Introduction
Colorectal cancer (CRC) is a serious malignant
disease, typically initiated by aberrant epithelial cell
proliferation in the colon or rectum (1). CRC is the third most frequently
diagnosed type of cancer worldwide (2), and is characterized by a high and
continuously increasing mortality rate, estimated to reach 60% by
2035 (3). The treatment options for
CRC currently include chemotherapy, targeted therapy and
immunotherapy (4). The
chemotherapeutic agents most widely used for CRC include
5-fluorouracil (5FU), irinotecan and oxaliplatin (4,5). 5FU
is a first-line drug that has been extensively used as a
monotherapy or as part of a combination regimen for the treatment
of CRC (4,5). However, the proportion of patients
with advanced CRC who respond to 5FU is limited to 10-15% (6), whereas ~50% of patients with
metastatic CRC display resistance to 5FU-based chemotherapies
(7,8). Therefore, the development of novel
therapeutic strategies for 5FU-resistant CRC is important.
Several quinazolinone compounds have been developed
and approved as novel anticancer agents, including nolatrexed,
idelalisib and raltitrexed (9–11). In
our laboratory, several quinazolinone compounds (such as MJ-29,
MJ-33 and LJJ-10) have been designed, synthesized and studied to
investigate their anticancer activities (12,13).
For example, the inhibitory effect of MJ-33 on AKT-mediated DU145
prostate cancer cell metastasis was previously reported (14). However, the cytotoxicity and
mechanisms underlying MJ-33 in chemoresistant cells are not
completely understood. Therefore, the present study aimed to
investigate the anticancer activities of MJ-33 in a CRC cell line
with acquired resistance to 5FU, namely HT-29/5FUR.
AKT is an important cancer-related regulator that is
responsible for cancer cell survival, proliferation and migration
(15,16). Moreover, AKT overactivation has been
reported to serve as a biomarker of tumorigenesis, tumor growth,
metastasis and resistance to cancer therapies (17). AKT/mTOR signaling-mediated control
of autophagy and apoptosis in chemoresistant cells has been
previously reported (18). In
addition, inhibiting the PI3K/AKT signaling pathway restores the
sensitivity of HT-29 cells to 5FU (19). A previous study also demonstrated
the inhibitory effects of MJ-33 on AKT phosphorylation, resulting
in antimetastatic activity (14).
The crosstalk between apoptosis and autophagy is a
novel therapeutic target in cancer (20). In the later stage, when autophagy no
longer serves a cytoprotective role, it leads to apoptosis
activation to induce programmed cell death (21). In a previous study, a novel
quinazolinone derivative induced autophagy-related apoptotic cell
death in lymphoblastic leukemia MOLT-4 cells (22). Therefore, it was hypothesized that
the mechanism underlying MJ-33-induced anticancer activity was
associated with the induction of apoptosis and autophagy. The
present study investigated the cytotoxic effects and mechanisms
underlying MJ-33, and explored the involvement of the AKT
downstream signaling pathway in HT-29/5FUR cells.
Materials and methods
Chemicals and reagents
MJ-33 was synthesized in our laboratory at the
School of Pharmacy, China Medical University. DAPI, 3-Methyladenine
(3-MA), chloroquine (CQ), Bafilomycin A1 (Baf.A1), AKT activator
(SC-79), Minimum Essential medium and MTT were purchased from
Sigma-Aldrich (Merck KGaA). L-glutamine, RPMI-1640 medium,
penicillin, streptomycin, trypsin-EDTA, acridine orange (AO),
LysoTracker Red, LC3-green fluorescent protein (GFP) and FBS were
purchased from Thermo Fisher Scientific, Inc. Pan-caspase inhibitor
(z-VAD-fmk), caspase-9 inhibitor (z-LEHD-fmk) and caspase-3
inhibitor (z-DEVD-fmk) were purchased from Merck KGaA. 5FU was
obtained from Pharmacia & Upjohn.
Cell lines and cell culture
The human colorectal cancer HT-29 cell line
(23) was obtained from the
Bioresource Collection and Research Center, Food Industry Research
and Development Institute. The cell line was authenticated via STR
profiling (Mission Biotech Co., Ltd. Cells were cultured in
75-cm2 tissue culture flasks in RPMI-1640 medium
supplemented with 2 mM L-glutamine, 10% FBS, 100 U/ml penicillin
and 100 µg/ml streptomycin at 37°C with 5% CO2.
5FU-resistant colorectal cancer cells (HT-29/5FUR) were established
according to the following protocol. Parental HT-29 cells were
exposed to an initial dose of 0.5 µg/ml (3.843 µM) at 37°C and
surviving cells were cultured to 90% confluence for four passages
(4 weeks). Surviving cells were exposed to 1 µg/ml 5FU (7.688 µM)
for four passages (4 weeks) and then 1.5 µg/ml 5FU (11.530 µM) for
four passages (4 weeks). Finally, surviving cells were exposed to 2
µg/ml 5FU (15.372 µM), the clinically relevant plasma
concentration, for four passages (4 weeks). Surviving resistant
cells were named 5FU-resistant colorectal cancer cells (HT-29/5FUR)
(24,25). The doubling time of parental HT-29
cells was 17 h and the doubling time of HT-29/5FUR cells was 34
h.
Prior to MJ-33 treatment, HT-29/5FUR cells were
treated with 10 mM 3-MA, 100 µM CQ or 10 nM Baf.A1 at 37°C for 1 h,
10 µM SC-79 at 37°C for 30 min, or 15 µM z-VAD-fmk, 15 µM
z-LEHD-fmk or 15 µM z-DEVD-fmk at 37°C for 2 h.
The normal human colon CCD 841 CoN cell line
(CRL-1790; American Type Culture Collection) was cultured in
Minimum Essential medium supplemented with 10% FBS, 100 U/ml
penicillin and 100 µg/ml streptomycin at 37°C with 5%
CO2.
Cell viability assay
HT-29/5FUR and CCD 841 CoN cells were seeded
(1×104 cells/well) into 96-well plates and treated with
different concentrations of MJ-33 (25, 50, 75 and 100 µM). Control
cells were treated with DMSO. Positive control cells were treated
with 30 µM oxaliplatin (Sanofi S.A.). Cells were incubated with
each treatment at 37°C for 24 or 48 h with 5% CO2. Then,
cell viability was assessed by performing the MTT assay as
previously described (18,26).
Cell death and morphological changes
on microscopic observation
HT-29/5FUR cells (1×104 cells/100 µl)
were treated with 25, 50, 75 and 100 µM MJ-33 or DMSO at 37°C for
48 h with 5% CO2. Subsequently, cultured cells were
observed using a phase-contrast light microscope (Leica
Microsystems GmbH; magnification, ×400) to visualize morphological
alterations characteristic of apoptotic or autophagic cell
death.
AO, LysoTracker Red and LC3-GFP
staining
HT-29/5FUR cells (1×105 cells/ml) were
treated with 75 µM MJ-33 or DMSO for 24 h at 37°C. After harvesting
using trypsin-EDTA, cells were fixed in 4% paraformaldehyde on ice
for 15 min. Subsequently, cells were stained with 1 µg/ml AO,
LysoTracker Red or LC3-GFP for 20 min at room temperature. Stained
cells were visualized using ImageXpress Micro Confocal High-Content
Image System (Molecular Devices, LLC; magnification, ×400) and
analyzed using MetaXpress (version 5.3.0.4; Molecular Devices, LLC)
to detect acidic vesicular organelles (for AO staining), lysosomal
function (for LysoTracker Red staining) or typical punctate pattern
(for LC3-GFP staining).
DAPI and TUNEL dual staining
HT-29/5FUR cells were seeded (1×105
cells/ml) into 12-well plates and treated with 75 µM MJ-33 or DMSO
for 48 h at 37°C with 5% CO2. Cells were harvested and
fixed with absolute ethanol at room temperature for 10 min.
Subsequently, cells were stained with 1 µg/ml DAPI solution for 30
min at room temperature as previously described (27). To detect DNA breaks, the In
Situ Cell Death Detection Kit, Fluorescein (Roche Diagnostics
GmbH) was used according to the manufacturer's protocol. After
applying mounting medium (10% glycerol in PBS), stained samples
were observed using a fluorescence microscope (Leica Microsystems
GmbH; magnification, ×400) as previously described (28). For each coverslip, 3 fields of view
with similar numbers of cells were photographed for counting and
further analysis.
Caspase-3 and caspase-7 activity
assays
The fluorochrome-labeled inhibitor of caspases assay
(FLICA) method was applied using the FAM-FLICA® Caspases
3 & 7 Assay Kit (ImmunoChemistry Technologies, LLC; cat. no.
93) and the NucleoCounter NC-3000 (ChemoMetec A/S) according to the
manufacturer's protocol. All samples were analyzed using NucleoView
NC-3000 software (version 1.4; ChemoMetec A/S). Briefly, HT-29/5FUR
cells were seeded (1×106 cells/well) into 6-well plates,
treated with 75 µM MJ-33 or DMSO for 12 h at 37°C. Cells were
resuspended in each well using 0.5 ml PBS. Samples were incubated
with diluted FLICA reagent and Hoechst 33342 for 1 h at 37°C.
Following washing twice with apoptosis wash buffer, cells were
resuspended in 100 µl apoptosis wash buffer supplemented with PI.
Subsequently, 30 µl cell suspension was immediately loaded into a
2-chamber slide (NC-Slide A2; ChemoMetec A/S) for analysis on the
NucleoCounter NC-3000 using the built-in caspase assay program.
Annexin assay and image cytometry
analysis
Experiments were conducted using the Annexin V-FITC
Apoptosis Detection Kit (cat. no. AVK050; Strong Biotech Corp.) and
the Counter NC-3000 cytometer (ChemoMetec A/S) according to the
manufacturer's protocol and the previously described protocol
(29). Briefly, HT-29/5FUR cells
were seeded (total 1×106 cells/well) into 6-well plates.
Samples were treated with 75 µM MJ-33 or DMSO at 37°C for 6 h.
Cells were resuspended in each well with 0.5 ml PBS. Subsequently,
samples were incubated with 100 µl Annexin V binding buffer as
previously described (29). Then,
samples were incubated with 2 µl Annexin V-CF 488A conjugate and 2
µl Hoechst 33342 (10 µg/ml) at 37°C for 15 min. Following
centrifugation at 200 × g at 37°C for 5 min, samples were washed
with Annexin V binding buffer. Cell pellets were resuspended using
100 µl Annexin V binding buffer supplemented with PI at room
temperature for 15 min. All samples were immediately analyzed using
NucleoView NC-3000 software (version 1.4; ChemoMetec A/S).
Subpopulations of stained cells were determined using scatterplots:
Healthy cells (Annexin V−/PI−); early
apoptosis (Annexin V+/PI−); late apoptosis
(Annexin V+/PI+); and necrosis (Annexin
V−/PI+).
Cell confluence assay
The IncuCyte S3 ZOOM System instrument (Essen
BioScience) was used to conduct the cell confluence assay.
HT-29/5FUR cells were seeded (1×104 cells/well) into a
96-well plate with 50 µM MJ-33 or DMSO for 48 h at 37°C. Cells were
visualized and photographed every 2 h as previously described
(30).
Western blotting
HT-29/5FUR cells were lysed with Trident RIPA Lysis
Buffer (GeneTex, Inc.). Protein concentrations were determined
using a Bio-Rad protein assay system (Bio-Rad Laboratories, Inc.).
