Introduction
The endoplasmic reticulum (ER) is an organelle that
receives various emergency stimuli and conducts diastolic signals.
When the function of the ER is impaired due to various factors, ER
stress becomes manifest, as the accumulation of misfolded and
unfolded proteins within the ER chamber, as well as the disturbance
of intracellular Ca2+ balance (1). Microenvironmental changes of
neoplastic cells, such as those due to glucose-depletion, hypoxia
or anti-tumor drugs, can induce ER stress, resulting in misfolded
and unfolded protein aggregation within the cavities of the ER and
intracellular imbalance of substances, which can then activate the
unfolded protein response (UPR) to correct misfolded proteins and
relieve stress. The subsequent elevated physiological demand for
protein folding can result in the accumulation of misfolded
proteins within the lumen of the ER's, which is termed ER stress
(2). Activation of the universal
periodic review triggers two temporarily different cellular events
to mitigate protein misfolding: i) An initial response to reduce
protein synthesis and boost the degradation of misfolded proteins;
and ii) a second transcriptional upregulation of hundreds of target
genes involved in the homeostasis of global proteins (3). Under normal physiological conditions,
the protein-folding machinery in the ER is capable of secretory
pathway requirements. However, whenever the accumulation of
misfolded proteins within the ER exceeds a tolerable threshold, the
local sensors in the ER trigger an UPR to transcriptionally and
translationally optimize its protein-folding capacity. Should these
corrective mechanisms prove to be insufficient, the cell will
undergo apoptosis (4).
Apoptosis is closely associated with the development
of neoplasms and is induced by two main pathways: The internal and
external pathways. The internal (endogenous) pathway is also known
as mitochondria-mediated apoptosis, which is mediated by cytokines
releasing and activating caspase-9 and caspase-3 (5). The external (exogenous) pathway refers
to death receptor (DR)-mediated apoptosis, which activates the
FAS-associated death domain (FADD) to form the death-inducing
signaling complex (DISC), resulting in the downstream activation of
caspase-8, −7, −6 and −3 (5,6).
Cancer cells proliferate because of their ability to evade
programmed cell death, or apoptosis (7).
ER stress is an important factor associated with
tumor growth and invasion (8).
Moreover, previous studies have provided in-depth mechanistic
insights into pathways via which ER stress disrupts the homeostasis
of the ER, which triggers an unresolvable UPR activation, and
impairs cellular metabolism and energetics, causing uncontrolled
cell death (8). With the
continuation or aggravation of ER stress, cancer cells are unable
to re-establish ER homeostasis via UPR, and ER stress thereby acts
as a pro-apoptotic factor (5,9,10).
Certain studies have confirmed that ER stress can also promote the
apoptosis of tumor cells (11–27).
Subsequently, the present study will summarize the effects of ER
stress on tumor cells in recent years, especially the role of ER
stress in tumor apoptosis. ER stress activation may represent a new
strategy for the synthesis and application of future
chemotherapeutic drugs.
UPR: A commonly defined endoplasmic
reticulum stress response
The ER is not only responsible for the synthesis and
folding of up to one third of cellular proteins (12) but is also an important organelle for
phospholipid synthesis and Ca2+ storage (13). Different ER subregions support
intracellular homeostasis and survival pathways (14), especially the smooth ER. The smooth
ER, which is responsible for lipid synthesis and metabolism, as
well as calcium storage, is often modified into specific domains,
including plasma-associated ER and mitochondria-associated membrane
formation, autophagosomes and lipid droplets (15). Under physiological or pathological
stimuli, or the action of certain drugs, the increased demand for
protein folding may result in the accumulation of misfolded
proteins within the ER, which triggers a UPR, expanding the folding
capacity of the ER and reducing its synthesis load (2,16).
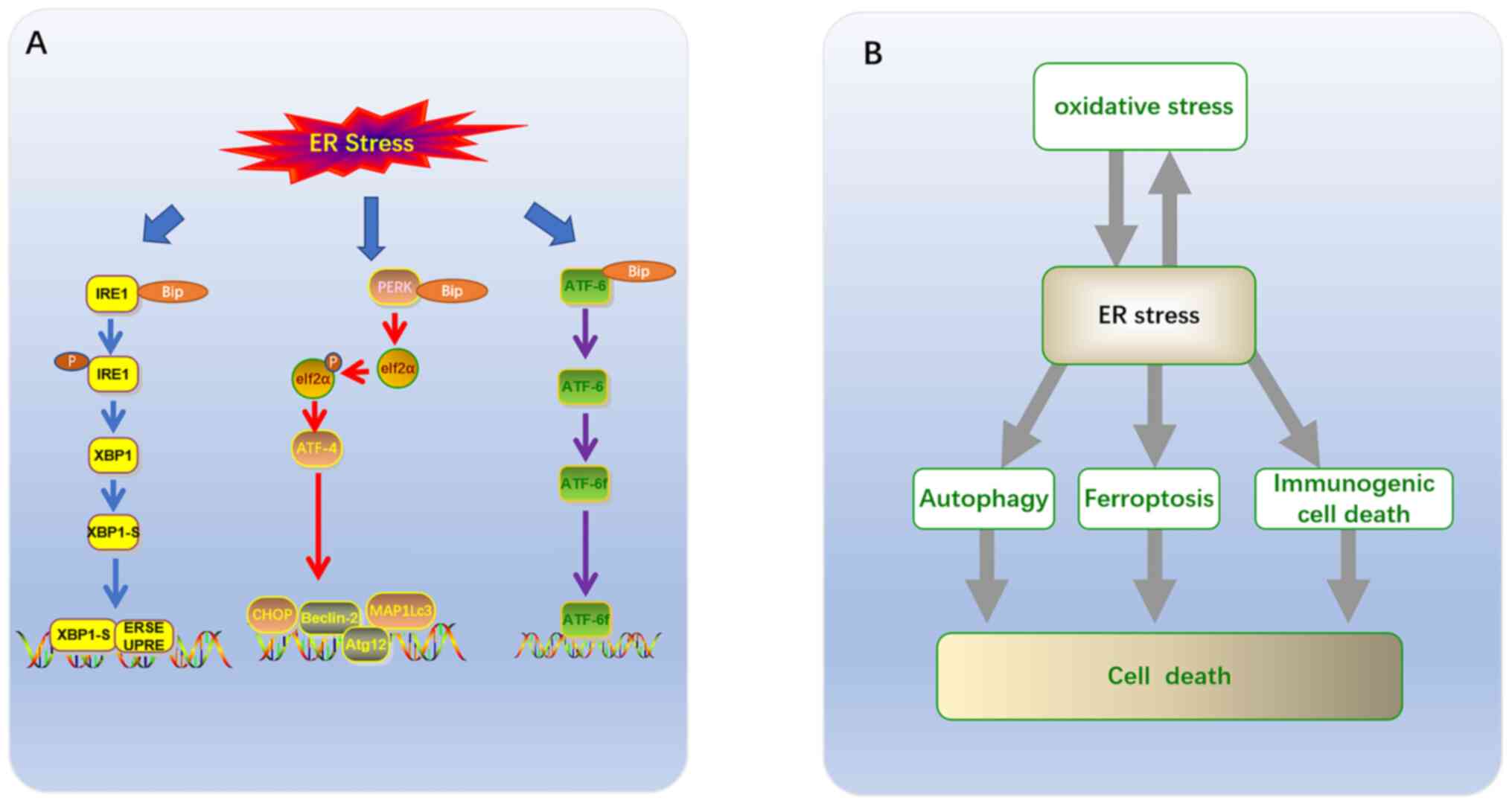
Three branches have been identified based on ER membrane-related ER
stress sensors (12,17). Each branch is defined by a class of
transmembrane ER-resident signaling components, which initiate
