Introduction
Lung cancer is the leading cause of cancer-related
death worldwide (1). Lung cancer is
classified into two major types; small cell lung cancer (SCLC)
accounting for 10–14% of all lung cancer cases and non-SCLC (NSCLC)
representing 85–90% of all lung cancer cases (2). NSCLC is further divided into three
subtypes according to histology: Squamous-cell carcinoma,
adenocarcinoma, and large cell carcinoma (3). Various clinical cancer therapies have
been used to treat lung cancer, but better efficacy is still
required. Many studies report inhibition of cell growth and
induction of apoptosis by many therapeutic agents (4,5), yet
novel agents that target specific intracellular targets of lung
cancer cells continue to be developed.
Apoptosis is a cellular response to anti-cancer
drugs. The mechanism of apoptosis mainly involves mitochondrial and
cell death receptor pathways (6).
The key element in the mitochondrial pathway is the efflux of
cytochrome c from mitochondria to cytosol. In the cytosol,
cytochrome c forms a complex (apoptosome) with apoptotic
protease-activating factor 1 (Apaf-1) and caspase-9, leading to the
activation of caspase-3 (6,7). The induction of apoptosis is
accompanied by increasing BAX and decreasing Bcl-2 levels, leading
to the loss of mitochondrial membrane potential (MMP; ∆Ψm)
(8). The cell death receptor
pathway is characterized by the binding of cell death ligands to
their death receptors with subsequent activation of caspase-8 and
−3 (9). Caspase-3 is a major
executioner caspase, whose activation can systematically dismantle
cells by cleaving key proteins, especially poly (ADP-ribose)
polymerase (PARP) (10). Thus,
targeted inhibition of anti-apoptotic pathways is an attractive
concept for the design of cancer treatments.
Auranofin, a thioredoxin reductase (TrxR) inhibitor,
was initially used for oral therapy for rheumatoid arthritis
(11). Originally, this agent was
considered an anti-inflammatory drug (12). Thioredoxin (Trx) and TrxR make a
coupled redox system, which plays a key role in maintaining redox
reactions in biosynthetic pathways and controlling redox
homeostasis. Trx, a redox regulatory protein, can be oxidized by
reactive oxygen species (ROS). Oxidative stress due to either
overproduction of ROS or accumulation thereof can initiate events
that lead to cell death (13,14).
Trx and TrxR are overexpressed in numerous cancer cells including
lung Cancer (15). Modulation of
the Trx system is thus a promising target for cancer therapy
(11). Trx and TrxR expression are
upregulated by nuclear factor-erythroid 2 p45-related factor 2
(16). Inhibition of TrxR increases
the efficacy of anti-cancer drugs in lung and colon cancer
(17–19). Downregulation of Trx by suberoyl
bis-hydroxamic acid is closely involved in lung cancer cell death
(20). Auranofin also induces
apoptosis in mesothelioma and cervical cancer cells via oxidative
stress (13,21).
Understanding of the anti-cancer effects of
auranofin in lung cancer cells remains poor. In the present study,
various lung cancer cells were used to investigate the molecular
basis of anti-cancer effects of auranofin, including cell death via
apoptosis or necrosis and cell cycle arrest.
Materials and methods
Cell culture
Human SCLC cell line (Calu-6), adenocarcinoma cell
lines (A549, SK-LU-1), and large cell carcinoma cell lines
(NCI-H460, NCI-H1299) were obtained from the American Type Culture
Collection (Manassas, VA). Normal human pulmonary fibroblast (HPF)
cells were obtained from PromoCell GmbH (C-12360, Heidelberg,
Germany). These cells were maintained in an incubator containing 5%
CO2 at 37°C. HPF and lung cancer cells were cultured in
RPMI-1640 containing 10% fetal bovine serum (Sigma-Aldrich Co., St.
Louis, MO) and 1% penicillin-streptomycin (Gibco BRL, Grand Island,
NY). Cells were grown in 100 mm plastic cell culture dishes (BD
Falcon. Franklin Lakes, NJ) and harvested with trypsin-EDTA (Gibco
BRL). HPF cells were used between passages of four to five.
Reagents
Auranofin was purchased from Sigma-Aldrich Co. and
was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich Co.) at 10
mM as a stock solution. Pan-caspase inhibitor
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-FMK),
caspase-3 inhibitor
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone (Z-DEVD-FMK),
caspase-8 inhibitor
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone (Z-IETD-FMK),
and caspase-9 inhibitor
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone (Z-LEHD-FMK)
were obtained from R&D Systems, Inc. (Minneapolis, MN,) and
dissolved in 10 mM DMSO as stock solutions. Cells were pretreated
with 15 µM of individual caspase inhibitors for 1 h prior to the
addition of auranofin. DMSO (0.01%) was used as a control vehicle
and did not affect cell growth or cell death.
Cell growth inhibition assay
The effects of auranofin on the proliferation of HPF
and lung cancer cells were determined by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT,
Sigma-Aldrich Co.) assays. Briefly, 3×104 cells were
seeded into 96-well microtiter plates (Nunc). After incubation with
the indicated doses of auranofin for 24 h, 20 µl of MTT solution [2
mg/ml in phosphate-buffered saline (PBS; GIBCO BRL)] was added to
each well. The plates were incubated for 4 h at 37°C. Medium in
plates was removed by pipetting, and 100–200 µl of DMSO was added
to each well to solubilize formazan crystals. Optical density was
measured at 570 nm using a microplate reader (Synergy™ 2, BioTekR
Instruments Inc. Winooski, VT).
Lactate dehydrogenase (LDH) release
assay
Necrosis in HPF and lung cancer cells treated with
auranofin were evaluated by LDH kit (Sigma-Aldrich Co.) Briefly,
1×106 cells in 60 mm culture dishes (BD Falcon) were
incubated with the indicated concentrations of auranofin for 24 h.
After treatment, cell culture media were collected and centrifuged
for 5 min at 200 × g at room temperature. Supernatants (50 µl) were
added to 96-lawell plates along with LDH assay reagent and
incubated at room temperature for 30 min. Absorbance values were
measured at 490 nm using a microplate reader (Synergy™ 2). LDH
release was expressed as the percentage of extracellular LDH
activity compared with untreated control cells.
Cell cycle and sub-G1 cell
analysis
Cell cycle and sub-G1 distributions of cells were
determined by propidium iodide (PI, Sigma-Aldrich Co.; Ex/Em=488
nm/617 nm) staining, as previously described (21). Briefly, 1×106 cells in 60
mm culture dishes (BD Falcon) were incubated with the indicated
concentrations of auranofin for 24 h. After washing whole cells
including floating cells with PBS, cells were fixed in 70% ethanol.
These cells were washed with PBS twice and then incubated with PI
(10 µg/ml) and RNase (Sigma-Aldrich) at 37°C for 30 min.
Proportions of cells in different phases of cell cycle or with
sub-G1 DNA content were measured and analyzed with a FAC Star flow
cytometer (BD Sciences, Franklin Lakes, NJ, USA).
Detection of apoptosis
Apoptosis was identified by staining with annexin
V-fluorescein isothiocyanate (FITC, Life Technologies;
Ex/Em=488/519 nm), as previously described (22). Briefly, 1106 cells in 60
mm culture dishes (BD Falcon) were incubated with the indicated
concentrations of auranofin for 24 h with or without individual
caspase inhibitors. Cells were washed twice with cold PBS and then
suspended in 200 µl of binding buffer (10 mM HEPES/NaOH pH 7.4, 140
mM NaCl, 2.5 mM CaCl2) at a concentration of
5×105 cells/ml at 37°C for 30 min. Annexin V-FITC (2 µl)
and PI (1 µg/ml) were added, and cells were analyzed with a FACStar
flow cytometer (BD Sciences).