Proteins were separated as previously described (18,31,32).
Briefly, proteins (35 µg) were separated via 10-12% SDS-PAGE and
transferred using the iBlot Dry Blotting System (Invitrogen; Thermo
Fisher Scientific, Inc.) to PVDF membranes. The membranes were
blocked with PBS containing 0.1% Tween-20 and 5% skimmed dry milk
for 2 h at room temperature. Subsequently, the membranes were
incubated overnight at 4°C with primary antibodies (all purchased
from Cell Signaling Technology, Inc.) targeted against: Bcl-2 (cat.
no. 4223; 1:1,000), Bax (cat. no. 5023; 1:1,000), BAD (cat. no.
9292; 1:1,000), phosphorylated (p)-BAD (cat. no. 5284; 1:1,000),
cytochrome c (cat. no. 4280; 1:1,000), apoptotic peptidase
activating factor-1 (Apaf-1; cat. no. 8969; 1:1,000), caspase-9
(cat. no. 9508; 1:1,000), caspase-3 (cat. no. 9662; 1:1,000),
autophagy related (ATG)-5 (cat. no. 12994; 1:1,000), ATG-7 (cat.
no. 8558; 1:1,000), ATG-12 (cat. no. 4180; 1:1,000), ATG-16 (cat.
no. 8089; 1:1,000), p62 (cat. no. 23214; 1:1,000), LC3 (cat. no.
12741; 1:1,000), AKT (cat. no. 9272; 1:1,000), p-AKT (cat. no.
4060; 1:1,000), mTOR (cat. no. 2972; 1:1,000), p-mTOR (cat. no.
5536; 1:1,000) and β-actin (cat. no. 8457; 1:1,000). Following
primary incubation, the membranes were incubated for 4 h at room
temperature with anti-rabbit IgG (cat. no. 7074; 1:10,000; Cell
Signaling Technology, Inc.) and anti-mouse IgG (cat. no. 7076;
1:10,000; Cell Signaling Technology, Inc.) HRP-conjugated secondary
antibodies. Protein bands were visualized using Immobilon Western
HRP Substrate (Merck KGaA) for 1 h.
Statistical analysis
Data are presented as the mean ± SD (n=3).
Comparisons among multiple groups were analyzed using one-way ANOVA
followed by Dunnett's or Tukey's post hoc test. Statistical
analyses were performed using SPSS software (version 16.0; SPSS,
Inc.). P<0.001 was considered to indicate a statistically
significant difference.
Results
MJ-33 selectively exerts cytotoxic
effects on HT-29/5FUR cells, which are associated with apoptosis
and autophagy
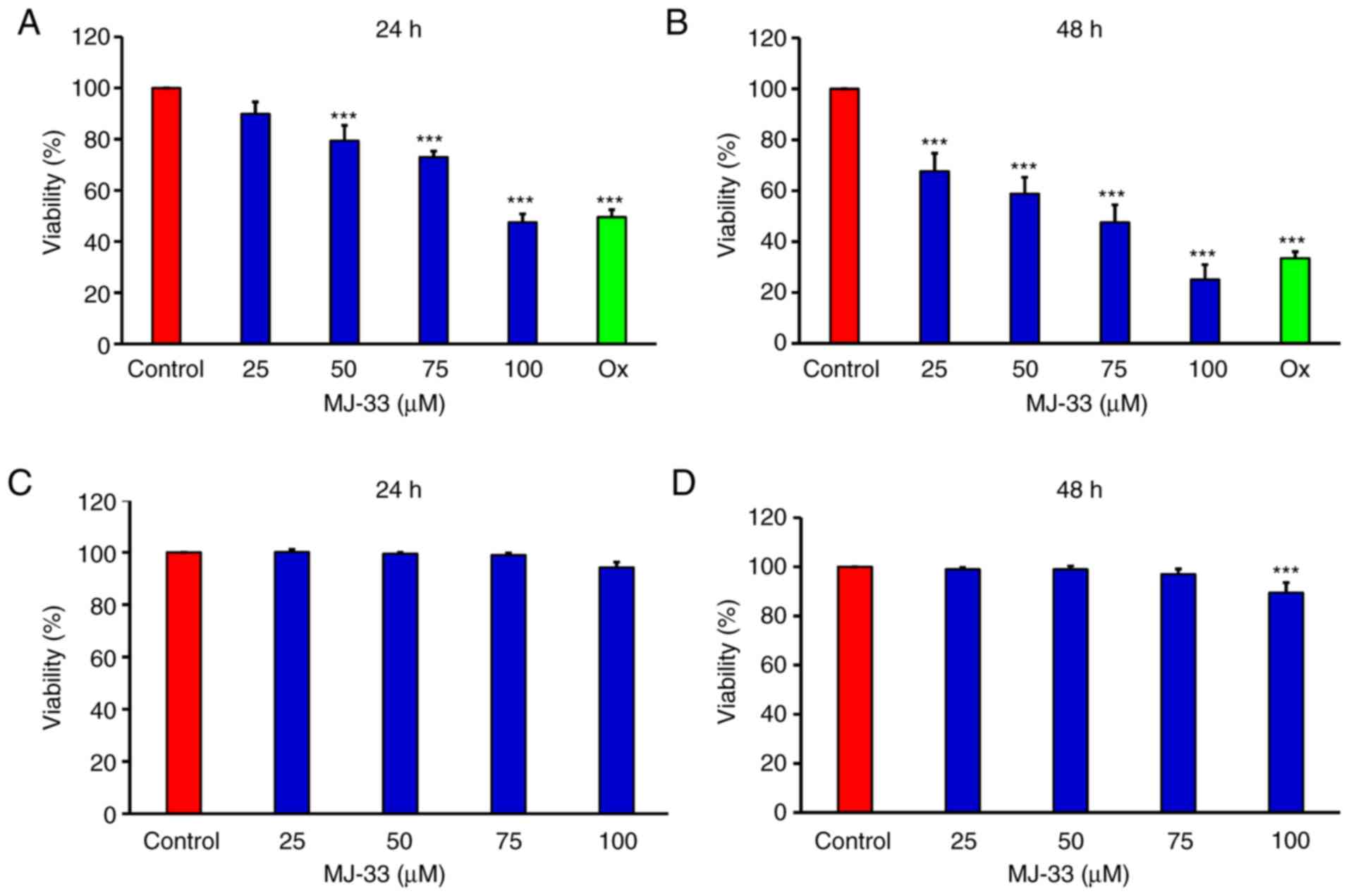
The MTT assay was performed to examine the
antiproliferative effects of MJ-33 on HT-29/5FUR cells in
vitro. The results demonstrated that cell viability was
significantly inhibited by MJ-33 in a concentration-dependent
manner compared with the control group (Fig. 1A and B). As the positive control,
oxaliplatin also significantly inhibited HT-29/5FUR cell viability
compared with the control group. In CCD 841 CoN cells, cell
viability was not significantly altered by MJ-33 treatment, except
for in the 100 µM MJ-33 for 48 h group, compared with the control
group (Fig. 1C and D).
The sensitivity of HT-29/5FUR cells and its parental
cell line (HT-29) to MJ-33, 5FU and Ox are presented in Table I. The IC50s of 5FU and Ox
in HT-29/5FUR cells were higher compared with in HT-29 cells.
Conversely, the IC50 of MJ-33 in HT-29/5FUR cells was
lower compared with HT-29 cells. Based on the IC50
values obtained, 75 µM MJ-33 was selected for subsequent
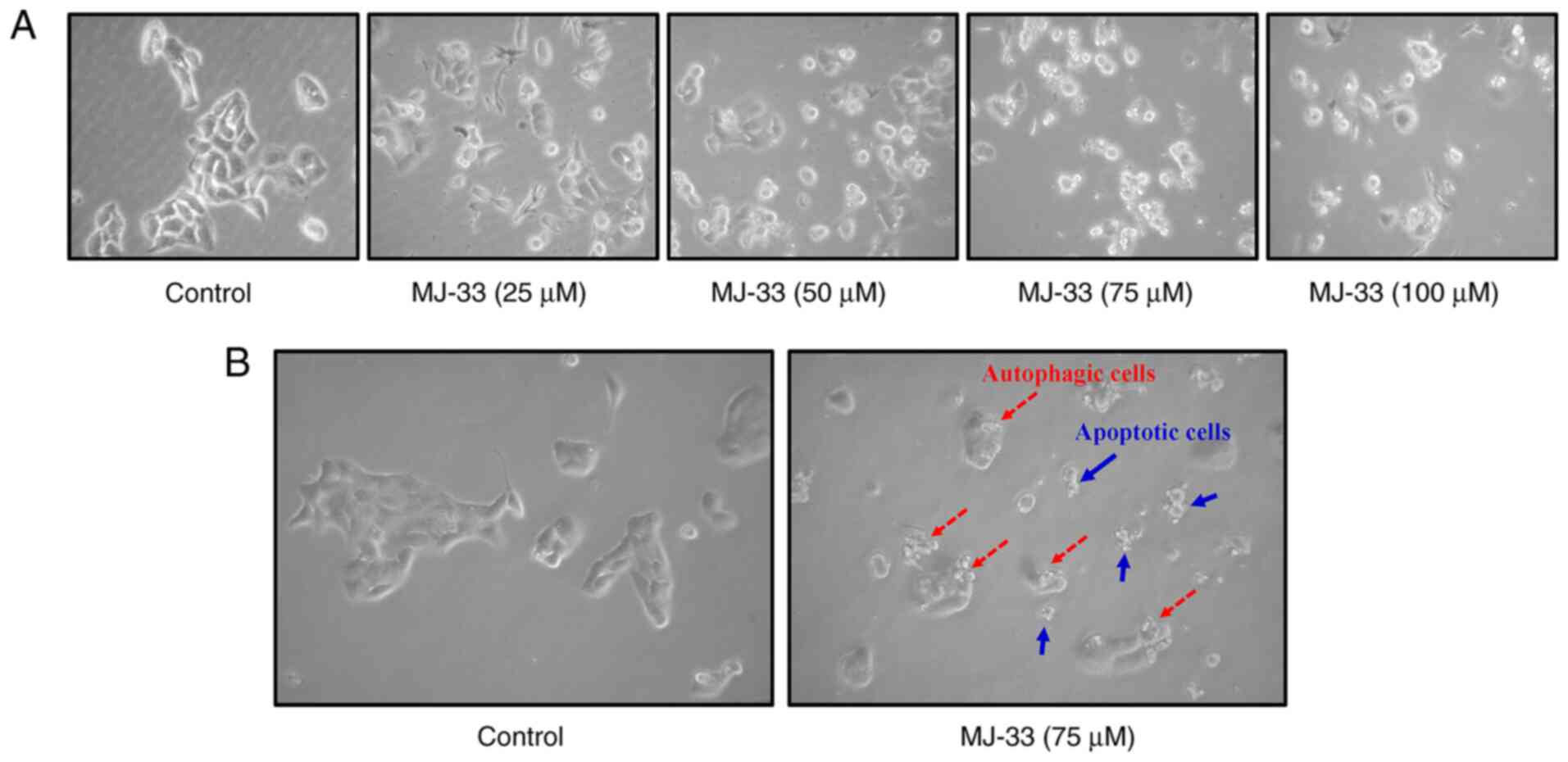
experiments. To further examine the effects of MJ-33 treatment on
HT-29/5FUR cell morphology, light microscopy and the IncuCyte S3
ZOOM system were used. Compared with the control group, MJ-33
treatment induced morphological alterations, including cell
shrinkage, and nuclear fragmentation and condensation, and cell
death (Fig. 2A, B and Video S1) suggested apoptotic phenomena.
