Introduction
Obesity is a major public health problem reaching
pandemic proportions. The prevalence of adult obesity, defined as a
body mass index (BMI) >30 kg/m2, tripled between 1975
and 2016, affecting 13% of the global adult population (>650
million individuals) (1). Obesity
is a risk factor for several diseases, including cardiovascular
disease and various types of cancer (2).
Gastric cancer can be caused by a variety of
environmental and genetic factors (3). As well as smoking and a high-salt
diet, obesity has been linked to gastric cancer (4,5). BMI
is used to evaluate obesity in epidemiology studies. However,
patients with the same BMI may have different body compositions.
Therefore, BMI is not suitable for evaluating the impact of obesity
on gastric cancer. One-third of women with a normal BMI have
symptoms characteristic of metabolic obesity, while 30% of obese
individuals can be classified as metabolically healthy (6,7).
Therefore, an improved marker of adiposity is required to
investigate the relationship between obesity and gastric
cancer.
Leptin, a multifunctional hormone, is a marker of
hyper-adiposity. It is primarily secreted by adipocytes, but also
by tissues in the stomach and placenta, for instance (8,9). The
main function of leptin, as the ligand of the leptin receptor, is
hypothalamic-mediated regulation of appetite, which modulates food
intake and energy expenditure (10,11).
Obesity-induced tumorigenesis is associated with dysregulation of
the adipokine signaling pathway, which includes leptin (7). Furthermore, the leptin receptor is
important in carcinogenesis (12).
Leptin-leptin receptor signaling promotes multiple processes
involved in cancer progression, including cell proliferation,
metastasis, angiogenesis and chemoresistance (13,14).
The mechanism by which leptin promotes cancer
progression is best reported in the field of breast cancer. Binding
of leptin to the leptin receptor activates various signaling
pathways including JAK/STAT, MAPK and PI3K pathways, which are
associated with cell proliferation (15). It has been reported that leptin is
an essential factor for mammary stem cell survival and maintenance
(16). In addition, leptin may
further activate malignant cell growth through stimulation of
angiogenesis and creation of vascular permeability (17,18).
Although gastric cardia cancer is reportedly
associated with obesity, in contrast to breast cancer, the
relationship between gastric cancer and leptin is unclear (2). The association between leptin and
gastric cancer has been evaluated in vitro (19,20).
The effect of leptin on cancer cell migration and invasion, and the
expression levels of angiogenesis-related growth factors were
investigated in vitro. The relationship between the serum
leptin level and clinical profile of patients with gastric cancer
was also explored.
Materials and methods
Patient selection
A total of 46 patients diagnosed with gastric
adenocarcinoma, who underwent curative gastrectomy between 2005 and
2007, were identified in the database of our institution. The
present study was approved (approval no. VCM07BR016) by the
Institutional Review Board of St. Vincent's Hospital, Catholic
University of Korea (Seoul, Republic of Korea). Written informed
consent was provided by all patients. None of the patients had
undergone prior surgery, chemotherapy, or radiation therapy. After
surgery, clinical follow-up data were collected at the outpatient
clinic. The cohort was initially classified based on BMI: normal,
BMI 18.5–25 kg/m2 and obese, BMI ≥25 kg/m2.
The clinicopathological characteristics of the cohort are listed in
Table I.
 | Table I.Clinicopathological characteristics
of the study population. |
Table I.
Clinicopathological characteristics
of the study population.
|
Characteristics | Normal (n=23) | Overweight
(n=23) | P-value |
|---|
| Age, years | 58.8±10.1 | 63±9.8 | 0.163 |
| Sex |
|
| 0.765 |
|
Male | 14 | 13 |
|
|
Female | 9 | 10 |
|
| Weight, kg | 53.8±6.5 | 66.44±8.4 | 0.001 |
| Body mass index
(kg/m2) | 20.3±1.4 | 25.83±3.4 | 0.001 |
| Histologic
type |
|
| 0.767 |
|
Differentiated | 11 (52.4%) | 12 (48%) |
|
|
Undifferentiated | 10 (47.6%) | 13 (52%) |
|
|
Tumor-node-metastasis stage |
|
| 0.005 |
| I | 14 (60.9%) | 6 (26.1%) |
|
| II | 7 (30.4%) | 5 (21.7%) |
|
|
III | 2 (8.7%) | 12 (52.2%) |
|
| Serum leptin level
(pg/ml) |
4,236.1±8,314.3 |
8,626.5±8,700.7 | 0.001 |
Cell culture
The gastric cancer cell lines AGS and MKN74 were
obtained from the Korean Cell Line Bank and cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS; both from
Invitrogen; Thermo Fisher Scientific, Inc.), 1% penicillin and 1%
streptomycin. The cells were incubated at 37°C in an atmosphere of
95% air and 5% CO2.
