Introduction
Colorectal cancer (CRC; cancer of the colon and
rectum) is a worldwide health concern. CRC is the second and third
most common cancer type in women and men, respectively (1). In 2018, there were more than 1.8 million
newly diagnosed CRC cases and 0.88 million deaths (1). In recent years, the incidence of CRC,
particularly rectal cancer, has demonstrated an increasing trend in
young individuals (2). Early
diagnosis and radical surgery effectively improves the 5-year
survival rate of patients; however, there are still a lack of
efficacious measures for patients at advanced stages of CRC (stages
III and IV) (3).
The activation of oncogenes and inactivation/defect
of tumor-suppressor genes are crucial for the tumorigenesis of CRC.
p53, a transcriptional factor, regulates cell cycle arrest,
apoptosis, DNA repair and tumor angiogenesis by mediating numerous
targeted genes (4,5). Importantly, p53 mutation occurs in
~40–50% of patients with CRC (6) and
is associated with resistance to current treatment regimens and a
poor prognosis. p53-reactivation and induction of massive
apoptosis-1, APR-017 (PRIMA-1), a small compound
(C9H5NO3), was determined to
restore the sequence-specific DNA binding domain of mutant p53 by
forming adducts with thiols for recovering wild-type structure and
function, thereby inducing cell apoptosis and thus selectively
killing cancer cells with mutant p53 (7,8).
PRIMA-1met (APR246) as a methylated analog of PRIMA-1,
is more effective in p53-mutant cells, and has been revealed to
inhibit the growth of cells without mutant p53 (8–12). It was
previously demonstrated that PRIMA-1met suppressed CRC
cell proliferation, migration, invasion and colony formation
independently of p53 status (13).
Additionally, PRIMA-1met induced robust apoptosis in
cells carrying mutant p53 by upregulating proapoptotic Noxa
(13). However, the mechanisms
underlying the cytotoxicity of PRIMA-1met in different
CRC cell lines are yet to be fully elucidated.
Autophagy is a ‘self-eating’ response to
intracellular and environmental stimuli, such as starvation,
nutrient deprivation and energy exhaustion. It occurs via
regulatory factors that consist of autophagy-related gene (ATG)
products produced by normal cells and cancer cells. Once receiving
nutrient or energy signals, regulators of autophagy induce the
process, forming a double-membraned autophagosome that fuses
lysosomes with cargoes, leading to degradation and recycling.
Through this process, intracellular proteins and organelles are
degraded in lysosomes and released into the cytoplasm for
biosynthesis and metabolism recycling (14). Autophagy was originally considered to
be a tumor-suppressive process, as autophagy gene Beclin-1 deletion
occurred in 40–75% of patients with breast, ovarian and prostate
cancer (15,16). A recent study indicated that autophagy
inhibits tumor initiation and progression by suppressing chronic
inflammation, DNA damage and genomic instability in the tumor
environment (17). Thus, key
molecules of autophagic pathways, including mTOR, AMPK, Akt, PI3K
class III, Beclin-1 and p53 have become promising targets for
anticancer studies.
The present study revealed the effect of
PRIMA-1met on autophagy in different CRC cell lines, and
further investigated the mechanisms underlying its inhibitory
effect in cells expressing different types of p53. The results
demonstrated that PRIMA-1met induced autophagy in
different CRC cells irrespective of p53 status by activating the
mTOR/AMPK-ULK1-Vps34 signaling cascade. Furthermore, upregulated
autophagy served a crucial role in the suppressive effect of
PRIMA-1met in cells carrying wild-type p53, which was
associated with Noxa upregulation. The results provide a deeper
understanding of the PRIMA-1met mechanism in patients
with CRC.
Materials and methods
Cell lines and drugs
The CRC cell lines used in the current study that
exhibited wild-type or mutant p53 were obtained from Dr Ting Zhang
(Cancer Institute, The Affiliated Hospital of Jiangnan University,
Wuxi, Jiangsu, China). The following cell lines were used: LOVO
(wild-type p53), RKO (wild-type p53), HCT116 (wild-type p53), DLD-1
(mutant p53-S241F), SW480 (mutant p53-R237H), SW620 (mutant
p53-R237H), HCT15 (mutant p53-P153A) and CaCO2 (mutant p53-E204X).
HCT116 null (p53−/−) cells were provided by Dr Jimmy
Chao (Bioprocessing Technology Institute, Singapore). Cells were
cultured in DMEM (HyClone; Cytiva) except for SW620, which was
cultured in RPMI-1640 medium (HyClone; Cytiva). Each medium was
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin/streptomycin (Invitrogen; Thermo Fisher
Scientific, Inc.) in a humidified incubator with 5% CO2
at 37°C. PRIMA-1met (APR-246; Santa Cruz Biotechnology,
Inc.) was dissolved in DMSO to a concentration of 50 mM.
Additionally, 3-methyladenine (3-MA; Absin Bioscience, Inc.) was
dissolved in PBS to a concentration of 67.04 mM (10 mg/ml; warmed).
Both were subsequently stored at −20°C. Working solutions were
diluted to appropriate concentrations with culture medium and the
same quantity of DMSO was used as a control.
Western blot analysis
CRC cell lines were seeded at a density of
2×106 cells per 100 mm overnight, and treated with DMSO,
PRIMA-1met or 3-MA (800 µM; Data S1). After incubation
at 37°C for 24 h, cells were harvested, lysed and analyzed via
western blotting. The following primary antibodies (all 1:1,000)
were used: β-actin (cat. no. AC026; ABclonal Biotech Co., Ltd.),
LC3 (cat. no. NB100-2220; Novus Biologicals, LLC), Noxa (cat. no.
OP180; EMD Millipore), p62 (cat. no. 8025; Cell Signaling
Technology, Inc.), Unc-51 like autophagy activating kinase 1 (ULK1;
cat. no. 8054; Cell Signaling Technology, Inc.), phospho-ULK1
(Ser757; cat. no. 14202; Cell Signaling Technology, Inc.), mTOR
(cat. no. 2983; Cell Signaling technology, Inc.), phospho-mTOR
(Ser2448; cat. no. 5536; Cell Signaling Technology, Inc.), AMPK
(cat. no. 2532; Cell Signaling Technology, Inc.), phospho-AMPK
(Thr172; cat. no. 2535; Cell Signaling Technology, Inc.),
phospho-PI3K Class III (Ser249; cat. no. 13857; Cell Signaling
Technology, Inc.) and PI3K Class III (cat. no. CY5322; Abways
Technology, Inc). Goat-anti-rabbit IgG HRP (cat. no. AS014;
1:5,000; ABclonal Biotech Co., Ltd.), and goat-anti-mouse IgG HRP
(cat. no. AS003; 1:5,000; ABclonal Biotech Co., Ltd.) secondary
antibodies were also used. The expression of protein was estimated
using the gray value, which was calculated using ImageJ version
1.8.0 software (National Institutes of Health). Data are presented
as a percentage normalized to β-actin.
Acridine orange (AO) staining
CRC cells with differing p53 statuses
(HCT116wt, RKO, CaCO2, SW480 and HCT116ko)
were seeded in 24-well plates at a density of 6×104
cells per well with 1 ml medium overnight. Samples were then
treated with DMSO or 30 µM PRIMA-1met for 24 h. AO
(Absin Bioscience, Inc.) solution was added to each well at a
concentration of 1 µg/ml, after which samples were incubated at
37°C for 15 min. After rinsing with PBS twice, slides were removed
and inverted onto carry sheet glass. Cell slides were then observed
and imaged under a confocal microscope (Hitachi, Ltd.). The filter
was excited to 488 nm and blocked at 515 nm.
Transmission electron microscopy
(TEM)
RKO and HCT15 cells were seeded overnight in 100-mm
dishes at a density of 2×106 cells per dish.