stress signaling pathways including: The protein kinase and
ribonuclease, inositol requiring enzyme 1 (IRE1), transcription
factor (TF), activating transcription factor 6 (ATF6), and protein
kinase RNA-like ER kinase (PERK) pathways (Fig. 1A) (18). Glucose-regulated protein 78
(GRP78)/binding immunoglobulin protein (BIP) is a type of
peptide-binding protein, which prevents aggregation and reduces the
rate of protein folding (1,16). GRP78/BIP has been demonstrated to be
a direct ER stress sensor leading to UPR activation (19). Changes in the Ca2+ levels
of the ER, or accumulation of either misfolded or mutant proteins,
can cause GRP78 to become separated from the sensors (IRE1, PERK
and ATF6), thus allowing for downstream signaling activation that
regulates transcription and translation in a manner that restores
homeostasis in the ER (20).
IRE1-XBP1
IRE1 is a transmembrane protein of the ER, which
consists of a serine/threonine kinase domain and an
endoribonuclease (RNase) domain. The mammalian genome encodes two
subtypes of IRE1: IRE1α and IRE1β (21–26).
The IRE1 signaling pathway is the most conservative of the three
UPR signaling pathways, and some studies have reported that the
IRE1 signaling pathway may be heavily involved in tumor cell
proliferation, invasion and migration (22–28).
Since the expression of IRE1α is ubiquitous, and the expression of
IRE1β is limited, most mammalian UPR research focuses on the IRE1α
pathway (23). Studies have
revealed that the IRE1α signaling pathway may serve a key role in
the proliferation, invasion and migration of tumors such as
multiple myeloma (24) and prostate
(25,26), breast (27) and colorectal cancer (28). Under normal physiological
conditions, IRE1α expression is the same as the other two signal
sensors (PERK and ATF-6), with luminal domains which bind chaperone
protein BIP (29). Upon the
occurrence of ER stress, IRE1α is separated from BIP, after which
BIP is transported to the unfolded or misfolded proteins located
within the ER, and IRE1 is activated into phosphorylated IRE1α
(Fig. 1A) (30). Phosphorylation of IRE1α activates
the corresponding RNA molecule, primes the mRNA encoding X
box-binding protein 1 (XBP1), and lyses it into splintered XBP1-S.
After XBP1-S binds to the UPR element (UPRE) and the ER
stress-response elements I and II (ERSE-I and ERSE-II) outside the
nucleus, it is able to cross the nuclear membrane and enter the
nucleus (31,32). The target gene initiated can
alleviate ER stress by coordinating protein folding, secretion,
ER-associated protein degradation (ERAD), lipid biosynthesis and ER
expansion (32).
PERK-eIF2α
PERK is a type I transmembrane, which oligomerizes
and transautophosphorylates upon BIP dissociation or following
reduced ER membrane fluidity (33).
Similar to IRE1, activated PERK is phosphorylated after separating
from BIP, secondary to the development of ER stress, inhibiting
general protein translation via phosphorylation of the eukaryotic
translation initiator factor-2 (eIF2a) at serine 51, which reduces
protein overload within the ER of a stressed cell (34). The growth-arrest and
DNA-damage-induced transcript 34 (GADD34) bound to the
serine/threonine protein phosphatase 1 (PP1) induce the
dephosphorylation of eIF2α, fine tuning mRNA translation to restore
general protein synthesis and promote cellular recovery from stress
(34). This event results in the
enhanced translation of cellular apoptotic inhibitors and ATF4
(Fig. 1A) (32). Furthermore, global translational
inhibition increases the selective translation of the mRNA sequence
encoding the transcription factor ATF4, which directly regulates
the expression of the transcription factor C/EBP-homologous protein
(CHOP). These two mechanisms work synergistically to induce the
transcription of multiple genes, predominantly involved in three
physiological activities: Amino acid biosynthesis, amino acid
transport and autophagy of the intracellular recycling system
(35). Among them, ATF4 upregulates
the expression of autophagy-related genes, such as MAP1LC3, Atg12
and Beclin1 (36). CHOP is a
recognized transcription factor that mediates cell death by
exacerbating oxidative stress, when a large number of proteins are
misfolded in the ER. Previous studies have demonstrated that PERK
and IRE1 signaling pathways regulate both cellular survival and
apoptosis, depending on the severity and duration of the ER stress
(24–28,35,37).