Proliferation assay
Cell proliferation assays were performed using the
Cell Counting Kit-8 (Sigma-Aldrich; Merch KGaA) according to the
manufacturer's protocol. Each cell line (5×103; 100 µl)
was seeded into a well of a 96-well plate and cultured in 100 µl of
RPMI-1640 supplemented with 10% FBS. After 24 h, seeded cells were
treated with 0, 40, 80 and 120 ng/ml leptin into the culture
medium. The cells were incubated for 2 h at 37°C with a total of
100 µl CCK-8 reagent. Absorbance was measured for each well at a
wavelength of 450 nm using an auto-microplate reader.
Wound healing assay
Cell migration was evaluated by wound healing assay.
AGS and MKN74 cells (100,000 cells per well) were seeded on 24-well
plates containing 500 µl of RPMI-1640 medium. The cells were washed
three times with phosphate-buffered saline (PBS) and a wound was
generated by removing the cells from the center of the well using a
sterile pipette tip. Detached cells were removed by washing with
PBS. The cells were incubated for 24 to 48 h. Cell migration was
studied by treating the cells with 4–80 ng/ml leptin, alone or
following 30-min pre-incubation with AG490 (50 µl) or U0126 (20 µl;
both from Sigma-Aldrich; Merck KGaA). Three images of the wound
were obtained using a TMS inverted light microscope connected to a
Coolpix 4500 camera (Nikon Corporation). Wound closure was
quantified by measuring the area (in pixels) between the edges of
the wound using the measurement tool in Adobe
Photoshop®, with a grid superimposed on the image as a
guide. Wound width was normalized to 100% at 0 h for each
condition; data are percentages of wound closure.
In vitro cell invasion assay
Invasion assay was performed using 24-well Transwell
plates with 8-µm isopore membranes (BD Biosciences). The upper side
of the membrane was coated with Matrigel (50 µg/well) and the
membranes were air-dried for 1-h incubation at 37°C. The lower side
of the membranes was coated with 5 µg of fibronectin (BD
Biosciences). To assess the effect of leptin (100 ng/ml for 24 h)
on cell invasion, treated or untreated gastric cancer cells
(2.5×104) in 200 µl of RPMI-1640 medium supplemented
with 5% charcoal-treated FBS were placed in the upper chamber. The
lower chamber was filled with 700 µl of RPMI-1640 medium, with 10%
FBS as a chemoattractant. The invasion chamber was incubated for 24
h at 37°C in 5% CO2. Cells on the upper surface of the
membrane were removed by gentle scrubbing using a cotton swab.
Membranes were fixed at room temperature in 95% ethanol and 5%
acetic acid for 30 min and stained for 30 min at room temperature
with 0.4% crystal violet. The number of cells on the lower surface
of the membrane in five randomly selected visual fields
(magnification, ×400) was counted using a bright-field light
microscope. Assays were performed in triplicate.
Cell line maintenance and isolation of
cancer-initiating cells (CICs)
The cell lines were authenticated using the
GeneMarker 10 Kit (Promega Corporation) and routinely screened for
mycoplasma infection by PCR. A total of 24 h before treatment, the
medium was exchanged for medium supplemented with 5%
charcoal-treated PBS. CICs were isolated from AGS and MKN74 cells
using stem-cell selection medium [Dulbecco's modified Eagle's
medium (DMEM)/F12 medium; Invitrogen; Thermo Fisher Scientific,
Inc.) supplemented with epidermal growth factor (20 ng/ml),
fibroblast growth factor-2 (10 ng/ml), bovine serum albumin (0.4%)
and insulin (5 µg/ml) without FBS under non-attachment conditions
(Corning, Inc.). After 6 days, spheroids (40–70 µm in diameter)
containing CICs were collected.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from gastric cancer cells
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). RNA was quantified by measuring the absorption
at 260 nm and stored at −80°C. PrimeScript RT kit (Takara
Biotechnology Co., Ltd.) and SYBR Select Master Mix (Invitrogen;
Thermo Fisher Scientific, Inc.) were used for reverse transcription
and qPCR. Briefly, RT-qPCR was performed under the following
conditions: 30 cycles of denaturation at 95°C for 30 sec, annealing
at 55°C for 30 sec, and extension at 72°C for 1 min, followed by 10
min normalization to the β-actin (forward,
5′-CACCATTGGCAATGAGCGGTTC-3′ and reverse,
5′-AGGTCTTTGCGGATGTCCACGT-3′) mRNA level. The mRNA levels of Snail
(forward, 5′-TGCCCTCAAGATGCACATCCGA-3′ and reverse,
5′-GGGACAGGAGAAGGGCTTCTC-3′), N-cadherin (forward,
5′-CCTCCAGAGTTTACTGCCATGAC-3′ and reverse,
5′-GTAGGATCTCCGCCACTGATTC-3′) and CD44 (forward,
5′-CCAGAAGGAACAGTGGTTTGGC-3′ and reverse,
5′-ACTGTCCTCTGGGCTTGGTGTT-3′) were measured by RT-qPCR. The
quantitative analysis was performed using the 2−ΔΔcq
method (21).