Subsequently, cells were treated with DMSO, 30 µM
PRIMA-1met or 60 µM PRIMA-1met and incubated
for 24 h. Cell pellets were harvested and fixed with 4%
glutaraldehyde solution at 4°C for 4 h, followed by fixation with
1% osmic acid for 2 h at 20°C. Specimens were dehydrated by 50–100%
ethanol or 100% acetone, and embedded with Epon 812. After slicing
to 60–80 nm sections, slides were stained with 2% uranyl acetate
and lead citrate to observe autophagic vesicles (AVs) with a double
membrane and autolysosomes under a TEM (Hitachi, Ltd.).
Cell Counting Kit-8 (CCK-8)
proliferation assay
After seeding cells in a 96-well plate at a density
of 7,000 cells/well with 100 µl medium for 24 h, cells were treated
with DMSO, PRIMA-1met (45 µM for HCT116wt, 40
µM for RKO and 55 µM for DLD-1 and HCT15), 3-MA (800 µM; Data S1)
alone or in combination for 48 h. A CCK-8 assay (Dojindo Molecular
Technologies, Inc.) was performed to measure cell proliferation by
quantifying WST-8 formazan dye, which is proportional to the number
of living cells according to previous research (18). Samples were calculated relative to
DMSO, which was set to 100%. Each cell line was assessed in three
independent experiments.
Colony formation assay
After seeding in a 6-well plate (6×105
cells/well) and incubating at 37°C overnight, HCT116wt
and DLD-1 cells were treated with DMSO, PRIMA-1met (50
µM for HCT116wt, 80 µM for DLD-1) and 3-MA (800 µM; Data
S1) for 48 h. Subsequently, cells were harvested and seeded a
second time into 6-well plates (1,000 cells per 2 ml medium per
well). Each sample was prepared in duplicate. After culturing at
37°C in a humidified incubator for 10 days, samples were fixed at
room temperature using 4% paraformaldehyde for 15 min and stained
with 0.5% crystal violet for 15 min. After rinsing three times,
colonies were observed and analyzed using ImageJ version 1.8.0
software (National Institutes of Health), in which colony size was
set to a value of 50 for counting.
Small interfering (si)RNA
transfection