In addition, autophagic cells with increasing volume of autophagic
vesicles were observed following MJ-33 treatment (Fig. 2B). The results suggested that
MJ-33-induced cell death was mediated via mechanisms involving
apoptosis and autophagy.
 | Table I.IC50s of MJ-33, 5FU and
oxaliplatin in parental HT-29 and HT-29/5FUR cells following
treatment for 24 or 48 h. |
Table I.
IC50s of MJ-33, 5FU and
oxaliplatin in parental HT-29 and HT-29/5FUR cells following
treatment for 24 or 48 h.
| A, Parental HT-29
cells |
|---|
|
|---|
| Time (h) | MJ-33 (µM) | 5FU (µM) | oxaliplatin
(µM) |
|---|
| 24 | 96.65±2.96 | 20.14±2.13 | 30.98±1.98 |
| 48 | 70.22±3.56 | 9.68±2.29 | 14.98±2.36 |
|
| B, HT-29/5FUR
cells |
|
| Time
(h) | MJ-33
(µM) | 5FU
(µM) | oxaliplatin
(µM) |
|
| 24 | 95.67±4.96 | 61.48±2.25 | 50.65±2.33 |
| 48 | 63.21±4.17 | 29.29±3.33 | 22.46±3.69 |
MJ-33 induces apoptosis via the
mitochondrial intrinsic signaling pathway in HT-29/5FUR cells
MJ-33-induced cell death was observed in HT-29/5FUR
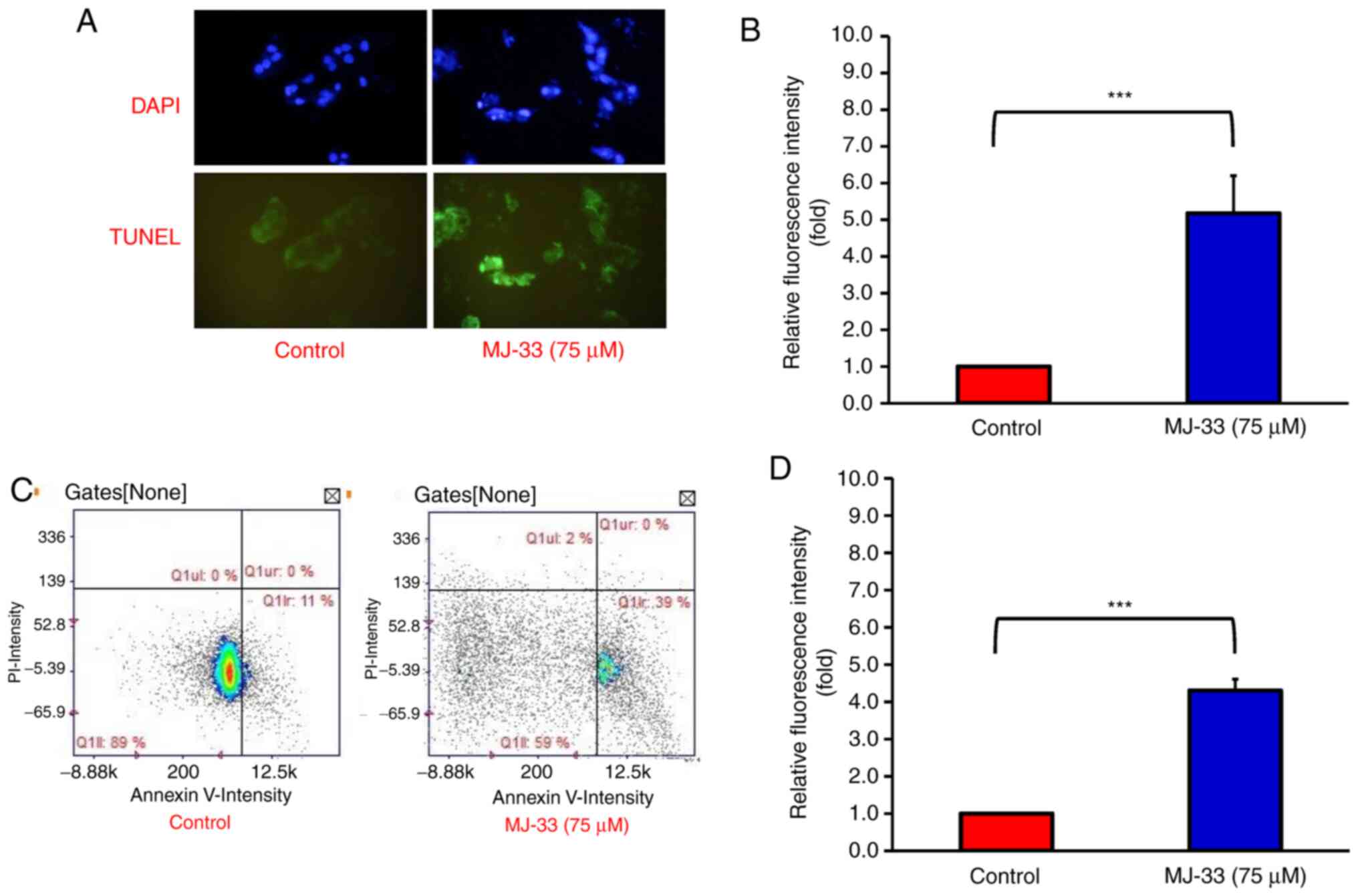
cells, thus the apoptotic bodies were subsequently quantified. By
performing DAPI and TUNEL staining on HT-29/5FUR cells, the
proportion of apoptotic cells after MJ-33 treatment was examined.
Under a fluorescence microscope, the nuclear morphology of dead
cells was observed, which displayed chromatin condensation, a
well-defined hallmark of apoptosis (Fig. 3A). The TUNEL-stained cells emitted
green fluorescence in response to DNA fragmentation, which is an
indicator of late-stage apoptosis (33). The results demonstrated that MJ-33
treatment significantly increased TUNEL-positive staining compared
with the control group (Fig. 3B).
Image cytometry analysis was conducted to determine the apoptotic
subpopulations of MJ-33-treated and control cells. The percentage
of early apoptotic cells was increased from 11% in control cells to
39% in MJ-33-treated cells (Fig.
3C). Moreover, MJ-33 treatment significantly increased the
number of Annexin-V-positive/PI-negative cells compared with the
control group (Fig. 3D). Therefore,
the results further indicated that HT-29/5FUR cell apoptosis was
induced by MJ-33 treatment.
To improve the current understanding of
MJ-33-induced apoptotic death in MJ-HT-29/5FUR cells, the possible
regulatory signaling pathways were investigated. It was
hypothesized that MJ-33 induced apoptotic cell death via the
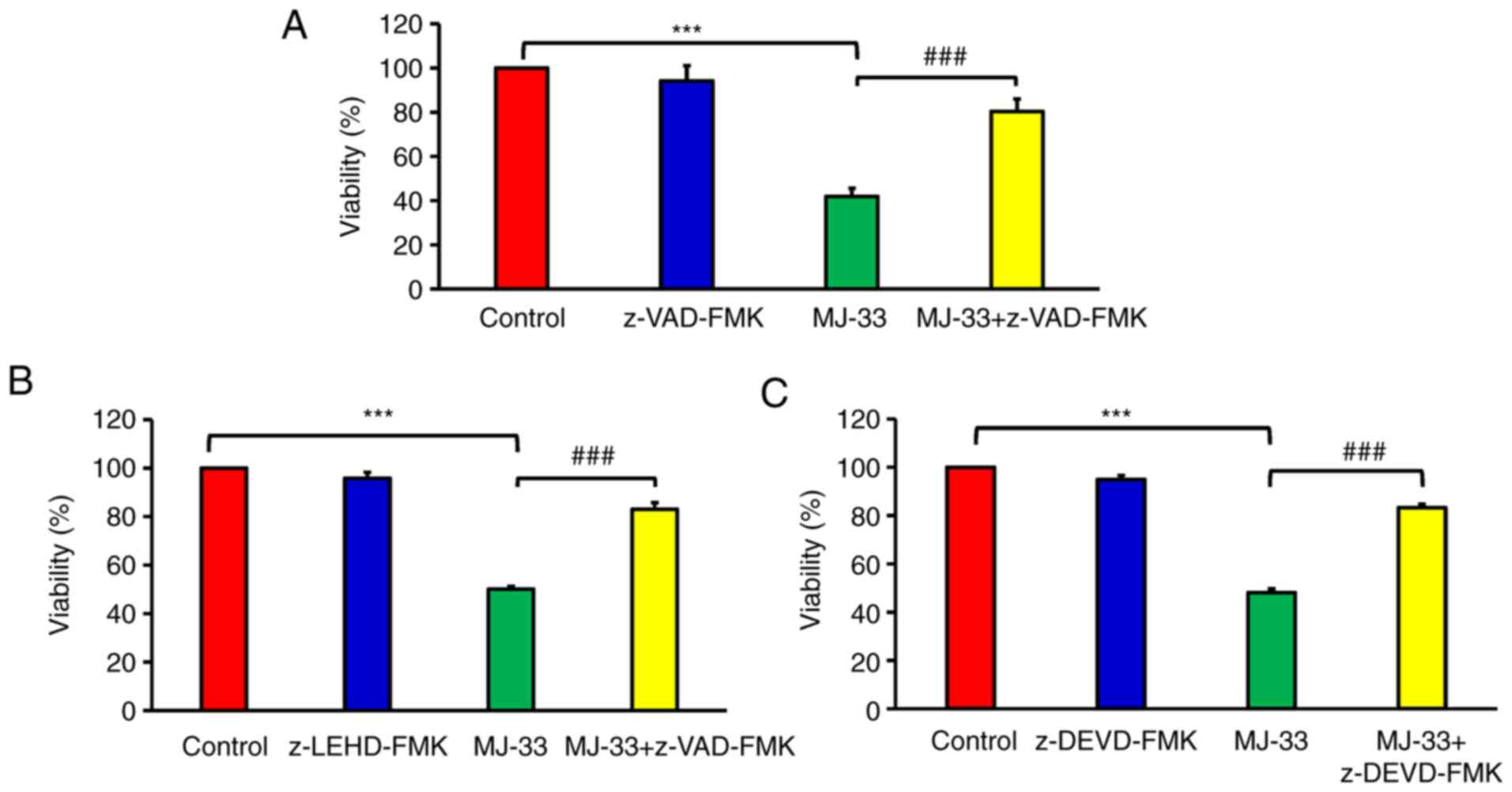
caspase-dependent signaling pathways. Therefore, cell viability was
examined following MJ-33 treatment with or without pan-caspase
inhibitor (z-VAD-FMK). Cell viability in the MJ-33-treated groups
was significantly lower compared with the control group (Fig. 4). Furthermore, cell viability was
significantly higher in the MJ-33 + z-VAD-FMK/MJ-33 group compared
with the MJ-33 group, suggesting that MJ-33-induced HT-29/5FUR cell
apoptosis was mediated via caspase activity. To determine the
effect of specific caspase enzymes on MJ-33-induced apoptosis,
cells were pretreated with z-LEHD-FMK (to inhibit caspase-9) or
z-DEVD-FMK (to inhibit caspase-3). The cell viability assay results
demonstrated MJ-33-induced cytotoxicity in HT-29/5FUR cells was
significantly inhibited by caspase-9 and caspase-3 inhibitors. The
results indicated that MJ-33-induced apoptosis was mediated via
caspase-9 and caspase-3 (Fig. 4B and
C). The aforementioned results suggested that MJ-33 induced
apoptotic cell death via the intrinsic signaling pathway in
HT-29/5FUR cells.
