Introduction
The molecular basis of chronic myeloid leukemia
(CML) is the BCR-ABL1 oncoprotein, which results from a chromosomal
translocation t(9; 22) (q34; q11) in hematopoietic stem cells
(1). The clinical course of CML
includes the chronic phase, accelerated phase, and blast crisis in
sequence. For patients with chronic phase CML, main molecular
remission (MMR) can be obtained in 74% of patients through
treatment with imatinib, a tyrosine kinase inhibitor (TKI)
targeting the BCR-ABL1 oncoprotein (2). However, 26% of chronic phase CML
patients experience disease progression and treatment failure due
to resistance and intolerance of treatment (3). For patients with accelerated phase or
blast crisis CML, their leukemia cells show significant resistance
to imatinib; complete cytogenetic remission (CCR) can be achieved
in only ~30% of patients through treatment with second-generation
TKI (4). In addition, patients who
respond effectively to drugs at the beginning of treatment may also
develop secondary resistance due to the evolution of leukemia cells
under the pressure of drug treatment (5). The updated TKI can effectively solve
the resistance caused by a BCR-ABL1 point mutation (6). However, primary and secondary
BCR-ABL1-independent resistance has become an prominent clinical
problem in the treatment of CML.
The mechanisms underpinning TKI resistance in CML
occur at multiple levels. First, at the cellular level, the
heterogeneity of leukemia stem cells (LSCs) and the evolution
driven by drug selection pressure lead to the formation of
drug-resistant dominant clones (7).
Progeny cells from these clones have a strong ability to
proliferate, and lose the ability to differentiate into relatively
mature blood cells. Second, genome instability leads to new
molecular abnormalities. For example, the formation of the
NUP98-HOXA9 fusion gene leads to rapid disease progression
(8). In addition, point mutations
in the kinase domain of the BCR-ABL1 fusion gene cause a
reduction in the drug binding efficiency (9). Third, there are abnormalities in the
regulation of molecular signaling pathways, such as those involved
in hematopoietic stem cell development (Wnt/β-catenin, Hif-1α)
(10–12), autophagy (ATG4B) (13) and epigenetic regulation (PRMT5,
SIRT1) (14,15). Moreover, the abnormal overexpression
of BCR-ABL1 (16) and drug efflux
mediated by ATP-binding cassette (ABC) transporters (ABCB1 or
ABCC2) (17,18) also play an essential role in TKI
resistance.
Transcription factor 7 (TCF7) is one of the members
of the TCF/LEF family (TCF7, TCF7L1, TCF7L2, LEF1), which functions
downstream of the Wnt/β-catenin signaling pathway. The protein
encoded by this gene contains a β-catenin binding domain (CBD) and
a high mobility group (HMG) domain. TCF7 can recognize and bind to
the DNA sequence called Wnt response element (WRE) through the HMG
domain, cause conformational changes of DNA and chromatin that lead
to further binding of other transcription complexes (19), and promote the expression of Wnt
target genes (20). Previous
studies have shown that TCF7 is closely related to the development
and progression of various malignancies, such as leukemia (21), chondrosarcoma (22), and prostate cancer (23,24).
In colorectal tumors, the transcription of Wnt target genes
mediated by TCF7 is necessary for the initial activity of tumor
stem cells (25). Studies
concerning tumor resistance have shown that targeting TCF7
by microRNA can inhibit the drug resistance in bladder and prostate
cancer cells (26,27). While the expression of TCF7 is
significantly increased in CML imatinib-resistant cells, the role
of TCF7 in CML imatinib-resistant cells is unclear.
In this study, we report that the expression of TCF7
is independent of BCR-ABL1 tyrosine kinase activity. TCF7
knockdown can inhibit the proliferation and restore imatinib
sensitivity of imatinib-resistant cells. Furthermore, we found that
TCF7 knockdown neutralized the upregulation trend of
Wnt/β-catenin and ABC transporter signaling pathways when
imatinib-resistant cells were treated with imatinib and confirmed
that TCF7 could transactivate ABCC2 transcription by binding
to the promoter region of ABCC2. Our findings revealed that
when CML imatinib-resistant cells are treated with imatinib, the
Wnt/β-catenin signaling pathway and ABC transporters play an
essential role in the formation of imatinib resistance. Thus,
targeting TCF7 to reduce the resistance of CML cells may be a
viable treatment approach.
Materials and methods
Cell culture
The CML imatinib-resistant cell line K562/G01 was a
kind gift from Professor Zhenlun Gu (Suzhou University, China). The
CML cell line, KCL22 and K562, and acute myeloid leukemia (AML)
cell lines, HL60 and NB4, were purchased from the Cell Bank of
Shanghai Institute of Cell Biology, Chinese Academy of Science
(Shanghai, China) and stored at our laboratory. All cell lines were
maintained in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% fetal bovine serum (FBS) (HyClone; GE
Healthcare) and 1% penicillin-streptomycin (Beyotime Institute of
Biotechnology) at 37°C in a 5% CO2 atmosphere.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The reagents and standard protocols used for the
extraction of total RNA (RNAiso Plus), RNA reverse transcription
into cDNA (PrimeScript™ RT reagent Kit), and RT-qPCR
(SYBR® Premix Ex Taq™ II) were obtained from Takara Bio.
Inc. The thermocycling conditions were as follows: Initial
denaturing step (95°C, 3 min), followed by 40 cycles of denaturing
(95°C, 10 sec), annealing (55°C, 30 sec) and extension (72°C, 30
sec). ACTB was used as an internal reference gene. The primers used
for RT-qPCR are listed in Table I.
Relative expression levels of mRNA were calculated using the
2−ΔΔCq method (28).
 | Table I.Sequences used for RT-qPCR. |
Table I.
Sequences used for RT-qPCR.
| Primers | Sequences
(5′-3′) |
|---|
| ACTB |
|
|
Forward |
ACTTAGTTGCGTTACACCCTT |
|
Reverse |
TGTCACCTTCACCGTTCC |
| ABCC2 |
|
|
Forward |
CCCTGCTGTTCGATATACCAATC |
|
Reverse |
TCGAGAGAATCCAGAATAGGGAC |
| BCR-ABL1 |
|
|
Forward |
ATCCGTGGAGCTGCAGATG |
|
Reverse |
TTCCAACGAGCGGCTTCACT |
| CCND1 |
|
|
Forward |
CATCCGCAAACACGC |
|
Reverse |
GGGCTCCTCAGGTTCA |
| TCF7 |
|
|
Forward |
CTGGCTTCTACTCCCTGACCT |
|
Reverse |
ACCAGAACCTAGCATCAAGGA |
Western blot analysis
Western blot analysis was performed according to a
standard protocol, as described previously (29). The following primary antibodies were
used: Anti-ACTB (cat. no. TA09 purchased from ZSGB-BIO/now OriGene
Technologies, Inc.), anti-ABCC2 (anti-MRP2) (cat. no. ab172630
purchased from Abcam, Inc.). Moreover, anti-BCR-ABL1 (cat. no.
2862), anti-p-BCR-ABL1 (cat. no. 2864), anti-CCND1 (cat. no. 2922),
anti-CTNNB1 (cat. no. 9562), anti-PARP1 (cat. no. 9532), anti-STAT5
(cat. no. 25656), anti-p-STAT5 (cat. no. 4322) and anti-TCF7 (cat.
no. 2203) were purchased from Cell Signaling Technology, Inc.
(CST). The antibodies were used at a dilution of 1:1,000, except
for anti-ACTB (1:2,000).
Lentiviral transduction
One scrambled negative control and two independent
TCF7-targeting short hairpin RNAs (shRNAs) were cloned into
the lentiviral vector GV248 (GeneChem, Shanghai, China) at the
AgeI and EcoRI sites. The shRNA sequences are
provided in Table II. K562/G01 and
K562 cells in logarithmic growth phase were plated into 96-well
plates (5,000 cells per well) and infected for 24 h with 50 IFU/ml
lentivirus and 10 µg/ml polybrene, and then replaced with the
normal medium and cultured for 48 h. Next, puromycin was added to
the plates at a final concentration of 2.0 µg/ml for reverse
selection of the stable cell lines. Medium containing puromycin was
replaced every 3 days. Transfection efficiency was monitored by
inverted fluorescence microscopy and flow cytometry. After stable
cell lines were produced, normal medium was used.
| Table II.shRNA sequences used for the scramble
and TCF7 knockdown. |
Table II.
shRNA sequences used for the scramble
and TCF7 knockdown.
| shRNA | Sequences
(5′>3′) |
|---|
| Scramble-F | CCGGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTG |
| Scramble-R |
AATTCAAAAATTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAA |
| TCF7-KD1F | CCGGCAACTCTCTCTCTACGAACATCTCGAGATGTTCGTAGAGAGAGAGTTGTTTTTG |
| TCF7-KD1R |
AATTCAAAAACAACTCTCTCTCTACGAACATCTCGAGATGTTCGTAGAGAGAGAGTTG |
| TCF7-KD2F |
CCGGGCGGGACAACTACGGGAAGAACTCGAGTTCTTCCCGTAGTTGTCCCGCTTTTTG |
| TCF7-KD2R |
AATTCAAAAAGCGGGACAACTACGGGAAGAACTCGAGTTCTTCCCGTAGTTGTCCCGC |
Immunofluorescence assay
Cells were smeared across a gelatin-coated slide to
form a cell monolayer, and then the cell smear was fixed with
methanol at −20°C for 20 min. The cell membranes were permeabilized
using 1% Triton X100-PBS at 37°C for 15 min. After washing the
fixed slides three times in PBS, non-specific antigens were blocked
with 10% goat serum at 37°C for 1 h. The anti-TCF7 primary antibody
(cat. no. 2203; CST) was diluted using 10% goat serum to 1:400,
applied to the slide to cover the cell smear, and incubated
overnight at 4°C. The slides were washed three times in 400 µl of
wash buffer (0.1% BSA in 1X PBS). The secondary antibody (goat
anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, cyanine 3;
cat. no. A10520; Invitrogen; Thermo Fisher Scientific, Inc.) was
diluted to 1:1,000, and 500 µl was added to the smear and incubated
at room temperature for 1 h. Slides were rinsed twice in 500 µl of
wash buffer, and the nuclei were stained using DAPI (Beyotime
Institute of Biotechnology) diluted with PBS to 1:1,000 for 5 min.