ATF6
ATF6 is also a well-researched ER transmembrane
protein that binds BIP. When ER stress is triggered, the activation
of ATF6 is isolated from BIP, in a similar manner to the activation
of PERK and IRE1 (16,29). Nonetheless, the activated ATF6
protein is translocated from the ER to the golgi apparatus in the
form of a cleaved protein domain under the action of regulated
intramembrane proteolysis (RIP), where it is cleaved sequentially
at the transmembrane site by site-1 protease (S1P) and site-2
protease (S2P) (38). This event
mobilizes a 50 kDa amino-terminal cytoplasmic fragment (ATF6f)
(Fig. 1A). The ATF6f transcription
factor is released into the nucleus, before binding the
aforementioned ER stress response elements (38,39).
ATF6 is a type II transmembrane glycoprotein which
has two isoforms in mammals: ATF6a (670 amino acids) and ATF6β (703
amino acids); the biochemical and physiological characteristics of
the former are significantly better documented than those of the
latter (39,40). ATF6α exhibits crosstalk with XBP1s
by forming heterodimers, which may drive specific gene expression
programs to upregulate a subset of XBP1-dependent chaperones,
oxidoreductases, as well as the quality control and degradation
machinery (41). ATF6α may promote
the survival and adaptation of carcinoma cells by regulating a
broad range of genes associated with transformation, such as
ATF6α-Rheb-mTOR signaling (39,42).
In addition, ATF6 protects the β-cells of the pancreas by reducing
the pressure on the ER (43).
Notwithstanding, the overlapping functions of ATF6 are required to
achieve transcriptional regulation of CHOP, which is a
transcription factor leading to apoptosis (40).
ER stress and the tumor cell apoptosis
Chronic ER stress and apoptosis
Several studies on a variety of cancers have
confirmed that ER stress and UPR activation in a stressful
microenvironment are part of a survival strategy in tumor cells
(4,24–28).
Nonetheless, if the adaptive response fails to restore the
protein-folding function of the ER, or if severe and sustained ER
stress occurs, the persistence of the UPR signal results in
apoptosis, this represents an alternate signaling program known as
the ‘terminal UPR’ (2,4). PERK and IRE1α are the two UPR kinases
which are also involved in the occurrence of apoptosis, resulting
in inevitable cell degeneration and death when ER stress cannot be
resolved (Table I) (44). Under continuous and severe ER
stress, IRE1α induces cell apoptosis via two mechanisms: i) IRE1α
recruits tumor necrosis factor α (TNFα) receptor-associated factor
2 (TRAF2) and apoptosis signal regulating kinase 1 (ASK1), then
activates c-Jun N-terminal kinase (JNK) which induces apoptosis
(45,46); and ii) IRE1α splices certain
microRNAs to inhibit the expression of caspase-2, thus inducing
cell apoptosis (47). Notably,
activation of IRE1α-induced JNK is key to the induction of
apoptosis in human pancreatic cells (48,49).
 | Table I.ER stress-related tumor
apoptosis. |
Table I.
ER stress-related tumor
apoptosis.
| ER stress-related
tumor apoptosis | Unfolded protein
response mediator(s) | ER stress-related
signaling pathways | (Refs.) |
|---|
| Chronic ER
Stress | TRAF2, CHOP,
caspase-12 | PERK/ATF4/CHOP and
ATF6/CHOP | (37,53,54)
(37,53,54) |
| Oxidative stress
induced | ATF4 | PERK/ATF4/CHOP | (32,90,91) |
|
| JNK | IRE1/JNK | (32,90,91) |
| Autophagy and
apoptosis | Elf2α | PERK/Elf2α | (32) |
|
| XBP1 | IRE1α/XBP1 | (32) |
|
|
| ATF6/DAPK1 | (32) |
|
|
| ISR | (32) |
|
Ferroptosis-dependent | ATF4 |
PERK/eIF2α/ATF4/CHOP | (67,68) |
During chronic ER stress, continuous activation of
PERK results in the phosphorylation of eIF2α and selective
induction of ATF4, which can enhance the expression of
pro-apoptotic CCAAT/enhancer binding protein (CHOP) (32,37).
Additionally, CHOP can induce apoptosis by downregulating the
anti-apoptotic protein Bcl-2 and upregulating the pro-apoptotic
proteins (BIM and PUMA), at the transcriptional level (50). These pro-apoptotic proteins are
capable of activating the transcription of a set of genes which
results in activation of caspases and eventually cell death
(51,52). As unfolded proteins accumulate,
PERK, IRE1 and GRP78 are dissolved and activated via
autophosphorylation; meanwhile, ATF6 is activated by proteolytic
enzymes in the golgi apparatus (53). Activated ATF6 molecules enhance the
transcriptional activity of caspase-3 by upregulating the
downstream CHOP molecules (54).
ATF6α has been demonstrated to perform key apoptotic functions
during late luteal regression in rats via the ATF6/CHOP and
caspase-12 pathways (55).
Moreover, in tunicamycin-exposed chondrocytes, celastrol prevented
osteoarthritis by inhibiting ER-mediated apoptosis via the
ATF6/CHOP signaling pathway (53).
Furthermore, ER stress-related proteins have been
shown to induce mitochondrial apoptosis in two principal ways. The
exogenous or death receptor pathway is characterized by
homo-oligomerization cleavage and activation of caspase-3, followed
by cleavage of caspase-8. In the mitochondrial pathway, cytochrome
c s released through the mitochondrial outer membrane pores
under the regulation of Bcl-2 family proteins. This activates
caspase-9 in the apoptotic corpuscle, which upregulates the
expression of downstream executioner caspases, such as caspase-3
and caspase-7 (56,57).
ER Stress and oxidative stress-induced
apoptosis
Oxidative stress is a major driver of tumor cell
proliferation, which may induce tumor survival and adaptation. The
production of reactive oxygen species (ROS) can increase the
mutation rate of tumor cells (58–60).
The UPR regulates the level of oxidative stress by reducing global
translation, while conferring cytoprotection against cellular
insults via inhibition of inflammatory and apoptotic signaling
pathways (32). Currently, the
oxidative stress pathway is considered as a balance between cell
survival and apoptosis (59,60).
Therefore, uncontrolled severe oxidative stress also triggers a
series of pro-apoptotic signaling pathways, including ER stress and
mitochondrial dysfunction, which ultimately results in cell
apoptosis (60). The level of
oxidative stress present in tumor cells is typically higher
compared with normal cells (60).