Enzyme-linked immunosorbent assay
(ELISA)
Gastric cancer cells were seeded into plates at
3×106 cells/well with 4 ml of RPMI-1640 medium
supplemented with 10% FBS. The cells were treated with 100 ng of
leptin or 100 ng of leptin + AG490 (50 µmol/ml) for 24, 48, or 72
h. Using a VEGF-C (H) ELISA kit (cat. no. BMS297-2; Thermo Fisher
Scientific, Inc.), performed according to the manufacturer's
protocol, vascular endothelial growth factor (VEGF) was quantified
in the supernatants of gastric cancer cells. A total of 200 µl of
supernatant were added to each well. The sensitivity of the ELISA
was 7.8 pg/ml. Absorbance was determined at 450 nm. Each test was
performed in triplicate.
Western blot analysis
After leptin or control treatment, cells were
harvested in cold PBS and the pellet was resuspended in lysis
buffer [20 mM Tris-HCl (pH 7.4), 137 mM NaCl, 2 mM EDTA, 1% Triton
X-100 and 10% glycerol] for 20 min at 4°C. The supernatant was then
collected. Protein concentrations were measured in duplicate by
Bradford assay. A total of 50–100 µg of protein was loaded in each
lane, resolved by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis, transferred to nitrocellulose membranes. Following
blocking (with 5% non-fat milk in TRIS-buffered saline with 0.1%
Tween 20 at room temperature for 1 h), the membrane was incubated
overnight at 4°C with primary antibodies against CD44 (1:1,000;
cat. no. sc-7297), phosphor-Erk (1:1,000; cat. no. sc-7383), Erk
(1:1,000; cat. no. sc-5294; all from Santa Cruz Biotechnology,
Inc.), AKT (1:1,000; cat. no. ab191606), phosphor-AKT (1:1,000;
cat. no. ab131443; both from Abcam), MMP-9 (1:1,000; cat. no.
sc-515876; Santa Cruz Biotechnology, Inc.), SNAIL (1:1,000; cat.
no. 3879; Cell Signaling Technology, Inc.), N-cadherin (1:1,000;
cat. no. 13116), E-cadherin (1:500; cat. no. 14472) and β-actin
(1:1,000; cat. no. A5441; all from Sigma-Aldrich; Merck KGaA).
HRP-conjugated goat anti-mouse/rabbit IgG secondary antibodies
(1:3,000; cat. no. 1662408; Bio-Rad Laboratories, Inc.) were
subsequently applied for 1 h at room temperature. Bands were
developed using the ECL Western Blot Analysis System (NEN, Western
Lightning; PerkinElmer, Inc.). The relative content of each protein
was detected by ImageQuant 10.1 TL software (GE Healthcare).
Statistical analysis
Data were analyzed using SPSS software (ver. 21.0;
IBM Corp.). Continuous variables are expressed as the mean ±
standard deviation (SD). Analysis of continuous variables was
conducted by independent two-sample t-test or ANOVA with post-hoc
analysis (Tukey's test). Categorical variables were compared with
the chi-squared test. The relationship between two variables was
analyzed using Pearson's correlation analysis. Survival curves were
generated by the Kaplan-Meier method and analyzed by the log-rank
test. P<0.05 was considered to indicate a statistically
significant difference and all tests were two-sided unless
otherwise indicated.
Results
BMI is associated with the serum
leptin level in patients with gastric cancer
The clinicopathological data of the study population
are presented in Table I. The
average BMI was 20.3±1.4 and 25.83±3.4 kg/m2 in the
normal and overweight groups, respectively (P=0.001). There were no
significant differences in age, sex, or histologic type between the
groups. In the normal group, the proportion of stage I patients was
60.9%, and in the overweight group, the proportion of stage III
patients was 52.2%. There were significantly more patients with
advanced disease in the overweight than normal group (P=0.005). The
average serum leptin level was 4,236.1±8,314.3 and 8,626.5±8,700.7
pg/ml in the normal and overweight groups, respectively (P=0.001).