DLD-1 and HCT116wt cells were seeded in
6-well plates (6×105 cells with 2 ml medium per well)
and cultured at 37°C for 24 h. A mixture containing 110 pmol Noxa
siRNA (cat. no. sc-37305; Santa Cruz Biotechnology, Inc.) or
control siRNA (cat. no. sc-36869; Santa Cruz Biotechnology, Inc.),
200 µl jetPRIME® buffer (Polyplus-transfection SA) and 4
µl jetPRIME® siRNA transfection reagent
(Polyplus-transfection SA) was vortexed for 10 sec, spun down and
incubated for 10 min at room temperature. The mixture was
subsequently added to each well of the 6-well plate and incubated
at 37°C for 24 h. Cells were then harvested for further
experiments.
Statistical analysis
Statistical analysis was performed using SPSS
version 19.0 (IBM Corp.) and data are presented as the mean + SEM.
Comparisons between groups were calculated using one-way ANOVA
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
PRIAM-1met induces
autophagy flux in different CRC cell lines
To investigate the effect of PRIMA-1met
on autophagy flux, different CRC cell lines were exposed to
PRIMA-1met and assessed via western blotting. Following
treatment for 24 h, the results demonstrated an increased
expression of LC3-II in two lines carrying wild-type p53, three of
the four p53-mutant cell lines (excluding DLD-1) and 1 p53-deleted
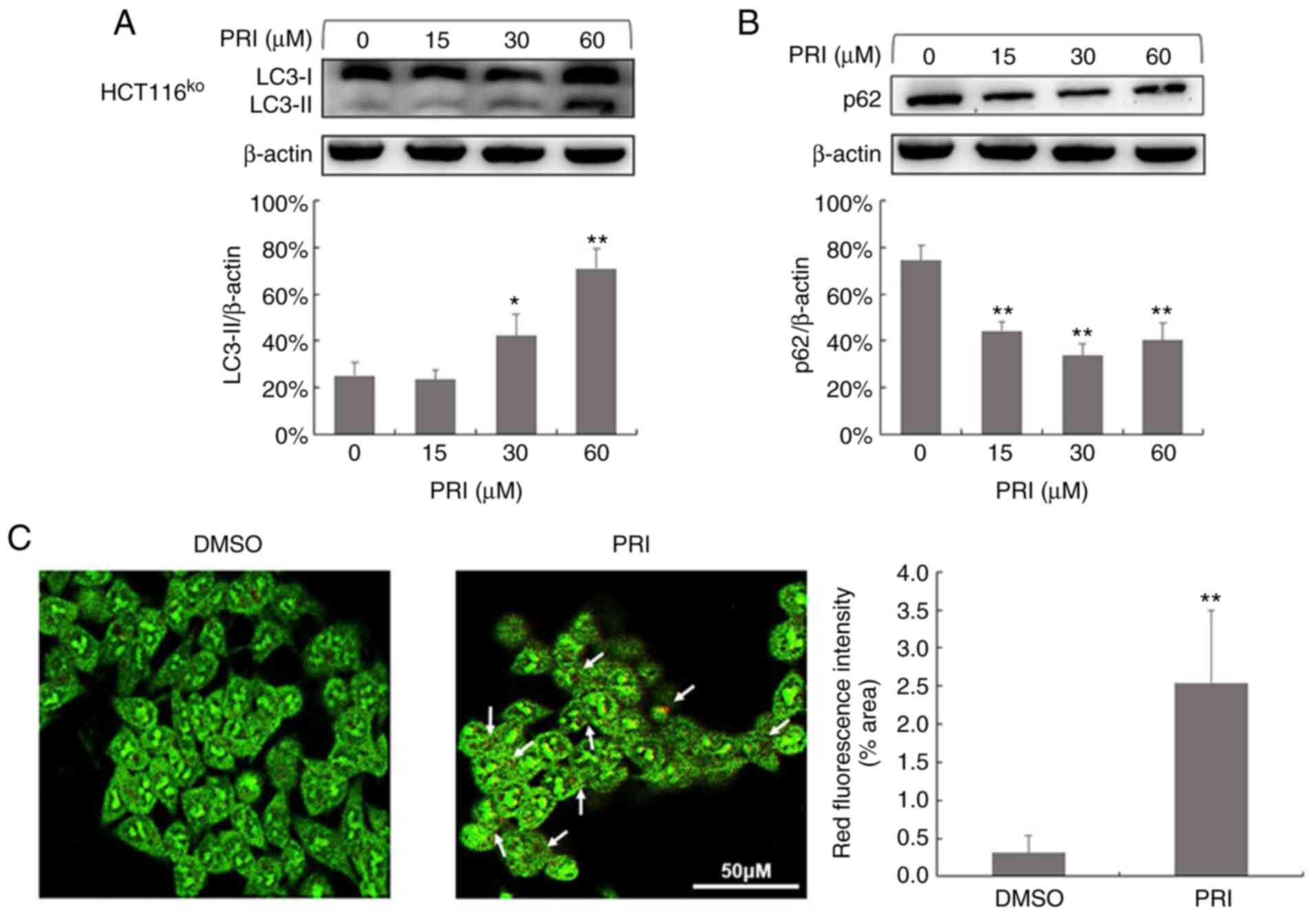
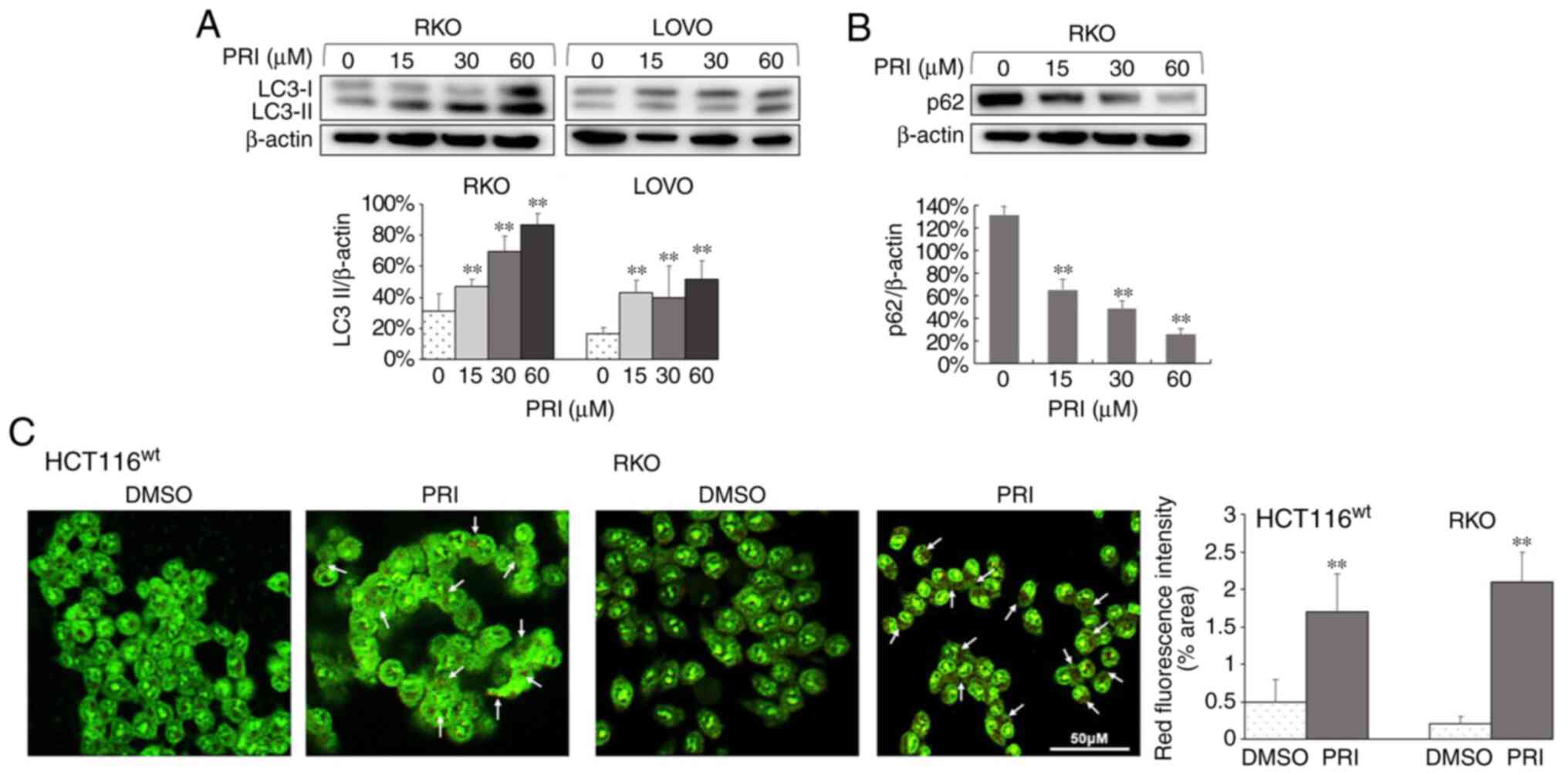
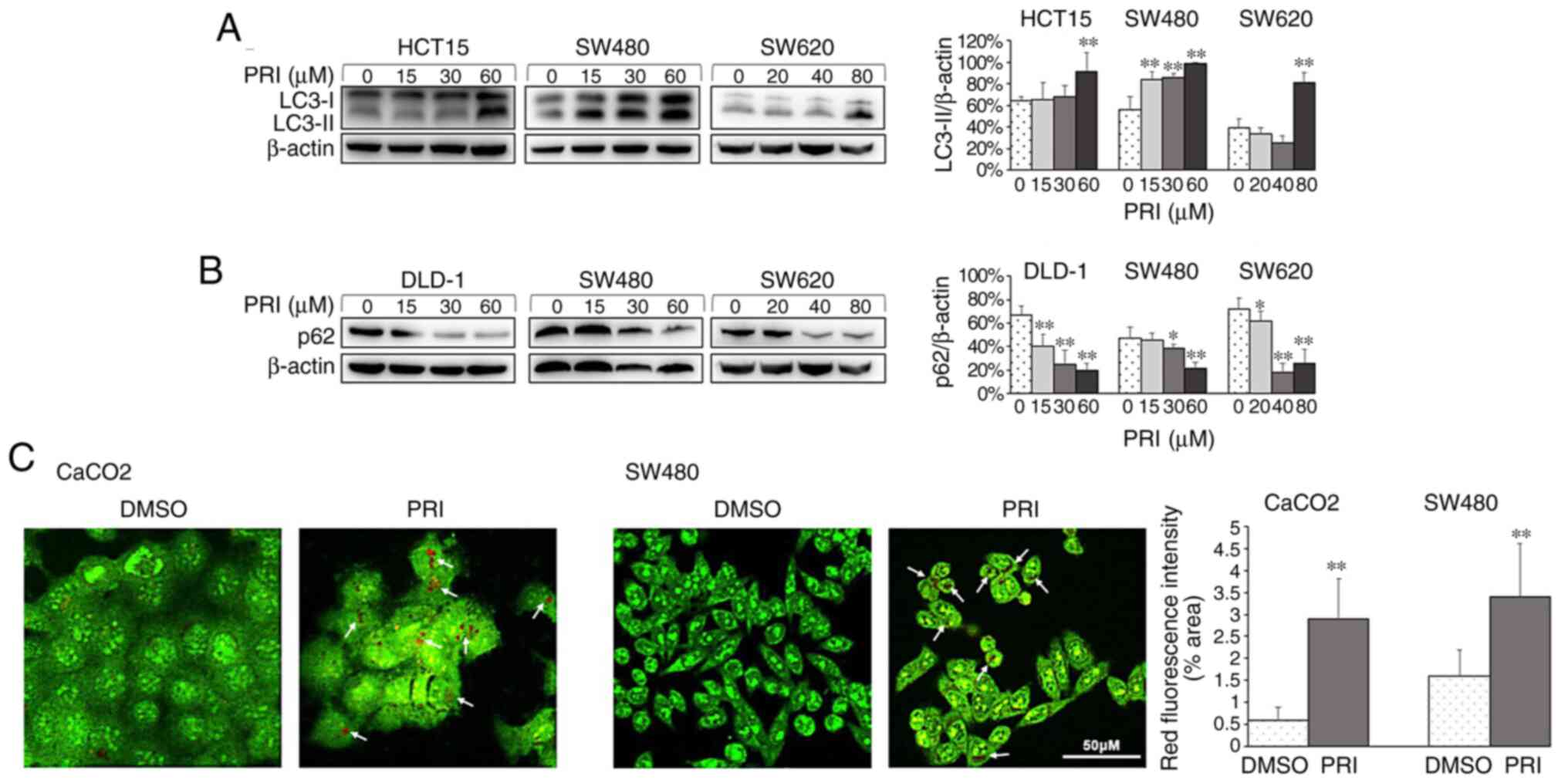
cell line (P<0.01; Figs. 1A,
2A and 3A). Conversely, reduced levels of p62 were
observed in HCT116ko, RKO, DLD-1, SW480 and SW620 cells
(P<0.01; Figs. 1B, 2B and 3B). The
results indicated that PRIMA-1met promoted the
dissociation of plasma LC3 (LC3-I) to membranous LC3 (LC3-II) for
AV-membrane extension and the degradation of cargo (p62) in
autolysosomes. To further determine whether PRIMA-1met
induced autophagy flux in CRC cells, AO staining was performed on
HCT116wt, RKO, SW480, CaCO2 and HCT116ko
cells following treatment for 24 h. The results revealed that