Measurement of mitochondrial membrane
potential (ΔΨm)
The mitochondrial membrane potential (MMP, ΔΨm) was
monitored using a fluorescent dye Rhodamine 123 (Sigma-Aldrich Co.;
Ex/Em=485/535 nm), a cell-permeable cationic dye, which
preferentially enters into mitochondria of their typical highly
negative MMP (∆Ψm). Depolarization of MMP (∆Ψm) results in the loss
of Rhodamine 123 from the mitochondria and decreases the
intracellular fluorescence of this dye, as previously described
(23). In brief, 1×106
cells in 60 mm culture dishes (Nunc) were incubated with the
designated doses of auranofin for 24 h with or without 15 µM
individual caspase inhibitors. Cells were washed twice with PBS and
incubated with Rhodamine 123 (0.1 mg/ml) at a concentration of
5×105 cells/ml at 37°C for 30 min. Rhodamine 123
staining intensities were determined using a FACStar flow
cytometer. Rhodamine 123 negative (−) cells indicated MMP (∆Ψm)
loss.
Western blot analysis
The protein expression levels were evaluated by
western blotting. Briefly, 5×106 cells in 100 mm culture
dishes (BD Falcon) were incubated condition with the indicated
concentrations of auranofin at 37°C for 24 h with or without
pan-caspase inhibitor, (Z-VAD). Cells were washed with PBS and
lysed for 30 min in RIPA buffer supplemented with protease and
phosphatase inhibitor cocktail (Intron Biotechnology, Seongnam
Korea). The samples were heated to 100°C for 5 min and placed on
ice. Total proteins (30 µg) were resolved using 8–15% SDS-PAGE gels
and then transferred to Immobilon-P PVDF membranes (Millipore) by
electroblotting. Membranes were probed with anti-PARP (no. 9543,
1:1,000 dilution), anti-cleaved PARP (no. 9541, 1:1,000 dilution),
anti-caspase-3 (no. 9662, 1:1,000 dilution), anti-caspase-8 (no.
9746, 1:1,000 dilution), anti-caspase-9 (no. 9502, 1:1,000
dilution), anti-cleaved caspase-3 (no. 9661, 1:1,000 dilution),
anti-cleaved caspase-8 (no. 9496, 1:1,000 dilution), anti-cleaved
caspase-9 (no. 9501, 1:1,000), anti-Bcl-2 (no. 2872, 1:1,000
dilution), anti-BAX (no. 2774, 1:1,000 dilution) (Cell Signaling
Technology); anti-Trx1 (SC-20146, 1:1,000 dilution) and anti-GAPDH
(SC-25778, 1:1,000 dilution) (Santa Cruz Biotechnology). Membranes
were incubated with horseradish peroxidase-conjugated secondary
antibodies (Santa Cruz Biotechnology) at 4°C for 1 h. Blots were
developed using an EZ-Western Lumi Pico ECL solution kit (DoGen Bio
Co, Seoul, Korea). All band intensities were quantified using the
Image J program (Fuji Film, Tokyo, Japan).
Detection of TrxR activity
The activity of TrxR was assessed using the
Thioredoxin Reductase assay kit according to the manufacturer's
instructions (Sigma-1Aldrich). In brief, 1×106 cells
were incubated in 60 mm culture dish (Nunc) with the indicated dose
of auranofin for 24 h. The cells were then washed in PBS and
suspended in five volumes of lysis buffer. Protein concentrations
were determined using the Bradford method. Supernatant samples
containing 30 µg total protein were used for the determination of
TrxR activity. These were added to each well in 96-well microtiter
plates (Nunc) with 5,5′-dithiobis (2-nitrobenzoic) acid at 25°C for
1 h. The optical density of each well was measured at 412 nm using
a microplate reader (Synergy™2).
Statistical analysis
The results are reported as the mean of at least two
or three independent experiments (mean ± SD). Data were analyzed
using Instat software (GraphPad Prism5). The Student's t-test or
one-way analysis of variance with post-hoc analysis using Tukey's
multiple comparison test was used for the parametric data.
Statistical significance was defined as P<0.05.
Results
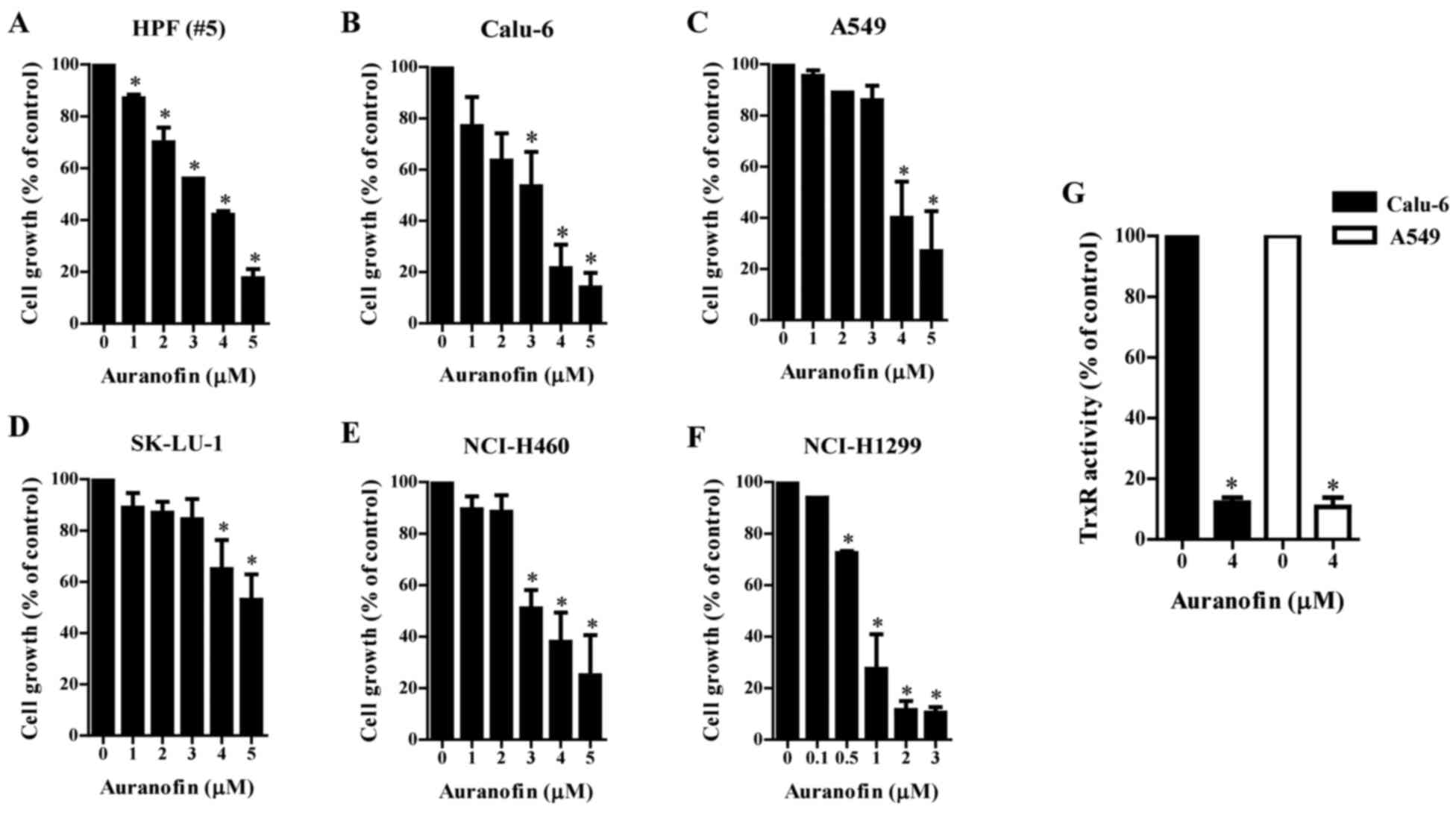
Effects of auranofin on cell growth
and TrxR activity in lung cancer cells
The effect of auranofin, a known inhibitor of TrxR,
on the growth of normal lung cell and lung cancer cell types was
examined using MTT assays. The growth of normal HPF cells showed
dose-dependent inhibition with an IC50 of ~3 µM
(Fig. 1A) after a 24-h incubation
with auranofin. The growth of Calu-6 cells was also
dose-dependently reduced with an IC50 of ~3 µM (Fig. 1B). The growth of A549 and SK-LU-1
cells was marginally reduced by 1–3 µM auranofin and significantly
decreased by 4–5 µM auranofin (Fig. 1C
and D). Auranofin inhibited the growth of NCI-H460 and
NCI-H1299 cells in a dose-dependent manner with IC50 of
~3 µM (Fig. 1E) and 1 µM (Fig. 1F). Furthermore, auranofin
significantly decreased the activity of TrxR in Calu-6 and A549
cells (Fig. 1G). Auranofin also
downregulated the expression of Trx1 protein in Calu-6 and A549
cells (Fig. S1).
