status, as well as on
primary AML cells.
After both pulsed and continuous treatments, a
higher sensitivity of the mutated cell lines compared with wt cells
was observed. Kevetrin exhibited a dose-dependent efficacy after a
48-h treatment, with a stronger effect on mutated models. We did
not observe cell cycle alterations in KASUMI-1 or MOLM-13 models,
whereas a significant accumulation of cells in the G0/G1 phase was
observed in OCI-AML3 and NOMO-1 cells. Only TP53-wt OCI-AML3
cells displayed an increase in the percentage of G2/M cells after
treatment, as previously reported in the TP53-wt A549 lung
carcinoma cell line (18). The
difference observed between KASUMI-1 and NOMO-1 mutated models may
be attributable to the predominant apoptotic effect induced by
kevetrin on KASUMI-1 cells and to the different TP53
mutational status. KASUMI-1 cells are characterized by a homozygous
mutation resulting in an inactive protein, whereas NOMO-1 cells
harbor a frameshift deletion (p.C242fs) leading to the synthesis of
a truncated protein. Of note, kevetrin was effective against
primary AML cells. Both TP53-wt and TP53-mutant cells
responded to ex vivo treatment, with mutant cells exhibiting
promising sensitivity. Kevetrin induced apoptosis in leukemic
blasts, whilst largely sparing the immune microenvironment, which
points to a less toxic drug profile.
Early response to kevetrin treatment, which was
assessed after a 6-h drug exposure, was characterized by
upregulation of the low-molecular weight cysteine-rich proteins MT1
and 2 in both cell lines. Several studies have reported that MTs
promote cell growth and protect cells from drug-induced oxidative
stress in cancer models (27).
Thus, increased MT expression shortly after kevetrin treatment may
represent an attempt to resist chemotherapeutic drugs (28). Although little is known on the role
of MTs in hematological malignancies, it was inferred that MT
upregulation exerted a tumor-suppressive function in our models, as
was recently suggested for MT3 overexpression in pediatric AML,
which inhibits proliferation and induces apoptosis (29). Of note, MTs are known to interact
with both wt and inactive p53 protein (30), and to modulate p53 transcriptional
activity through modification of its conformation (31), as reported in breast cancer and
osteosarcoma cell lines (31,32).
Downregulation of STAT5, a mediator of KIT-driven
leukemogenesis, only occurred in the mutated model and may be
correlated with the downregulation of the signaling pathway
activated by KIT mutation in KASUMI-1 cells (33).
With regard to TP53 interactors, the mutated
model exhibited downregulation of E2F4, which is known to be
involved in p53-dependent gene repression (34), and GRWD1, which appears to
interact with p53, negatively regulating its transcriptional
activity (35). The downregulation
of another member of the E2F transcription factor family, E2F1, was
documented by Kumar et al in MDA-MB-231 breast cancer cells,
MIA PaCa-2 pancreatic cancer cells and K-562 TP53-mutant
leukemic cells (19,36). A reduction in ELANE levels, a target
highly expressed in leukemia patients and negatively correlated
with TP53, was also observed (37).
The p53 pathway was modulated by a 48-h kevetrin
treatment, with transcriptional upregulation of several p53 targets
in TP53-wt and -mutant models. In the wt cell lines,
kevetrin induced a dose-dependent upregulation of the p21 protein,
in line with the trend observed in TP53-wt solid tumor cells
(18,20). In the TP53-mutant models, p21
upregulation was observed at an intermediate kevetrin dose, with
increased p53 nuclear localization (also detected at the highest
dose), together with a high fraction of p53-positive cells with
nuclear fragmentation. The differences between western blot
analysis and immunofluorescence may be partly attributed to the
strong effects of the treatment on cell viability (with loss of
apoptotic cells in the washing steps required for pellet
preparation). Moreover, immunofluorescence analysis is
characterized by higher sensitivity. Overall, the data of the
present study revealed that kevetrin exerted a therapeutic effect
against both TP53-wt and -mutant AML cells, the latter being
more sensitive in terms of apoptosis and p53 induction. The results
obtained from wt lines, i.e., no significant p53 nuclear
translocation and few p53-related transcriptional changes at early
timepoints, suggest an indirect kevetrin effect on p53-wt AML
cells.
Several cellular processes, including glycolysis,
DNA repair and UPR, were deregulated in response to high-dose
kevetrin treatment. Kevetrin exposure targeted a common core
transcriptional program including genes involved in myelopoiesis
and leukemogenesis. Among these, JUN and enhancer of zeste
homolog 1 (EZH1) were upregulated. JUN, which plays a
key role in cellular proliferation, differentiation and apoptosis,
is highly expressed in AML (38).