Slides were rinsed thrice with PBS and once with water. Finally,
the smears with a drop of 70% glycerin were covered with
coverglasses. The expression and distribution of fluorescence were
observed using a fluorescence microscope (magnification, ×1,000;
Nikon Corporation).
Cell viability and colony formation
assay
Cells were plated into 96-well flat-bottomed plates
with 2×103 cells per well and treated with or without
imatinib at the indicated concentrations. After cell culture for
12, 24, 48, 72, 96 and 120 h, cell viability was determined using a
CCK-8 kit (Solarbio, Inc.). For the colony formation assay, cells
were seeded into 24-well flat-bottomed plates with 200 cells per
well and grown in semi-solid medium containing 1.35%
methylcellulose. After 9 days, the colonies were counted using an
inverted fluorescence microscope (magnification, ×40; Nikon
Corporation).
Flow cytometric analysis
Cell cycle, apoptosis, cell counts, and GFP
fluorescence were detected using flow cytometry (FCM). Cells were
collected after treatment with or without imatinib at the indicated
concentrations. To examine cell cycle dynamics, cells were
subjected to serum starvation for 64 h to obtain synchronized
cells, following which the serum supply was restored. Cell cycle
status was monitored at 0, 8, 16, 24, and 32 h. Cell cycle
profiling was delineated by the FL2 fluorescence generated by the
binding of propidium iodide (PI) to DNA, and the percentages of
cells in different phases of the cell cycle were analyzed by FlowJo
VX.0.7 software (FlowJo LLC). To detect cell apoptosis, the cells
were double-labeled with Annexin V-APC and DAPI and measured by FCM
according to the manufacturer's protocol. Given that flow cytometry
records the volume of fluid and the fluorescence parameters of
particles simultaneously, if the sample is thoroughly mixed, an
accurate cell count and GFP fluorescence can be obtained.
RNA sequencing (RNA-seq) and
bioinformatic analysis
Total RNAs from four groups of K562/G01 cells with
scramble, imatinib, TCF7_KD, and TCF7_KD+imatinib treatment were
extracted using an RNeasy kit (Qiagen, Inc.), and treated with
DNase I (Qiagen Inc.). Imatinib was used at 1 µM in K562/G01 cells.
Shanghai Lifegenes Biotechnology performed RNA quantification,
quality appraisal, library preparation, and sequencing. Raw data
(raw reads) of fastq format were firstly processed through in-house
perl scripts. HTSeq v0.6.1 (https://htseq.readthedocs.io/en/master/) was used to
count the read number mapped to each gene. Gene fragment per
kilobase of exon per million reads (FPKMs) were computed by summing
the FPKMs of transcripts in each gene group. Gene set enrichment
analysis (GSEA) (30) software
v4.0.3 was used to analyze RNA-seq data. Cytoscape software v3.6.0
was used to visualize the GSEA reasults (31). The cut-offs of differentially
expressed genes (DEGs) were set as |log2 (fold change)|
>0.5 and FPKM >0.3, and consequently 1,034 DEGs were
obtained. The Gene Ontology (GO) enrichment analysis of DEGs was
executed using the R package clusterProfiler v3.11.1 (32). All sequencing data were used in
principal component analysis (PCA). Gene expression heatmaps and
PCA were performed using the web tool ClustVis (https://biit.cs.ut.ee/clustvis/) (33). Three-dimensional plots were produced
using the R package lattice v0.20-38. Venn diagrams were calculated
and drawn using a web tool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
The visualization of RNA-seq and chromatin immunoprecipitation
sequence (ChIP-seq) data were performed using Integrative Genomics
Viewer software v2.6.3 (http://software.broadinstitute.org/software/igv/)
(34).
Chromatin immunoprecipitation-qPCR
(ChIP-qPCR)
Chromatin immunoprecipitation kit (cat. no. 9005)
was purchased from CST, and anti-TCF7 (cat. no. bs1987) was
purchased from Bioword, Inc. The ChIP experiment was performed
according to the manufacturer's instructions. Immunoprecipitated
DNA fragments were purified by phenol extraction and then
quantified by qPCR. The primer sequences are listed in Table III.
| Table III.Sequences used for RT-qPCR. |
Table III.
Sequences used for RT-qPCR.
| Primers | Sequences
(5′-3′) |
|---|
| Negative
control |
|
|
Forward |
TTGGAATCATACAGTATGTAGCC |
|
Reverse |
CTATTGAGCCATGAAAAGATGTG |
| pmABCC2 |
|
|
Forward |
ACTGTGCACTCTTGATTTGTTGG |
|
Reverse |
AGGAGTGGCCATACATAAAAGG |
Statistical analysis
Results of column charts and line charts are
presented as the mean ± standard deviation and were analyzed by
GraphPad (Prism 5) (GraphPad Software, Inc.). Each experiment was
performed at least three times. Statistical analysis were performed
using the Student's t-test or one-way analysis of variance (ANOVA)
with Tukey's post hoc test. Statistical significance levels were as
follows: *P<0.05, **P<0.01, ***P<0.001 (as shown in the
figure legends with the respective symbols).
Results
TCF7 is highly expressed in CML
imatinib-resistant cells and independent of tyrosine kinase
activity of BCR-ABL1
In the chronic phase, BCR-ABL1 is recognized as an
effective target for CML treatment, but other targets need to be
explored when resistance develops. We first investigated gene
expression microarray datasets GSE47927 (35) and GSE4170 (36), and results of the analysis revealed
that the Wnt/β-catenin signaling pathway was activated in all
phases of CML and in the event of imatinib resistance (Fig. 1A). TCF7 expression was higher
in blast crisis and imatinib-resistant samples, when compared with
chronic phase and imatinib-sensitive samples, respectively.
(Fig. 1B and C). Futhermore, we
analyzed dataset GSE76312 (37) and
the results were consistent with the results in GSE47927 and
GSE4170 (Fig. S1E). To assess the
expression of TCF7 in leukemia cell lines, we tested CML cell lines
(K562, K562/G01 and KCL22), and acute myeloid leukemia (AML) cell
lines (HL60 and NB4) using RT-qPCR and western blot analyses. The
results showed that TCF7 was significantly overexpressed in blast
crisis (K562, KCL22) and imatinib-resistent (K562/G01) CML cell
lines (Fig. 1D and E). The K562/G01
cell line is evolved from the K562 cell line by long-term treatment
with imatinib, and it has the characteristic of imatinib
resistance.