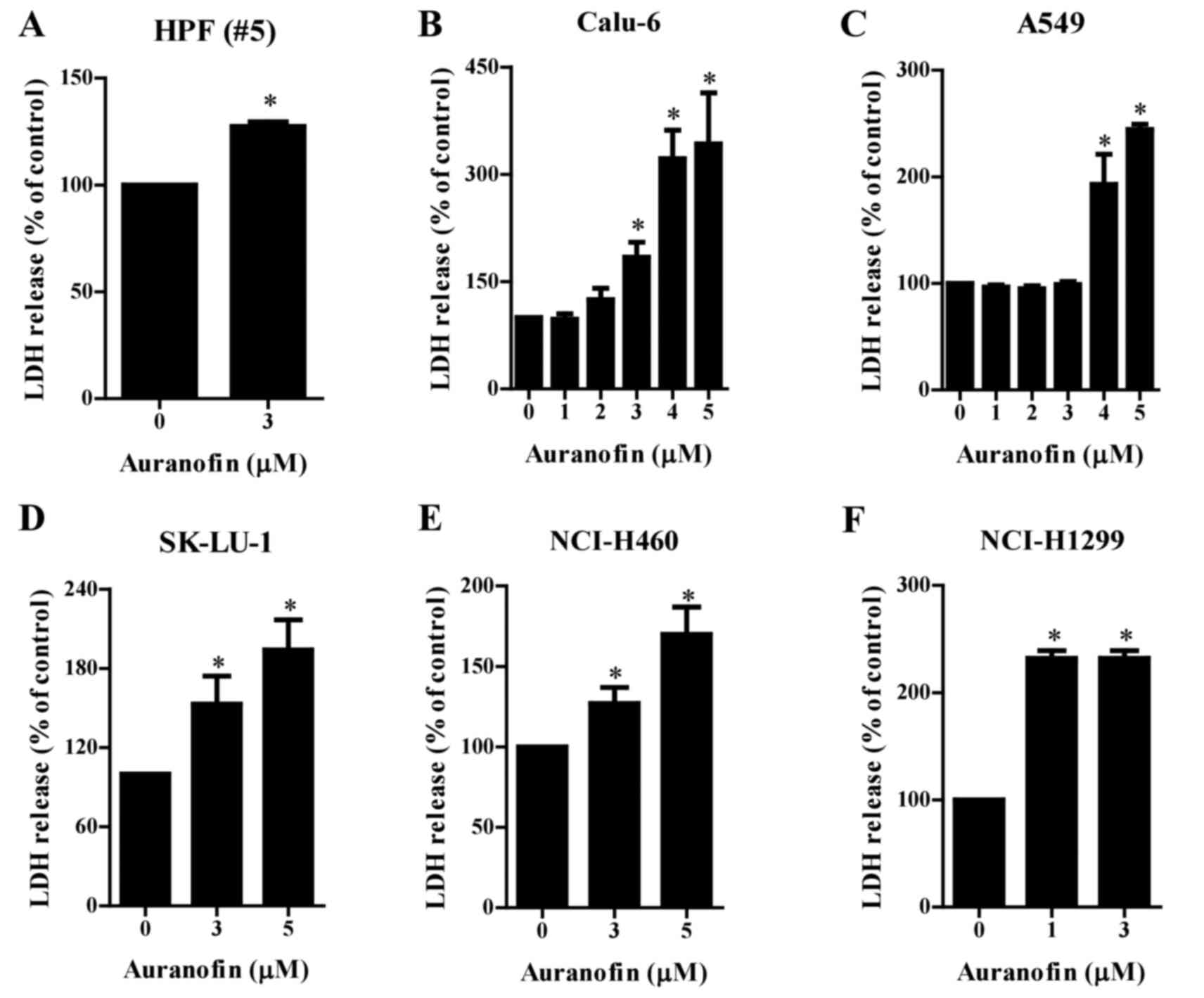
Effects of auranofin on cell death in
lung cancer cells
LDH release was measured to determine whether
auranofin causes cell necrosis. Treatment increased the release of
LDH in the normal HPF cells after a 24 h incubation with 3 µM
auranofin (Fig. 2A). Auranofin (3–5
µM) induced significant LDH release in Calu-6, SK-LU-1, and
NCI-H460 cells in a dose-dependent manner (Fig. 2B, D and E), at 4–5 µM triggered LDH
release in A549 cells (Fig. 2C),
and at 1 and 3 µM concentrations increased LDH release in NCI-H1299
cells (Fig. 2F).
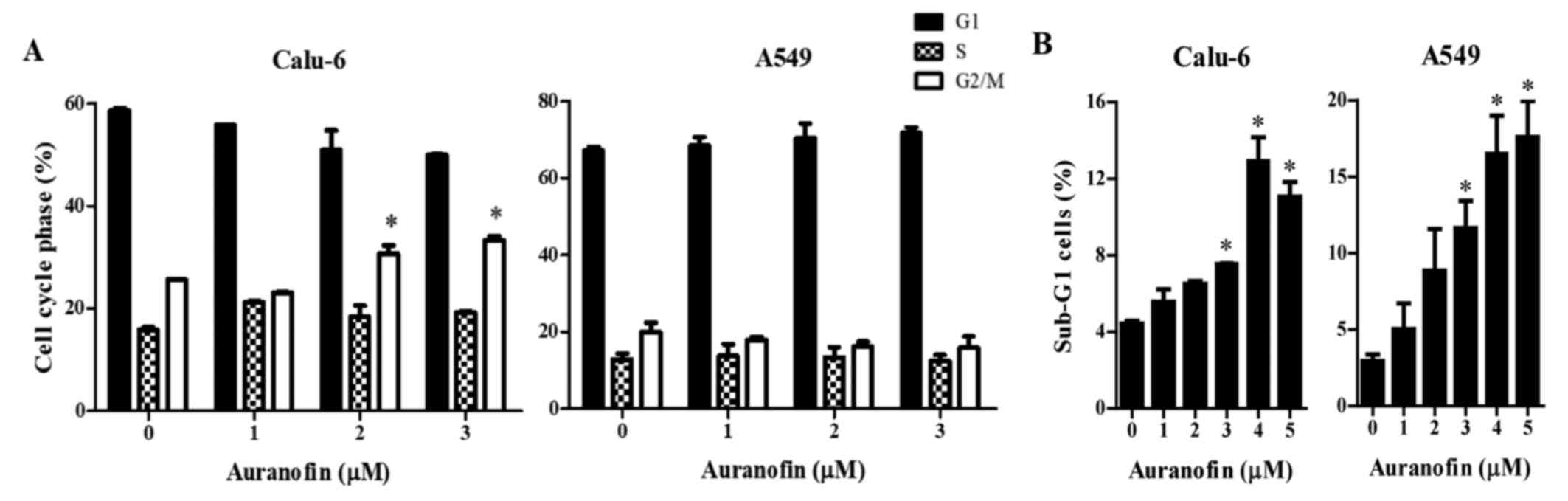
Effects of auranofin on the cell cycle
distributions in Calu-6 and A549 lung cancer cells
As growth inhibition of Calu-6 and A549 cells by
auranofin could be explained by an arrest during cell cycle
progression, distribution of the cells in different stages of the
cell cycle were examined after a 24 h incubation with auranofin.