Kevetrin-induced upregulation of JUN is in line with
previous findings indicating that c-JUN represses p53 transcription
and downregulates its protein levels, thereby controlling cell
cycle progression (39).
EZH1 is involved in the methylation of the histone H3 and
may be mutated in AML (3). Its
increased expression in our models may have been caused by
deregulated chromatin modification following drug exposure.
Moreover, JUN or EZH1/2 inhibition exerts a
therapeutic effect against leukemic cells, suggesting that these
genes may represent potential targets for kevetrin-based
combination strategies (40,41).
Kevetrin modulates several oncogenic pathways,
including those involving c-MYC, MYB and B-cell lymphoma/leukemia
11A (BCL11A). c-MYC, a gene playing a key role in
hematopoiesis, is frequently overexpressed and activated in AML and
is often associated with leukemogenesis (42,43).
Hoffman et al documented the importance of c-MYC in
apoptosis induction, highlighting its crosstalk with p53 (44). c-MYB plays an important role in
cellular proliferation, while BCL11A is involved in normal
hematopoiesis and lymphoid malignancies (45) and inhibits p53 activity (46). We also observed downregulation of
two additional key leukemia-related genes: Mitochondrial
HK2, frequently upregulated by the presence of internal
tandem duplications of the FLT3 gene, which, in turn,
promotes glycolysis, and the isocitrate dehydrogenase 2
(IDH2), which is commonly mutated in AML, with a consequent
impact on DNA methylation and 2-hydroxiglutarate metabolism
(47). This evidence, along with
the repression of a glycolysis-related gene set in drug-treated
cells, suggests that kevetrin altered cellular metabolism in our
models.
Given the importance of the TP53 gene in
cancer and the frequency of its mutation, several compounds have
been tested with the aim of restoring wt p53 function or of
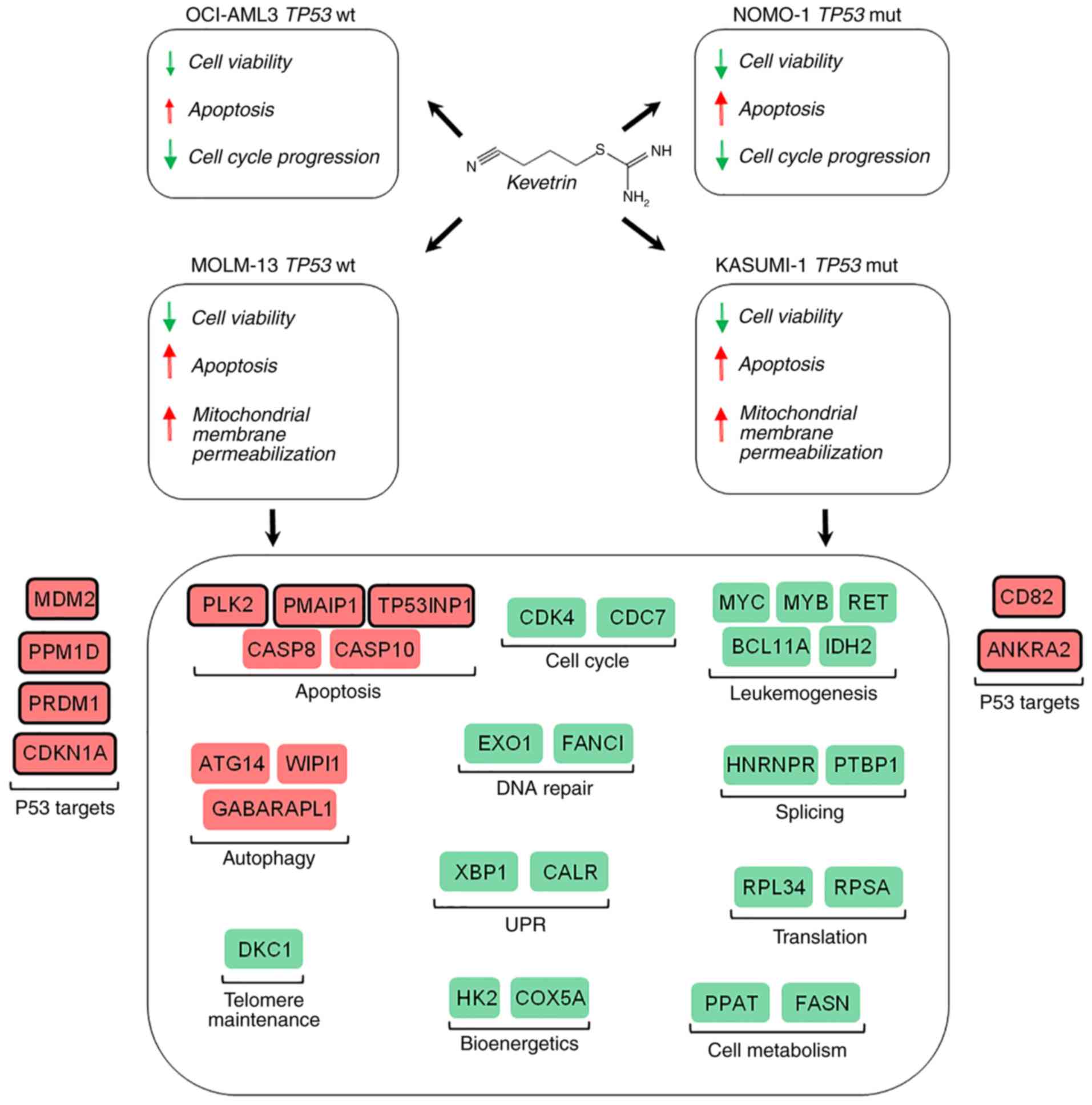
degrading the mutated protein to promote apoptosis. Despite the
lack of in vivo experiments, which represents a limitation
to the present study, the results suggest that kevetrin may be a
promising novel drug for the treatment of AML patients carrying
either wt or mutant TP53, with the latter representing an
imperative medical need due to its associated dismal prognosis
(Fig. 6) (9). A phase I clinical trial evaluating
kevetrin activity in advanced solid tumors has been successfully
completed, and its results indicate good tolerability and the
potential for therapeutic response (NCT01664000) (36,48).
The data presented herein provide a rationale for an experimental
trial in AML patients, particularly those carrying TP53
mutation, for whom the therapeutic options are currently
limited.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets generated during the current study are