treated cells exhibited stronger red fluorescence than DMSO
controls (P<0.01; Figs. 1C,
2C and 3C). However, AO demonstrated an affinity for
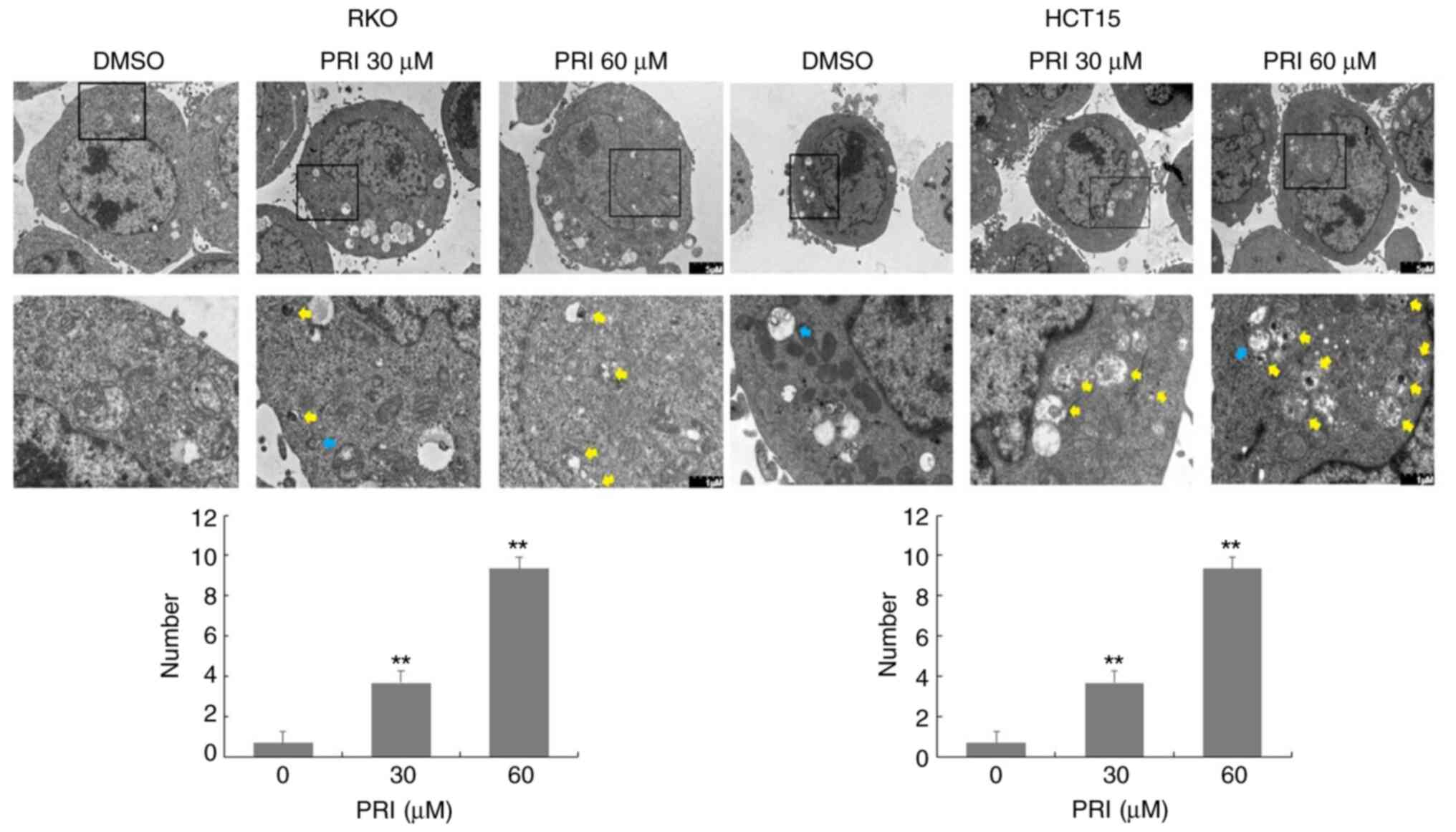
AV organelles within the autolysosome and lysosome. Therefore, to
distinguish these results further, autolysosomes were observed in
PRIMA-1met-treated RKO and HCT15 cells via TEM. As
presented in Fig. 4, numbers of
autolysosomes and autophagosomes were increased significantly
following PRIMA-1met administration in each of the cell
lines compared with the DMSO control. This effect was more
significant at higher concentrations (P<0.01). Taken together,
PRIMA-1met treatment promoted AV formation, cargo
removal and cargo degradation in autophagy flux, irrespective of
p53 status.
PRIMA-1met regulates the
mTOR/AMPK-ULK1-Vps34 autophagic signaling cascade in different CRC
cell lines
Several molecules involved in autophagic signaling
were assessed by western blotting to determine the distinct
mechanism of PRIMA-1met in CRC cell autophagy. The
results revealed decreased levels of phospho-mTOR (a nutrient
sensor) in PRIMA-1met-treated HCT116wt and
RKO cells with wild-type p53. Increased levels of phospho-AMPK (an
energy sensor) were also revealed in HCT116wt, RKO and
HCT15 cells (P<0.01; Fig. 5A).
Furthermore, PRIMA-1met administration upregulated the
expression of phospho-ULK1 in RKO, DLD-1 and HCT15 cell lines, and
phospho-PI3K Class III (the mammalian homolog of Vps34) in RKO,
HCT116wt and HCT15 cells (P<0.01; Fig. 5B). The results indicated that
PRIMA-1met mediated the autophagic mTOR/AMPK-ULK1-Vps34
signaling cascade in CRC cells independently of p53 status.
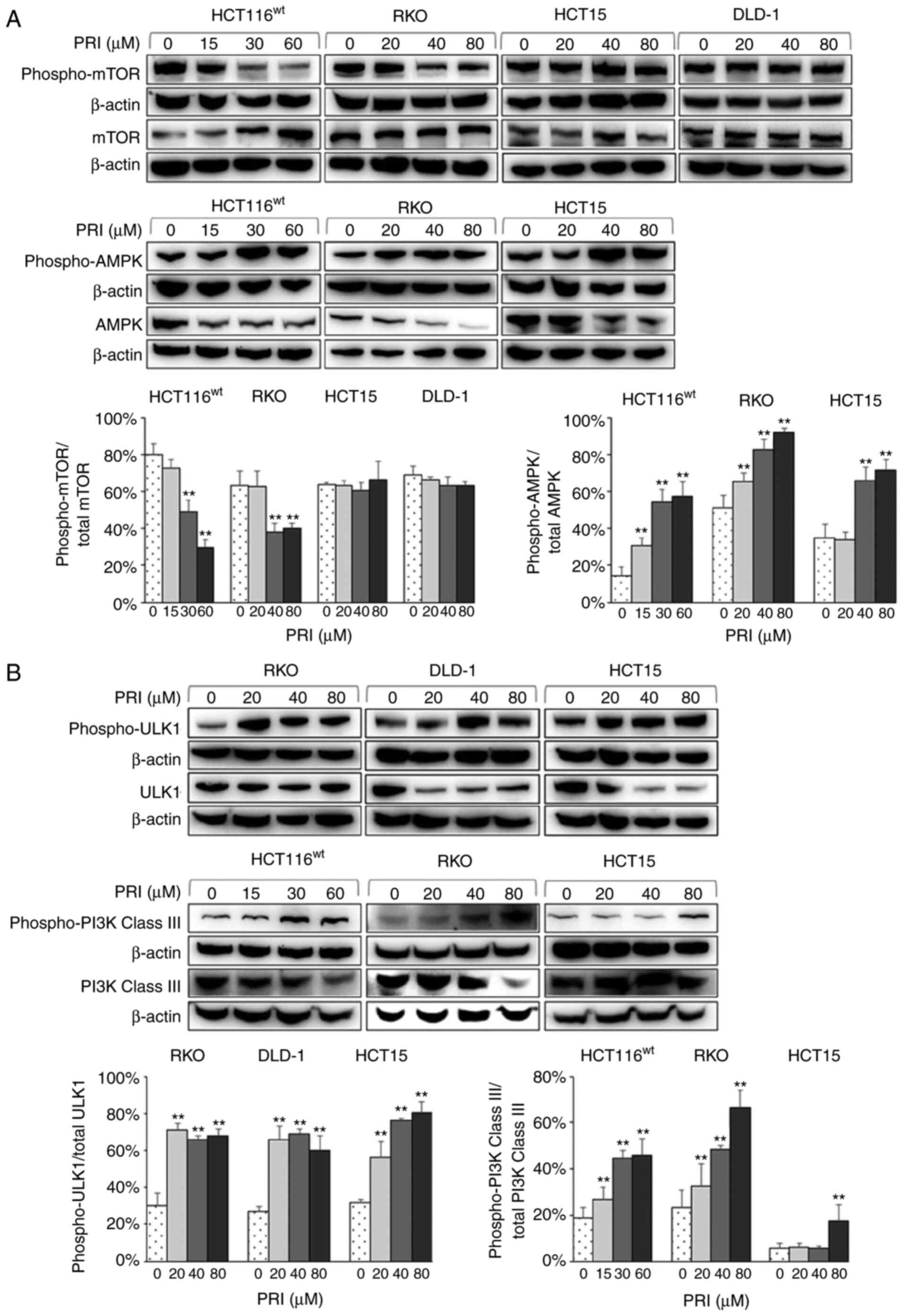 | Figure 5.Western blot analysis of colorectal
cancer cell lysates treated with different concentrations of PRI
for 24 h. (A) Phospho-mTOR and mTOR levels were increased and
decreased respectively in HCT116wt and RKO cells
carrying wild-type p53. However, there were no significant
differences among the p53-mutant cell lines (HCT15 and DLD-1).