DNA flow cytometric analysis indicated that 2 and 3 µM auranofin
induced a G2/M phase arrest of the cell cycle in Calu-6 cells and
that 1 µM auranofin did not affect cell cycle distributions. In
addition, auranofin did not show specific cell cycle arrest in A549
cells (Fig. 3A and B). Moreover,
auranofin significantly increased the percentages of sub-G1 cells
in Calu-6 and A549 cells at 24 h (Fig.
3B).
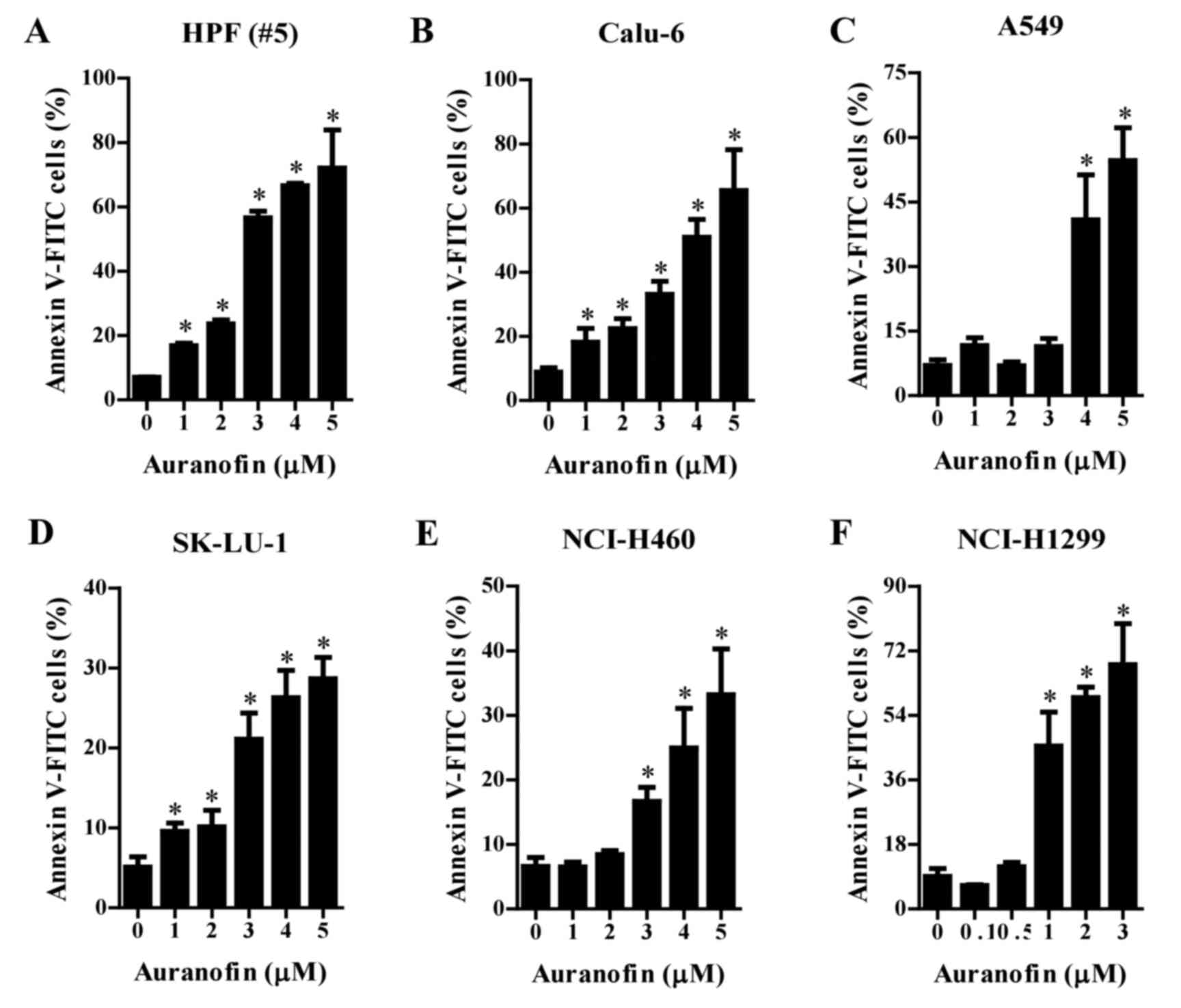
Effects of auranofin on apoptosis in
lung cancer cells
Whether auranofin induces apoptosis in cells was
assessed using an annexin V-staining assay. The number of annexin
V-positive normal HPF and Calu-6 cells significantly increased in a
dose-dependent manner after treatment with 1–5 µM auranofin
(Fig. 4A and B). At 4–5 µM, the
number of annexin V-positive A549 cells was greatly increased
(Fig. 4C). Similarly, treatment
with 1–5 µM auranofin increased the number of annexin V-positive
SK-LU-1 cells (Fig. 4D). The number
of annexin V-positive NCI-H460 and NCI-H1299 cells were increased
after incubation in 3–5 and 1–3 µM concentrations of auranofin,
respectively (Fig. 4E and F).
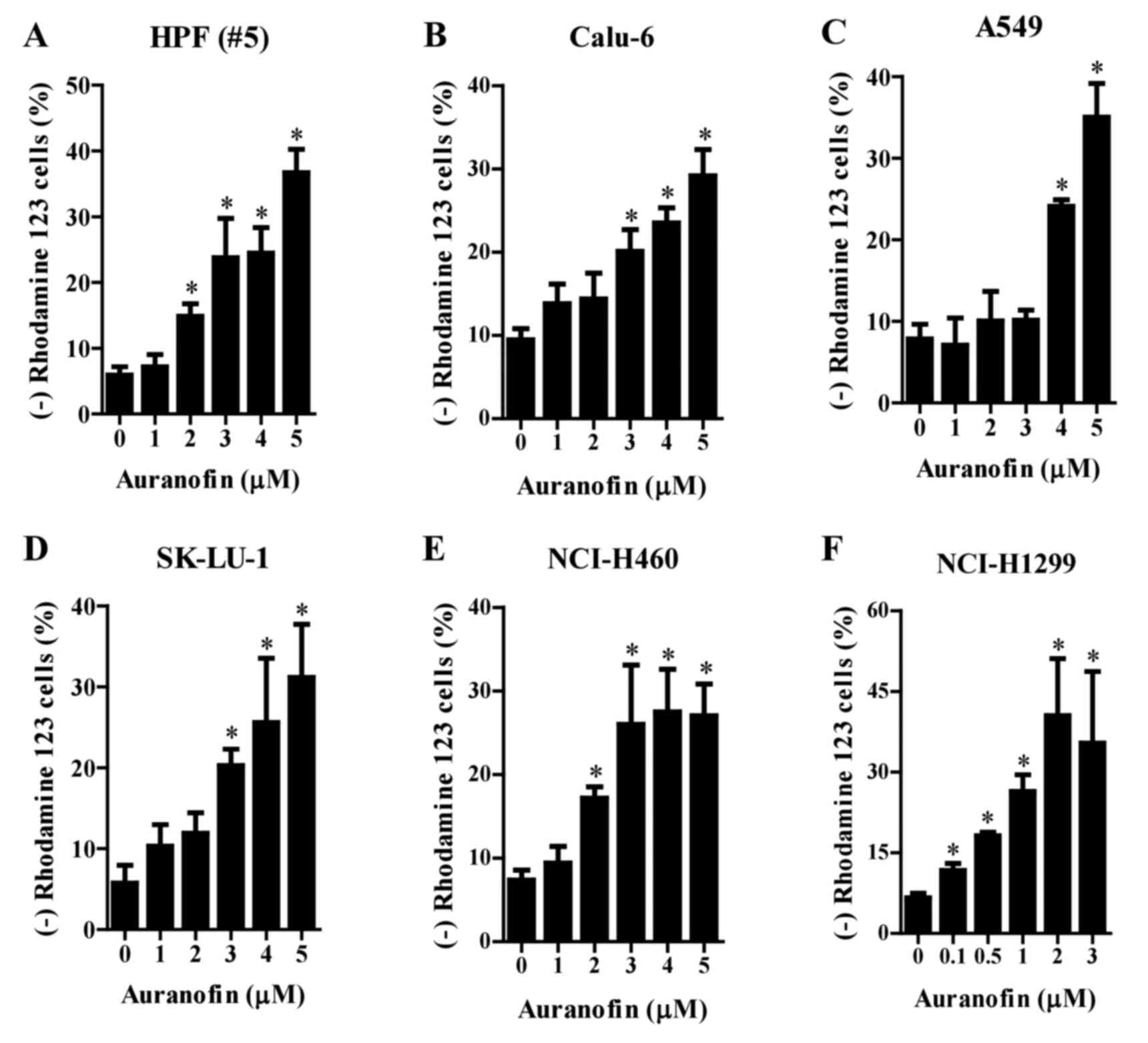
Effects of auranofin on mitochondrial
membrane potential (MMP; ∆Ψm) in lung cancer cells
Since apoptosis is closely related to the collapse
of MMP (∆Ψm), loss of MMP (∆Ψm) in auranofin-treated cells was
assessed using Rhodamine 123 dye. Loss of MMP (∆Ψm) in the normal
HPF cells was dose-dependently induced by auranofin at
concentrations of 2–5 µM (Fig. 5A).