available in the GEO repository, under the accession number
GSE137574.
Authors' contributions
Conceptualization, RN, SC, GS, AC, GMu and GMa;
Data curation, SDM, SC, DC and GS; formal analysis, RN, SB, GA, LC,
MG, MTB, CL, LM and GS; investigation, RN, SDM and KM; supervision,
AC and GMa; writing of the original draft, RN, SDM and GS; review
and editing, SC, KM, GMu and GMa. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Primary samples were obtained after informed
consent, as approved by the Institutional Ethics Committees in
accordance with the Declaration of Helsinki (protocol
112/2014/U/Tess of Policlinico Sant'Orsola-Malpighi).
Patient consent for publication
Not applicable.
Competing interests
KM is a co-founder of Innovation Pharmaceuticals
and he is the inventor of the patent (US 8338454 B2) for the
kevetrin molecule used in this study. GM has received honoraria
from Novartis, BNS, Roche, Pfizer, Ariad and EMSB. All other
authors declare that they have no competing interests.
References
|
1
|
Döhner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Döhner H, Estey E, Grimwade D, Amadori S,
Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA,
et al: Diagnosis and management of AML in adults: 2017 ELN
recommendations from an international expert panel. Blood.
129:424–447. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cancer Genome Atlas Research Network, ;
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson AG,
Hoadley K, Triche TJ Jr, Laird PW, et al: Genomic and epigenomic
landscapes of adult de novo acute myeloid leukemia. N Engl J Med.
368:2059–2074. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haferlach C, Dicker F, Herholz H,
Schnittger S, Kern W and Haferlach T: Mutations of the TP53 gene in
acute myeloid leukemia are strongly associated with a complex
aberrant karyotype. Leukemia. 22:1539–1541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rücker FG, Schlenk RF, Bullinger L, Kayser
S, Teleanu V, Kett H, Habdank M, Kugler CM, Holzmann K, Gaidzik VI,
et al: TP53 alterations in acute myeloid leukemia with complex
karyotype correlate with specific copy number alterations,
monosomal karyotype, and dismal outcome. Blood. 119:2114–2121.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leung GMK, Zhang C, Ng NKL, Yang N, Lam
SSY, Au CH, Chan TL, Ma ESK, Tsui SP, Ip HW, et al: Distinct
mutation spectrum, clinical outcome and therapeutic responses of
typical complex/monosomy karyotype acute myeloid leukemia carrying
TP53 mutations. Am J Hematol. 94:650–657. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fontana MC, Marconi G, Feenstra JDM, Fonzi
E, Papayannidis C, Ghelli Luserna di Rorá A, Padella A, Solli V,
Franchini E, Ottaviani E, et al: Chromothripsis in acute myeloid
leukemia: Biological features and impact on survival. Leukemia.