Thus, tumor cells are more likely to produce high levels of ROS,
resulting in significant changes in the cytoskeleton, cell cycle
arrest, DNA damage and apoptosis of tumor cells (61). Sustained production of high levels
of ROS can trigger ER stress by interfering with the folding of
amino acids into proteins, modifying chaperone proteins or ERAD
(62). When intracellular oxidative
stress levels become very high or adaptation is insufficient,
apoptosis is initiated by inducing UPR-related apoptotic molecules
(63).
The UPR is a commonly defined pathway in cells that
regulates oxidative stress in two ways. Firstly, the UPR regulates
oxidative stress by reducing the translation of transcription
factors, thus inhibiting inflammatory and apoptotic signaling
pathways (64). Secondly, if the
stress is severe and prolonged, or if homeostasis cannot be
restored, the sustained UPR will be converted to a terminal UPR,
which results in cell death (65).
Nude mouse tumor models were used to demonstrate that turmeric can
induce the production of ROS by non-small cell lung cancer cells,
activate JNK and the relevant factors of the ER stress pathway
(ATF4, ATF6 and CHOP), block cell proliferation and induce cell
apoptosis (Table I) (63). A study on human truncated APC colon
cancer cells reported that truncated APC selective inhibitor-1 can
induce ER stress-dependent JNK activation accompanied by oxidative
stress, resulting in cell death (66). Besides, ROS may induce the
transcription of ER oxidative reducing factor 1 (ERO1α) via the
ATF4/CHOP axis, leading to cell death due to the resulting high
oxidative environment in the ER (56,67).
Overactivated ERO1α can increase the production of ROS (50,63).
ERO1α can result in inositol-1,4,5-trisphosphate receptor-mediated
Ca2+ leakage in the ER, which activates Ca2+
sensing kinase CaMKII in the cytoplasm. This induces oxidative
stress caused by the NADPH oxidase subunit NOX2 and leads to
PER-dependent CHOP induction as a positive feedback loop in ER
stress (50). Moreover, CaMKII
causes activation of pro-apoptotic pathways, including Fas and
mitochondrial membrane permeability transformation (50).
ER stress induced autophagy and
apoptosis
Activation of the UPR is generally known to induce
protective autophagy to promote cell survival during ER stress
(32). PERK/Elf2α, IRE1α/XBP1,
ATF6/DAPK1 and the integrated stress response (ISR) can all promote
the conversion of LC3I to LC3II and form autophagosomes either
directly or indirectly (Table I)
(32,68). In tumor cells, autophagy is widely
regarded as a self-protective strategy, employed in hypoxic and
nutrient-deficient microenvironments (68). In vitro and in vivo
experiments confirmed that the ER stress mediated by the
Brigetinib-induced autophagy response was simultaneously induced by
the ER-phage as a protective mechanism to relieve excessive ER
stress. Furthermore, the combination of Brigitinib and autophagy
inhibitors was shown to significantly enhance the anti-colorectal
cancer effects of Brigitinib (69).
The researchers established a xenograft model of nude mice using
human CRC DLD-1 cells that were subcutaneously inoculated. The
study confirmed that the combinatorial treatment of Brigatinib with
chloroquine resulted in a a further reduction in xenograft tumor
size, growth rate and weight compared with that of Brigatinib
treatment alone (69).
Consequently, inhibition of autophagy (69) and acute ER stress (52) in tumor cells is currently recognized
as a potential anti-tumor strategy. At present, clinical studies
have proven that inhibitors of ER stress are helpful in the
treatment of pancreatic cancer (70,71).
Nevertheless, it has been confirmed that triggering excessive ER
stress induces autophagy in tumor cells, which ultimately leads to
cell death (72). Accordingly,
through the establishment of animal models of prostate cancer and
in vitro experiments, anacardic acid was demonstrated to
activate DAPK via ER stress, causing an increase in the expression
of AKT and preventing its phosphorylation. This signaling pathway
can upregulate the expression of LC3, Beclin-1 and Atg7 via mTOR,
consequently inducing the expression of the autophagy-related
protein known as Beclin-1, via suppression of mTOR phosphorylation.
This results in the activation of the pro-apoptotic protein named
Bax, which causes in apoptosis (73,74).
ER stress-dependent immunogenic cell
death in tumor cells
In response to stressors such as hypoxia and drugs,
tumor cells become immunogenic, which can result in a specific
cellular immune response (75). The
main anticancer effector cells of the immune system are the
differentiation group (CD8+ T cells), which can
differentiate into cytotoxic T lymphocytes that kill tumor cells;
an event known as tumor immunogenic death (ICD) (76,77).
ICD is associated with a series of characteristic molecular events,
including the translocation of calcium netting protein (CRT) from
the cytoplasm to the cell membrane surface, the release of
extracellular ATP and the secretion of the high mobility group
protein 1. Immunogenic tumor cells can induce ER stress in order to
trigger apoptotic cell death; moreover, it has long been
hypothesized that apoptotic cell death is primarily non-immune
(78). However, ER stress leads to
the release of damage-associated molecular patterns (DAMPs) from
the ER lumen to the plasma membrane, which can act as opsonins to
facilitate the uptake of tumor associated antigens by dendritic
cells (DCs) (76). During cellular
stress (such as ER stress), cells can induce an immunogenic
response via expression of pro-apoptotic DAMPs, including
calreticulin (CALR) exposure to the cell surface, as well as the
release of HMBG1 and ATP. During ER stress, Elf2α is phosphorylated
by the activation of PERK to induce surface exposure of CALR and
heat-shock proteins (HSPs) (79,80).
Surface exposure to CALR is considered a critical marker of the
immunogenicity of apoptosis (76).
Certain specific receptors (CD91, the purinergic receptors P2Y2 and
P2X7, and TLR4) recognize these DAMPs, which facilitates the
phagocytosis of dying cells, attraction of DCs into the tumor bed,
production of IL-1b and tumor cell antigen presentation (79). These cytokines stimulate the
proliferation of CD8+ T cells, allowing the body to
launch an anti-tumor immune response (81). Notably, the binding of DAMPs to
immune cell receptors activates the UPR in immune cells resulting
in an anti-tumor immune response (79).