Similar loss of MMP (∆Ψm) was observed after treatment of Calu-6
and SK-LU-1 cells (Fig. 5B and D),
A549 cells (Fig. 5C), and NCI-H460
cells (Fig. 5E) with auranofin at
3–5, 4–5, and 2–5 µM, respectively. Concentrations of 1–3 µM
auranofin did not show this effect in A549 cells (Fig. 5C). Furthermore, auranofin at
concentrations of 0.1–0.5 µM significantly increased loss of MMP
(∆Ψm) in NCI-H1299 cells (Fig.
5F).
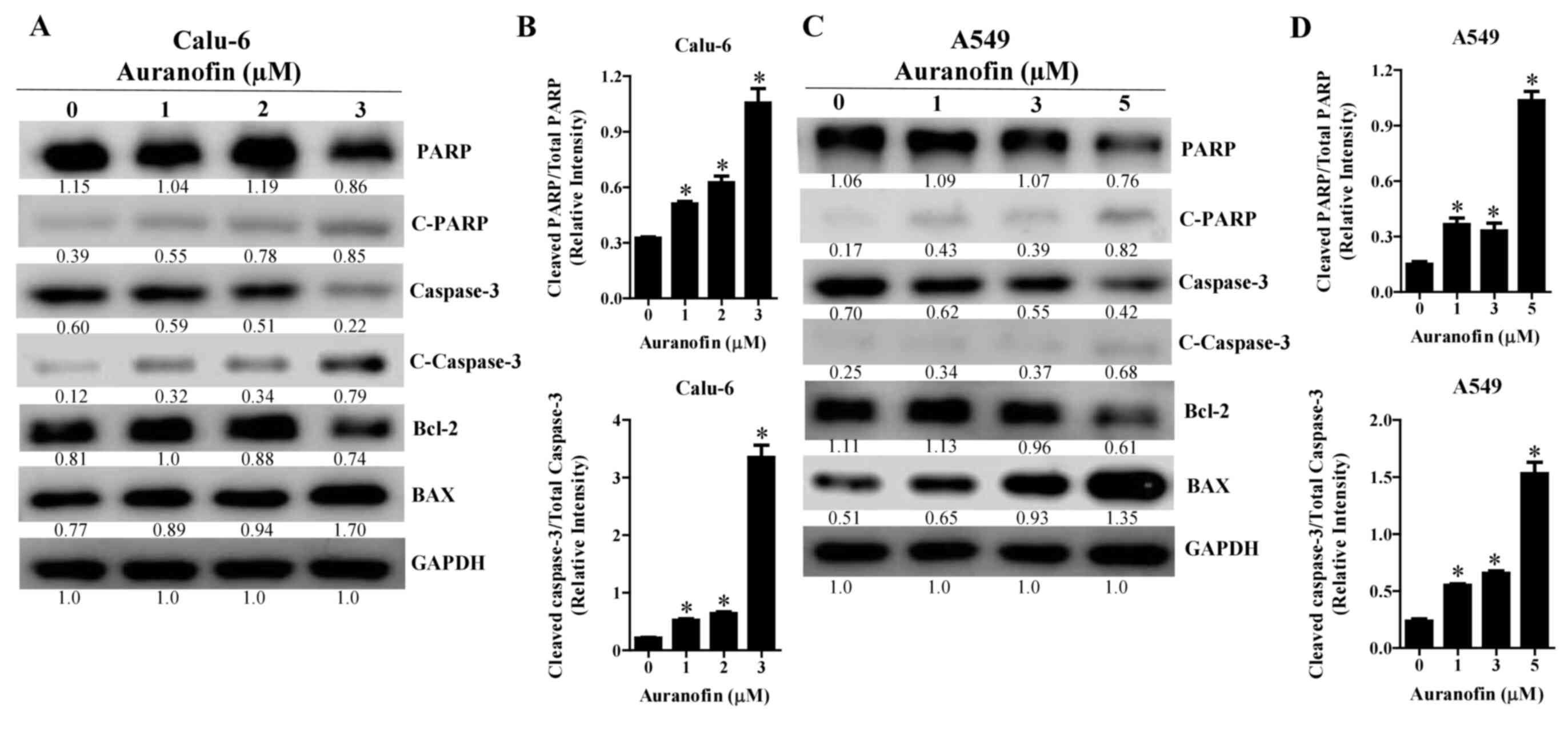
Effects of auranofin on
apoptosis-related protein levels in Calu-6 and A549 cells
As auranofin increased the number of annexin
V-positive cells, levels of apoptosis-related proteins were
evaluated by western blot analysis. Intact forms of PARP decreased
in auranofin-treated Calu-6 and A549 cells whereas the cleavage
forms of PARP increased in these cells (Fig. 6A and C). In addition, the levels of
cleaved caspase-3 were dose-dependently upregulated in
auranofin-treated Calu-6 and A549 cells (Fig. 6A and C). Auranofin also decreased
the levels of Bcl-2 and increased the levels of BAX in Calu-6 and
A549 cells (Fig. 6A and C). The
ratios of cleaved PARP/total PARP and cleaved caspase-3/total
caspase-3 were increased in auranofin-treated Calu-6 and A549 cells
(Fig. 6B and D). All blots
presented together were probed from the same membrane.
Effects of caspase inhibitors on cell
death, MMP (∆Ψm), and apoptosis-related protein levels in
auranofin-treated Calu-6 and A549 cells
To determine which caspases were involved in
auranofin-induced apoptosis, cells were pretreated with various
caspase inhibitors before treatment with auranofin. Z-VAD (a
pan-caspase inhibitor) significantly decreased the number of
annexin V-positive Calu-6 cells treated with 3 µM auranofin
(Fig. 7A). Furthermore, all of the
tested caspase inhibitors (Z-DVED for caspase-3, Z-IETD for
caspase-8, and Z-LEHD for caspase-9) significantly reduced the
death of Calu-6 cells following auranofin treatment (Fig. 7A). In addition, all caspase
inhibitors significantly protected against the loss of MMP (∆Ψm) in
Calu-6 cells caused by auranofin (Fig.