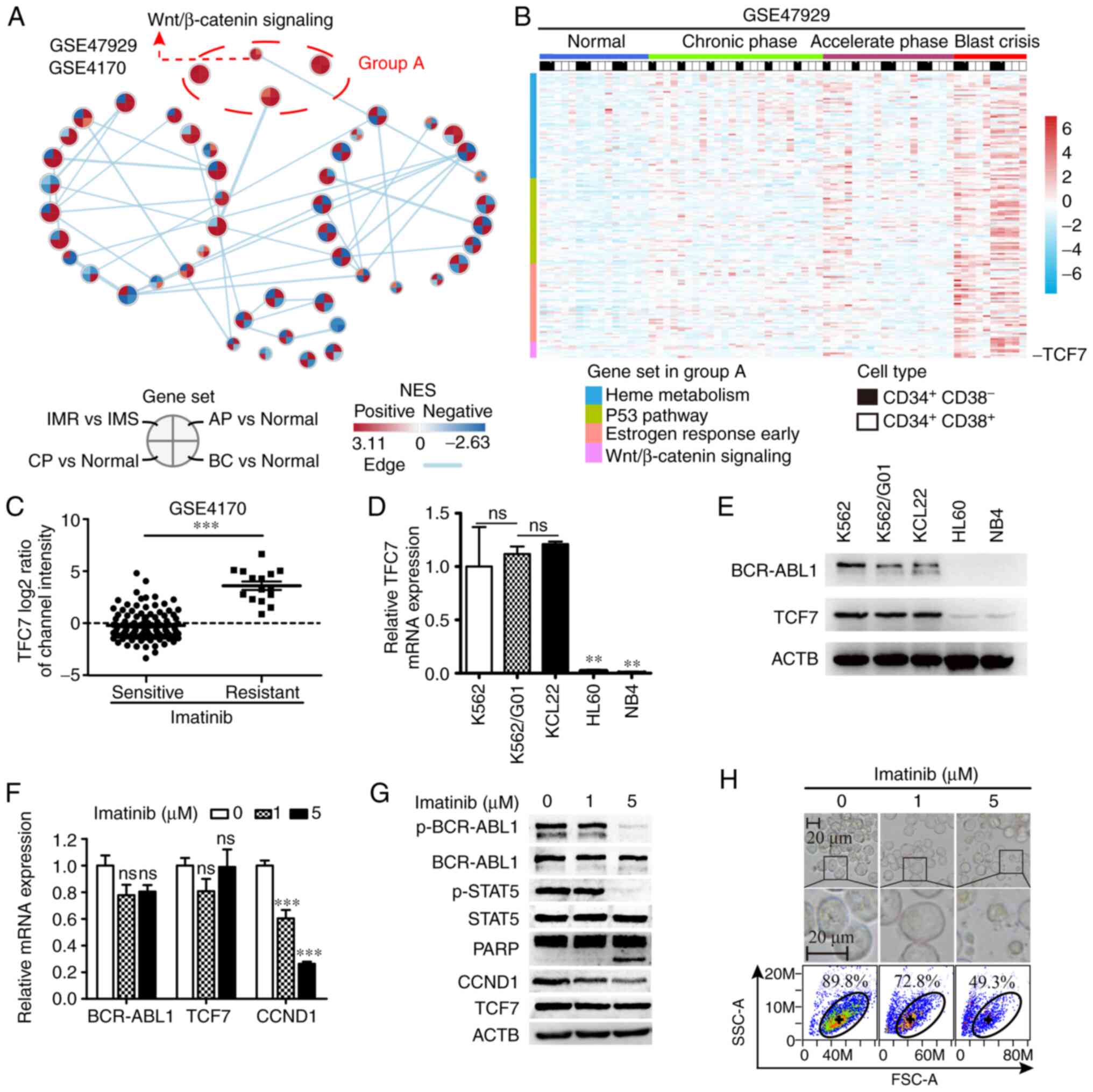 | Figure 1.Expression of TCF7 in CML and its
relationship with BCR-ABL1. (A) Cytoscape visualizes the changes in
signaling pathways during progression (GSE47927) and IM resistance
(GSE4170) of CML. IMR, imatinib resistant; IMS, imatinib sensitive;
BC, blast crisis; CP, chronic phase; AP, accelerated phase. (B)
Heatmap showing expression levels of 727 genes in group A signaling
pathway in the GSE47927 dataset. (C) Grouped scatter plot showing
levels of TCF7 expression in CML cells from
imatinib-sensitive (n=104) and imatinib-resistant (n=15) patient
samples in the GSE4170 dataset. (D and E) Expression of TCF7
mRNA (B) and protein (C) in CML cell lines, K562, K562/G01 and
KCL22, and AML cell lines, HL60, and NB4. (F) RT-qPCR analysis
showing mRNA expression of BCR-ABL1, TCF7, and CCND1.
(G) Western blot analysis showing protein expression of BCR-ABL1
and STAT5, and the expression of TCF7, CCND1, and PARP. (H) Light
microscopic images and flow cytometry scatter plots showing
K562/G01 morphological changes under increasing concentrations of
imatinib treatment. Two-tailed Student's t-test was used for C,
one-way ANOVA with Tukey's post hoc test were performed for D and
E. **P<0.01, ***P<0.001; ns, not significant. TCF7,
transcription factor 7; CML, chronic myeloid leukemia; AML, acute
myeloid leukemia; CCND1, cyclin D1; STAT5, signal transducer and
activator of transcription 5; PARP, poly(ADP) ribose polymerase;
ACTB, β-actin; p-, phosphorylated. |
To investigate whether the expression of TCF7 in
imatinib-resistant cells was affected by the activity of BCR-ABL1,
K562/G01 cells were treated with imatinib at concentrations of 1
and 5 µM, and the mRNA expression levels of BCR-ABL1, TCF7,
and CCND1 were detected using RT-qPCR. Next, the activation
status of BCR-ABL1, STAT5, and the protein expression levels of
BCR-ABL1, STAT5, PARP, CCND1, and TCF7 were detected using western
blot analysis. In response to imatinib, the results of RT-qPCR
showed that there was no significant change in the mRNA expression
of TCF7 and BCR-ABL1 except CCND1 (Fig. 1F). Consistent with this, the results
of western blot analysis showed that the expression of CCND1 was
gradually decreased while the expression of BCR-ABL1, STAT5, and
TCF7 did not change. In addition, when cells were exposed to a high
concentration of imatinib (5 µM), PARP1 began cleaving into
fragments, and the activity of BCR-ABL1 and its downstream target
STAT5, in the form of phosphorylated (p)-BCR-ABL1 and p-STAT5, were
significantly inhibited (Fig. 1G).
These results indicate that BCR-ABL1 activity has no significant
effect on the regulation of TCF7. In addition, changes in cell
morphology suggested that increasing drug concentrations led to
increased death of imatinib-resistant cells to a certain extent
(Fig. 1H).
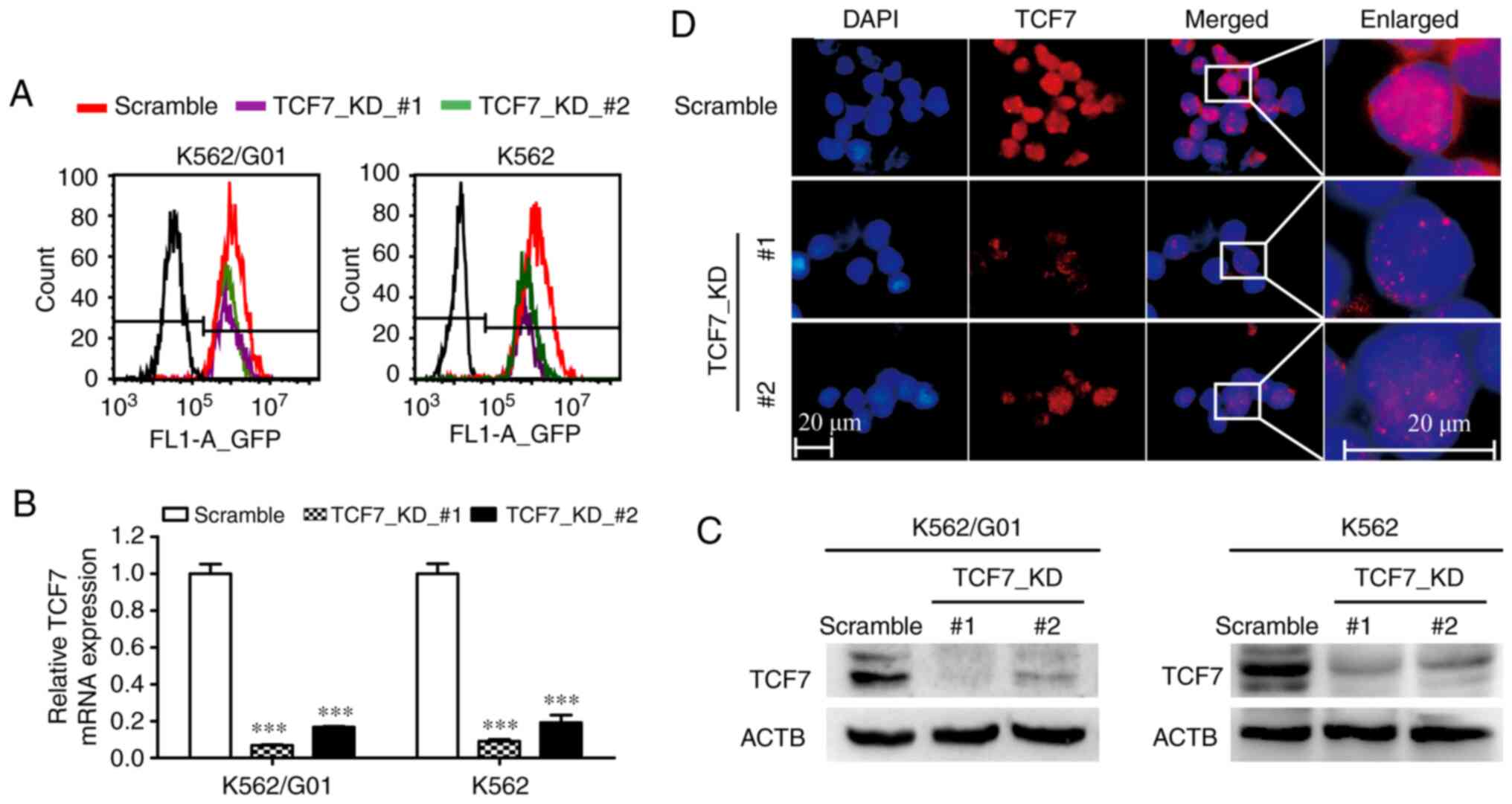
TCF7 knockdown in K562 and K562/G01
cells
K562 and K562/G01 cells were transduced with two
LV-TCF7-RNAi recombinant lentiviruses and one LV-Scramble
lentivirus, respectively. After puromycin treatment, stably
transduced cells were obtained. The results of flow cytometry
indicated that transduction efficiency was close to 100% (Fig. 2A). Next, The RT-qPCR results showed
that the knockdown efficiencies of the designed shRNAs were all
greater than 80% (Fig. 2B).