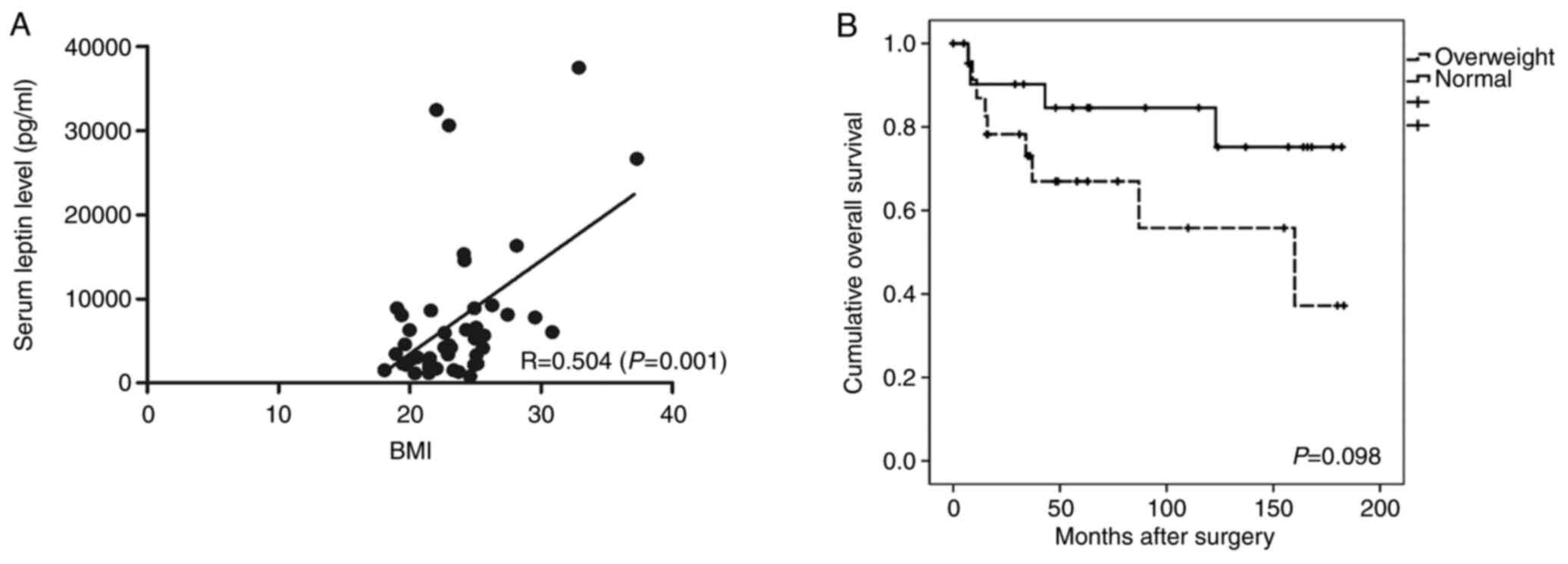
There was a positive correlation between BMI and the serum leptin
level (R=0.504, P=0.001; Fig. 1A).
The 5-year overall survival (OS) rate was 58.4% in the overweight
group and 82.6% in the normal group; the difference was not
significant (Fig. 1B).
Leptin induces gastric cancer cell
proliferation, migration and invasion
To investigate the effect of leptin on gastric
cancer cell migration, the leptin level in AGS and MKN74 cells was
evaluated. RT-qPCR and western blot analysis revealed that leptin
and leptin receptors were expressed in both cell lines (data not
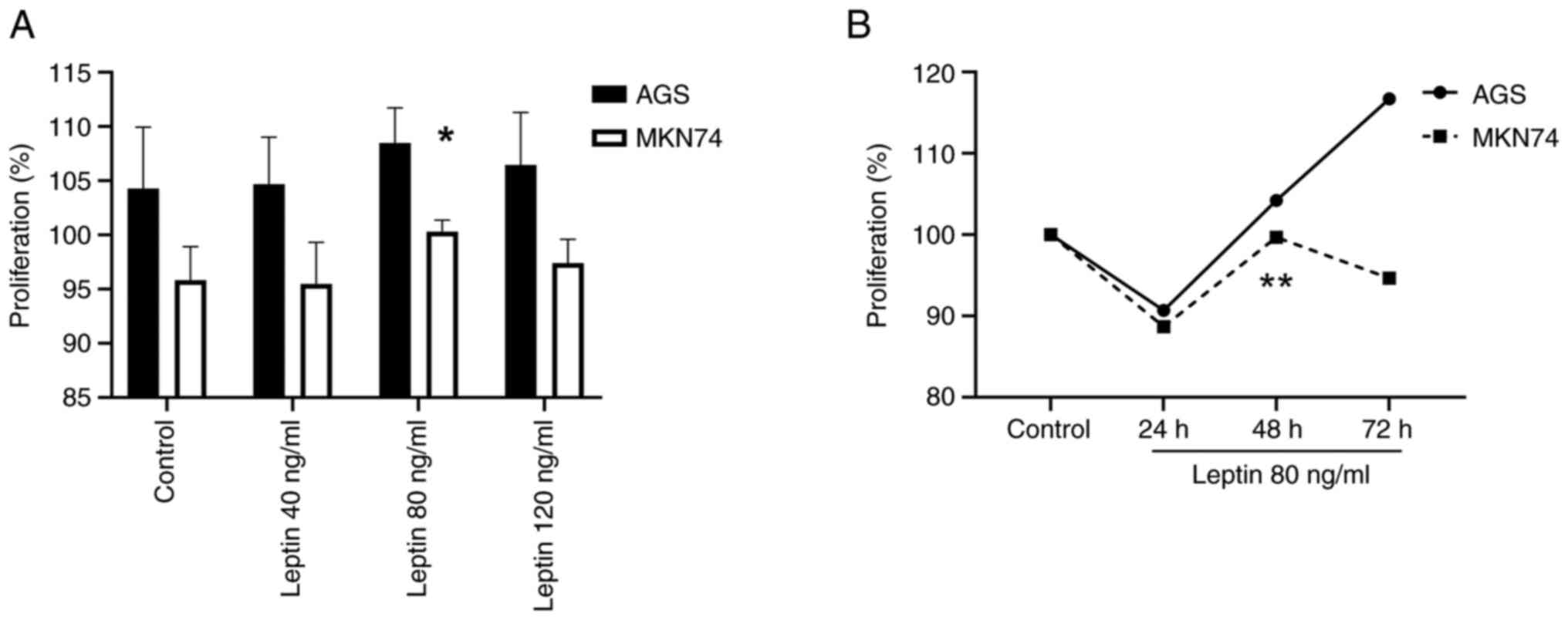
shown). Leptin was associated with the proliferation of gastric
cancer cells (Fig. 2A and B).