Phospho-mTOR was normalized to total mTOR levels. PRI treatment
also upregulated the expression of phospho-AMPK, and downregulated
AMPK in HCT116wt, RKO and HCT15 cells. Phospho-AMPK was
normalized to that of total AMPK and each experiment was performed
in triplicate. Data are presented as the mean + SEM (n=3).
**P<0.01, compared with the PRI untreated group. (B)
Phospho-ULK1 and ULK1 expression was increased and decreased
respectively in RKO, DLD-1 and HCT15 cell lines after PRI
treatment. Phospho-ULK1 was normalized to total ULK1 levels. The
expression of phospho-PI3K Class III was upregulated following PRI
treatment in HCT116wt, RKO and HCT15 cells. Phospho-PI3K
Class III was normalized to that of total PI3K Class III. Data are
presented as the mean + SEM (n=3). **P<0.01, compared with the
PRI untreated group. PRI, p53-reactivation and induction of massive
apoptosis-1, APR-017 methylated; phosphor, phosphorylated; ULK1,
Unc-51 like autophagy activating kinase 1. |
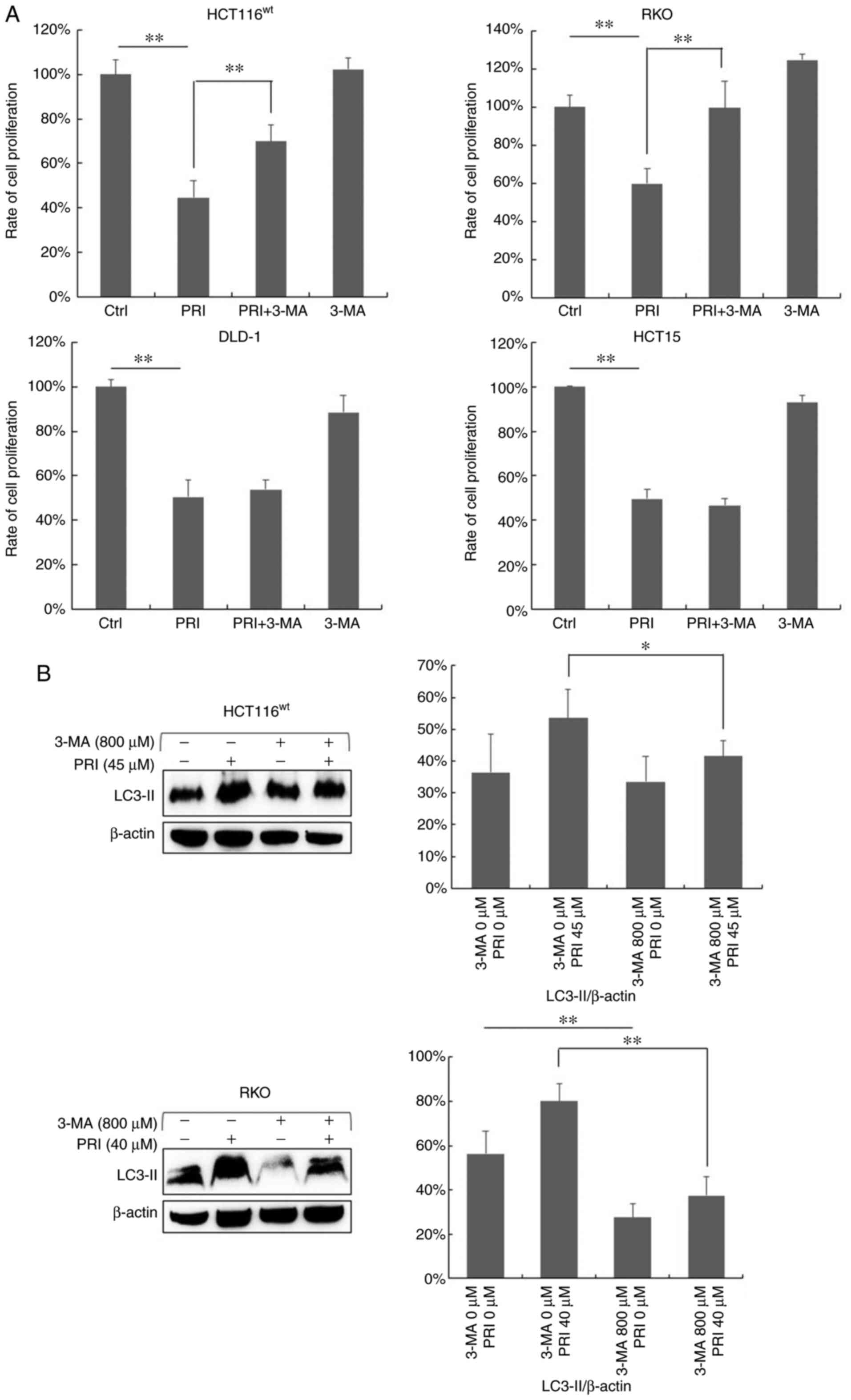
PI3K inhibitor suppresses the
inhibitory effect of PRIMA-1met in CRC cells with
wild-type p53
To determine whether PRIMA-1met-induced
autophagy influenced cell growth, the PI3K inhibitor (3-MA) was
applied to cells. HCT116wt, RKO, DLD-1 and HCT15 cells
were exposed to PRIMA-1met and 3-MA either alone or in
combination for 48 h, after which a CCK-8 assay was performed. As
predicted, cell proliferation decreased in all cell lines following
PRIMA-1met treatment (P<0.01; Fig. 6A), while 3-MA exposure did not affect
cell proliferation (Fig. 6A).
However, when administered in combination, the inhibition of cell
growth was markedly weakened compared with PRIMA-1met
treatment alone in HCT116wt and RKO cells expressing
wild-type p53 (P<0.01; Fig. 6A).