The purpose of the present study was to explore the
impact of ER-related signals in the pathogenesis and survival of
tumor cells. ER stress is essential for activating intracellular
signaling pathways that regulate ICD (81). Certain chemotherapeutic agents, such
as anthracyclines and oxaliplatin have been demonstrated to induce
CRT exposure pathways, which are activated by pre-apoptotic ER
stress and phosphorylation of eIF2 kinase via PERK (Table I) (76). Subsequently, the anterograde
transport of CRT from the ER to the golgi apparatus and exocytosis
of CRT-containing vesicles results in the transport of CRT to the
plasma membrane surface (76).
Additionally, previous studies have confirmed that ROS production
induces ICD via ER stress (81,82).
In conclusion, further investigation is imperative prior to the
application of ICD induction as a therapeutic strategy against
tumors.
Ferroptosis and ER stress
Ferroptosis is a type of programmed cell death
distinct from apoptosis, pyroptosis and necrosis. It is caused by
failure of the antioxidant defense mechanism involving glutathione
(GSH/GSSG) (83). Cells that
undergo ferroptosis are typically characterized by mitochondrial
atrophy, increased mitochondrial membrane density and reduced
intracellular NADH levels (84–86).
It is generally believed that the accumulation of iron produced by
lipid peroxidation and the subsequent increase in ROS levels are
the key factors triggering ferroptosis (84). There is a large amount of evidence
indicating that ER stress and ROS production typically interact and
interfere with each other. The PERK/eIF2α signaling pathway was
found to regulate the production of ROS during ferroptosis
(83,87). After treatment with GSK414 in a
mouse model of colitis, the number of necrotic cells of colonic
intestinal epithelial cells, the malondialdehyde and iron contents,
and the levels of ferritin light chain (FTL) and ferritin heavy
chain (FTH) proteins were all reduced, suggesting that inhibition
of ER stress resulted downregulation of ferroptosis, which is
consistent with the findings observed from in vitro
experiments (88). This study
revealed that HCoEpiC cells, that were treated with RSL3 (a
canonical inducer of ferroptosis), resulting in apparent necrotic
cell death and ROS accumulation and increased levels of FTL, FTH,
and PTGS2 in the RSL3-treated cells, which implied that ferroptosis
had occurred (88).
Ferroptosis-dependent apoptosis
ATF4 is a basic leucine zipper transcription factor
that regulates the expression levels of several UPR TARGET genes
through PERK-eIF2α-ATF4 (89).
Studies have shown that inhibition of the cystine glutamate
exchange by ferroptotic agents can lead to activation of the ER
stress response. Among them, the ferroptotic agent APT can lead to
upregulation of ATF-4 dependent related genes, such as CHOP
(90,91). CHOP binds to the promoter site of
the pro-apoptotic protein PUMA (p53 upregulating apoptotic
regulator) during ER stress to induce the expression of PUMA
(44,90). Withal, this study only confirmed the
correlation between ferroptosis and ER stress; reporting that
ferroptotic agents induce ER stress and increase the expression of
the pro-apoptotic molecular protein named PUMA through the ER
stress-mediated PERk-eIF2α-ATF4-CHOP pathway, without inducing
apoptosis (Table I) (44).
Ferroptosis-dependent
no-apoptosis
In tumor cells, GSH intake is reduced and the
production of ROS is increased, thus the sensitivity of tumor cells
to ferroptosis is higher than that of normal cells due to p53
mutation and high expression of the RAS-RAF-MEK pathway (88,92).
ER stress has been suggested to contribute to ferroptosis via the
UPR. Ferroptotic agents cause activation of an ER stress response
and upregulation of the glutathione-specific
γ-glutamylcyclotransferase 1 gene via inhibition of the cystine
glutamate exchange. In glioma cells, ER stress has been
demonstrated to attenuate docosahexaenoic acid (DHA)-induced
ferroptosis by the UPR (93).
DHA-induced ER stress activates the PERK/ATF4/HSPA5 pathway to
resist ferroptosis and protect the cells (93). Drug resistance of tumor cells is
related to the activation of ATF4, which participates in the
development of chemical resistance by transcriptional regulation of
membrane transporters and enzymes required for the biosynthesis of
GSH in cancer cells (93).
Furthermore, the activation of PERK-mediated ATF4 has a protective
effect on erastin-induced ferroptosis in pancreatic cancer cells
(93,94). At present, studies have confirmed
that ferroptosis and ER stress can be induced by ferroptotic agents
(such as ART) or cystine deficiency in tumor cells due to abnormal
lipid metabolism (92–94). Notwithstanding, further
investigation is necessary to determine whether an upstream or
downstream correlation exists between ER stress and
ferroptosis.
Conclusion
Despite the fact that inhibitors targeting ER stress
are being considered for clinical adjunctive therapy against
tumors, reactivation of ER stress in tumor cells has become a new
basis for the treatment of neoplasms, owing to deepening
understanding of ER stress. ER stress with concurrent ferroptosis,
ER stress leading to autophagy, as well as the synergistic effects
of oxidative and ER stress can alter the homeostasis of tumor
cells, which induces apoptosis (Table
I). Numerous studies have published results supporting the
hypothesis that ER stress promotes ferroptosis and ICD in tumor
cells. Moreover, there is also evidence that ER stress is
associated with other cellular death pathways caused by ICD and
ferroptosis, which may represent an attractive field for the
development of effective pharmacotherapies in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shandong Key
Research and Development Program Project (grant no.
2018GSF118124).
Availability of data and materials
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
Authors' contributions
XF and JC wrote the manuscript, organized materials
and provided concepts for the study. XM, PJ, QZ and WZ were
involved in the conception of the study. XC designed the study and
revised the manuscript. All authors have read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sitia R and Braakman I: Quality control in
the endoplasmic reticulum protein factory. Nature. 426:891–894.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lu M, Lawrence DA, Marsters S,
Acosta-Alvear D, Kimmig P, Mendez AS, Paton AW, Paton JC, Walter P
and Ashkenazi A: Opposing unfolded-protein-response signals
converge on death receptor 5 to control apoptosis. Science.