Particularly, the effect of leptin at 80 ng/ml in MKN74 cells was
significantly different compared with the controls (P=0.034). In
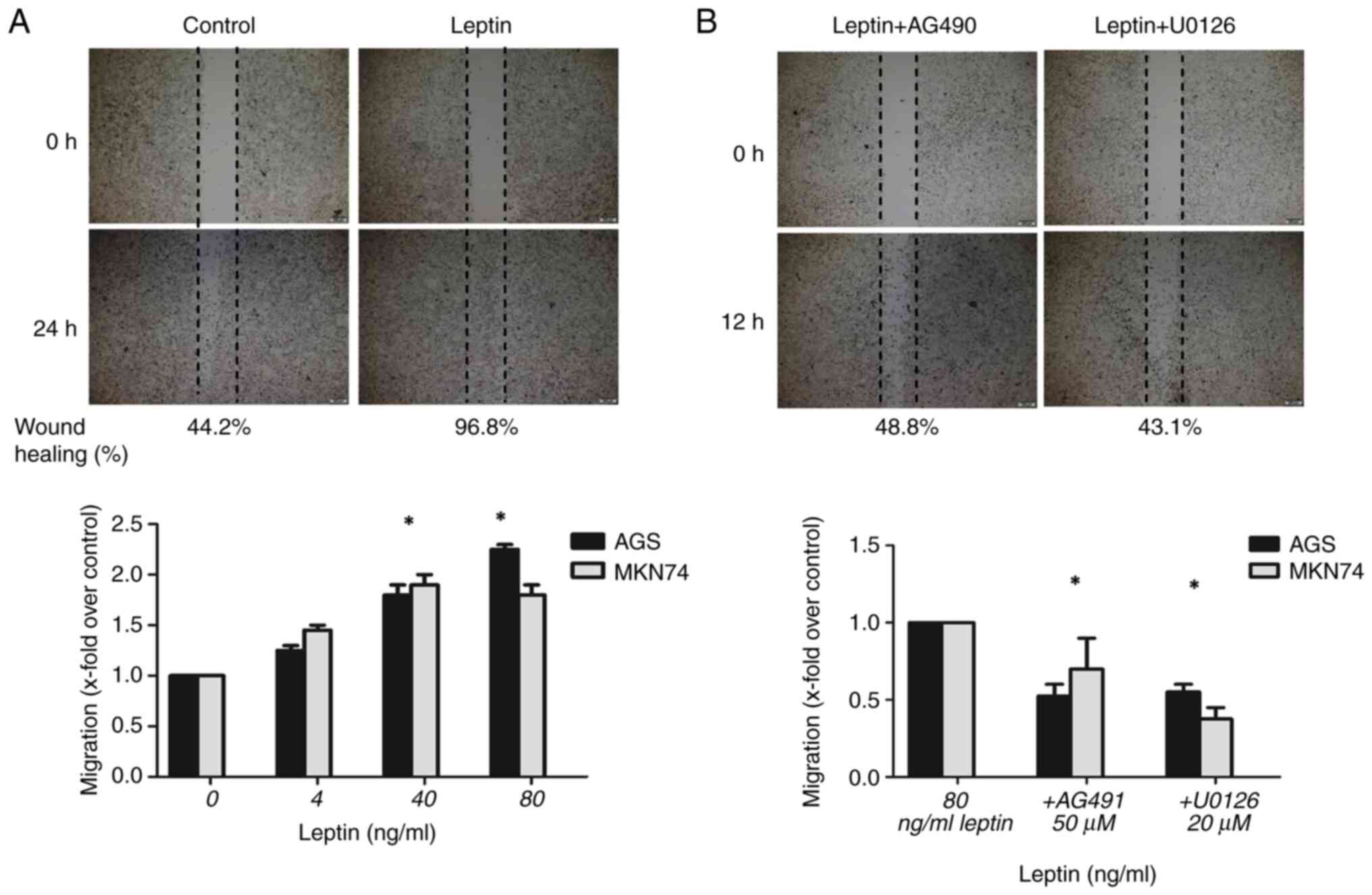
addition, leptin (4–80 ng/ml) significantly enhanced cell
migration, with a maximal response at 80 ng/ml in AGS cells and 40
ng/ml in MKN74 cells compared with the controls (P<0.001 for
both). The number of migratory AGS and MKN74 cells was 2.3-fold
(P=0.01) and 1.8-fold higher, respectively, in the presence of 80
ng/ml leptin (Figs. 3A and
S1A). The movement of
leptin-treated cells (80 ng/ml) was inhibited by the JAK-STAT
inhibitor AG490 and MEK inhibitor U0126. Moreover, leptin-induced
migration was inhibited by 48% in AGS cells by 50 µM AG490