7B). Likewise, all tested caspase inhibitors slightly decreased
apoptotic A549 cell death following incubation with 5 µM auranofin
(Fig. 7C). However, these decreases
were not statistically significant. Caspase inhibitors marginally
reduced the loss of MMP (∆Ψm) in auranofin-treated A549 cells
(Fig. 7D). The expression of
apoptosis-related proteins showed an increase in the intact form of
PARP in auranofin-treated Calu-6 cells in the presence of Z-VAD and
a decrease in the cleavage form of PARP in those cells (Fig. 7E). Furthermore, Z-VAD reduced
cleavage forms of caspase-3, −8, and −9 in auranofin-treated Calu-6
cells (Fig. 7E). Finally, the
expression of Bcl-2 in auranofin-treated cells was clearly
upregulated in the presence of Z-VAD, and the levels of BAX in
those cells were downregulated (Fig.
7E). The ratio of cleaved PARP/total PARP, cleaved
caspase-3/total caspase-3, cleaved caspase-8/total caspase-8 and
cleaved caspase-9/total caspase-9 were increased in
auranofin-treated Calu-6 cells (Fig.
7F). However, these were decreased in auranofin and Z-VAD
treated Calu-6 cells (Fig. 7F). All
blots presented together were probed from the same membrane.
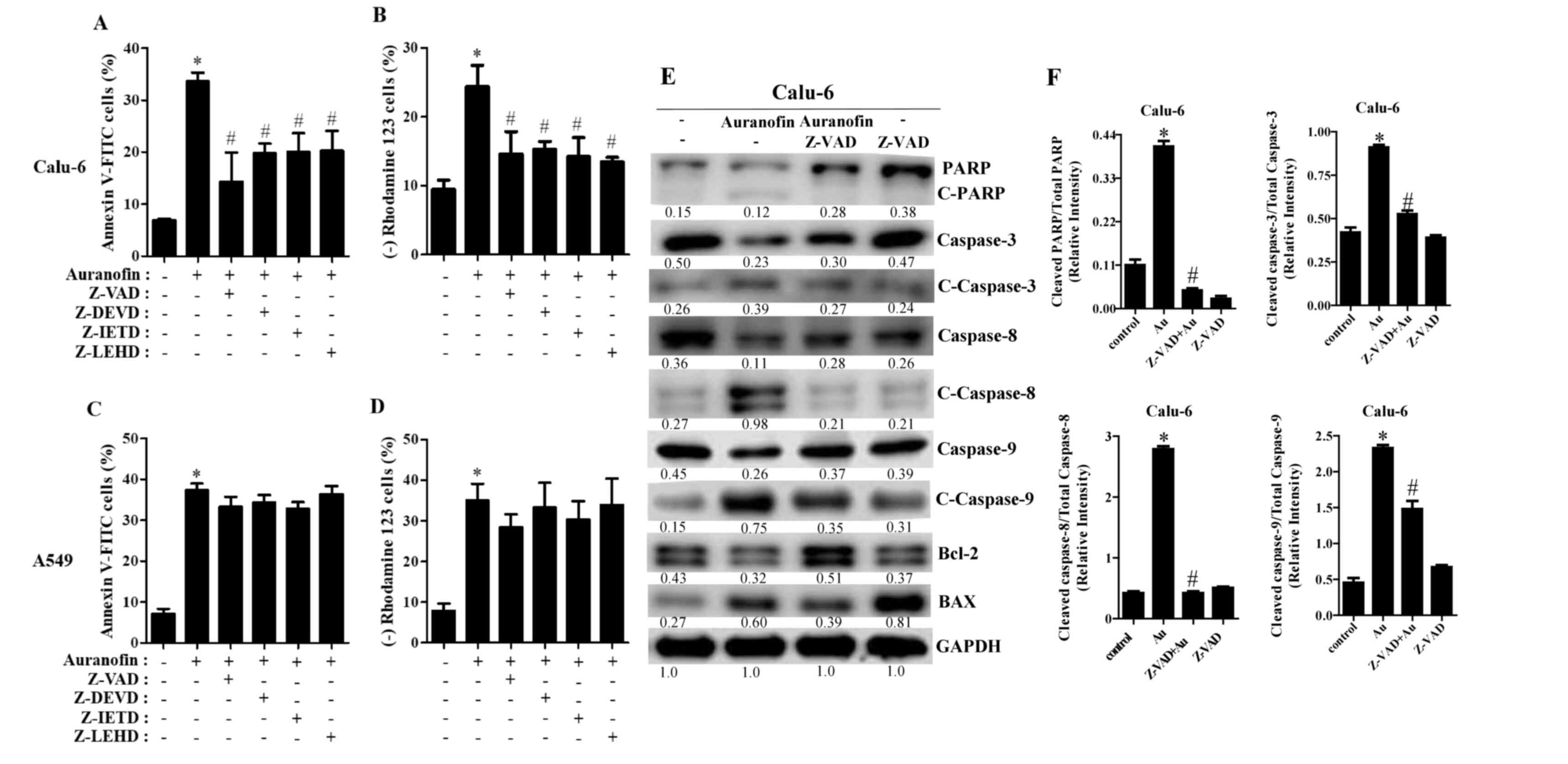 | Figure 7.Effects of caspase inhibitors on cell
death, MMP (∆Ψm) loss, and apoptosis-related proteins in
auranofin-treated cells. Exponentially growing Calu-6 and A549
cells were incubated in the presence of 3 and 5 µM auranofin for 24
h, respectively, following 1 h preincubation with 15 µM of
individual caspase inhibitors. Graphs show the percentages of
annexin V-positive cells (A) and Rhodamine 123-negative [MMP (∆Ψm)
loss] cells as assessed using FACStar flow cytometer with Calu-6 (A
and B) and A549 cells (C and D). Protein extracts were resolved by
8–15% SDS-PAGE gel, transferred to PVDF membranes, and
immunoblotted with the indicated antibodies. Western blot results
show the levels of PARP; cleaved caspase-3, −8, −9; Bcl-2; BAX; and
GAPDH (E). Graphs show ratio of Cleaved PARP/Total PARP, Cleaved
caspase-3/Total caspase-3, Cleaved caspase-8/Total caspase-8 and
Cleaved caspase-9/Total caspase-9 in auranofin-treated Calu-6 cells
(F), and band intensities were quantified using the Image J
program. *P<0.05 compared with auranofin-untreated control
cells. #P<0.05 compared with cells treated with
auranofin only. |
Discussion
Although auranofin was approved by the U.S. Food and
Drug Administration for the treatment of rheumatoid arthritis, this
agent has recently been studied as a possible therapeutic drug for
various human diseases, including cancer (11). According to the current result,
auranofin inhibited the activity of TrxR in Calu-6 and A549 cells,
supporting that auranofin is a TrxR inhibitor. This study
demonstrated that auranofin significantly and efficiently decreased
the growth of lung cancer cells in a dose-dependent manner. The
sensitivities of lung cancer cells to auranofin treatment are
generally lower than those of prostate, leukemia, and ovarian
cancer cell lines (24–26). However, they are similar to those of
cervical cancer and mesothelioma cancer cell lines (13,21).
Interestingly, NCI-H1299 cell growth was inhibited by a lower dose
of auranofin (0.5 µM) after a 24 h incubation. This result suggests
high sensitivity of these cells. The growth of normal HPF cells was
dose dependently reduced by auranofin with an IC50 of
approximately 3 µM. Survival and proliferation of TrxR1-deficient
tumors strictly depend on a functional glutathione system (27). Those results suggest that
sensitivity to auranofin depends on the varying capacity of
antioxidation pathways in different cell types.