32:1609–1620. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rücker FG, Dolnik A, Blätte TJ, Teleanu V,
Ernst A, Thol F, Heuser M, Ganser A, Döhner H, Döhner K and
Bullinger L: Chromothripsis is linked to TP53 alteration, cell
cycle impairment, and dismal outcome in acute myeloid leukemia with
complex karyotype. Haematologica. 103:e17–e20. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Papaemmanuil E, Gerstung M, Bullinger L,
Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F,
Bolli N, et al: Genomic classification and prognosis in acute
myeloid leukemia. N Engl J Med. 374:2209–2221. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Simonetti G, Padella A, do Valle IF,
Fontana MC, Fonzi E, Bruno S, Baldazzi C, Guadagnuolo V, Manfrini
M, Ferrari A, et al: Aneuploid acute myeloid leukemia exhibits a
signature of genomic alterations in the cell cycle and protein
degradation machinery. Cancer. 125:712–725. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brosh R and Rotter V: When mutants gain
new powers: News from the mutant p53 field. Nat Rev Cancer.
9:701–713. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 502:333–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Scoumanne A and Chen X: Protein
methylation: A new mechanism of p53 tumor suppressor regulation.
Histol Histopathol. 23:1143–1149. 2008.PubMed/NCBI
|
|
14
|
Honda R, Tanaka H and Yasuda H:
Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53.
FEBS Lett. 420:25–27. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nat Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bieging KT, Mello SS and Attardi LD:
Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev
Cancer. 14:359–370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lakin ND and Jackson SP: Regulation of p53
in response to DNA damage. Oncogene. 18:7644–7655. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kumar A, Hiran T, Holden SA, Chafai-Fadela
K, Rogers S, Ram S and Menon K: Kevetrin™, a novel small molecule,
activates p53, enhances expression of p21, induces cell cycle
arrest and apoptosis in a human cancer cell line. Cancer Res.
71:44702011.
|
|
19
|
Kumar A, Holden SA, Chafai-Fadela K, Ram S
and Menon KE: Kevetrin targets both MDM2-p53 and Rb-E2F pathways in
tumor suppression. Cancer Res. 72:28742012.
|
|
20
|
Kumar A, Brennan DP, Chafai-Fadela K,
Holden SA, Ram S, Shapiro GI and Menon GI: Kevetrin induces
p53-dependent and independent cell cycle arrest and apoptosis in
ovarian cancer cell lines representing heterogeneous histologies.
Cancer Res. 77:3222017.
|
|
21
|
Huang DW, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mootha VK, Lindgren CM, Eriksson KF,
Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E,
Ridderstråle M, Laurila E, et al: PGC-1alpha-responsive genes
involved in oxidative phosphorylation are coordinately
downregulated in human diabetes. Nat Genet. 34:267–273. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fischer M: Census and evaluation of p53
target genes. Oncogene. 36:3943–3956. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Muller PA and Vousden KH: P53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Prokocimer M, Molchadsky A and Rotter V:
Dysfunctional diversity of p53 proteins in adult acute myeloid
leukemia: Projections on diagnostic workup and therapy. Blood.
130:699–712. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takahashi S: Molecular functions of
metallothionein and its role in hematological malignancies. J
Hematol Oncol. 5:412012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Basu A and Krishnamurthy S: Cellular
responses to cisplatin-induced DNA damage. J Nucleic Acids.