345:98–101. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hetz C and Papa FR: The unfolded protein
response and cell fate control. Mol Cell. 69:169–181. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang M and Kaufman RJ: The impact of the
endoplasmic reticulum protein-folding environment on cancer
development. Nat Rev Cancer. 14:581–597. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim C and Kim B: Anti-cancer natural
products and their bioactive compounds inducing ER stress-mediated
apoptosis: A review. Nutrients. 10:10212018. View Article : Google Scholar
|
|
6
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reed JC: Dysregulation of apoptosis in
cancer. J Clin Oncol. 17:2941–2953. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schleicher SM, Moretti L, Varki V and Lu
B: Progress in the unraveling of the endoplasmic reticulum
stress/autophagy pathway and cancer: Implications for future
therapeutic approaches. Drug Resist Updat. 13:79–86. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ferri KF and Kroemer G: Organelle-specific
initiation of cell death pathways. Nat Cell Biol. 3:E255–E263.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yadav RK, Chae SW, Kim HR and Chae HJ:
Endoplasmic reticulum stress and cancer. J Cancer Prev. 19:75–88.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park IJ, Kim MJ, Park OJ, Choe W, Kang I,
Kim SS and Ha J: Cryptotanshinone induces ER stress-mediated
apoptosis in HepG2 and MCF7 cells. Apoptosis. 17:248–257. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pastor-Cantizano N, Ko DK, Angelos E, Pu Y
and Brandizzi F: Functional diversification of ER stress responses
in Arabidopsis. Trends Biochem Sci. 45:123–136. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anelli T and Sitia R: Protein quality
control in the early secretory pathway. EMBO J. 27:315–327. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lynes EM and Simmen T: Urban planning of
the endoplasmic reticulum (ER): How diverse mechanisms segregate
the many functions of the ER. Biochim Biophys Acta. 1813:1893–1905.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
English AR, Zurek N and Voeltz GK:
Peripheral ER structure and function. Curr Opin Cell Biol.
21:596–602. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang M and Kaufman RJ: Protein misfolding
in the endoplasmic reticulum as a conduit to human disease. Nature.
529:326–335. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hayashi T, Rizzuto R, Hajnoczky G and Su
TP: MAM: More than just a housekeeper. Trends Cell Biol. 19:81–88.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Adams CJ, Kopp MC, Larburu N, Nowak PR and
Ali MMU: Structure and molecular mechanism of ER stress signaling
by the unfolded protein response signal activator IRE1. Front Mol
Biosci. 6:112019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hollien J and Weissman JS: Decay of
endoplasmic reticulum-localized mRNAs during the unfolded protein
response. Science. 313:104–107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin JH, Li H, Yasumura D, Cohen HR, Zhang
C, Panning B, Shokat KM, Lavail MM and Walter P: IRE1 signaling
affects cell fate during the unfolded protein response. Science.
318:944–949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim H, Bhattacharya A and Qi L:
Endoplasmic reticulum quality control in cancer: Friend or foe.
Semin Cancer Biol. 33:25–33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen Y and Brandizzi F: IRE1: ER stress
sensor and cell fate executor. Trends Cell Biol. 23:547–555. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cross BC, Bond PJ, Sadowski PG, Jha BK,
Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D
and Harding HP: The molecular basis for selective inhibition of
unconventional mRNA splicing by an IRE1-binding small molecule.
Proc Natl Acad Sci USA. 109:E869–E878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu J, Xiao M, Li J, Wang D, He Y, He J,
Gao F, Mai L, Li Y, Liang Y, et al: Activation of UPR signaling
pathway is associated with the malignant progression and poor
prognosis in prostate cancer. Prostate. 77:274–281. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sheng X, Nenseth HZ, Qu S, Kuzu OF,
Frahnow T, Simon L, Greene S, Zeng Q, Fazli L, Rennie PS, et al:
IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC
signaling. Nat Commun. 10:3232019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rajapaksa G, Nikolos F, Bado I, Clarke R,
Gustafsson JÅ and Thomas C: ERβ decreases breast cancer cell
survival by regulating the IRE1/XBP-1 pathway. Oncogene.
34:4130–4141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Niederreiter L, Fritz TM, Adolph TE,
Krismer AM, Offner FA, Tschurtschenthaler M, Flak MB, Hosomi S,
Tomczak MF, Kaneider NC, et al: ER stress transcription factor Xbp1
suppresses intestinal tumorigenesis and directs intestinal stem
cells. J Exp Med. 210:2041–2056. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rutkowski DT and Kaufman RJ: A trip to the
ER: Coping with stress. Trends Cell Biol. 14:20–28. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pincus D, Chevalier MW, Aragón T, Van
Anken E, Vidal SE, El-Samad H and Walter P: BiP binding to the
ER-stress sensor Ire1 tunes the homeostatic behavior of the
unfolded protein response. PLoS Biol. 8:e10004152010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang D, Hou C, Cao Y, Cheng Q, Zhang L, Li
H, Feng L and Shen Y: XBP1 activation enhances MANF expression via
binding to endoplasmic reticulum stress response elements within
MANF promoter region in hepatitis B. Int J Biochem Cell Biol.
99:140–146. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bhardwaj M, Leli NM, Koumenis C and
Amaravadi RK: Regulation of autophagy by canonical and
non-canonical ER stress responses. Semin Cancer Biol. 66:116–128.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Volmer R, Van Der Ploeg K and Ron D:
Membrane lipid saturation activates endoplasmic reticulum unfolded
protein response transducers through their transmembrane domains.
Proc Natl Acad Sci USA. 110:4628–4633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choy MS, Yusoff P, Lee IC, Newton JC, Goh
CW, Page R, Shenolikar S and Peti W: Structural and functional
analysis of the GADD34:PP1 eIF2α phosphatase. Cell Rep.
11:1885–1891. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
B'chir W, Maurin AC, Carraro V, Averous J,
Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P and Bruhat
A: The eIF2α/ATF4 pathway is essential for stress-induced autophagy
gene expression. Nucleic Acids Res. 41:7683–7699. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Luhr M, Torgersen ML, Szalai P, Hashim A,
Brech A, Staerk J and Engedal N: The kinase PERK and the
transcription factor ATF4 play distinct and essential roles in
autophagy resulting from tunicamycin-induced ER stress. J Biol
Chem. 294:8197–8217. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rozpedek W, Pytel D, Mucha B, Leszczynska
H, Diehl JA and Majsterek I: The role of the PERK/eIF2α/ATF4/CHOP
signaling pathway in tumor progression during endoplasmic reticulum
stress. Curr Mol Med. 16:533–544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shen J, Chen X, Hendershot L and Prywes R:
ER stress regulation of ATF6 localization by dissociation of
BiP/GRP78 binding and unmasking of Golgi localization signals. Dev
Cell. 3:99–111. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hillary RF and Fitzgerald U: A lifetime of
stress: ATF6 in development and homeostasis. J Biomed Sci.