Auranofin induces apoptosis in normal and lung
cancer cells. In particular, Calu-6 and A549 cells treated with
auranofin appear to show a decrease in Bcl-2 levels and an increase
in BAX levels, along with increases in the cleavage forms of
caspase-3 and PARP. In addition, auranofin dose-dependently
triggered necrosis in these cells, as evidenced by the release of
LDH. This agent also increased the percentages of sub-G1 cells in
Calu-6 and A549 cells. Thus, auranofin induced lung cancer cell
death via apoptosis and/or necrosis, depending on its
concentrations. DNA flow cytometry indicates that auranofin induced
arrest at the G2/M phase of the cell cycle in Calu-6 cells.
Similarly, a TrxR-1 inhibitor, Chaetocin, induced G2/M phase arrest
in gastric cancer cells (28).
Thus, G2/M phase arrest is a plausible underlying mechanism for the
inhibition of cell proliferation. Of note, auranofin led to G1
phase arrest in SK-LU-1 cells (data not shown) and, in A549 cells,
auranofin did not induce arrest in any specific phase of the cell
cycle. These results indicate that specificity of cell cycle arrest
depends on both auranofin concentration and cell type. Use of
auranofin for cancer therapy should be subject to consideration of
the various mechanisms involved in the anti-cancer effects of
auranofin as well as the specificity of cells in the target
tumor.
Apoptosis is closely associated with the collapse of
MMP (∆Ψm), and auranofin can cause a breakdown in MMP (∆Ψm)
(29). Similarly, auranofin induced
the loss of MMP (∆Ψm) in both normal and lung cancer cells. The
degree of MMP (∆Ψm) loss in auranofin-treated lung cells was very
similar to that of annexin V-positive cells. For example,
concentrations of 1–3 µM auranofin that did not induce apoptosis in
A549 cells also did not significantly increase the loss of MMP
(∆Ψm). Interestingly, although lower doses of auranofin did not
induce apoptosis in large cell carcinoma cells (NCI-H460 and
NCI-H1299), such doses did trigger the loss of MMP (∆Ψm). These
results suggest that auranofin initially impacts mitochondrial
membranes, especially large cell carcinoma cells, which precedes
the next step in apoptosis. Additionally, differences in
sensitivity to auranofin in relation to MMP (∆Ψm) and apoptosis are
probably due to the different basal activities of mitochondria,
which vary by cell type, tissue origin, and species (30).
Apoptosis involves cell death receptor (extrinsic)
and mitochondrial (intrinsic) pathways (6). When auranofin-treated Calu-6 and A549
cells were treated with various caspase inhibitors, these
inhibitors, including Z-VAD, significantly decreased the
percentages of annexin V-stained Calu-6 cells and MMP (∆Ψm) loss
following auranofin treatment in cells. In addition, Z-VAD reduced
cleavage forms of caspase-3, −8, and −9 in these cells, upregulated
the expression of Bcl-2, and downregulated the levels of BAX. All
caspase inhibitors decreased to some extent the numbers of annexin
V-stained A549 cells and MMP (∆Ψm) loss following auranofin
treatment. Thus, auranofin-induced apoptosis in lung cancer cells
may involve both extrinsic and intrinsic pathways.
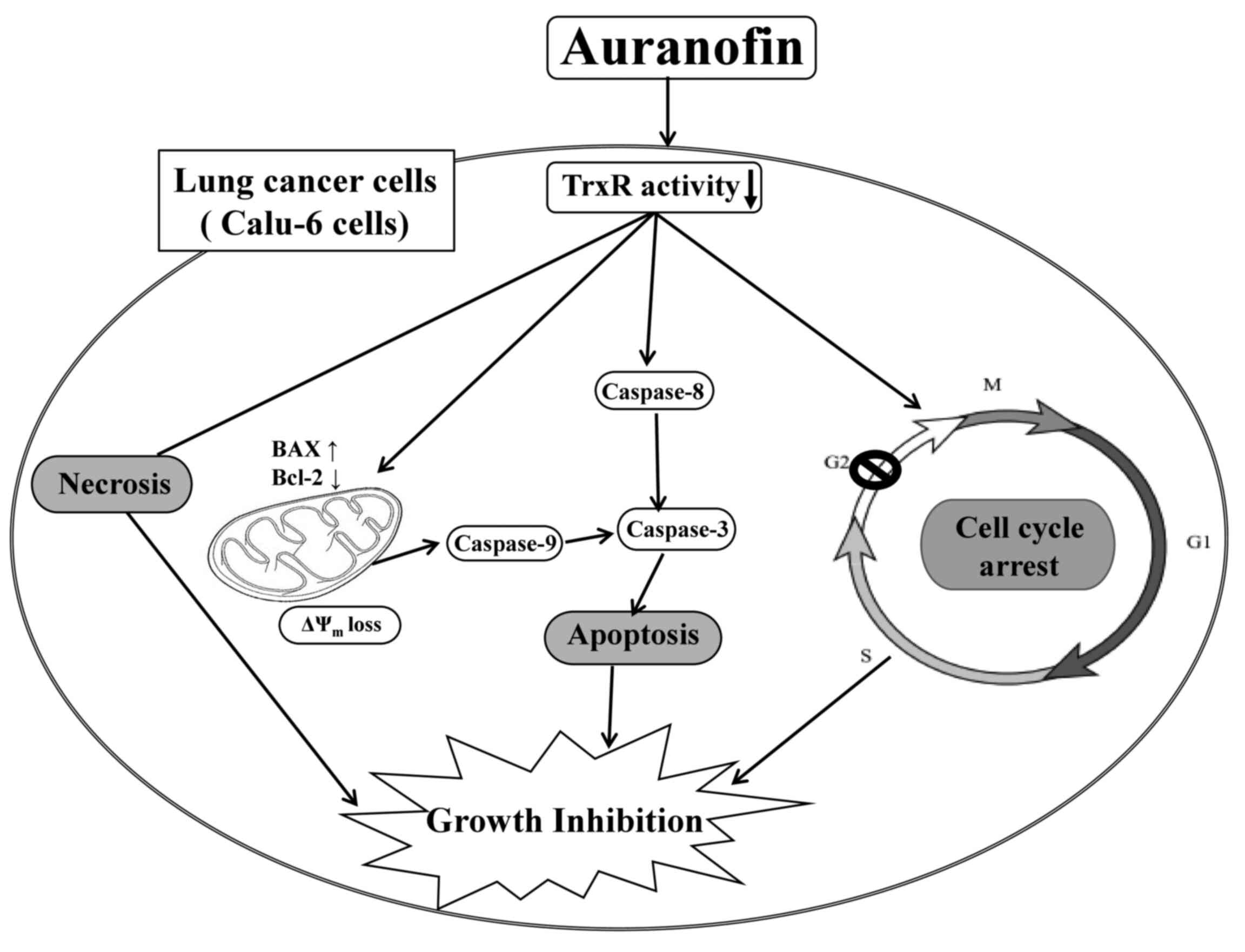
In conclusion, auranofin efficiently inhibits lung
cancer cell proliferation, especially in Calu-6 cells. This
inhibition is mediated by cell cycle arrest and cell death due to
necrosis and caspase-dependent apoptosis (Fig. 8). The present data provide useful
information for understanding cellular and molecular anti-cancer
mechanisms of auranofin in lung cancer cells.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by ‘Research Base
Construction fund Support Program’ funded by Jeonbuk National
University in 2020 and the Basic Science Research Program through
the National Research Foundation of Korea (NRF) funded by the
Ministry of Education (2019R1I1A2A01041209).
Availability of data and materials
Data collected during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
WHP and XYC designed the study. XYC mainly conducted
experiments and wrote early version of the manuscript.