Furthermore, the western blot results confirmed the results of the
RT-qPCR (Fig. 2C). In addition,
immunofluorescence assays results visualized TCF7 expression
changes and nuclear localization in K562/G01 cells (Fig. 2D).
TCF7 knockdown inhibits the
proliferation of K562/G01 cells
To test the effect of TCF7 knockdown on the
proliferation of K562 and K562/G01 cells, we first performed a cell
viability test. The results showed that the cell viability of
K562/G01cells was significantly inhibited in the TCF7_KD groups
compared with the Scramble groups (Fig.
3A). Interestingly, in contrast to K562 cells, the inhibition
of cell viability caused by TCF7 knockdown was more pronounced in
the K562/G01 cells (Fig. 3B). In
addition, cell count results showed a lower cell counts in the
TCF7_KD groups of K562/G01 cells (Fig.
3F).
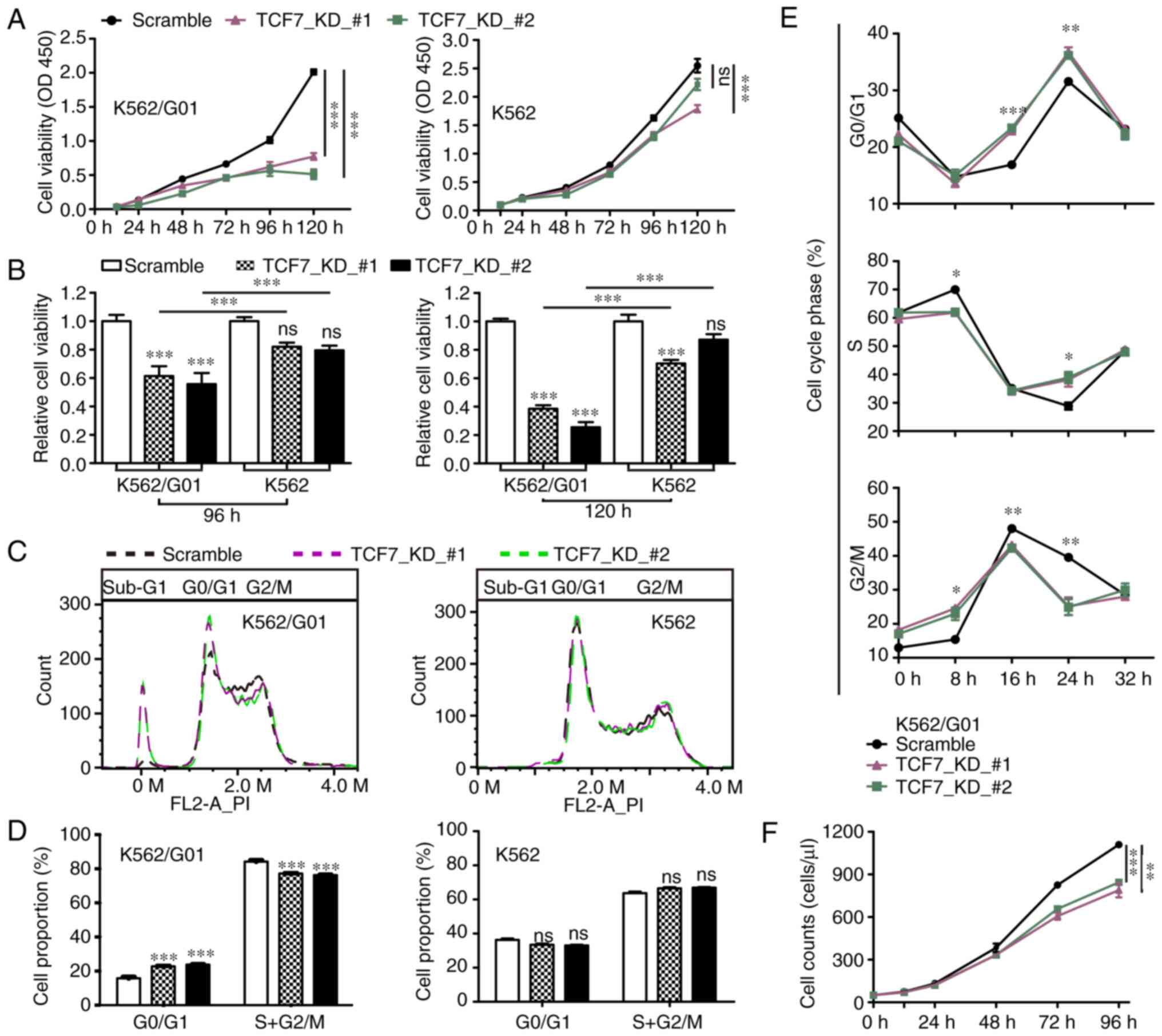 | Figure 3.TCF7 knockdown reduces the
proliferation and survival of CML K562/G01 cells. (A and B) CCK-8
assays were used to detect the cell viability of the Scramble and
TCF7_KD groups at 12, 24, 48, 72, 96 and 120 h. (C and D) Cell
cycle distribution of the Scramble and TCF7_KD groups during the
logarithmic growth phase. (E) Cell cycle distribution of the
Scramble and TCF7_KD groups following serum starvation and release
at 0, 8, 16, 24, and 32 h. (F) Flow cytometry cell counts of the
Scramble and TCF7_KD groups at 0, 12, 24, 48, 72, and 96 h. One-way
ANOVA with Tukey's post hoc test were performed. *P<0.05,
**P<0.01, ***P<0.001. TCF7, transcription factor 7; CML,
chronic myeloid leukemia; KD, knockdown. |
We further performed cell cycle assays. The results
showed that compared with the Scramble group, the TCF7_KD groups
consisted of a higher proportion of G0/G1 phase cells, and less
S+G2/M phase cells in the K562/G01 cells but not in the K562 cells
(Fig. 3C and D). In addition, the
TCF7_KD groups showed a significant increase in the number of
sub-G1 phase cells (Fig. 3C). The
serum starvation release test showed that after restoring serum to
the serum-free medium, cells in the TCF7_KD group re-entered the
cell cycle more slowly (Fig. 3E).
The above results suggest that TCF7 knockdown led to an
inhibition of proliferation, particularly in the CML
imatinib-resistant cells.
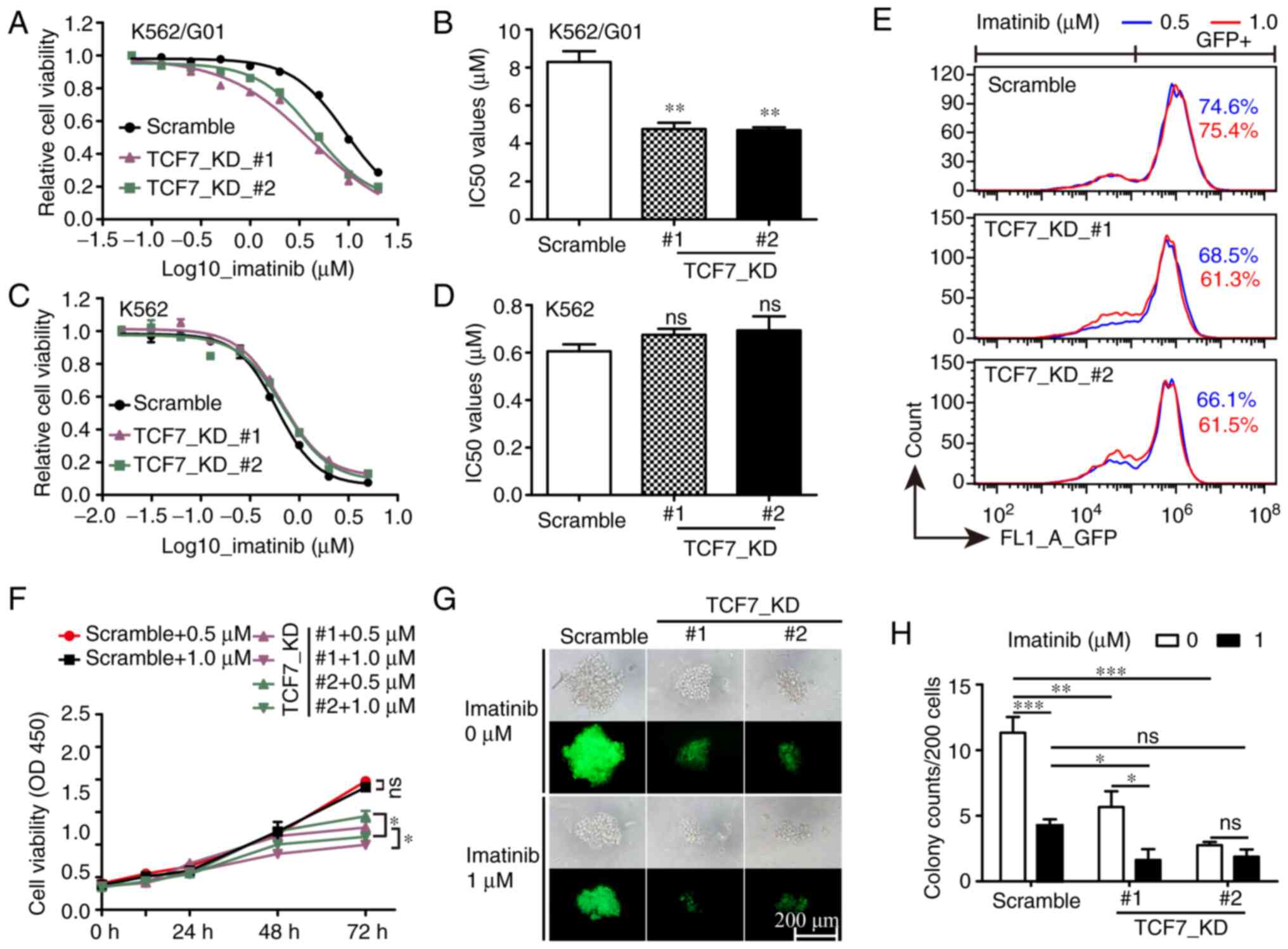
TCF7 knockdown improves the
sensitivity of K562/G01 cells to imatinib
Compared with the parental K562 cells, K562/G01
cells exhibit significant resistance to imatinib, and previous
reports have shown that TCF7 may affect the drug resistance of
tumor cells (26,27). Therefore, we investigated whether
TCF7 knockdown can increase imatinib sensitivity in CML
cells. The results of the drug sensitivity test showed that, in
K562/G01 cells, the half maximal inhibitory concentration
(IC50) value of the Scramble group was 8.3 µM, while the
IC50 value of the TCF7_KD group was 4.7 µM (Fig. 4A and B). In comparison, no
significant change in imatinib sensitivity was observed in K562
cells (Fig. 4C and D). Cell
viability and GFP-positive cell count results showed that, in
K562/G01 cells, cell proliferation in the TCF7_KD group was
inhibited while imatinib concentration increased from 0.5 to 1.0
µM, but not in the Scramble group (Fig.