(P<0.001), and by 68% in MKN74 cells by 20 µM U0126 (P<0.001)
(Figs. 3B and S1B).
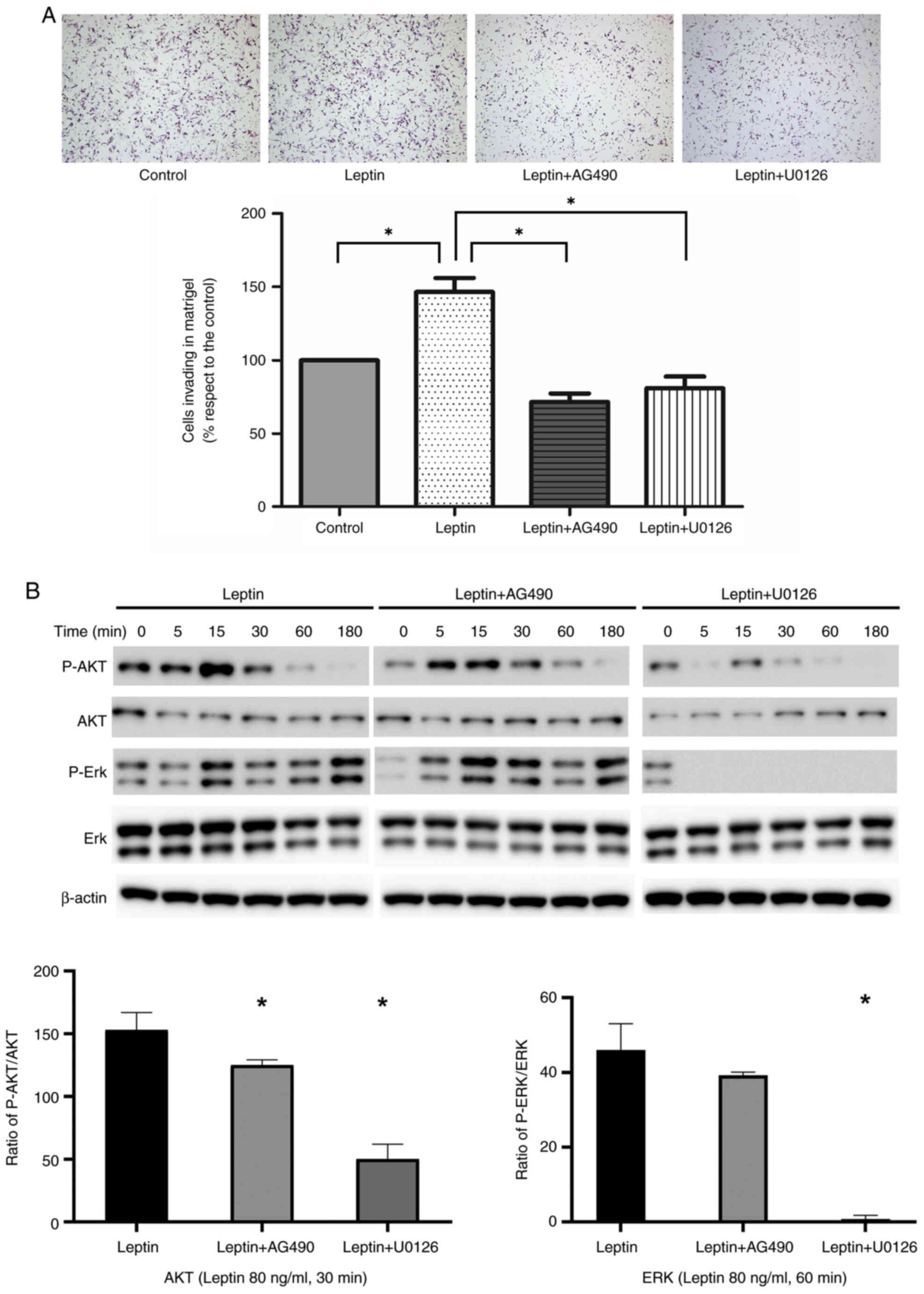
In a Matrigel Boyden chamber assay, leptin promoted
AGS cell invasion of Matrigel (P<0.001). Invasion by
leptin-treated cells was inhibited by AG490 and U0126 (P<0.001;
Fig. 4A). Next, AGS cells were
pre-incubated with JAK and MEK inhibitors. Western blotting and
quantitative analysis suggested that the effect of leptin-induced
phosphorylation of AKT and ERK proteins in AGS cells was
significantly suppressed by AG491 or U0126. β-actin was used as the
internal control (Fig. 4B).
Similarly, the MMP-9 protein levels were slightly upregulated by
leptin in AGS cell line and this effect was suppressed by AG490 or