Specifically, XYC retained cells and performed flow cytometry. SHP
assisted with providing resources for cell culture, flow cytometry,
and other experiments. XYC and SHP completed statistical analysis.
WHP and CXY reviewed and edited the final manuscript. All authors
have read and approved the final version of the manuscript, and
have verified that the accuracy and integrity of all parts of the
work have been properly investigated and addressed.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Trx
|
thioredoxin
|
|
TrxR
|
thioredoxin reductase
|
|
NSCLC
|
non-small cell lung cancer
|
|
SCLC
|
small cell lung cancer
|
|
MMP
|
mitochondrial membrane potential
(∆Ψm)
|
|
PARP
|
anti-poly ADP-ribose polymerase
|
|
FITC
|
fluorescein isothiocyanate
|
|
LDH
|
lactate dehydrogenase
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
Z-VAD
|
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
|
|
Z-DEVD
|
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone
|
|
Z-IETD
|
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone
|
|
Z-LEHD
|
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone
|
References
|
1
|
Hu Z, Li M, Chen Z, Zhan C, Lin Z and Wang
Q: Advances in clinical trials of targeted therapy and
immunotherapy of lung cancer in 2018. Transl Lung Cancer Res.
8:1091–1106. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dela Cruz CS, Tanoue LT and Matthay RA:
Lung cancer: Epidemiology, etiology, and prevention. Clin Chest
Med. 32:605–644. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carter BW, Lichtenberger JP III,
Benveniste MK, de Groot PM, Wu CC, Erasmus JJ and Truong MT:
Revisions to the TNM staging of lung cancer: Rationale,
significance, and clinical application. Radiographics. 38:374–391.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park HK, Han BR and Park WH: Combination
of arsenic trioxide and valproic acid efficiently inhibits growth
of lung cancer cells via G2/M-phase arrest and apoptotic cell
death. Int J Mol Sci. 21:26492020. View Article : Google Scholar
|
|
5
|
You BR and Park WH: Suberoyl bishydroxamic
acid inhibits the growth of A549 lung cancer cells via
caspase-dependent apoptosis. Mol Cell Biochem. 344:203–210. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chung C: Restoring the switch for cancer
cell death: Targeting the apoptosis signaling pathway. Am J Health
Syst Pharm. 75:945–952. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Würstle ML, Laussmann MA and Rehm M: The
central role of initiator caspase-9 in apoptosis signal
transduction and the regulation of its activation and activity on
the apoptosome. Exp Cell Res. 318:1213–1220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huska JD, Lamb HM and Hardwick JM:
Overview of BCL-2 family proteins and therapeutic potentials.
Methods Mol Biol. 1877:1–21. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu X, Yue P, Zhou Z, Khuri FR and Sun SY:
Death receptor regulation and celecoxib-induced apoptosis in human
lung cancer cells. J Natl Cancer Inst. 96:1769–1780. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Onodera T, Momose I and Kawada M:
Potential anticancer activity of auranofin. Chem Pharm Bull
(Tokyo). 67:186–191. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Isakov E, Weisman-Shomer P and Benhar M:
Suppression of the pro-inflammatory NLRP3/interleukin-1β pathway in
macrophages by the thioredoxin reductase inhibitor auranofin.
Biochim Biophys Acta. 1840:3153–3161. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
You BR and Park WH: Auranofin induces
mesothelioma cell death through oxidative stress and GSH depletion.
Oncol Rep. 35:546–551. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Park WH and You BR: Antimycin A induces
death of the human pulmonary fibroblast cells via ROS increase and
GSH depletion. Int J Oncol. 48:813–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fernandes AP, Capitanio A, Selenius M,
Brodin O, Rundlöf AK and Björnstedt M: Expression profiles of
thioredoxin family proteins in human lung cancer tissue:
Correlation with proliferation and differentiation. Histopathology.
55:313–320. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hawkes HJ, Karlenius TC and Tonissen KF:
Regulation of the human thioredoxin gene promoter and its key
substrates: A study of functional and putative regulatory elements.
Biochim Biophys Acta. 1840:303–314. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yan X, Zhang X, Wang L, Zhang R, Pu X, Wu
S, Li L, Tong P, Wang J, Meng QH, et al: Inhibition of
thioredoxin/thioredoxin reductase induces synthetic lethality in
lung cancers with compromised glutathione homeostasis. Cancer Res.
79:125–132. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fu JN, Li J, Tan Q, Yin HW, Xiong K, Wang
TY, Ren XY and Zeng HH: Thioredoxin reductase inhibitor ethaselen
increases the drug sensitivity of the colon cancer cell line LoVo
towards cisplatin via regulation of G1 phase and reversal of G2/M
phase arrest. Invest New Drugs. 29:627–636. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Poerschke RL and Moos PJ: Thioredoxin
reductase 1 knockdown enhances selenazolidine cytotoxicity in human
lung cancer cells via mitochondrial dysfunction. Biochem Pharmacol.
81:211–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
You BR and Park WH: Down-regulation of
thioredoxin1 is involved in death of Calu-6 lung cancer cells
treated with suberoyl bishydroxamic acid. J Cell Biochem.
117:1250–1261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
You BR, Shin HR, Han BR, Kim SH and Park
WH: Auranofin induces apoptosis and necrosis in HeLa cells via
oxidative stress and glutathione depletion. Mol Med Rep.
11:1428–1434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park WH: Hydrogen peroxide inhibits the
growth of lung cancer cells via the induction of cell death and
G1-phase arrest. Oncol Rep. 40:1787–1794. 2018.PubMed/NCBI
|
|
23
|
Park WH, Seol JG, Kim ES, Hyun JM, Jung
CW, Lee CC, Kim BK and Lee YY: Arsenic trioxide-mediated growth
inhibition in MC/CAR myeloma cells via cell cycle arrest in
association with induction of cyclin-dependent kinase inhibitor,
p21, and apoptosis. Cancer Res. 60:3065–3071. 2000.PubMed/NCBI
|
|
24
|
Marzano C, Gandin V, Folda A, Scutari G,
Bindoli A and Rigobello MP: Inhibition of thioredoxin reductase by
auranofin induces apoptosis in cisplatin-resistant human ovarian
cancer cells. Free Radic Biol Med. 42:872–881. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shin DW, Kwon YJ, Ye DJ, Baek HS, Lee JE
and Chun YJ: Auranofin suppresses plasminogen activator inhibitor-2
expression through annexin a5 induction in human prostate cancer
cells. Biomol Ther (Seoul). 25:177–185. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fidyt K, Pastorczak A, Goral A, Szczygiel
K, Fendler W, Muchowicz A, Bartlomiejczyk MA, Madzio J, Cyran J,
Graczyk-Jarzynka A, et al: Targeting the thioredoxin system as a
novel strategy against B-cell acute lymphoblastic leukemia. Mol
Oncol. 13:1180–1195. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mandal PK, Schneider M, Kölle P,
Kuhlencordt P, Förster H, Beck H, Bornkamm GW and Conrad M: Loss of
thioredoxin reductase 1 renders tumors highly susceptible to
pharmacologic glutathione deprivation. Cancer Res. 70:9505–9514.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wen C, Wang H, Wu X, He L, Zhou Q, Wang F,
Chen S, Huang L, Chen J, Wang H, et al: ROS-mediated inactivation
of the PI3K/AKT pathway is involved in the antigastric cancer
effects of thioredoxin reductase-1 inhibitor chaetocin. Cell Death
Dis. 10:8092019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Radenkovic F, Holland O, Vanderlelie JJ
and Perkins AV: Selective inhibition of endogenous antioxidants
with Auranofin causes mitochondrial oxidative stress which can be
countered by selenium supplementation. Biochem Pharmacol.
146:42–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mejia EM and Hatch GM: Mitochondrial
phospholipids: Role in mitochondrial function. J Bioenerg Biomembr.
48:99–112. 2016. View Article : Google Scholar : PubMed/NCBI
|