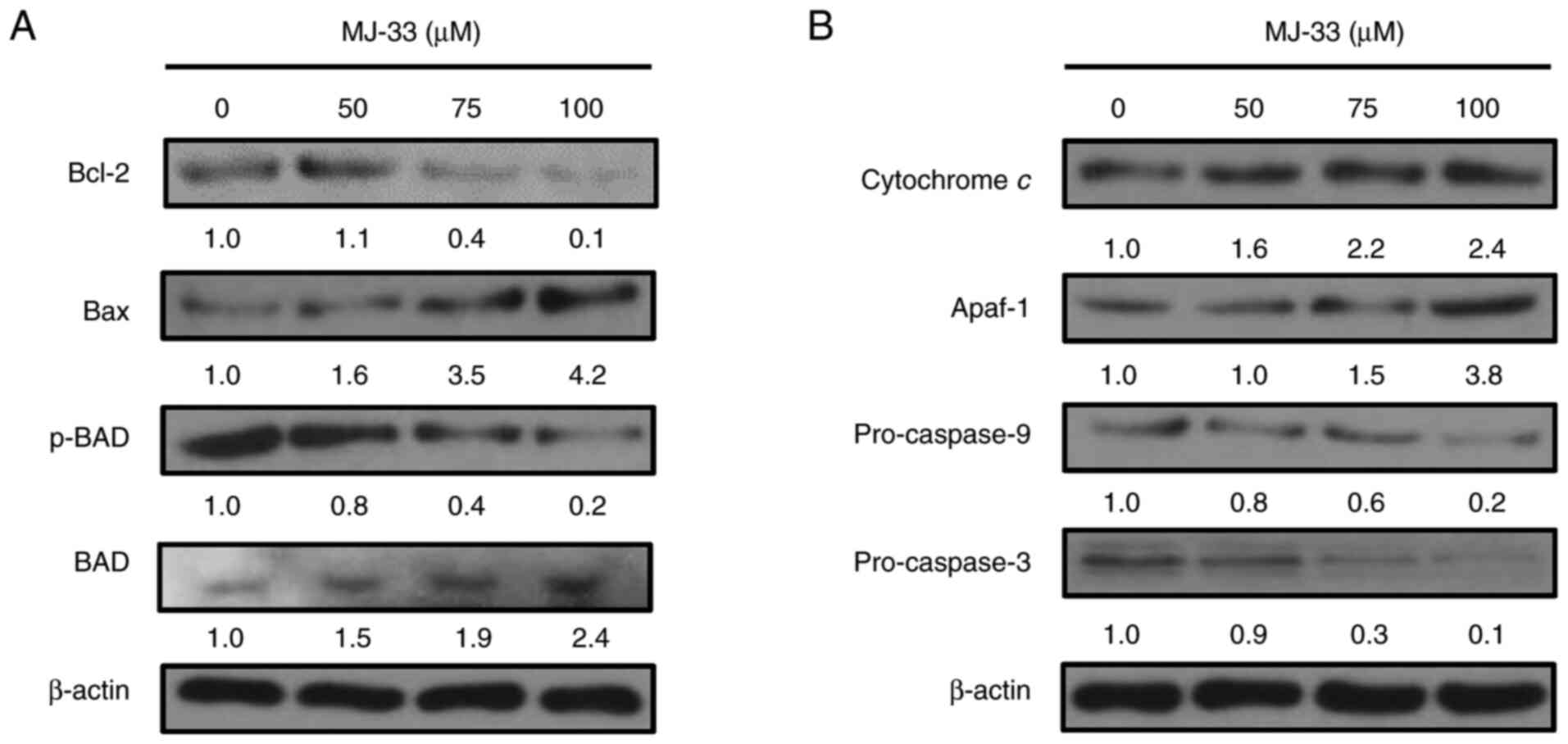
To identify the roles of proapoptotic proteins in
the molecular mechanisms underlying MJ-33-induced apoptosis, the
expression levels of proapoptotic proteins, including Bax, Bcl-2
and p-BAD, were investigated in MJ-33-treated HT-29/5FUR cells.
Compared with the control group, Bax and BAD protein expression
levels were markedly increased in a concentration-dependent manner
following treatment with MJ-33 (Fig.
5A). By contrast, p-BAD protein expression levels were markedly
decreased by MJ-33 treatment compared with the control group, and
reversely associated with the protein expression levels of Bax.
Compared with the control group, Bcl-2 expression levels were
slightly increased following treatment with 50 µM MJ-33, but
notably decreased following treatment with 75 and 100 µM MJ-33.
Similarly, the protein expression levels of
cytochrome c, Apaf-1, pro-caspase-9 and pro-caspase-3 were
examined in HT-29/5FUR cells following treatment with MJ-33. The
protein expression levels of cytochrome c and Apaf-1 were
notably increased by MJ-33 treatment in a concentration-dependent
manner compared with the control group (Fig. 5B). Conversely, the protein
expression levels of pro-caspase-9 and pro-caspase-3 were decreased
by MJ-33 treatment compared with the control group. The results
indicated that apoptotic cell death was promoted by MJ-33 treatment
via the mitochondria-mediated apoptotic signaling pathway.
MJ-33 activates an autophagic
mechanism in HT-29/5FUR cells
The aforementioned results prompted further
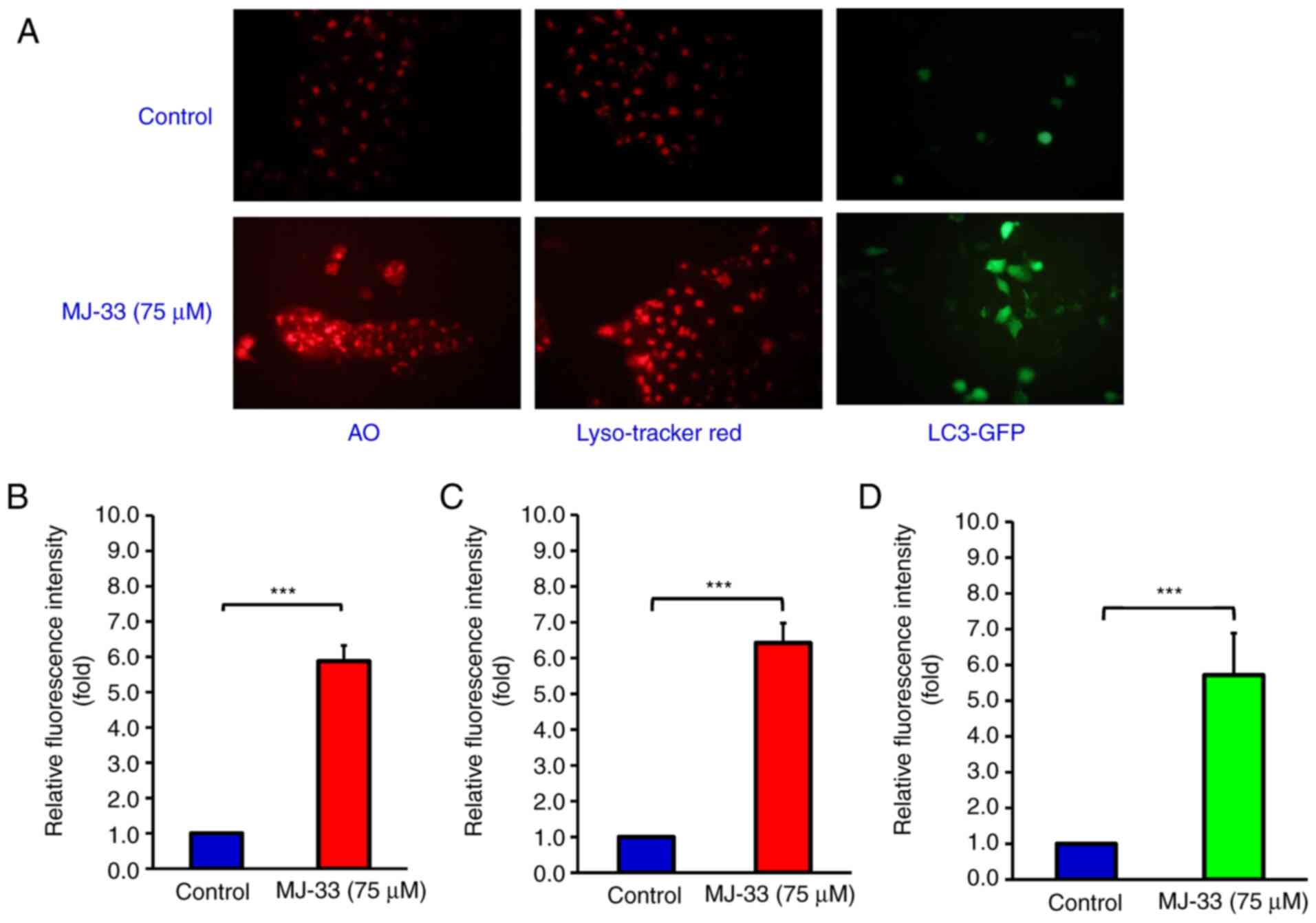
investigation into MJ-33-induced autophagy in HT-29/5FUR cells.
Cells were treated with 75 µM MJ-33 and then stained with AO,
LysoTracker Red or LC3-GFP (Fig.
6A). The relative fluorescence intensity emitted in the red
range indicated significantly increased uptake of AO in the
MJ-33-treated group compared with the control group, which
suggested increased formation of acidic vesicles (Fig. 6B). LysoTracker Red staining results
suggested significantly increased lysosomal activity and
autophagosome maturation in the MJ-33 treatment group compared with
the control group (Fig. 6C).
Additionally, the green fluorescence emitted in the LC3-GFP stained
group was significantly higher in the MJ-33 treatment group
compared with the control group, indicating punctate formation,
which is typically observed in autophagic cells (Fig. 6D). Collectively, the aforementioned
results suggested that MJ-33 treatment activated an autophagic
mechanism in HT-29/5FUR cells.
MJ-33 inhibits AKT/mTOR activity and
elevates the expression of autophagy-related proteins in HT-29/5FUR
cells
It was previously reported that MJ-33 treatment
regulated the MAPK, AKT, NF-κB and activator protein-1 signaling
pathways in DU145 cells (14).