4E and F). In addition, the colony formation assay showed that
TCF7 knockdown combined with imatinib could significantly
inhibit the colony formation rate of K562/G01 cells (Fig. 4G and H). The above results showed
that TCF7 knockdown can increase imatinib sensitivity in
imatinib-resistant cells and that TCF7 knockdown combined
with imatinib can inhibit imatinib-resistant cells more
effectively.
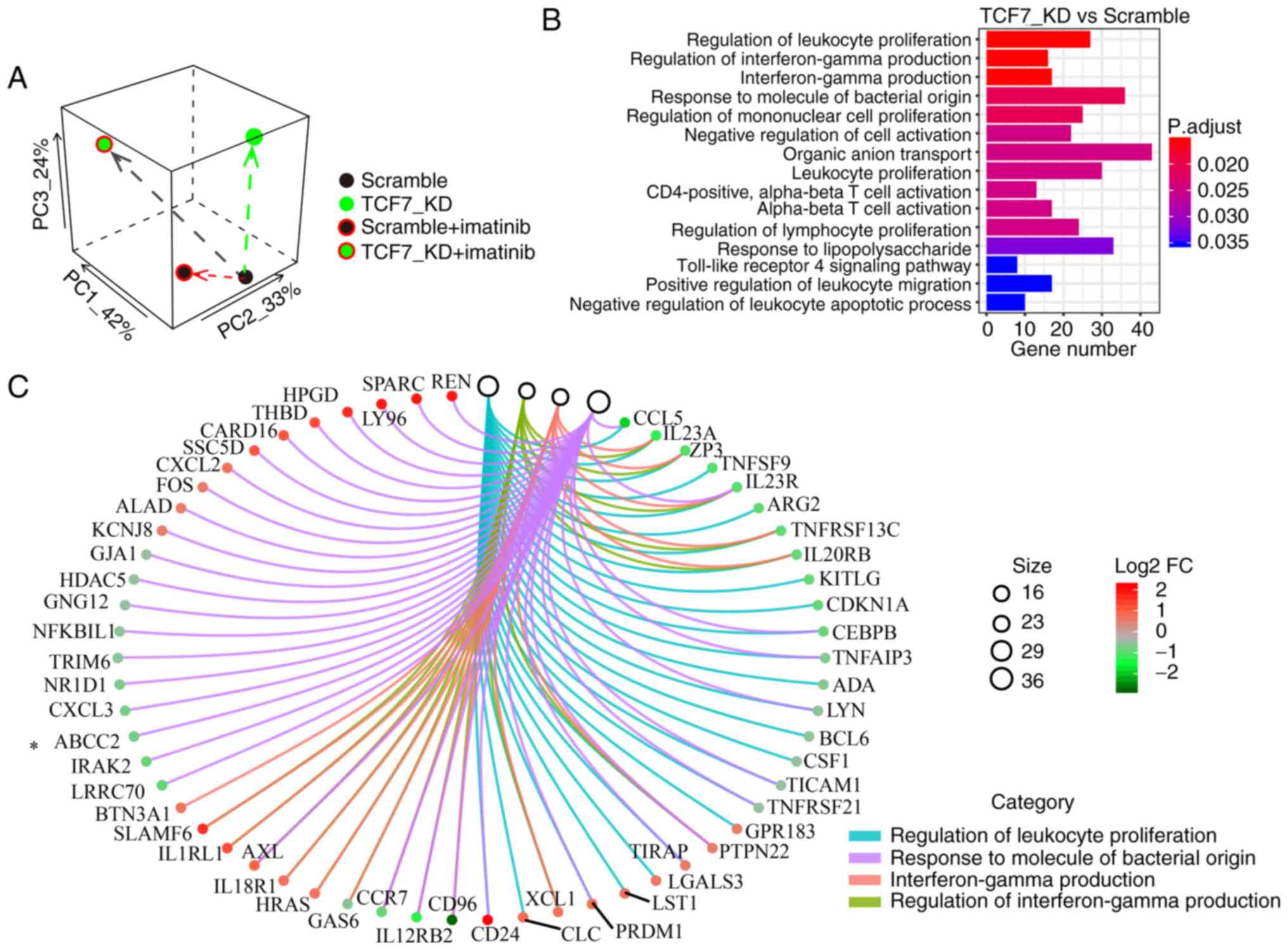
Principal component analysis (PCA) and
Gene Ontology (GO) enrichment analysis of RNA-seq data
To investigate why TCF7 knockdown affects
proliferation and drug resistance of K562/G01 cells, we obtained
RNA-seq data (GSE152220) from the Scramble, TCF7_KD,
Scramble+Imatinib, and TCF7_KD+Imatinib groups. PCA result showed
that the combination group underwent more intervention on the
transcriptome (Fig. 5A). GO
enrichment analysis showed that differentially expressed genes
(DEGs) in the TCF7_KD group were particularly enriched in the term
of leukocyte proliferation (Fig.
5B). These results explain our findings that TCF7
knockdown can affect proliferation of K562/G01 cells. Next, the GO
Chord plot lists the core genes such as ABCC2 (Fig. 5C). ABCC2 is a member of the
ATP-binding cassette (ABC) transporter superfamily, and this family
is often associated with multidrug resistance of tumors (38,39).
TCF7 knockdown neutralizes upregulated
ABC transporters and Wnt/β-catenin signaling during imatinib
treatment
Given the critical role of the ABC transporter
family in chemotherapy resistance of tumor cells (40,41),
we used RNA-seq data to analyze the changes in the ABC transporter
signaling pathway in CML cells. Using single-cell RNA-seq data of
1,062 BCR-ABL1+ LSCs from the GSE76312 dataset (37) for TCF7 single-gene GSEA
analysis, it was found that the expression of TCF7 was
positively correlated with the gene expression of ABC transporter
signaling pathway (Fig. S1A). We
subsequently set the Scramble group as the control and compared it
with the TCF7_KD, Scramble+Imatinib, and TCF7_KD+Imatinib groups
using GSEA analysis. The results showed that although the ABC
transporter signaling pathway was upregulated in K562/G01 cells
following imatinib treatment, TCF7 knockdown caused its
expression to be downregulated. When imatinib was used after
TCF7 knockdown, the upregulated trend of the ABC transporter
signaling pathway was neutralized (Fig. S1A).
Furthermore, we analyzed the Wnt (Fig. S1B) and Wnt/β-catenin signaling
pathway (Fig. 6A), and the results
showed that the trend in the changes in each group was consistent
with the changes in ABC transporters. The expression levels of core
enrichment genes representing ABC transporters and Wnt signaling
pathway in the TCF7_KD group are displayed in the heatmap (Fig. 6B). Next, the expression levels of
ABCC2 and CCND1 were verified by RT-qPCR and western blot analysis.
The results confirmed that the expression levels of ABCC2 and CCND1
were decreased when TCF7 was silenced in the CML
imatinib-resistant cells (Fig. 6C and
D). In addition, the expression level of CTNNB1, a key protein
of the canonical Wnt signaling pathway, was also decreased
(Fig. 6C).