There was no significant difference in proliferation between
PRIMA-1met and combinational regimens in DLD-1 and HCT15
cell lines carrying mutant p53 (Fig.
6A). LC3-II expression was assessed via western blotting after
3-MA treatment in HCT116wt and RKO cells (Fig. 6B). To further investigate the
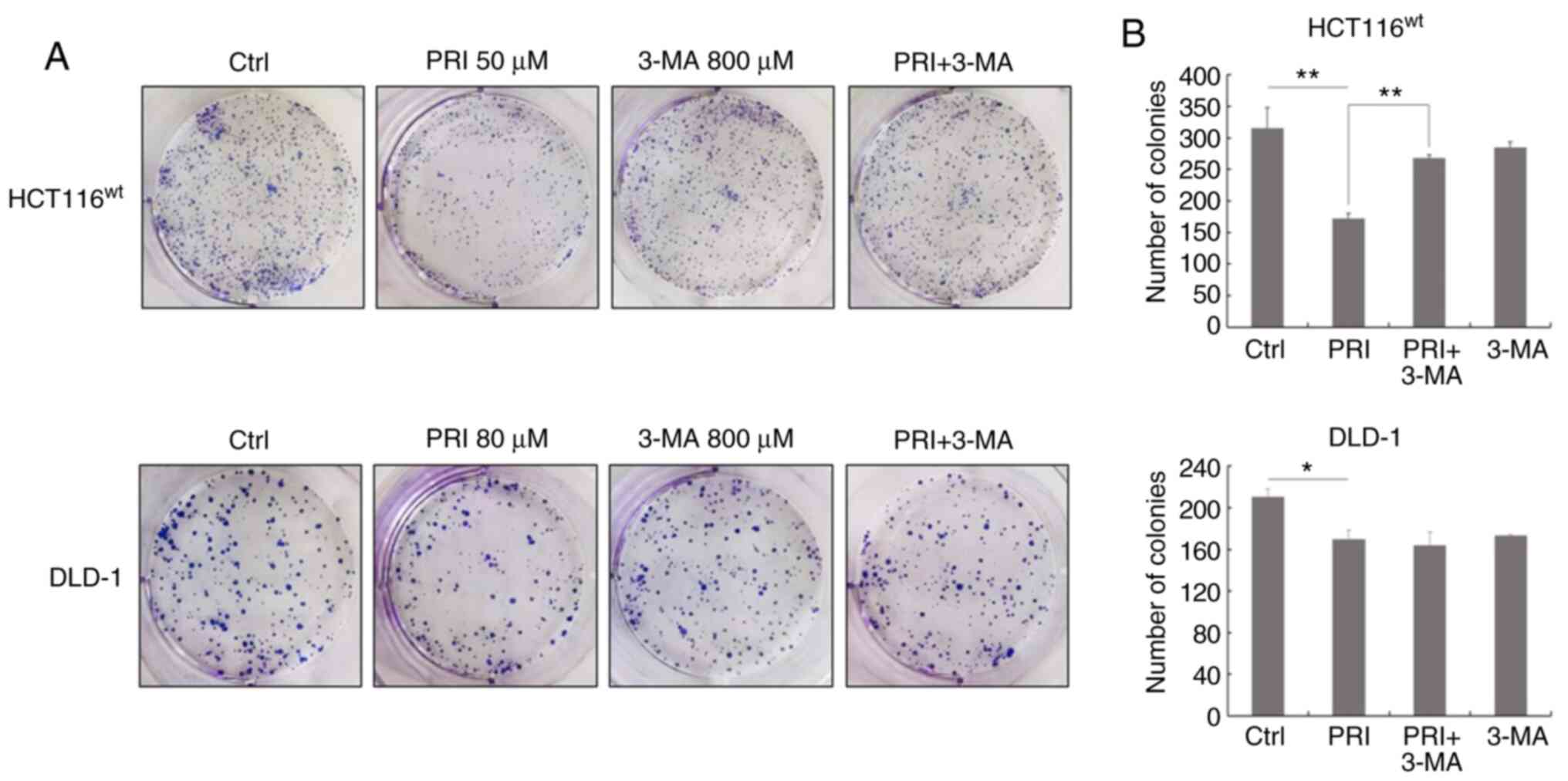
influence of the PI3K inhibitor on malignant transformation, cell
colonies were observed and counted after PRIMA-1met and
3-MA treatment for 10 days (Fig. 7A).
The number of colonies was significantly decreased following
PRIMA-1met treatment compared with DMSO treatment alone
in each cell line (P<0.01 in HCT116 cells; P<0.05 in DLD-1
cells; Fig. 7B). However,
co-treatment of PRIMA-1met and 3-MA reversed this effect
in HCT116wt cells (P<0.01; Fig. 7B). The results indicated that the PI3K
inhibitor suppressed the inhibitory effect of PRIMA-1met
on the growth of CRC cell lines expressing wild-type p53.
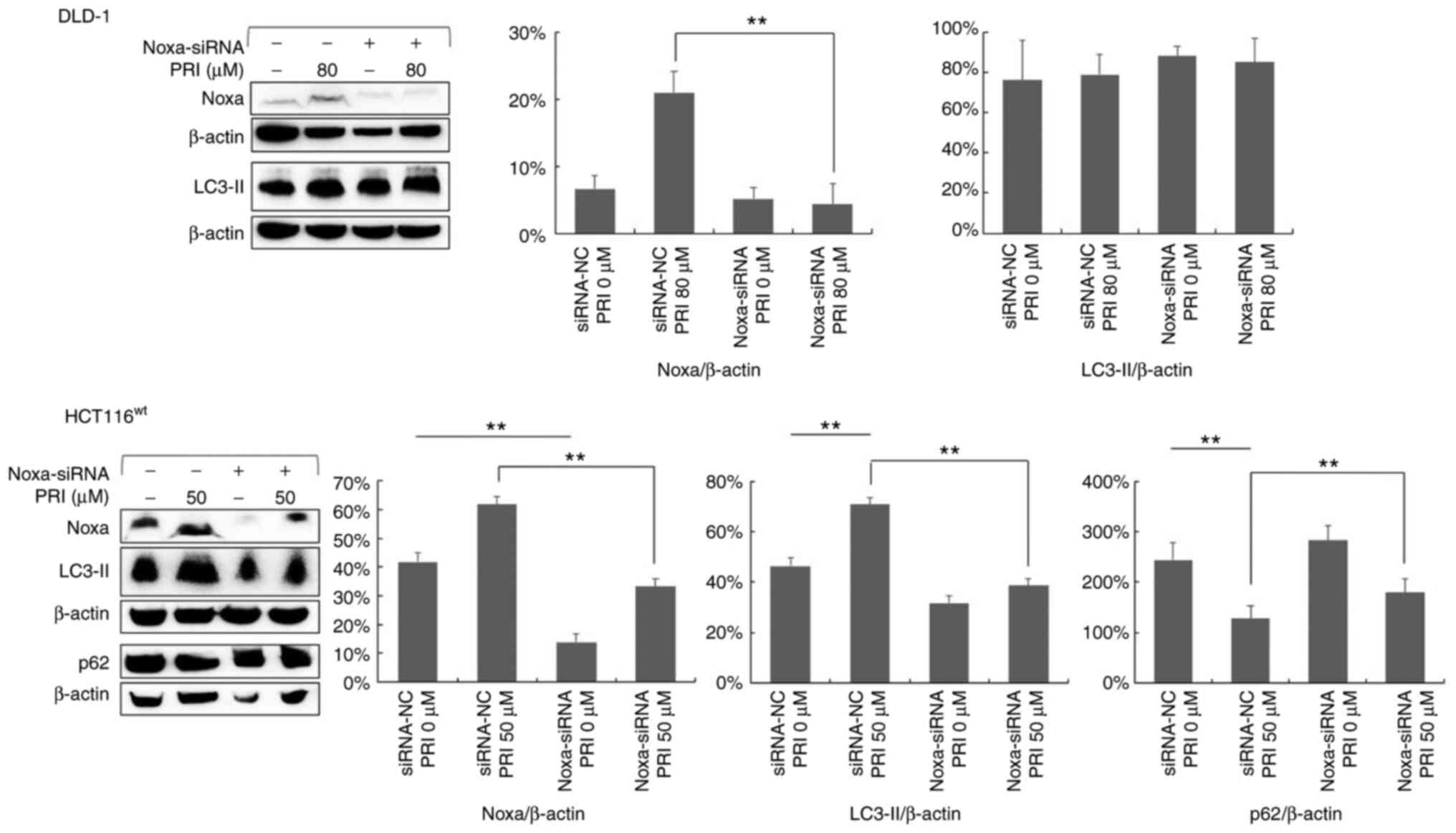
PRIMA-1met-induced
autophagy in CRC cells expressing wild-type p53 is positively
correlated with Noxa
It was previously reported that Noxa upregulation, a
pro-apoptotic molecule, is crucial for the PRIMA-1met
induction of apoptosis in CRC cell lines with mutant p53 (10). To investigate the role of Noxa in
PRIMA-1met-induced autophagy, Noxa was knocked down
using siRNA in DLD-1 and HCT116wt cell lines. The
results of western blotting revealed a decreased expression of Noxa
(Fig. 8). In addition, LC3-II
expression was reduced in HCT116wt cells after Noxa
knockdown (P<0.01), whereas there was no significant effect in
the p53-mutant DLD-1 cell line (Fig.