Since AKT/mTOR signaling may mediate both apoptosis and autophagy
(16,34), the effect of MJ-33 treatment on AKT
and mTOR protein expression levels in HT-29/5FUR cells was
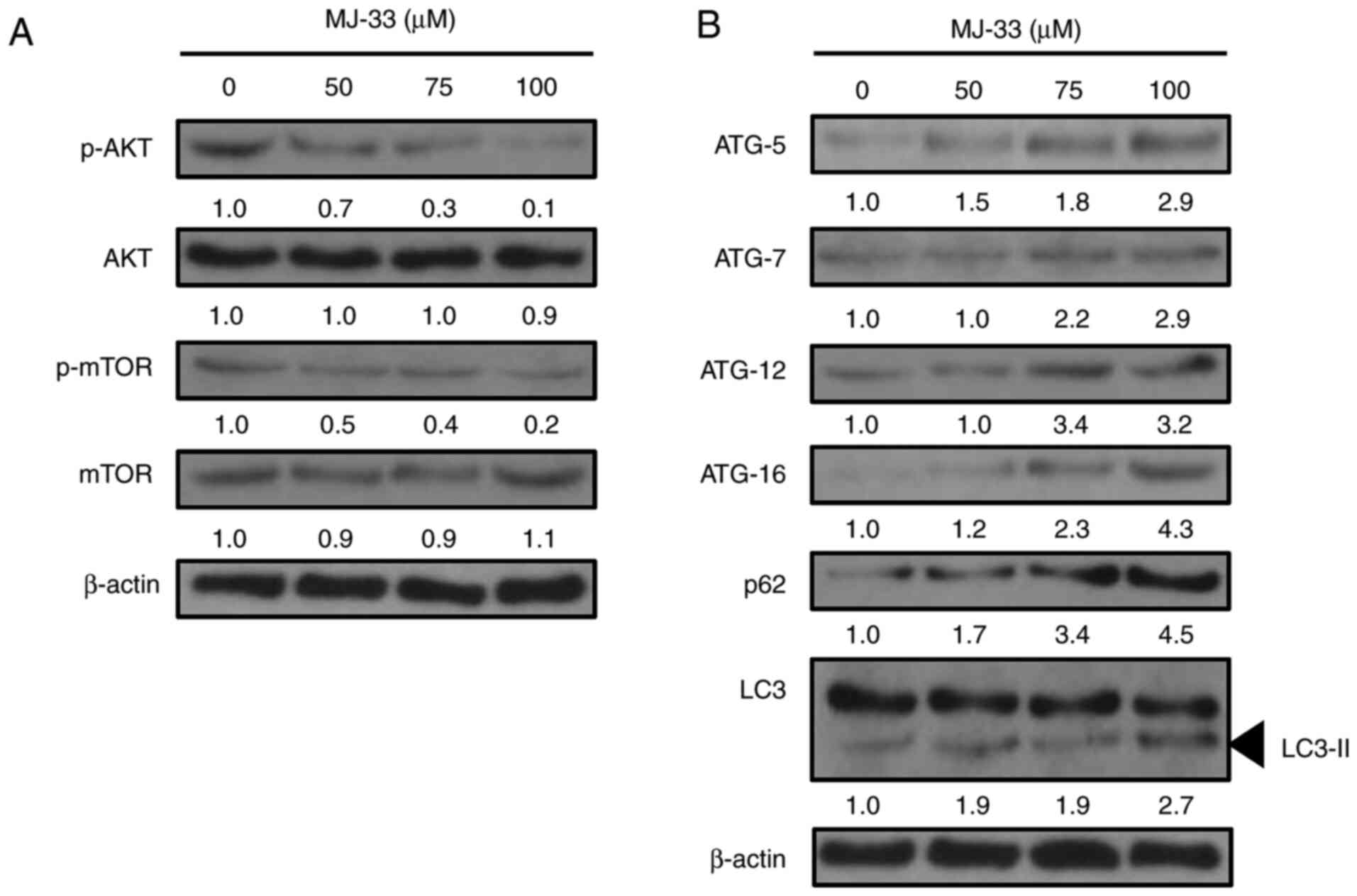
evaluated. The western blotting results demonstrated that the
ratios of p-AKT/AKT and p-mTOR/mTOR protein were notably decreased
by MJ-33 treatment in a concentration-dependent manner compared
with the control group (Fig. 7A).
Therefore, the results suggested that the activity of the AKT/mTOR
axis was inhibited by MJ-33 treatment in HT-29/5FUR cells.
According to previous studies, inhibition of mTOR
activity may induce autophagy via upregulating the expression of
the ATG protein family (15,18,35).
To further elucidate the molecular mechanisms underlying
MJ-33-induced autophagy in HT-29/5FUR cells, the expression levels
of autophagy-related proteins, including ATG-5, ATG-7, ATG-12, and
ATG-16, p62 and LC3-II, were examined in MJ-33-treated HT-29/5FUR
cells. The protein expression levels of all ATG proteins and p62
were notably increased, whereas the ratio of LC3/LC3-II was
markedly decreased by MJ-33 treatment in a concentration-dependent
manner compared with the control group (Fig. 7B). The results suggested that
MJ-33-induced autophagy may be triggered at the vesicle nucleation
step, resulting in ATG protein involvement, reductions in the
LC3/LC3-II ratio and increased p62 expression, ultimately
supporting autophagosome formation and the overall progression of
autophagy.
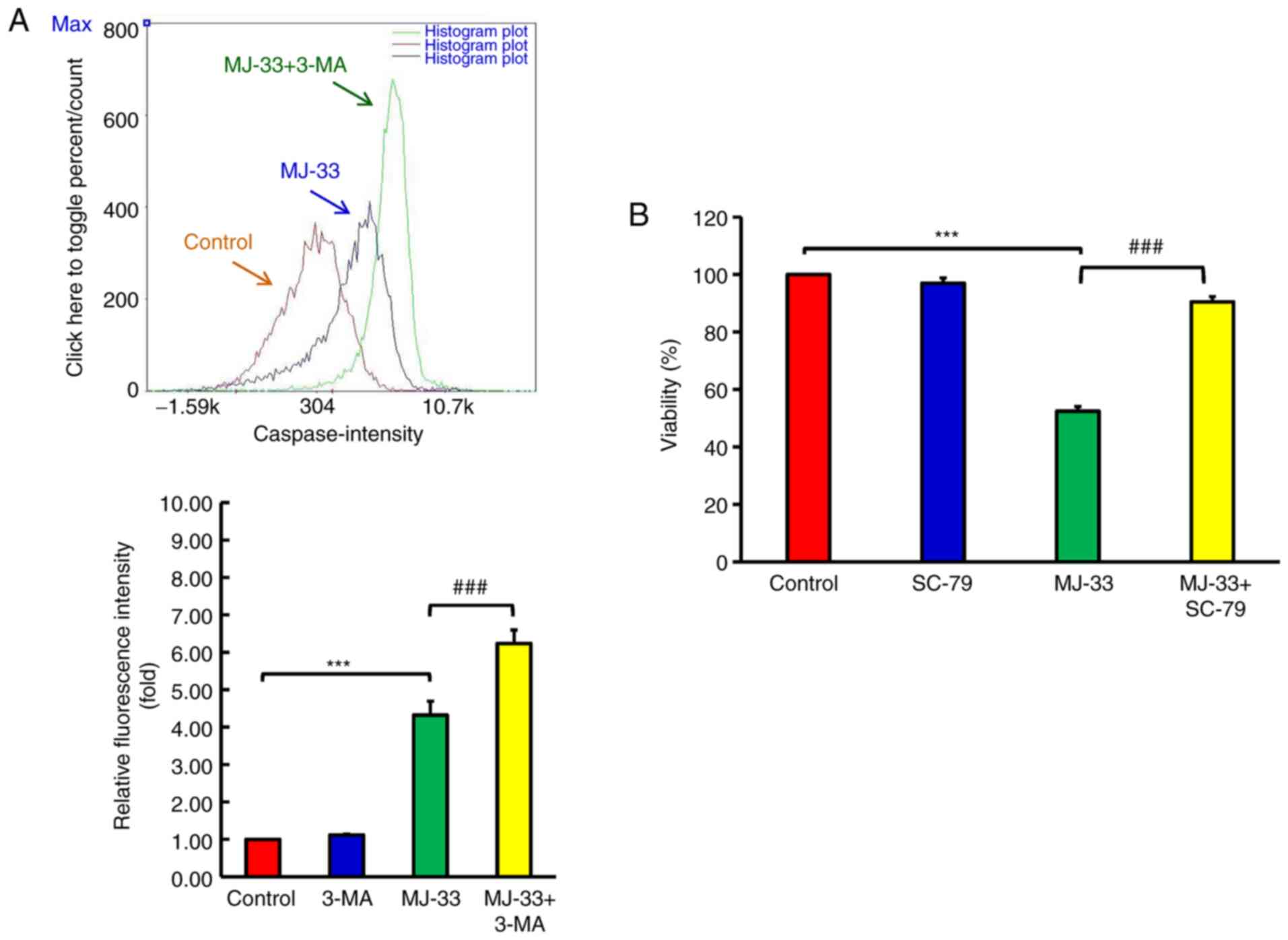
Inhibition of autophagy enhances
MJ-33-induced apoptosis in HT-29/5FUR cells
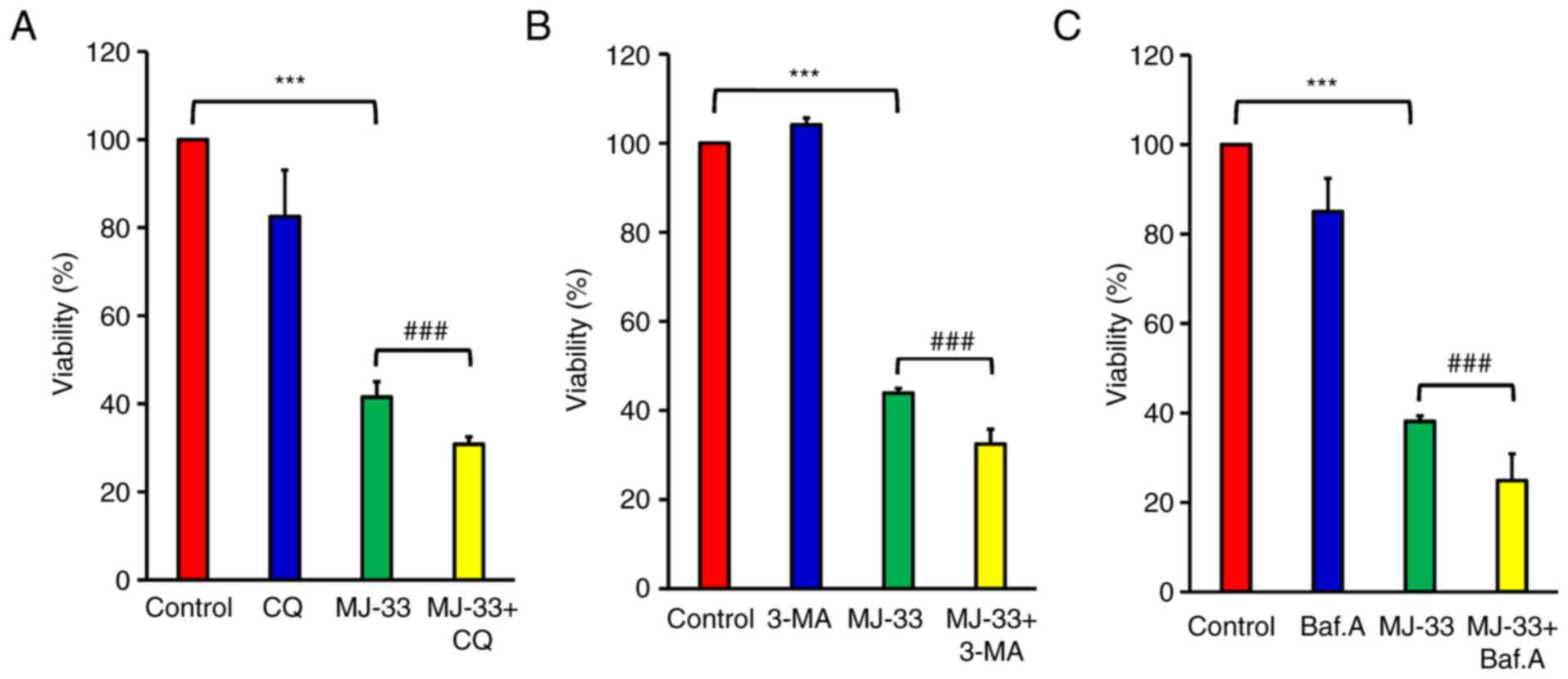
To further examine the contribution of autophagy
signaling to MJ-33-induced cell death, HT-29/5FUR cells were
treated with different autophagy inhibitors, including CQ, 3-MA and
Baf.A1. HT-29/5FUR cells were treated with autophagy inhibitor
and/or MJ-33. Combined treatments significantly decreased cell
viability compared with treatment with MJ-33 alone (Fig. 8A-C). To clarify the effect of
autophagy inhibitors on MJ-33-induced apoptosis, the effects of
MJ-33 and 3-MA treatment on caspase-3 and caspase-7 activities were
assessed by performing FLICAs. MJ-33 treatment alone significantly
elevated caspase-3 and caspase-7 activities compared with the
control group (Fig. 9A). However,
the combination of MJ-33 and 3-MA treatment induced significantly
higher caspase-3 and caspase-7 activities compared with the MJ-33
group. Collectively, the results suggested that the autophagy
mechanism in HT-29/5FUR cells was associated with MJ-33-induced
cell death, indicating that autophagy may serve a cytoprotective
role by inhibiting the apoptosis mechanism.