2010:2013672010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tao YF, Xu LX, Lu J, Cao L, Li ZH, Hu SY,
Wang NN, Du XJ, Sun LC, Zhao WL, et al: Metallothionein III (MT3)
is a putative tumor suppressor gene that is frequently inactivated
in pediatric acute myeloid leukemia by promoter hypermethylation. J
Transl Med. 12:1822014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ostrakhovitch EA, Olsson PE, Jiang S and
Cherian MG: Interaction of metallothionein with tumor suppressor
p53 protein. FEBS Lett. 580:1235–1238. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Méplan C, Richard MJ and Hainaut P:
Metalloregulation of the tumor suppressor protein p53: Zinc
mediates the renaturation of p53 after exposure to metal chelators
in vitro and in intact cells. Oncogene. 19:5227–5236. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Habel N, Hamidouche Z, Girault I,
Patiño-García A, Lecanda F, Marie PJ and Fromigué O: Zinc
chelation: A metallothionein 2A's mechanism of action involved in
osteosarcoma cell death and chemotherapy resistance. Cell Death
Dis. 4:e8742013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chatterjee A, Ghosh J, Ramdas B, Mali RS,
Martin H, Kobayashi M, Vemula S, Canela VH, Waskow ER, Visconte V,
et al: Regulation of Stat5 by FAK and PAK1 in oncogenic FLT3- and
KIT-driven leukemogenesis. Cell Rep. 9:1333–1348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Benson EK, Mungamuri SK, Attie O,
Kracikova M, Sachidanandam R, Manfredi JJ and Aaronson SA:
p53-dependent gene repression through p21 is mediated by
recruitment of E2F4 repression complexes. Oncogene. 33:3959–3969.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kayama K, Watanabe S, Takafuji T, Tsuji T,
Hironaka K, Matsumoto M, Nakayama KI, Enari M, Kohno T, Shiraishi
K, et al: GRWD1 negatively regulates p53 via the RPL11-MDM2 pathway
and promotes tumorigenesis. EMBO Rep. 18:123–137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shapiro G, Mier JW, Hilton JF, Gandhi L,
Chau NG, Bullock AJ, Supko JG, Verselis SJ, Murgo K, Sze C, et al:
A phase 1, dose-escalation, safety, pharmacokinetic,
pharmacodynamic study of thioureidobutyronitrile, a novel p53
targeted therapy, in patients with advanced solid tumors. J Clin
Oncol. 33 (Suppl 15):TPS26132015. View Article : Google Scholar
|
|
37
|
Zhao Y, Si L, Zhang W, Huang W and Wang R:
ELANE is highly expressed in leukemia patients and predicts poor
survival. Int J Clin Exp Med. 12:3153–3160. 2019.
|
|
38
|
Rangatia J, Vangala RK, Singh SM, Zada
AAP, Elsässer A, Kohlmann A, Haferlach T, Tenen DG, Hiddemann W and
Behre G: Elevated c-Jun expression in acute myeloid leukemias
inhibits C/EBPalpha DNA binding via leucine zipper domain
interaction. Oncogene. 22:4760–4764. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schreiber M, Kolbus A, Piu F, Szabowski A,
Möhle-Steinlein U, Tian J, Karin M, Angel P and Wagner EF: Control
of cell cycle progression by c-Jun is p53 dependent. Genes Dev.
13:607–619. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fujita S, Honma D, Adachi N, Araki K,
Takamatsu E, Katsumoto T, Yamagata K, Akashi K, Aoyama K, Iwama A
and Kitabayashi I: Dual inhibition of EZH1/2 breaks the quiescence
of leukemia stem cells in acute myeloid leukemia. Leukemia.
32:855–864. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou C, Martinez E, Di Marcantonio D,
Solanki-Patel N, Aghayev T, Peri S, Ferraro F, Skorski T, Scholl C,
Fröhling S, et al: JUN is a key transcriptional regulator of the
unfolded protein response in acute myeloid leukemia. Leukemia.
31:1196–1205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dolores Delgado M and León J: Myc roles in
hematopoiesis and leukemia. Genes Cancer. 1:605–616. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hoffman B, Amanullah A, Shafarenko M and
Liebermann DA: The proto-oncogene c-myc in hematopoietic
development and leukemogenesis. Oncogene. 21:3414–3421. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hoffman B and Liebermann DA: Apoptotic
signaling by c-MYC. Oncogene. 27:6462–6472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu P, Keller JR, Ortiz M, Tessarollo L,
Rachel RA, Nakamura T, Jenkins NA and Copeland NG: Bcl11a is
essential for normal lymphoid development. Nat Immunol. 4:525–532.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu Y, Wang J, Khaled W, Burke S, Li P,
Chen X, Yang W, Jenkins NA, Copeland NG, Zhang S and Liu P: Bcl11a
is essential for lymphoid development and negatively regulates p53.
J Exp Med. 209:2467–2483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Im AP, Sehgal AR, Carroll MP, Smith BD,
Tefferi A, Johnson DE and Boyiadzis M: DNMT3A and IDH mutations in
acute myeloid leukemia and other myeloid malignancies: Associations
with prognosis and potential treatment strategies. Leukemia.
28:1774–1783. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shapiro G, Supko JG, Cho DC, Hilton JF,
Hadfield M, Pruitt-Thompson S, Bordoli-Trachsela E, Zvereva N,
Wolanski A, Sato-DiLorenzo A, et al: A phase I, dose-escalation,
safety, pharmacokinetic, pharmacodynamic study of
thioureidobutyronitrile, a novel p53 targeted therapy, in patients
with advanced solid tumors. J Clin Oncol. 31 (Suppl
15):TPS26272013. View Article : Google Scholar
|