25:482018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Correll RN, Grimes KM, Prasad V, Lynch JM,
Khalil H and Molkentin JD: Overlapping and differential functions
of ATF6α versus ATF6β in the mouse heart. Sci Rep. 9:20592019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shoulders MD, Ryno LM, Genereux JC,
Moresco JJ, Tu PG, Wu C, Yates JR III, Su AI, Kelly JW and Wiseman
RL: Stress-independent activation of XBP1s and/or ATF6 reveals
three functionally diverse ER proteostasis environments. Cell Rep.
3:1279–1292. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schewe DM and Aguirre-Ghiso JA:
ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor
cells in vivo. Proc Natl Acad Sci USA. 105:10519–10524. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Usui M, Yamaguchi S, Tanji Y, Tominaga R,
Ishigaki Y, Fukumoto M, Katagiri H, Mori K, Oka Y and Ishihara H:
Atf6α-null mice are glucose intolerant due to pancreatic β-cell
failure on a high-fat diet but partially resistant to diet-induced
insulin resistance. Metabolism. 61:1118–1128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Urra H, Dufey E, Lisbona F, Rojas-Rivera D
and Hetz C: When ER stress reaches a dead end. Biochim Biophys
Acta. 1833:3507–3517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen L, Xu S, Liu L, Wen X, Xu Y, Chen J
and Teng J: Cab45S inhibits the ER stress-induced IRE1-JNK pathway
and apoptosis via GRP78/BiP. Cell Death Dis. 5:e12192014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Upton JP, Wang L, Han D, Wang ES, Huskey
NE, Lim L, Truitt M, Mcmanus MT, Ruggero D, Goga A, et al: IRE1α
cleaves select microRNAs during ER stress to derepress translation
of proapoptotic Caspase-2. Science. 338:818–822. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Brozzi F, Nardelli TR, Lopes M, Millard I,
Barthson J, Igoillo-Esteve M, Grieco FA, Villate O, Oliveira JM,
Casimir M, et al: Cytokines induce endoplasmic reticulum stress in
human, rat and mouse beta cells via different mechanisms.
Diabetologia. 58:2307–2316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Brozzi F, Gerlo S, Grieco FA, Juusola M,
Balhuizen A, Lievens S, Gysemans C, Bugliani M, Mathieu C,
Marchetti P, et al: Ubiquitin D regulates IRE1α/JNK-dependent
apoptosis in pancreatic beta cells. J Biol Chem. 291:12040–12056.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang Z, Zhang L, Zhou L, Lei Y, Zhang Y
and Huang C: Redox signaling and unfolded protein response
coordinate cell fate decisions under ER stress. Redox Biol.
25:1010472019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ma Y and Hendershot LM: Delineation of a
negative feedback regulatory loop that controls protein translation
during endoplasmic reticulum stress. J Biol Chem. 278:34864–34873.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jaud M, Philippe C, Di Bella D, Tang W,
Pyronnet S, Laurell H, Mazzolini L, Rouault-Pierre K and Touriol C:
Translational regulations in response to endoplasmic reticulum
stress in cancers. Cells. 9:5402020. View Article : Google Scholar
|
|
53
|
Liu DD, Zhang BL, Yang JB and Zhou K:
Celastrol ameliorates endoplasmic stress-mediated apoptosis of
osteoarthritis via regulating ATF-6/CHOP signalling pathway. J
Pharm Pharmacol. 72:826–835. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tang YH, Yue ZS, Zheng WJ, Shen HF, Zeng
LR, Hu ZQ and Xiong ZF: 4-Phenylbutyric acid presents therapeutic
effect on osteoarthritis via inhibiting cell apoptosis and
inflammatory response induced by endoplasmic reticulum stress.
Biotechnol Appl Biochem. 65:540–546. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yang Y, Sun M, Shan Y, Zheng X, Ma H, Ma
W, Wang Z, Pei X and Wang Y: Endoplasmic reticulum stress-mediated
apoptotic pathway is involved in corpus luteum regression in rats.
Reprod Sci. 22:572–584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Iurlaro R and Muñoz-Pinedo C: Cell death
induced by endoplasmic reticulum stress. FEBS J. 283:2640–2652.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Masud A, Mohapatra A, Lakhani SA,
Ferrandino A, Hakem R and Flavell RA: Endoplasmic reticulum
stress-induced death of mouse embryonic fibroblasts requires the
intrinsic pathway of apoptosis. J Biol Chem. 282:14132–14139. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Clarke HJ, Chambers JE, Liniker E and
Marciniak SJ: Endoplasmic reticulum stress in malignancy. Cancer
Cell. 25:563–573. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang X, Chen M, Zou P, Kanchana K, Weng
Q, Chen W, Zhong P, Ji J, Zhou H, He L and Liang G: Curcumin analog
WZ35 induced cell death via ROS-dependent ER stress and G2/M cell
cycle arrest in human prostate cancer cells. BMC Cancer.
15:8662015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kong N, Ji X, Wang J, Sun X, Chen G, Fan
T, Liang W, Zhang H, Xie A, Farokhzad OC and Tao W: ROS-Mediated
selective killing effect of black phosphorus: Mechanistic
understanding and its guidance for safe biomedical applications.
Nano Lett. 20:3943–3955. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Geraghty P, Wallace A and D'armiento JM:
Induction of the unfolded protein response by cigarette smoke is
primarily an activating transcription factor 4-C/EBP homologous
protein mediated process. Int J Chron Obstruct Pulmon Dis.
6:309–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ma J, Liu J, Lu C and Cai D: Pachymic acid
induces apoptosis via activating ROS-dependent JNK and ER stress
pathways in lung cancer cells. Cancer Cell Int. 15:782015.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cubillos-Ruiz JR, Bettigole SE and
Glimcher LH: Tumorigenic and immunosuppressive effects of
endoplasmic reticulum stress in cancer. Cell. 168:692–706. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bravo R, Parra V, Gatica D, Rodriguez AE,
Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E,
et al: Endoplasmic reticulum and the unfolded protein response:
Dynamics and metabolic integration. Int Rev Cell Mol Biol.