AKT serves a pivotal role in the
mechanism underlying MJ-33-induced cytotoxicity in HT-29/5FUR
cells
MJ-33 inhibits AKT activity in a
concentration-dependent manner, thus further experiments to
determine the role of AKT in MJ-33-induced cytotoxicity were
performed. Following treatment with MJ-33 and/or SC-79 (AKT
specific activator), HT-29/5FUR cell viability was assessed. SC-79
did not significantly alter cell viability compared with the
control group (Fig. 9B).
Conversely, SC-79 significantly restored HT-29/5FUR cell viability
in MJ-33-treated cells. The results suggested that AKT may serve as
a critical regulator of MJ-33-induced cytotoxicity in HT-29/5FUR
cells.
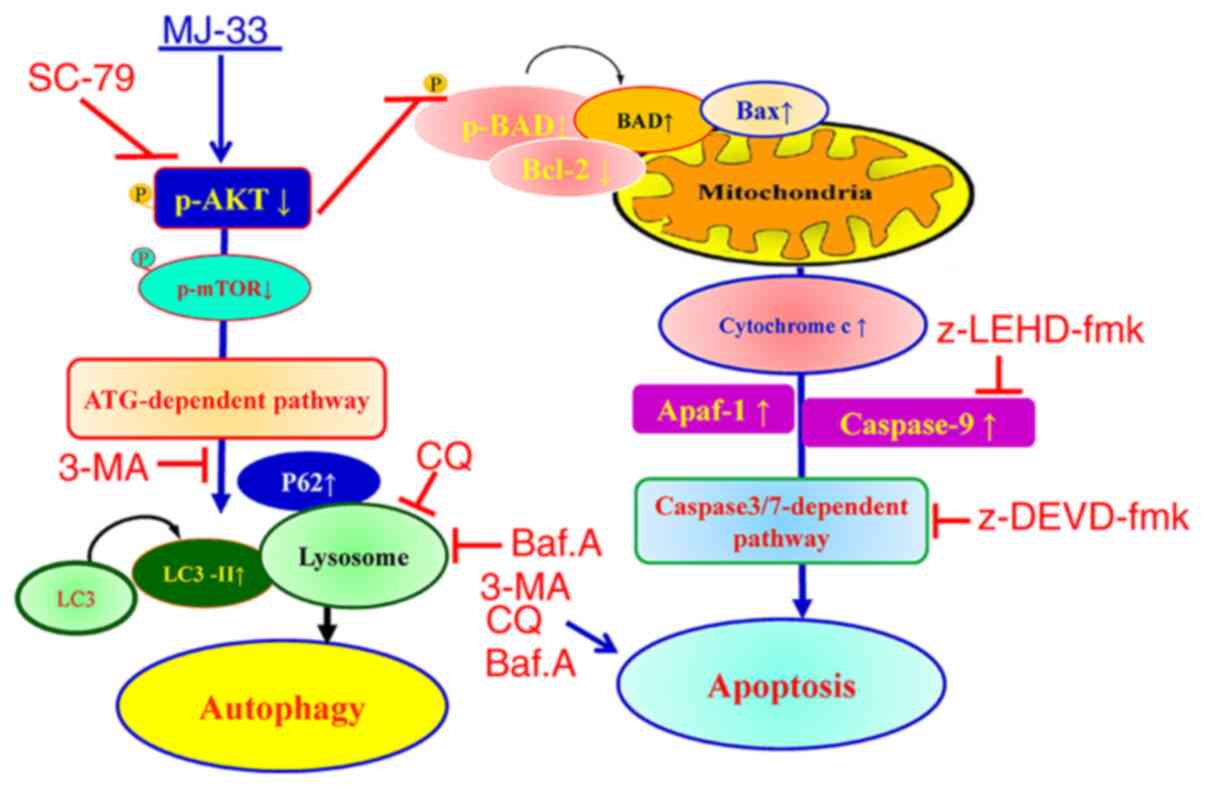
Collectively, the results indicated that MJ-33
selectively induced cytotoxicity in HT-29/5FUR cells via mediating
the AKT/mTOR signaling pathway. The proposed molecular mechanism
underlying MJ-33 in HT-29/5FUR cells is presented in Fig. 10. The results indicated that MJ-33
inhibited AKT activity, which triggered apoptosis via the
caspase-dependent signaling pathway and induced autophagy via the
ATG-dependent pathway.
Discussion
Quinazolinone compounds may possess diverse
bioactivities, including anticancer properties (22,28,36).
The anticancer mechanisms underlying apoptosis induction and
antiproliferative activity have been previously reported (28). However, the autophagy-associated
mechanisms of action of these compounds, particularly in
chemoresistant cancer cells, have not been extensively investigated
(36,37). The present study examined the
cytotoxic effect of MJ-33 on a chemoresistant CRC cell line. The
results indicated that MJ-33-induced cytotoxicity was associated
with apoptosis and autophagy. Since the antimetastatic effects of
MJ-33 were previously reported (14), the findings of the present study
provided further understanding of the effects of MJ-33,
contributing to establishing the therapeutic profile of MJ-33,
which may be useful for further exploration of the anticancer
effects of MJ-33 in 5FU-resistant cancer cells.
Apoptosis is a programmed cell death mechanism that
serves a key role in the inhibition of tumor growth by limiting the
number of cells (33). 5FU is an
apoptosis-inducing agent, which is widely utilized for CRC
treatment, despite its low reported response rates (6–8).
Chemoresistant cancer cells are generally characterized by the
absence of response to treatment, maintaining their ability to
survive and continuous proliferation under chemotherapy treatment
(38). In cancer cells exhibiting
resistance to 5FU, the inactivation of apoptosis was reported in
several studies (39,40). In the present study, the results
suggested that MJ-33 induced cytotoxicity by reactivating the
apoptotic mechanism in HT-29/5FUR cells. DNA condensation and
fragmentation were observed following MJ-33 treatment, suggesting
induction of HT-29/5FUR cell apoptosis. The significantly increased
number of early apoptotic cells (Annexin V positive/PI negative) in
the MJ-33 treatment group compared with the control group further
indicated that MJ-33 induced apoptosis. Moreover, the results
suggested that MJ-33-induced apoptosis was mediated via a
caspase-dependent signaling pathway. Since AKT activity is
responsible for cell survival and apoptosis (15), the effect of MJ-33 on AKT activity
and possible downstream signaling pathways were evaluated in
HT-29/5FUR cells. Compared with the control group, the p-AKT/AKT
ratio was notably decreased following MJ-33 treatment, suggesting
that AKT activity was inhibited via downregulation of AKT
phosphorylation. Moreover, compared with the control group, AKT
activator treatment alone did not enhance cell viability, but in
combination with MJ-33 treatment, AKT activator significantly
restored cell viability, suggesting that MJ-33-induced apoptosis
may be controlled via AKT signaling. In addition, the Bcl-2/BAX and
p-BAD/BAD ratios were markedly decreased in the MJ-33 treatment
group compared with the control group. The Bcl-2/BAX ratio is a
proapoptotic indicator and the activity of BAD is regulated via
phosphorylation, both of which control the process of cell
apoptosis (13,29). Moreover, compared with the control
group, MJ-33 treatment markedly upregulated proapoptotic protein
expression, promoting programmed cell death. Furthermore, the
notably increased expression levels of cytochrome c and
Apaf-1, and markedly decreased expression levels of pro-caspase-9
and pro-caspase-3 in the MJ-33 treatment group compared with the
control group suggested that MJ-33-induced apoptosis was triggered
and controlled via the intrinsic mitochondria-dependent signaling
pathway.
Autophagy is also a mechanism closely associated
with cell death and survival (21).
The interaction between cytotoxicity and autophagy displays dual
roles as dependent on the level of the signal, stimuli or stress
can induce autophagy as an offensive or defensive mechanism
(41,42). The antiproliferative effect
associated with autophagy of several quinazolinone compounds was
previously reported (22,43). In the present study, MJ-33-induced
HT-29/5FUR cell autophagy was detected by observing morphological
alterations via microscopic examination. Furthermore, the formation
of acidic vesicles, increasing volume of autophagic vesicles,
acidic vesicular organelles and increased lysosomal activity
indicated autophagy activation following MJ-33 treatment. Moreover,
the punctate patterns suggested autophagosome maturation in
MJ-33-treated cells compared with control cells, which also
indicated autophagy induction. The ATG12-ATG5 conjugate interacts
with ATG16 to form a complex that catalyzes the expanding
autophagosome membrane, promoting early-stage autophagy (18,41).
The conversion of LC3 to LC3-II via lipidation is required for
autophagosome membrane maturation during the process of autophagy
(44). In addition, the p62 protein
is an autophagy substrate and an indicator for cargo recognition,
serving an important role in delivering ubiquitinated proteins to
the proteasome for degradation (45). In the present study, compared with
the control group, MJ-33 treatment markedly increased p62 protein
expression levels, which indicated induction of later-stage
autophagy via triggering of autophagy-related protein expression.
Collectively, the results indicated that MJ-33-induced HT-29/5FU
autophagy was triggered and processed via inhibiting mTOR
phosphorylation, and subsequently upregulating the expression of
autophagy-related proteins.