U0126 (Fig. S2).
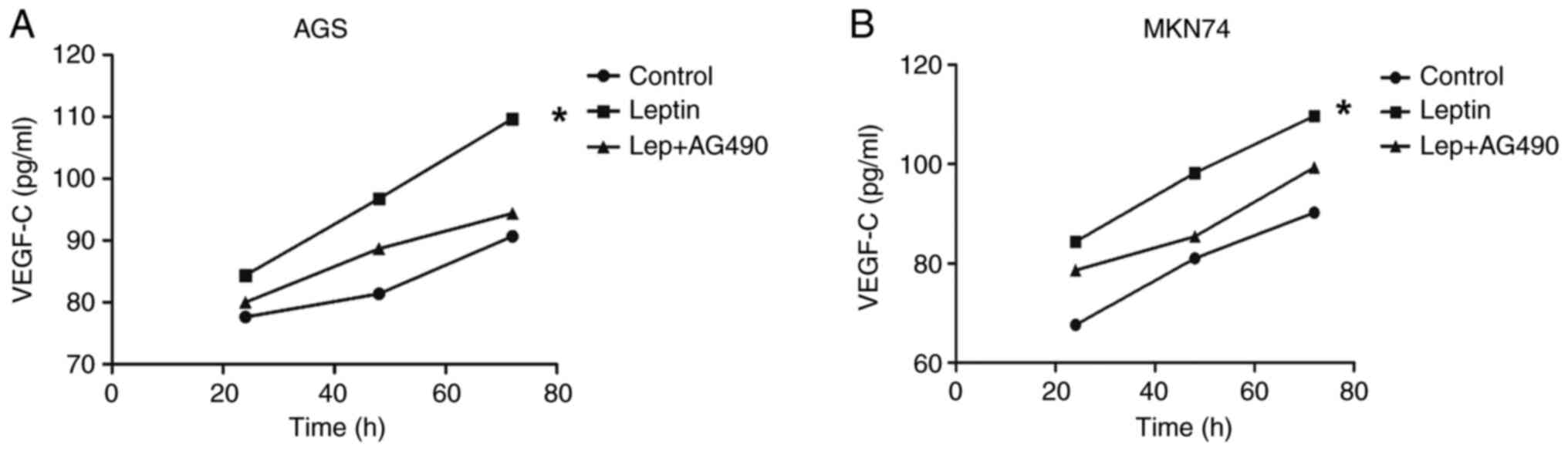
Leptin promotes VEGF-C secretion by
AGS and MKN74 cells
AGS and MKN74 cells did not express VEGF-C in the
presence or absence of leptin (data not shown). The effect of
leptin on the VEGF-C level in AGS cell supernatant was analyzed.
Initially, both AGS and MKN74 cells expressed low levels of VEGF-C
in response to leptin. Significant time-dependent effects were
observed in AGS and MKN cells (P<0.001). ELISA revealed that
leptin increased the VEGF-C levels in AGS and MKN74 cell
supernatants compared with the control. Moreover, AG490
significantly decreased the VEGF-C levels in AGS and MKN74 cell
supernatants compared with the leptin group (P=0.008; Fig. 5A and B).