8). Furthermore, the results revealed that the decreased
expression of p62 following PRIMA-1met treatment was
reversed in HCT116wt cells treated with Noxa siRNA
(P<0.05; Fig. 8).
Discussion
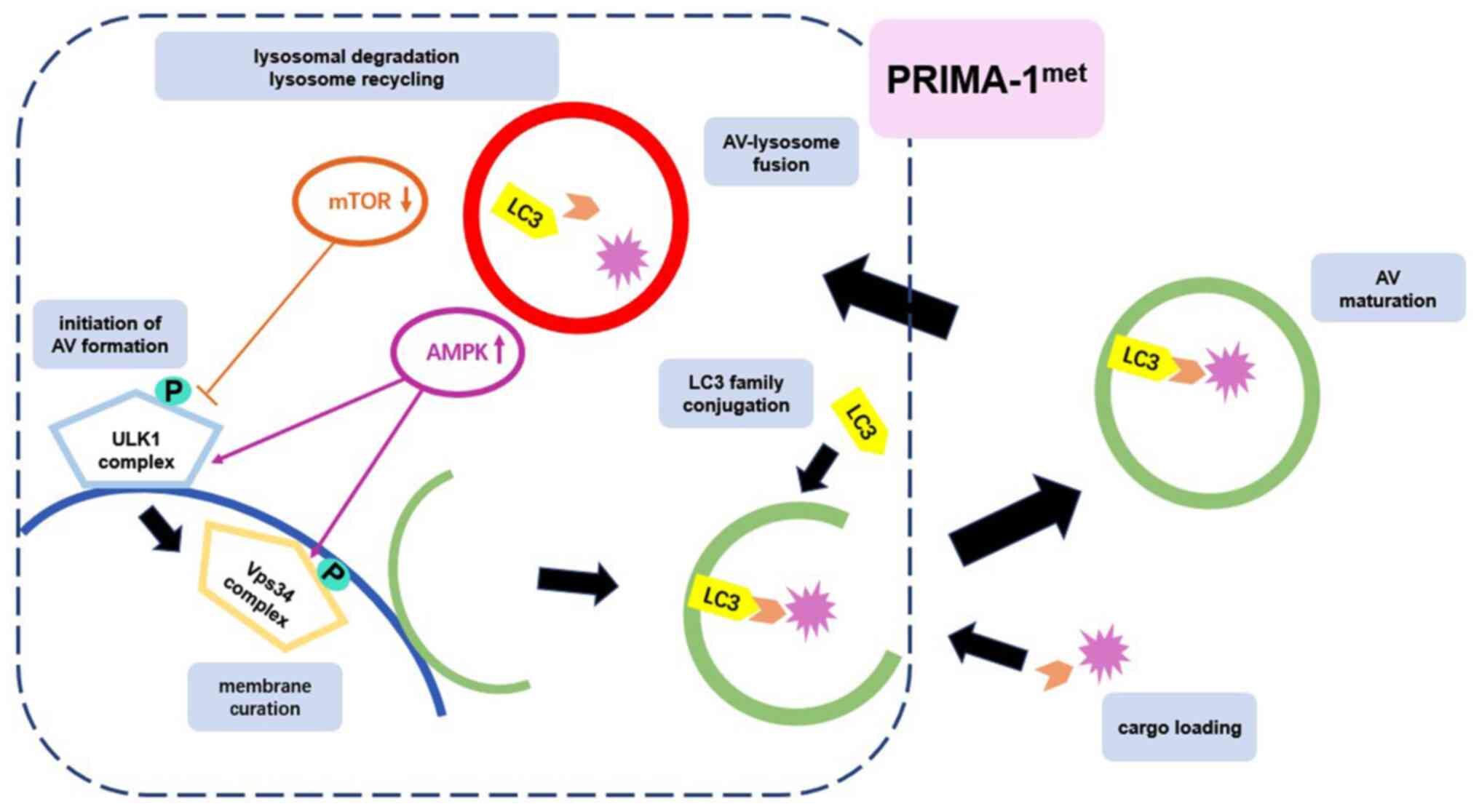
Autophagy occurs through several phases, including
initiation (in which the membrane is prepared to form AVs),
membrane curation, LC3 family conjugation, cargo loading, AV
maturation, AV-lysosome fusion, lysosomal degradation and lysosome
recycling (19). This dynamic process
is also called autophagy flux. In the present study, several
methods, such as western blotting, AO staining and TEM, were used
to determine that PRIMA-1met treatment induced autophagy
in CRC cell lines expressing different p53 statuses.
PRIMA-1met treatment upregulated LC3 conversion,
resulting in an increase in AVs and promoting the degradation of
cargo through combination with receptor p62. This effect was
exhibited regardless of p53 status but was relative to cell type.
Furthermore, the results of the current study determined that the
molecular mechanism underlying PRIMA-1met-induced
autophagy in CRC cells involved the mediation of the
mTOR/AMPK-ULK1-Vps34 signaling cascade.
ULK1, the mammalian homolog of ATG1, serves a
convergent role in multiple signals that regulate autophagy,
receiving nutrient and energy signals from its two upstream factors
(AMPK and mTOR) (20). Additionally,
AMPK that is activated under low energy conditions with an elevated
AMP/ATP ratio phosphorylates ULK1 (21). mTOR, as an autophagy inhibitor,
reduces the phosphorylation of ULK1 during nutrient stress and
disrupts the interaction of ULK1 and AMPK (20). Once activated, the ULK1 complex,
consisting of ULK1, ATG13, FIP200 and ATG101, promotes AV formation
(22), leading to Vps34 complex
activation, which includes Vps34 (PI3K Class III), Beclin-1, Vps15
and ATG14 for AV membrane curvature (23). The present study revealed that
PRIMA-1met upregulated the phosphorylation of AMPK, ULK1
and PI3K Class III in CRC cells expressing both wild-type p53 and
mutant p53. However, a decrease in the phosphorylation of mTOR was
only observed in cells expressing wild-type p53.
PRIMA-1met also increased the phosphorylation of PI3K
Class III, but did not affect ULK1 in the HCT116wt cell
line, which may be due direct activation by AMPK. The results
indicated an association between PRIMA-1met and
autophagy in different CRC cell lines mediated by the
mTOR/AMPK-ULK1-Vps34 signaling cascade.