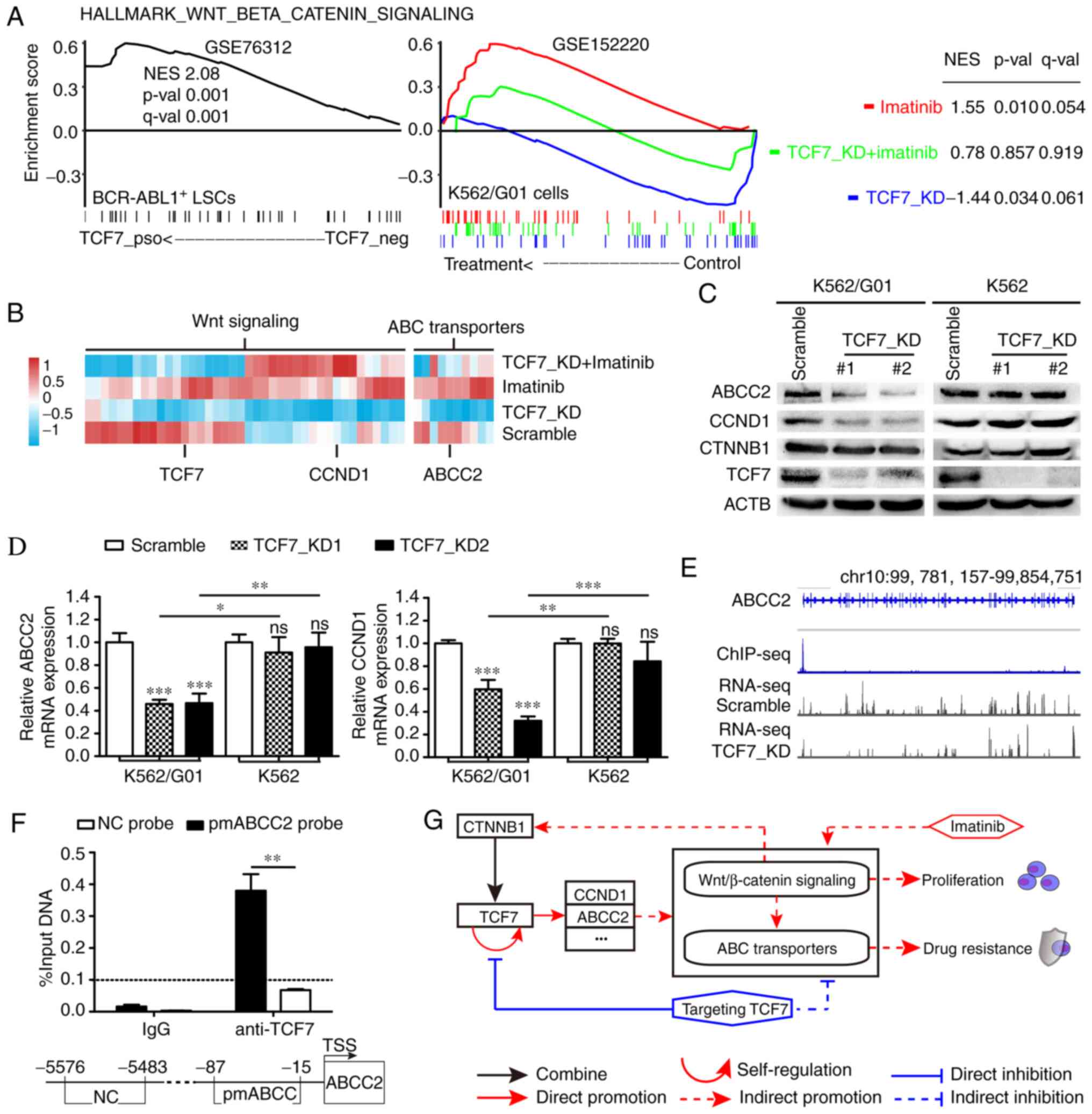 | Figure 6.TCF7 knockdown neutralizes the
intensity of ABC transporters and Wnt/β-catenin signal in response
to imatinib. (A) GSEA analysis shows the effects of TCF7 and
imatinib on Wnt/β-catenin signaling pathways in CML
imatinib-resistant cells. (B) Heatmap of the genes in TCF7_KD core
enriched ABC transporters and Wnt/β-catenin signaling pathways. (C)
Western blot analysis showing the expression of ABCC2, CCND1,
CTNNB1 and TCF7 proteins. (D) RT-qPCR analysis showing the
expression of ABCC2 and CCND1 mRNA in the Scramble
and TCF7_KD groups. (E) Integrative genomics viewer (IGV) showing
the recruitment of TCF7 to the ABCC2 promoter region and the
transcription level of ABCC2 in K562/G01 cells with or
without TCF7_KD treatment. (F) Recruitment of TCF7 in the
ABCC2 promoter region shown by ChIP-qPCR. (G) Role of TCF7
and imatinib on the Wnt and ABC transporters signaling pathway in
imatinib-resistant CML. One-way ANOVA with Tukey's post hoc test
were performed for F. Two-tailed Student's t-test was used for I.
*P<0.05, **P<0.01, ***P<0.001. ns, not significant. GSEA,
Gene Set Enrichment Analysis; TCF7, transcription factor 7; CML,
chronic myeloid leukemia; KD, knockdown; CCND1, cyclin D1; CTNNB1,
catenin β1; ACTB, β-actin. |
ABCC2 is a TCF7 target gene
As a transcription factor, TCF7 promotes the
transcription of many genes by binding to motifs. TCF7 target genes
were calculated from ChIP-seq data in the GTRD database (42) and collated into a gene set, named
TCF7_targets. We then analyzed the single-cell RNA-seq data of
BCR-ABL1+ LSCs in the dataset GSE76312 (37) and our four groups of RNA-seq dataset
GSE152220. The results showed that the expression of TCF7 was
positively correlated with TCF7_targets gene set in
BCR-ABL1+ LSCs cells (Fig.
S1C). On the other hand, TCF7 knockdown resulted in its
downregulation in K562/G01 cells (Fig.
S1C).
ABCC2 was identified by screening a
intersection of four gene, namely TCF7_targets, TCF7
correlated core enrichment genes, TCF7_KD downregulated genes, and
ABC transporters (Fig. S1D).
Integrative genomics viewer (IGV) was used to visualize the
processed ChIP-seq data ENCFF476IUK, and it was found that TCF7 had
a binding peak in the promoter region of ABCC2 (Fig. 6E). In addition, by integrating our
RNA-seq data into IGV, it can be seen intuitively that the
transcription level of ABCC2 was lower in the TCF7_KD group
compared with the Scramble group (Fig.
6E). Furthermore, ChIP-qPCR results showed that TCF7 was
recruited to the promoter region of ABCC2 in K562/G01 cells
(Fig. 6F). These results indicate
that TCF7 is a direct transcriptional regulator of ABCC2 in
K562/G01 cells. In summary, the roles of TCF7 and imatinib in CML
imatinib-resistant cells are shown in a graphical abstract
(Fig. 6G).
Discussion
Since the application of first-generation tyrosine
kinase inhibitor (TKI), imatinib, in clinical practice, the problem
of drug resistance with complex mechanisms has emerged. TKIs can
effectively solve the drug resistance caused by BCR-ABL1 point
mutations (6), while
BCR-ABL1-independent drug resistance has become a new urgent
concern. The results of the present study indicate that the
expression of transcription factor 7 (TCF7) is independent of the
tyrosine kinase activity of BCR-ABL1 in imatinib-resistant cells.
TCF7 knockdown can significantly inhibit the proliferation
and improve imatinib sensitivity of imatinib-resistant cells. In
addition, GSEA analysis indicated that ABC transporters and the
Wnt/β-catenin signaling pathways are upregulated during imatinib
treatment in imatinib-resistant cells, while TCF7 knockdown
can neutralize this trend.
Wnt signaling is involved in regulating embryonic
development and adult tissue homeostasis, and components of Wnt
signaling pathway aberrant regulation are closely linked to the
development of various tumors (43). Genome-wide ChIP-Seq results show
that the TCF/LEF family is the most critical transcription factor
group mediating Wnt/β-catenin signaling function (44). Previous studies have shown that
overexpression of TCF7 is often associated with disease progression
and poor prognosis in nasopharyngeal cancer (45), gastric cancer (46), and astroglioma (47). Consistent with these finding, TCF7
expression was significantly increased in imatinib-resistant
patients compared with imatinib-sensitive patients. These results
indicate that TCF7 may play a vital role in the development of drug
resistance in chronic myeloid leukemia (CML) cells. An increasing
number of studies have shown that replacing or combining other
targets to conquer leukemia drug resistance has become a feasible
strategy (48,49). In the present study, even when
BCR-ABL1 activity was inhibited entirely, TCF7 expression was not
significantly altered, indicating that TCF7 expression is
BCR-ABL1-independent and combined targets of TCF7 and BCR-ABL1 may
have a synergistic effect on the inhibition of CML
imatinib-resistant cells.
In bladder and prostate cancers, targeting TCF7 can
increase the sensitivity of cancer cells to chemotherapy (26,27).