Alterations in AKT activity are crucial for cell
survival and strongly regulate apoptosis (15). The effect of MJ-33 on the regulation
of AKT in HT-29/5FUR cells was investigated. The results
demonstrated a markedly reduced p-AKT/AKT ratio in MJ-33-treated
cells compared with control cells, indicating that AKT activity was
inhibited by suppressing its phosphorylation. It was inferred that
MJ-33-induced apoptosis may be controlled via AKT signaling. mTOR
is an important molecule downstream of AKT, which serves critical
roles in autophagy pre-initiation and progression (15). As the main component of the mTOR
complex 1, it inhibits Unc-51 like autophagy activating kinase
(46) and also regulates lysosome
activity by suppressing transcription factor EB (47). Therefore, the present study
investigated the effects of MJ-33 treatment on AKT/mTOR activity in
HT-29/5FUR cells, and the association between the effects and
MJ-33-induced autophagy. Compared with the control group, MJ-33
treatment inhibited the AKT/mTOR axis, thereby promoting autophagy.
In previous studies investigating quinazolinone compounds, the
anticancer effects against CRC cells have been reported via a
variety of mechanisms, including apoptosis, antiangiogenesis and
antimetastasis mechanisms (48,49).
The molecular mechanisms associated with the anticancer properties
of these compounds have been reported, including thymidylate
synthase (TS) inhibition (50) and
PI3K inactivation (48,51). The findings of the present study
were consistent with previously reported results on the inhibitory
effect of MJ-33 on the AKT/mTOR/AMPK axis in DU145 cells (14), which resulted in NF-κB
downregulation. Therefore, exploring the relationships among NF-κB
activity, the mechanism underlying 5FU resistance and the activity
of MJ-33 should be investigated in future studies. Constitutive
nuclear NF-κB activity is highly activated in TS
inhibitor-resistant cells (52),
and inhibiting NF-κB translocation may lower TS levels in CRC
(53), causing higher TS expression
in 5FU-resistant HT-29 cells (54).
Since TS is the primary target of 5FU, the aforementioned studies
also indicated that MJ-33 may serve as a potential therapeutic
agent among quinazolinone class compounds for developing drugs
against 5FU-resistant CRC.
The interplay between autophagy and apoptosis is
complicated, with multiple processes, connections and crosstalk
(20,21). Autophagy typically occurs before
apoptosis in cells, serving an important role in determining cell
survival (20,55). However, depending on the stress
level, autophagy can result in autophagic cell death or a
cytoprotective effect (41,42). In the present study, the association
between MJ-33-induced apoptosis and autophagy was examined in
HT-29/5FUR cells by using different autophagy inhibitors (3-MA,
Baf.A1 and CQ). HT-29/5FUR cells were treated with autophagy
inhibitors and/or MJ-33 to evaluate the association between
MJ-33-induced autophagy and apoptosis. The three autophagy
inhibitors used in the present study interfered with different
stages of the autophagic process. At the early stages, 3-MA
inhibits class-III-PI3K to block the formation of autophagosomes
(56); at later stages, Baf.A1
inhibits vacuolar-type H+-ATPase to prevent fusion of
the lysosome with the autophagosome (57), whereas CQ serves as a lysosomal
deacidification agent (58). Each
autophagy inhibitor combined with MJ-33 treatment significantly
decreased cell viability compared with MJ-33 treatment alone.
Moreover, 3-MA treatment combined with MJ-33 treatment
significantly increased caspase-3 and caspase-7 activities compared
with MJ-33 treatment alone, suggesting a cytoprotective role of the
autophagy mechanism in HT-29/5FUR cells. The results indicated the
protective function of autophagy under these conditions via the
‘recycling’ mechanism, a function of lysosomes to produce amino
acids from dysfunctional cellular components. Moreover, the results
of the present study were consistent with previous studies
reporting that autophagy inhibition may induce apoptosis or enhance
chemotherapy-induced apoptosis via several mechanisms involving
apoptosis-autophagy crosstalk (59–61).
As previously reported, autophagy is a ‘double-edged sword’,
resulting in an offensive or defensive mechanism dependent on the
stress level (41,42). Based on the results of the present
study, it was hypothesized that MJ-33-induced autophagy served a
protective role in HT-29/5FUR cells, protecting against
stress-induced cell death. MJ-33-induced autophagy-dependent cell
death is considered as a result of autophagy-apoptosis interaction,
which was recently categorized as ‘autophagy-associated cell death’
(21). Recent views have emerged,
suggesting an improved cancer treatment strategy by combining
autophagy-inducing anticancer drugs with an autophagy inhibitor
compared with monotherapy (57,58,61,62).
Future studies should investigate the aspects of MJ-33-induced
autophagy, as well as the protective function of MJ-33-induced
autophagy in other types of cancer cells, and MJ-33 combination
strategies.
The present study had a number of limitations. The
proliferative inhibitory concentration of MJ-33 in HT-29/5FUR cells
was considerably high, which might be attributed to the
chemoresistant properties of the cell line, which could be
considered as an immortalized cell line. CRC chemoresistance has
been suggested to occur as a result of alterations in drug targets
(38,52,54);
therefore, higher treatment doses or therapeutic with increased
selectivity are required. The insignificant cytotoxicity of MJ-33
on normal colon cells supports further investigation of the
anti-CRC effect of MJ-33. Future studies should aim to design
suitable combination regimens with possible synergistic effects in
order to lower the effective concentration of MJ-33. In the present
study, the results suggested that the combination of MJ-33 with an
autophagy inhibitor may serve as a useful strategy for improved
cytotoxicity with lower treatment doses.
The proposed molecular mechanism underlying MJ-33 in
HT-29/5FUR cells that was identified in the present study is
presented in Fig. 10. Consistent
with a previous study that investigated the antimetastasis effects
of MJ-33 (14), the interaction of
MJ-33 and the AKT/mTOR pathway was further indicated in the present
study. The results of the present study may provide novel insights
into the cytotoxicity of MJ-33, MJ-33-induced autophagy and
apoptosis, as well as the underlying molecular mechanisms.
Additional effects of MJ-33 on different hallmarks of cancer, such
as angiogenesis, metastasis and molecular targets, require further
investigation.
In conclusion, the present study demonstrated that
MJ-33 treatment significantly inhibited HT-29/5FUR cell viability
compared with the control group in vitro. Moreover, the
results indicated that MJ-33-induced cytotoxicity in HT-29/5FUR
cells was mediated via the inhibitory effect of MJ-33 on the
AKT/mTOR signaling pathway, which subsequently triggered apoptosis
and autophagy. Therefore, MJ-33 may serve as a promising anticancer
drug for 5FU-resistant CRC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Mr Lai-Hsiang Chang
and Mr Chin-Chen Lin (Tekon Scientific Corp.) for providing
assistance and equipment for the present study.
Funding
The present study was supported by the Ministry of
Science and Technology, Taiwan (grant no. MOST 109-2320-B-039-041)
and China Medical University Hospital (grant no. DMR-108-106).
Availability of data and materials
The datasets generated during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
MJH, HAH and JSY contributed to designing the study.
JHC, DTB, YNJ and YHL performed the experiments. FJT and JSY
analyzed the data. HAH, JSY and FJT wrote and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interest.
References
|
1
|
Pan P, Yu J and Wang LS: Colon cancer:
What we eat. Surg Oncol Clin N Am. 27:243–267. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Araghi M, Soerjomataram I, Jenkins M,
Brierley J, Morris E, Bray F and Arnold M: Global trends in
colorectal cancer mortality: Projections to the year 2035. Int J
Cancer. 144:2992–3000. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Y, Chen Z and Li J: The current
status of treatment for colorectal cancer in China: A systematic
review. Medicine (Baltimore). 96:e82422017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van Cutsem E, Cervantes A, Adam R, Sobrero
A, Van Krieken JH, Aderka D, Aranda Aguilar E, Bardelli A, Benson
A, Bodoky G, et al: ESMO consensus guidelines for the management of
patients with metastatic colorectal cancer. Ann Oncol.
27:1386–1422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pardini B, Kumar R, Naccarati A, Novotny
J, Prasad RB, Forsti A, Hemminki K, Vodicka P and Bermejo JL:
5-Fluorouracil-based chemotherapy for colorectal cancer and
MTHFR/MTRR genotypes. Br J Clin Pharmacol. 72:162–163. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Douillard JY, Cunningham D, Roth AD,
Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J,
Alakl M, et al: Irinotecan combined with fluorouracil compared with
fluorouracil alone as first-line treatment for metastatic
colorectal cancer: A multicentre randomised trial. Lancet.
355:1041–1047. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Giacchetti S, Perpoint B, Zidani R, Le
Bail N, Faggiuolo R, Focan C, Chollet P, Llory JF, Letourneau Y,
Coudert B, et al: Phase III multicenter randomized trial of
oxaliplatin added to chronomodulated fluorouracil-leucovorin as
first-line treatment of metastatic colorectal cancer. J Clin Oncol.
18:136–147. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khan M, Saif A, Sandler S and Mirrakhimov
AE: Idelalisib for the treatment of chronic lymphocytic leukemia.
ISRN Oncol. 2014:9318582014.PubMed/NCBI
|
|
10
|
Zhao H, Zhang Y, Sun J, Zhan C and Zhao L:
Raltitrexed inhibits HepG2 cell proliferation via G0/G1 cell cycle
arrest. Oncol Res. 23:237–248. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pivot X, Wadler S, Kelly C, Ruxer R,
Tortochaux J, Stern J, Belpomme D, Humblet Y, Domenge C, Clendeninn
N, et al: Result of two randomized trials comparing nolatrexed
(Thymitaq™) versus methotrexate in patients with recurrent head and
neck cancer. Ann Oncol. 12:1595–1599. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen KT, Hour MJ, Tsai SC, Chung JG, Kuo
SC, Lu CC, Chiu YJ, Chuang YH and Yang JS: The novel synthesized
6-fluoro-(3-fluorophenyl)-4-(3-methoxyanilino)quinazoline (LJJ-10)
compound exhibits anti-metastatic effects in human osteosarcoma U-2
OS cells through targeting insulin-like growth factor-I receptor.
Int J Oncol. 39:611–619. 2011.PubMed/NCBI
|
|
13
|
Yang JS, Hour MJ, Huang WW, Lin KL, Kuo SC
and Chung JG: MJ-29 inhibits tubulin polymerization, induces
mitotic arrest, and triggers apoptosis via cyclin-dependent kinase
1-mediated Bcl-2 phosphorylation in human leukemia U937 cells. J
Pharmacol Exp Ther. 334:477–488. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hour MJ, Tsai SC, Wu HC, Lin MW, Chung JG,
Wu JB, Chiang JH, Tsuzuki M and Yang JS: Antitumor effects of the
novel quinazolinone MJ-33: Inhibition of metastasis through the
MAPK, AKT, NF-κB and AP-1 signaling pathways in DU145 human
prostate cancer cells. Int J Oncol. 41:1513–1519. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
O'Donnell JS, Massi D, Teng MWL and
Mandala M: PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux.
Semin Cancer Biol. 48:91–103. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nitulescu GM, Van De Venter M, Nitulescu
G, Ungurianu A, Juzenas P, Peng Q, Olaru OT, Grădinaru D, Tsatsakis
A, Tsoukalas D, et al: The Akt pathway in oncology therapy and
beyond (Review). Int J Oncol. 53:2319–2331. 2018.PubMed/NCBI
|
|
17
|
West KA, Castillo SS and Dennis PA:
Activation of the PI3K/Akt pathway and chemotherapeutic resistance.
Drug Resist Updat. 5:234–248. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chang CH, Lee CY, Lu CC, Tsai FJ, Hsu YM,
Tsao JW, Juan YN, Chiu HY, Yang JS and Wang CC: Resveratrol-induced
autophagy and apoptosis in cisplatin-resistant human oral cancer
CAR cells: A key role of AMPK and Akt/mTOR signaling. Int J Oncol.