3-MA targets Vps34 and PI3Kγ by inhibiting PI3K
Class I permanently and PI3K Class III temporarily, and as such is
a useful reagent to block autophagosome formation. In the current
study, the combined treatment of 3-MA and PRIMA-1met
suppressed inducible autophagy and further reduced the inhibitory
effect of PRIMA-1met on cell proliferation and colony
formation in CRC cells expressing wild-type p53 compared with
PRIMA-1met treatment alone. The results supported the
notion that the mechanism underlying PRIMA-1met
cytotoxicity in cells with wild-type p53 was associated with the
induction of autophagy.
Previous studies have demonstrated that the
crosstalk between autophagy and apoptosis, particularly in the
Bcl-2 family, was crucial to regulating cell growth and survival
(24,25). The present study revealed that the
reduced expression of Noxa induced by siRNA suppressed LC3
conversion and p62 degradation in the HCT116wt cell
line. However, this effect was not demonstrated in the DLD-1 cell
line. Noxa upregulation therefore influenced cell autophagy
following PRIMA-1met treatment in CRC cells carrying
wild-type p53. The results confirmed that different mechanisms were
involved in the inhibitory effect of PRIMA-1met in CRC
cells expressing different types of p53. PRIMA-1met
mainly promoted apoptosis in cells harboring mutant p53, whereas
PRIMA-1met induced autophagy in cells carrying wild-type
p53 or null p53. Inducible autophagy, including autophagic
proteins, was determined to participate in apoptosis regulation
through the complex network of molecular interactions in p53-mutant
cells after PRIMA-1met treatment.
In conclusion, the present study elucidated that
PRIMA-1met induced autophagy in CRC cells by activating
the mTOR/AMPK-ULK1-Vps34 signaling cascade in a p53-independent
manner (Fig. 9). Furthermore, induced
autophagy was closely associated with the cytotoxicity of
PRIMA-1met and the upregulation of Noxa in cells
expressing wild-type p53. According to the complex interaction
between autophagy and apoptosis, a deeper insight into the
association between the two processes following
PRIMA-1met induction, particularly in p53-mutant cells,
is required for further study. The results of the current study
demonstrated a further understanding of the mechanisms underlying
the antitumor activity of PRIMA-1met in CRC, supporting
PRIMA-1met-based therapy as a novel strategy for
treating patients with advanced CRC.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Jun-dong Zhou
(The Affiliated Suzhou Hospital of Nanjing Medical University,
China) and Dr Fang Lin (Soochow University, China) for providing
the laboratory and experimental equipment during the current
study.
Funding
The current study was supported by a grant from the
National Natural Science Foundation of China (NSFC; grant no.
81603128), which funded the design of the study, experiments, data
analysis and writing of the manuscript. The current study was also
supported by the Gusu Health Layer Training Program for Young Top
Talent (grant no. GSWS2019059) and the Science and Technology
Project of Suzhou (grant nos. SYSD2018136 and SS201717), which
contributed to completion of the experiments.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XLL, JZ and ZRC designed the current study. XLL and
JZ wrote the manuscript. ZRC revised the manuscript. XLL, CJX, HM
and ZKL performed the experiments. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kyaw M and Sung JJ: Young-onset colorectal
cancer in the Asia-Pacific region. Med J Aust. 205:450–451. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ryan KM, Phillips AC and Vousden KH:
Regulation and function of the p53 tumor suppressor protein. Curr
Opin Cell Biol. 13:332–337. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bieging KT, Mello SS and Attardi LD:
Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev
Cancer. 14:359–370. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takayama T, Miyanishi K, Hayashi T, Sato Y
and Niitsu Y: Colorectal cancer: Genetics of development and
metastasis. J Gastroenterol. 41:185–192. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li XL, Zhou J, Chen ZR and Chng WJ: P53
mutations in colorectal cancer-molecular pathogenesis and
pharmacological reactivation. World J Gastroenterol. 21:84–93.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lambert JM, Moshfegh A, Hainaut P, Wiman
KG and Bykov VJ: Mutant p53 reactivation by PRIMA-1MET induces
multiple signaling pathways converging on apoptosis. Oncogene.
29:1329–1338. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoshikawa N, Kajiyama H, Nakamura K,
Utsumi F, Niimi K, Mitsui H, Sekiya R, Suzuki S, Shibata K, Callen
D and Kikkawa F: PRIMA-1MET induces apoptosis through accumulation
of intracellular reactive oxygen species irrespective of p53 status
and chemo-sensitivity in epithelial ovarian cancer cells. Oncol
Rep. 35:2543–2552. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu T, Zou Y, Xu G, Potter JA, Taylor GL,
Duan Q, Yang Q, Xiong H, Qiu H, Ye D, et al: PRIMA-1Met suppresses
colorectal cancer independent of p53 by targeting MEK. Oncotarget.
7:83017–83030. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sobhani M, Abdi J, Manujendra SN, Chen C
and Chang H: PRIMA-1Met induces apoptosis in Waldenstrom's
Macroglobulinemia cells independent of p53. Cancer Biol Ther.
16:799–806. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tessoulin B, Descamps G, Moreau P, Maïga
S, Lodé L, Godon C, Marionneau-Lambot S, Oullier T, Le Gouill S,
Amiot M and Pellat-Deceunynck C: PRIMA-1Met induces myeloma cell
death independent of p53 by impairing the GSH/ROS balance. Blood.
124:1626–1636. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li XL, Zhou J, Chan ZL, Chooi JY, Chen ZR
and Chng WJ: PRIMA-1met (APR-246) inhibits growth of colorectal
cancer cells with different p53 status through distinct mechanisms.
Oncotarget. 6:36689–36699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
White E, Mehnert JM and Chan CS:
Autophagy, metabolism, and cancer. Clin Cancer Res. 21:5037–5046.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aita VM, Liang XH, Murty VV, Pincus DL, Yu
W, Cayanis E, Kalachikov S, Gilliam TC and Levine B: Cloning and
genomic organization of beclin 1, a candidate tumor suppressor gene
on chromosome 17q21. Genomics. 59:59–65. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Karantza-Wadsworth V, Patel S, Kravchuk O,
Chen G, Mathew R, Jin S and White E: Autophagy mitigates metabolic
stress and genome damage in mammary tumorigenesis. Genes Dev.
21:1621–1635. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang KK, Ramnarayanan K, Zhu F,
Srivastava S, Xu C, Tan ALK, Lee M, Tay S, Das K, Xing M, et al:
Genomic and epigenomic profiling of high-risk intestinal metaplasia
reveals molecular determinants of progression to gastric cancer.
Cancer Cell. 33:137–150.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Amaravadi RK, Kimmelman AC and Debnath J:
Targeting autophagy in cancer: Recent advances and future
directions. Cancer Discov. 9:1167–1181. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hardie DG: The AMP-activated protein
kinase pathway-new players upstream and downstream. J Cell Sci.
117((Pt 23)): 5479–5487. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kumar S, Gu Y, Abudu YP, Bruun JA, Jain A,
Farzam F, Mudd M, Anonsen JH, Rusten TE, Kasof G, et al:
Phosphorylation of Syntaxin 17 by TBK1 controls autophagy
initiation. Dev Cell. 49:130–144.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Behrends C, Sowa ME, Gygi SP and Harper
JW: Network organization of the human autophagy system. Nature.
466:68–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou F, Yang Y and Xing D: Bcl-2 and
Bcl-xL play important roles in the crosstalk between autophagy and
apoptosis. FEBS J. 278:403–413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang J, Cui D, Gu S, Chen X, Bi Y, Xiong X
and Zhao Y: Autophagy regulates apoptosis by targeting NOXA for
degradation. Biochim Biophys Acta Mol Cell Res. 1865:1105–1113.
2018. View Article : Google Scholar : PubMed/NCBI
|