In CML, silencing of β-catenin or inhibition of β-catenin with the
small molecule drug C82 can also have the same effect of reducing
drug resistance (10). Consistent
with the above studies, our results showed that knockdown of
TCF7 resulted in impaired cell proliferation and enhancement
of imatinib sensitivity in CML imatinib-resistant cells. Thus,
combined target therapy can more effectively inhibit the viability
of imatinib-resistant cells. Interestingly, although there are four
members of the TCF/LEF family that interact with β-catenin in the
Wnt signaling pathway, the fact that TCF7 knockdown can
function alone suggests that the Wnt/β-catenin/TCF7 signaling axis
is involved in the initiation of drug resistance during TKI
treatment.
The molecular events specifically affected by
TCF7 knockdown are the vital clues revealing the mechanism
of phenotype generation. In a previous report, ABCC2 overexpression
conferred tumor cell resistance to multiple chemotherapeutic drugs
such as vincristine, cisplatin, etoposide, doxorubicin, and
methotrexate (38). Previous
studies have shown that the ABCC2
T−24G1249T3972 haplotype is
related to imatinib resistance (50). Its expression is relatively higher
in imatinib-resistant patients compared to imatinib-sensitive
patients, and its knockdown can restore the sensitivity of
resistant cells to imatinib (18).
The above data indicate that ABCC2 contributes to CML resistance.
In this study, we found that TCF7 is recruited to the promoter
region of ABCC2 and transactivates ABCC2
transcription. Furthermore, TCF7 knockdown can weaken the
intensity of ABC transporter signaling.
Interestingly, a recent study by Trojani et
al (51) demonstrated that
long-term use of second-generation TKI (nilotinib) in CML patients
can induce the upregulation of ABC transporters (ABCC4, ABCC5,
ABCD3) in bone marrow CD34+/lin− cells.
Another independent study by Mehrvar et al (52) demonstrated the changes in expression
pattern of ABCC transporters in peripheral blood leukocytes of
patients with acute lymphoblastic leukemia (ALL) recurrence. In
particular, the expression of ABCC2 was significantly increased and
could be used as a predictor of ALL hematologic relapse. Based on
the abovementioned studies, we can speculate regarding the
following two points: One is that under long-term chemotherapy, ABC
transporters in leukemia cells will be abnormally expressed, and
the second is that the abnormal expression of ABC transporters will
be related to leukemia hematologic relapse. In CML, the therapeutic
regimen involves TKI administration, and the basis for relapse is
TKI resistance. Thus, TKIs can lead to abnormal expression of ABC
transporters, which in turn can lead to the generation of TKI
resistance in CML cells. However, the samples consisted of bone
marrow CD34+/lin− cells and peripheral blood
leukocytes used in the previous studies. Because the proportion of
leukemia cells is unknown, it is ambiguous whether the appearance
of abnormal indicators originates from leukemia cells. Our study
has answered this question. When imatinib-resistant cells were
treated with imatinib, the intensity of ABC transporter signaling
was significantly increased. More importantly, in
BCR-ABL1+ LSCs, TCF7 expression was positively
correlated with ABC transporters. TCF7 knockdown can lead to
its downregulation, which is contrary to the effect of imatinib on
imatinib-resistant cells. In addition, we found that imatinib
induced the upregulation of the Wnt/β-catenin signaling pathway in
imatinib-resistant cells, and TCF7 knockdown could partially
offset this trend. To the best of our knowledge, this is the first
study to show these effects of TKI and TCF7 on Wnt/β-catenin and
ABC transporter signaling pathways in imatinib-resistant cells.
One limitation of this study is that RNA-seq data at
the cell population level cannot characterize the various subsets
contained in the whole tumor. Moreover, even when
imatinib-resistant cells are exposed to high concentrations of
imatinib, some cells could still survive, which will become a major
hidden danger that blocks CML patients to achieve full recovery. A
deep understanding of the existence and formation of
imatinib-resistant cells is critical to overcoming CML recurrence.
Further research should be performed at the level of single cells
to achieve more detailed data of subsets of imatinib-resistant
cells, and then exploration of the mechanism of protective feedback
during cellular stress must be carried out. Moreover, our results
could be further generalized if we conducted our investigations
using primary tumor cells.
In summary, our study found that imatinib treatment
induced protective upregulation of Wnt/β-catenin and ABC
transporter signals, and TCF7 knockdown neutralized this
effect and restored imatinib sensitivity in imatinib-resistant
cells. Additionally, this study showed that TCF7 konckdown
could decrease the expression of CCND1 and ABCC2. Finally, our
study revealed that regulation of the Wnt/β-catenin/TCF7/ABC
transporter signaling axis through TCF7 may become an effective
strategy for overcoming imatinib resistance.
Supplementary Material
Supporting Data
Acknowledgements
We thank Dr Jiwei Li for contributing to the RNA-seq
analysis.
Funding
This research study was supported by the National
Natural Science Foundation of China (no. 81772255).
Availability of data and materials
The sequencing data was deposited in the GEO
database with accession code GSE152220.
Authors' contributions
WF conceived and supervised the study. HZ performed
the experiments and wrote the manuscript. YW and HY participated in
analyses of the experimental results. ZH and XW made substantial
contributions to the conception and design of the study. All
authors read and approved this manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rowley JD: Letter: A new consistent
chromosomal abnormality in chronic myelogenous leukaemia identified
by quinacrine fluorescence and Giemsa staining. Nature.
243:290–293. 1973. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goldman JM and Melo JV: Targeting the
BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med.
344:1084–1086. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Holyoake TL and Vetrie D: The chronic
myeloid leukemia stem cell: Stemming the tide of persistence.
Blood. 129:1595–1606. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pasic I and Lipton JH: Current approach to
the treatment of chronic myeloid leukaemia. Leukemia Res. 55:65–78.
2017. View Article : Google Scholar
|
|
5
|
Hehlmann R, Lauseker M, Sausele S,
Pfirrmann M, Krause SW, Kolb HJ, Neubauer A, Hossfeld DK, Nerl C,
Gratwohl A, et al: Assessment of imatinib as first-line treatment
of chronic myeloid leukemia: 10-year survival results of the
randomized CML study IV and impact of non-CML determinants.
Leukemia. 31:2398–2406. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Redaelli S, Mologni L, Rostagno R, Piazza
R, Magistroni V, Ceccon M, Viltadi M, Flynn D and
Gambacorti-Passerini C: Three novel patient-derived BCR/ABL mutants
show different sensitivity to second and third generation tyrosine
kinase inhibitors. Am J Hematol. 87:E125–E128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eide CA and Druker BJ: Understanding
cancer from the stem cells up. Nat Med. 23:656–657. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ashton JM, Balys M, Neering SJ, Hassane
DC, Cowley G, Root DE, Miller PG, Ebert BL, McMurray HR, Land H and
Jordan CT: Gene sets identified with oncogene cooperativity
analysis regulate in vivo growth and survival of leukemia stem
cells. Cell Stem Cell. 11:359–372. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Soverini S, Branford S, Nicolini FE,
Talpaz M, Deininger MW, Martinelli G, Müller MC, Radich JP and Shah
NP: Implications of BCR-ABL1 kinase domain-mediated resistance in
chronic myeloid leukemia. Leuk Res. 38:10–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou H, Mak PY, Mu H, Mak DH, Zeng Z,
Cortes J, Liu Q, Andreeff M and Carter BZ: Combined inhibition of
β-catenin and Bcr-Abl synergistically targets tyrosine kinase
inhibitor-resistant blast crisis chronic myeloid leukemia blasts
and progenitors in vitro and in vivo. Leukemia. 31:2065–2074. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Grassi S, Palumbo S, Mariottit V, Liberati
D, Guerrini F, Ciabatti E, Salehzadeh S, Baratè C, Balducci S,
Ricci F, et al: The WNT pathway is relevant for the
BCR-ABL1-independent resistance in chronic myeloid leukemia. Front
Oncol. 9:5322019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheloni G, Tanturli M, Tusa I, Ho DeSouza
N, Shan Y, Gozzini A, Mazurier F, Rovida E, Li S and Dello Sbarba
P: Targeting chronic myeloid leukemia stem cells with the
hypoxia-inducible factor inhibitor acriflavine. Blood. 130:655–665.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rothe K, Lin H, Lin KB, Leung A, Wang HM,
Malekesmaeili M, Brinkman RR, Forrest DL, Gorski SM and Jiang X:
The core autophagy protein ATG4B is a potential biomarker and
therapeutic target in CML stem/progenitor cells. Blood.
123:3622–3634. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jin Y, Zhou J, Xu F, Jin B, Cui L, Wang Y,
Du X, Li J, Li P, Ren R and Pan J: Targeting methyltransferase
PRMT5 eliminates leukemia stem cells in chronic myelogenous
leukemia. J Clin Invest. 126:3961–3980. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Abraham A, Qiu S, Chacko BK, Li H,
Paterson A, He J, Agarwal P, Shah M, Welner R, Darley-Usmar VM and
Bhatia R: SIRT1 regulates metabolism and leukemogenic potential in
CML stem cells. J Clin Invest. 129:2685–2701. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chandran RK, Geetha N, Sakthivel KM,
Aswathy CG, Gopinath P, Raj TV, Priya G, Nair J and Sreedharan H:
Genomic amplification of BCR-ABL1 fusion gene and its impact on the
disease progression mechanism in patients with chronic myelogenous
leukemia. Gene. 686:85–91. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen JR, Jia XH, Wang H, Yi YJ and Li YJ:
With no interaction, knockdown of Apollon and MDR1 reverse the
multidrug resistance of human chronic myelogenous leukemia K562/ADM
cells. Oncol Rep. 37:2735–2742. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He B, Bai Y, Kang W, Zhang X and Jiang X:
LncRNA SNHG5 regulates imatinib resistance in chronic myeloid
leukemia via acting as a CeRNA against MiR-205-5p. Am J Cancer Res.