50:873–882. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu J, Zhang S, Wang R, Wu X, Zeng L and Fu
Z: Knockdown of PRDX2 sensitizes colon cancer cells to 5-FU by
suppressing the PI3K/AKT signaling pathway. Biosci Rep.
37:BSR201604472017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marino G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Denton D and Kumar S: Autophagy-dependent
cell death. Cell Death Differ. 26:605–616. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kumar S, Guru SK, Pathania AS, Mupparapu
N, Kumar A, Malik F, Bharate SB, Ahmed QN, Vishwakarma RA and
Bhushan S: A novel quinazolinone derivative induces cytochrome c
interdependent apoptosis and autophagy in human leukemia MOLT-4
cells. Toxicol Rep. 1:1013–1025. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kawai K, Viars C, Arden K, Tarin D,
Urquidi V and Goodison S: Comprehensive karyotyping of the HT-29
colon adenocarcinoma cell line. Genes Chromosomes Cancer. 34:1–8.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sartore-Bianchi A, Martini M, Molinari F,
Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P,
De Dosso S, Mazzucchelli L, et al: PIK3CA mutations in colorectal
cancer are associated with clinical resistance to EGFR-targeted
monoclonal antibodies. Cancer Res. 69:1851–1857. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Plasencia C, Abad A, Martinez-Balibrea E
and Taron M: Antiproliferative effects of ZD0473 (AMD473) in
combination with 5-Fluorouracil or SN38 in human colorectal cancer
cell lines. Invest New Drugs. 22:399–409. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee HP, Chen PC, Wang SW, Fong YC, Tsai
CH, Tsai FJ, Chung JG, Huang CY, Yang JS, Hsu YM, et al: Plumbagin
suppresses endothelial progenitor cell-related angiogenesis in
vitro and in vivo. J Functional Foods. 52:537–544. 2019. View Article : Google Scholar
|
|
27
|
Huang TY, Peng SF, Huang YP, Tsai CH, Tsai
FJ, Huang CY, Tang CH, Yang JS, Hsu YM, Yin MC, et al:
Combinational treatment of all-trans retinoic acid (ATRA) and
bisdemethoxycurcumin (BDMC)-induced apoptosis in liver cancer Hep3B
cells. J Food Biochem. 44:e131222020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lu CC, Yang JS, Chiang JH, Hour MJ, Lin
KL, Lee TH and Chung JG: Cell death caused by quinazolinone HMJ-38
challenge in oral carcinoma CAL 27 cells: Dissections of
endoplasmic reticulum stress, mitochondrial dysfunction and tumor
xenografts. Biochim Biophys Acta. 1840:2310–2320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Beberok A, Wrzesniok D, Rok J, Rzepka Z,
Respondek M and Buszman E: Ciprofloxacin triggers the apoptosis of
human triple-negative breast cancer MDA-MB-231 cells via the
p53/Bax/Bcl-2 signaling pathway. Int J Oncol. 52:1727–1737.
2018.PubMed/NCBI
|
|
30
|
Gelles JD and Chipuk JE: Robust
high-throughput kinetic analysis of apoptosis with real-time
high-content live-cell imaging. Cell Death Dis. 7:e24932016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee CF, Chiang NN, Lu YH, Huang YS, Yang
JS, Tsai SC, Lu CC and Chen FA: Benzyl isothiocyanate (BITC)
triggers mitochondria-mediated apoptotic machinery in human
cisplatin-resistant oral cancer CAR cells. Biomedicine (Taipei).
8:152018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin CC, Chen KB, Tsai CH, Tsai FJ, Huang
CY, Tang CH, Yang JS, Hsu YM, Peng SF and Chung JG: Casticin
inhibits human prostate cancer DU 145 cell migration and invasion
via Ras/Akt/NF-κB signaling pathways. J Food Biochem.
43:e129022019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang JH and Xu M: DNA fragmentation in
apoptosis. Cell Res. 10:205–211. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nitulescu GM, Margina D, Juzenas P, Peng
Q, Olaru OT, Saloustros E, Fenga C, Spandidos DA, Libra M and
Tsatsakis AM: Akt inhibitors in cancer treatment: The long journey
from drug discovery to clinical use (Review). Int J Oncol.
48:869–885. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lu CC, Chiang JH, Tsai FJ, Hsu YM, Juan
YN, Yang JS and Chiu HY: Metformin triggers the intrinsic apoptotic
response in human AGS gastric adenocarcinoma cells by activating
AMPK and suppressing mTOR/AKT signaling. Int J Oncol. 54:1271–1281.
2019.PubMed/NCBI
|
|
36
|
Dohle W, Jourdan FL, Menchon G, Prota AE,
Foster PA, Mannion P, Hamel E, Thomas MP, Kasprzyk PG, Ferrandis E,
et al: Quinazolinone-based anticancer agents: Synthesis,
antiproliferative SAR, antitubulin activity, and tubulin co-crystal
structure. J Med Chem. 61:1031–1044. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rahman MU, Jeyabalan G, Saraswat P,
Parveen G, Khan S and Yar MS: Quinazolines and anticancer activity:
A current perspectives. Synthetic Communications. 47:379–408. 2017.
View Article : Google Scholar
|
|
38
|
Housman G, Byler S, Heerboth S, Lapinska
K, Longacre M, Snyder N and Sarkar S: Drug resistance in cancer: An
overview. Cancers (Basel). 6:1769–1792. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
He L, Zhu H, Zhou S, Wu T, Wu H, Yang H,
Mao H, SekharKathera C, Janardhan A, Edick AM, et al: Wnt pathway
is involved in 5-FU drug resistance of colorectal cancer cells. Exp
Mol Med. 50:1012018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Maiuthed A, Ninsontia C, Erlenbach-Wuensch
K, Ndreshkjana B, Muenzner JK, Caliskan A, Husayn AP, Chaotham C,
Hartmann A, Roehe AV, et al: Cytoplasmic p21 mediates
5-fluorouracil resistance by inhibiting pro-apoptotic Chk2. Cancers
(Basel). 10:3732018. View Article : Google Scholar
|
|
41
|
Ravanan P, Srikumar IF and Talwar P:
Autophagy: The spotlight for cellular stress responses. Life Sci.
188:53–67. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sever ON and Demir OG: Autophagy: Cell
death or survive mechanism. J Oncol Sci. 3:37–44. 2017. View Article : Google Scholar
|
|
43
|
Xia X, Wang L, Zhang X, Wang S, Lei L,
Cheng L, Xu Y, Sun Y, Hang B, Zhang G, et al: Halofuginone-induced
autophagy suppresses the migration and invasion of MCF-7 cells via
regulation of STMN1 and p53. J Cell Biochem. 119:4009–4020. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu
HL, Yang C and Liu HF: p62 links the autophagy pathway and the
ubiqutin-proteasome system upon ubiquitinated protein degradation.
Cell Mol Biol Lett. 21:292016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang JS, Lu CC, Kuo SC, Hsu YM, Tsai SC,
Chen SY, Chen YT, Lin YJ, Huang YC, Chen CJ, et al: Autophagy and
its link to type II diabetes mellitus. Biomedicine (Taipei).
7:82017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhao E and Czaja MJ: Transcription factor
EB: A central regulator of both the autophagosome and lysosome.
Hepatology. 55:1632–1634. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hussain A, Qazi AK, Mupparapu N, Guru SK,
Kumar A, Sharma PR, Singh SK, Singh P, Dar MJ, Bharate SB, et al:
Modulation of glycolysis and lipogenesis by novel PI3K selective
molecule represses tumor angiogenesis and decreases colorectal
cancer growth. Cancer Lett. 374:250–260. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jackman AL, Kimbell R, Brown M, Brunton L,
Harrap KR, Wardleworth JM and Boyle FT: The antitumour activity of
ZD9331, a non-polyglutamatable quinazoline thymidylate synthase
inhibitor. Adv Exp Med Biol. 370:185–188. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Benepal TS and Judson I: ZD9331: Discovery
to clinical development. Anticancer Drugs. 16:1–9. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hussain A, Qazi AK, Mupparapu N, Kumar A,
Mintoo MJ, Mahajan G, Sharma PR, Singh SK, Bharate SB, Zargar MA,
et al: A novel PI3K axis selective molecule exhibits potent tumor
inhibition in colorectal carcinogenesis. Mol Carcinog.
55:2135–2155. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang W and Cassidy J: Constitutive nuclear
factor-kappa B mRNA, protein overexpression and enhanced
DNA-binding activity in thymidylate synthase inhibitor-resistant
tumour cells. Br J Cancer. 88:624–629. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rajitha B, Belalcazar A, Nagaraju GP,
Shaib WL, Snyder JP, Shoji M, Pattnaik S, Alam A and El-Rayes BF:
Inhibition of NF-κB translocation by curcumin analogs induces G0/G1
arrest and downregulates thymidylate synthase in colorectal cancer.
Cancer Lett. 373:227–233. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Qiu T, Xiang X, Lei W, Hao C, Zhang L,
Feng M, Yu F, Li J and Xiong J: Establishment and biological
characterization of 5-fluorouracil-resistant human colon cancer
HT-29/5-FU cell line. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi.
31:328–332. 2015.(In Chinese). PubMed/NCBI
|
|
55
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–1906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ito S, Koshikawa N, Mochizuki S and
Takenaga K: 3-Methyladenine suppresses cell migration and invasion
of HT1080 fibrosarcoma cells through inhibiting phosphoinositide
3-kinases independently of autophagy inhibition. Int J Oncol.
31:261–268. 2007.PubMed/NCBI
|
|
57
|
Kawaguchi T, Miyazawa K, Moriya S, Ohtomo
T, Che XF, Naito M, Itoh M and Tomoda A: Combined treatment with
bortezomib plus bafilomycin A1 enhances the cytocidal effect and
induces endoplasmic reticulum stress in U266 myeloma cells:
Crosstalk among proteasome, autophagy-lysosome and ER stress. Int J
Oncol. 38:643–654. 2011.PubMed/NCBI
|
|
58
|
Liu X, Sun K, Wang H and Dai Y: Inhibition
of autophagy by chloroquine enhances the antitumor efficacy of
sorafenib in glioblastoma. Cell Mol Neurobiol. 36:1197–1208. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zheng X, Jin X, Li F, Liu X, Liu Y, Ye F,
Li P, Zhao T and Li Q: Inhibiting autophagy with chloroquine
enhances the anti-tumor effect of high-LET carbon ions via ER
stress-related apoptosis. Med Oncol. 34:252017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Masud Alam M, Kariya R, Kawaguchi A,
Matsuda K, Kudo E and Okada S: Inhibition of autophagy by
chloroquine induces apoptosis in primary effusion lymphoma in vitro
and in vivo through induction of endoplasmic reticulum stress.
Apoptosis. 21:1191–1201. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li J, Hou N, Faried A, Tsutsumi S,
Takeuchi T and Kuwano H: Inhibition of autophagy by 3-MA enhances
the effect of 5-FU-induced apoptosis in colon cancer cells. Ann
Surg Oncol. 16:761–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu T, Zhang J, Li K, Deng L and Wang H:
Combination of an autophagy inducer and an autophagy inhibitor: A
smarter strategy emerging in cancer therapy. Front Pharmacol.
11:4082020. View Article : Google Scholar : PubMed/NCBI
|