301:215–290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang L, Kim SB, Luitel K and Shay JW:
Cholesterol depletion by TASIN-1 induces apoptotic cell death
through the ER stress/ROS/JNK signaling in colon cancer cells. Mol
Cancer Ther. 17:943–951. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Rouschop KM, Van den Beucken T, Dubois L,
Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W,
Voncken JW, et al: The unfolded protein response protects human
tumor cells during hypoxia through regulation of the autophagy
genes MAP1LC3B and ATG5. J Clin Invest. 120:127–141. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang Z, Gao W, Zhou L, Chen Y, Qin S,
Zhang L, Liu J, He Y, Lei Y, Chen HN, et al: Repurposing brigatinib
for the treatment of colorectal cancer based on inhibition of
ER-phagy. Theranostics. 9:4878–4892. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wang J, Qi Q, Zhou W, Feng Z, Huang B,
Chen A, Zhang D, Li W, Zhang Q, Jiang Z, et al: Inhibition of
glioma growth by flavokawain B is mediated through endoplasmic
reticulum stress induced autophagy. Autophagy. 14:2007–2022. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Atkins C, Liu Q, Minthorn E, Zhang SY,
Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N, et al:
Characterization of a novel PERK kinase inhibitor with antitumor
and antiangiogenic activity. Cancer Res. 73:1993–2002. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Rah B, Ur Rasool R, Nayak D, Yousuf SK,
Mukherjee D, Kumar LD and Goswami A: PAWR-mediated suppression of
BCL2 promotes switching of 3-azido withaferin A (3-AWA)-induced
autophagy to apoptosis in prostate cancer cells. Autophagy.
11:314–331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Fujiwara N, Usui T, Ohama T and Sato K:
Regulation of beclin 1 protein phosphorylation and autophagy by
protein phosphatase 2A (PP2A) and death-associated protein kinase 3
(DAPK3). J Biol Chem. 291:10858–10866. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tan J, Jiang X, Yin G, He L, Liu J, Long
Z, Jiang Z and Yao K: Anacardic acid induces cell apoptosis of
prostatic cancer through autophagy by ER stress/DAPK3/Akt signaling
pathway. Oncol Rep. 38:1373–1382. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang YJ, Fletcher R, Yu J and Zhang L:
Immunogenic effects of chemotherapy-induced tumor cell death. Genes
Dis. 5:194–203. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zitvogel L, Kepp O, Senovilla L, Menger L,
Chaput N and Kroemer G: Immunogenic tumor cell death for optimal
anticancer therapy: The calreticulin exposure pathway. Clin Cancer
Res. 16:3100–3104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Radogna F and Diederich M: Stress-induced
cellular responses in immunogenic cell death: Implications for
cancer immunotherapy. Biochem Pharmacol. 153:12–23. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Obeid M, Tesniere A, Ghiringhelli F, Fimia
GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T,
Casares N, et al: Calreticulin exposure dictates the immunogenicity
of cancer cell death. Nat Med. 13:54–61. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Obacz J, Avril T, Rubio-Patiño C,
Bossowski JP, Igbaria A, Ricci JE and Chevet E: Regulation of
tumor-stroma interactions by the unfolded protein response. FEBS J.
286:279–296. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Panaretakis T, Kepp O, Brockmeier U,
Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N,
Pierron G, Van Endert P, et al: Mechanisms of pre-apoptotic
calreticulin exposure in immunogenic cell death. EMBO J.
28:578–590. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Li W, Yang J, Luo L, Jiang M, Qin B, Yin
H, Zhu C, Yuan X, Zhang J, Luo Z, et al: Targeting photodynamic and
photothermal therapy to the endoplasmic reticulum enhances
immunogenic cancer cell death. Nat Commun. 10:33492019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Deng H, Zhou Z, Yang W, Lin LS, Wang S,
Niu G, Song J and Chen X: Endoplasmic reticulum targeting to
amplify immunogenic cell death for cancer immunotherapy. Nano Lett.
20:1928–1933. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lee YS, Lee DH, Choudry HA, Bartlett DL
and Lee YJ: Ferroptosis-induced endoplasmic reticulum stress:
Cross-talk between ferroptosis and apoptosis. Mol Cancer Res.
16:1073–1076. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Hassannia B, Vandenabeele P and Vanden
Berghe T: Targeting ferroptosis to iron out cancer. Cancer Cell.
35:830–849. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hartman ML: Non-apoptotic cell death
signaling pathways in melanoma. Int J Mol Sci. 21:29802020.
View Article : Google Scholar
|
|
87
|
Park EJ, Park YJ, Lee SJ, Lee K and Yoon
C: Whole cigarette smoke condensates induce ferroptosis in human
bronchial epithelial cells. Toxicol Lett. 303:55–66. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Xu M, Tao J, Yang Y, Tan S, Liu H, Jiang
J, Zheng F and Wu B: Ferroptosis involves in intestinal epithelial
cell death in ulcerative colitis. Cell Death Dis. 11:862020.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Hong SH, Lee DH, Lee YS, Jo MJ, Jeong YA,
Kwon WT, Choudry HA, Bartlett DL and Lee YJ: Molecular crosstalk
between ferroptosis and apoptosis: Emerging role of ER
stress-induced p53-independent PUMA expression. Oncotarget.
8:115164–115178. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Su N and Kilberg MS: C/EBP homology
protein (CHOP) interacts with activating transcription factor 4
(ATF4) and negatively regulates the stress-dependent induction of
the asparagine synthetase gene. J Biol Chem. 283:35106–35117. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ghosh AP, Klocke BJ, Ballestas ME and Roth
KA: CHOP potentially co-operates with FOXO3a in neuronal cells to
regulate PUMA and BIM expression in response to ER stress. PLoS
One. 7:e395862012. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. 66:89–100. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang
L, Wang D, Xing J, Hou B, Li H, et al: Dihydroartemisinin-induced
unfolded protein response feedback attenuates ferroptosis via
PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res.
38:4022019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of Cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3:e025232014. View Article : Google Scholar : PubMed/NCBI
|