7:1704–1713. 2017.PubMed/NCBI
|
|
19
|
Mosimann C, Hausmann G and Basler K:
Beta-catenin hits chromatin: Regulation of Wnt target gene
activation. Nat Rev Mol Cell Biol. 10:276–286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vlad A, Röhrs S, Klein-Hitpass L and
Müller O: The first five years of the Wnt targetome. Cell Signal.
20:795–802. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu SY, Li FY, Xing SJ, Zhao TY, Peng WQ
and Xue HH: Hematopoietic and leukemic stem cells have distinct
dependence on Tcf1 and Lef1 transcription factors. J Biol Chem.
291:11148–11160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu XL, Tang XD, Guo W, Yang K and Ren TT:
TCF-1 participates in the occurrence of dedifferentiated
chondrosarcoma. Tumor Biol. 37:14129–14140. 2016. View Article : Google Scholar
|
|
23
|
Yin H, Sheng Z, Zhang X, Du Y, Qin C, Liu
H, Dun Y, Wang Q, Jin C, Zhao Y and Xu T: Overexpression of SOX18
promotes prostate cancer progression via the regulation of TCF1,
c-Myc, cyclin D1 and MMP-7. Oncol Rep. 37:1045–1051. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen WY, Liu SY, Chang YS, Yin JJ, Yeh HL,
Mouhieddine TH, Hadadeh O, Abou-Kheir W and Liu YN: MicroRNA-34a
regulates WNT/TCF7 signaling and inhibits bone metastasis in
Ras-activated prostate cancer. Oncotarget. 6:441–457. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shiokawa D, Sato A, Ohata H, Mutoh M,
Sekine S, Kato M, Shibata T, Nakagama H and Okamoto K: The
induction of selected Wnt target genes by Tcf1 mediates generation
of tumorigenic colon stem cells. Cell Rep. 19:981–994. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu X, Liu X, Wu Y, Fang Z, Wu Q, Wu C,
Hao Y, Yang X, Zhao J, Li J, et al: MicroRNA-34a attenuates
metastasis and chemoresistance of bladder cancer cells by targeting
the TCF1/LEF1 axis. Cell Physiol Biochem. 48:87–98. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Siu MK, Chen WY, Tsai HY, Chen HY, Yin JJ,
Chen CL, Tsai YC and Liu YN: TCF7 is suppressed by the androgen
receptor via microRNA-1-mediated downregulation and is involved in
the development of resistance to androgen deprivation in prostate
cancer. Prostate Cancer Prostatic Dis. 20:172–178. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang T, Huang Z, Huang N, Peng Y, Gao M,
Wang X and Feng W: Inhibition of KPNB1 inhibits proliferation and
promotes apoptosis of chronic myeloid leukemia cells through
regulation of E2F1. Onco Targets Ther. 12:10455–10467. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Metsalu T and Vilo J: ClustVis: A web tool
for visualizing clustering of multivariate data using Principal
Component Analysis and heatmap. Nucleic Acids Res. 43:W566–W570.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Thorvaldsdóttir H, Robinson JT and Mesirov
JP: Integrative Genomics Viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cramer-Morales K, Nieborowska-Skorska M,
Scheibner K, Padget M, Irvine DA, Sliwinski T, Haas K, Lee J, Geng
H, Roy D, et al: Personalized synthetic lethality induced by
targeting RAD52 in leukemias identified by gene mutation and
expression profile. Blood. 122:1293–1304. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Radich JP, Dai H, Mao M, Oehler V,
Schelter J, Druker B, Sawyers C, Shah N, Stock W, Willman CL, et
al: Gene expression changes associated with progression and
response in chronic myeloid leukemia. Proc Natl Acad Sci USA.
103:2794–2799. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Giustacchini A, Thongjuea S, Barkas N,
Woll PS, Povinelli BJ, Booth CAG, Sopp P, Norfo R, Rodriguez-Meira
A, Ashley N, et al: Single-cell transcriptomics uncovers distinct
molecular signatures of stem cells in chronic myeloid leukemia. Nat
Med. 23:692–702. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
König J, Nies AT, Cui YH, Leier I and
Keppler D: Conjugate export pumps of the multidrug resistance
protein (MRP) family: Localization, substrate specificity, and
MRP2-mediated drug resistance. Biochim Biophys Acta. 1461:377–394.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lian G, Yuan J and Gao Y: In vitro
transport ability of ABCC2 (G1249A) polymorphic variant towards
anticancer drugs. Onco Targets Ther. 13:1413–1419. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Marchetti S, de Vries NA, Buckle T, Bolijn
MJ, van Eijndhoven MA, Beijnen JH, Mazzanti R, van Tellingen O and
Schellens JH: Effect of the ATP-binding cassette drug transporters
ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva)
disposition in in vitro and in vivo pharmacokinetic studies
employing Bcrp1(−/-)/Mdr1a/1b(−/-) (triple-knockout) and wild-type
mice. Mol Cancer Ther. 7:2280–2287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kiyotani K, Mushiroda T, Imamura CK,
Hosono N, Tsunoda T, Kubo M, Tanigawara Y, Flockhart DA, Desta Z,
Skaar TC, et al: Significant effect of polymorphisms in CYP2D6 and
ABCC2 on clinical outcomes of adjuvant tamoxifen therapy for breast
cancer patients. J Clin Oncol. 28:1287–1293. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yevshin I, Sharipov R, Kolmykov S,
Kondrakhin Y and Kolpakov F: GTRD: A database on gene transcription
regulation-2019 update. Nucleic Acids Res. 47:D100–D105. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ng LF, Kaur P, Bunnag N, Suresh J, Sung
ICH, Tan QH, Gruber J and Tolwinski NS: WNT signaling in disease.
Cells. 8:8262019. View Article : Google Scholar
|
|
44
|
Schuijers J, Mokry M, Hatzis P, Cuppen E
and Clevers H: Wnt-induced transcriptional activation is
exclusively mediated by TCF/LEF. EMBO J. 33:146–156. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhan Y, Feng J, Lu J, Xu L, Wang W and Fan
S: Expression of LEF1 and TCF1 (TCF7) proteins associates with
clinical progression of nasopharyngeal carcinoma. J Clin Pathol.
72:425–430. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xu XG, Liu ZX, Tian F, Xu J and Chen YM:
Clinical significance of transcription factor 7 (TCF7) as a
prognostic factor in gastric cancer. Med Sci Monit. 25:3957–3963.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kafka A, Bačić M, Tomas D, Žarković K,
Bukovac A, Njirić N, Mrak G, Krsnik Ž and Pećina-Šlaus N: Different
behaviour of DVL1, DVL2, DVL3 in astrocytoma malignancy grades and
their association to TCF1 and LEF1 upregulation. J Cell Mol Med.
23:641–655. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Schneeweiss-Gleixner M, Byrgazov K,
Stefanzl G, Berger D, Eisenwort G, Lucini CB, Herndlhofer S,
Preuner S, Obrova K, Pusic P, et al: CDK4/CDK6 inhibition as a
novel strategy to suppress the growth and survival of
BCR-ABL1(T315I)+ clones in TKI-resistant CML. EBioMedicine.
50:111–121. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Eide CA, Zabriskie MS, Savage Stevens SL,
Antelope O, Vellore NA, Than H, Schultz AR, Clair P, Bowler AD,
Pomicter AD, et al: Combining the allosteric inhibitor asciminib
with ponatinib suppresses emergence of and restores efficacy
against highly resistant BCR-ABL1 mutants. Cancer Cell.
36:431–443.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Au A, Baba AA, Azlan H, Norsa'adah B and
Ankathil R: Clinical impact of ABCC1 and ABCC2 genotypes and
haplotypes in mediating imatinib resistance among chronic myeloid
leukaemia patients. J Clin Pharm Ther. 39:685–690. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Trojani A, Pungolino E, Dal Molin A,
Lodola M, Rossi G, D'Adda M, Perego A, Elena C, Turrini M, Borin L,
et al: Nilotinib interferes with cell cycle, ABC transporters and
JAK-STAT signaling pathway in CD34+/lin−
cells of patients with chronic phase chronic myeloid leukemia after
12 months of treatment. PLoS One. 14:e02184442019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mehrvar N, Abolghasemi H, Rezvany MR,
Esmaeil Akbari M, Saberynejad J, Mehrvar A, Ehsani MA, Nourian M,
Qaddoumi I and Movafagh A: Pattern of ABCC transporter gene
expression in pediatric patients with relapsed acute lymphoblastic
leukemia. Rep Biochem Mol Biol. 8:184–193. 2019.PubMed/NCBI
|