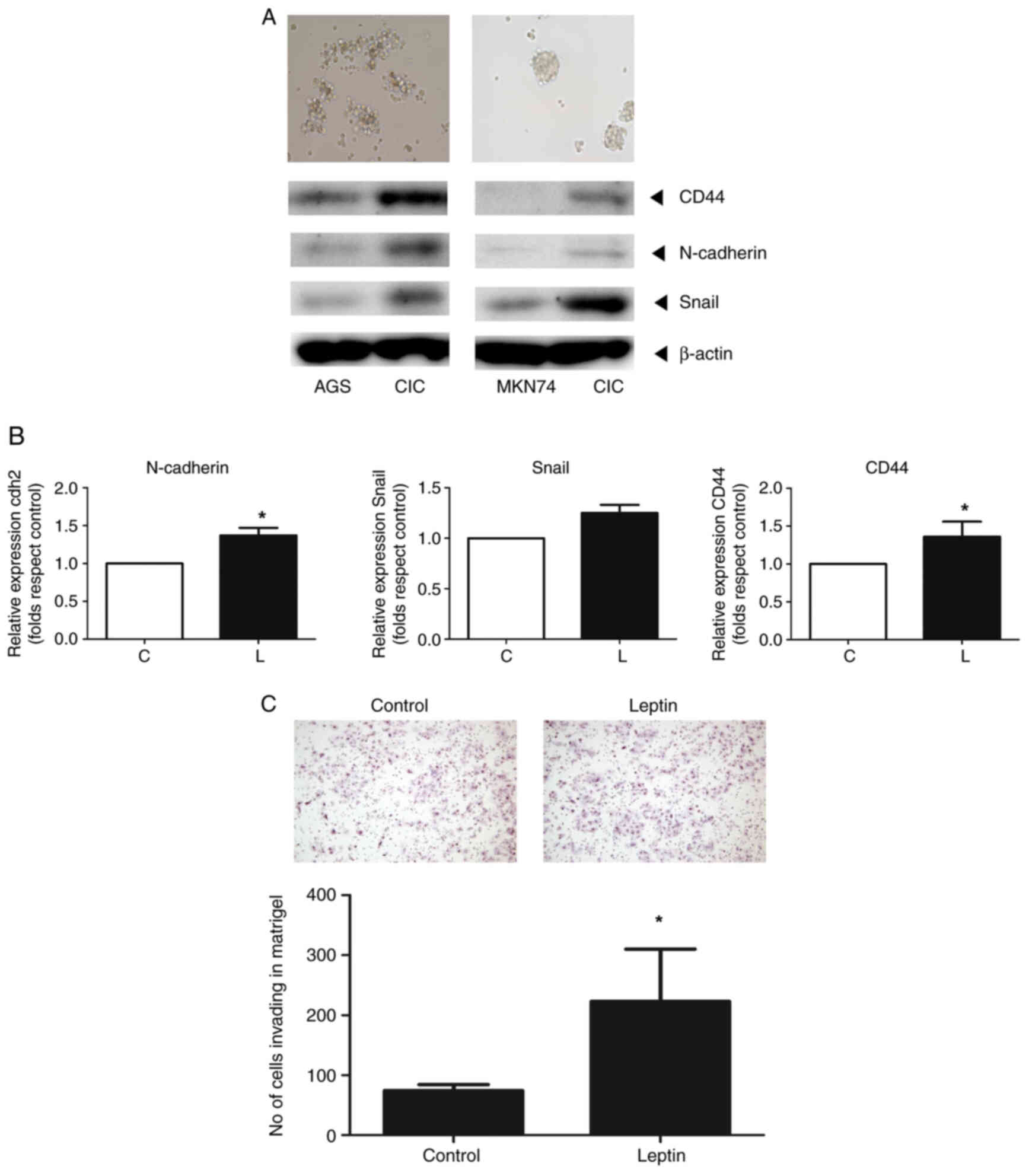
Leptin induces stemness and
epithelial-mesenchymal transition (EMT)
CICs were isolated from AGS and MKN74 gastric cells
using stem cell selection medium. Western blotting showed that the
protein levels of markers for CD44, N-cadherin and Snail increased
in both cell types (Fig. 6A).
Leptin increased the mRNA levels of markers of stemness (CD44) and
EMT (Snail and N-cadherin) one- to two-fold compared with the
controls, as determined by RT-qPCR (Fig. 6B). Finally, leptin increased the
invasion of CICs 2.1-fold (Fig.
6C).
Discussion
According to a nationwide survey by the Korean
Gastric Cancer Association, the rate of obese patients with gastric
cancer increased from 28.7% in 2014 to 35.3% in 2019 (22). In the present study, leptin
increased the invasion of AGS and MKN74 cells and activated the
STAT and MEK signaling pathways, which are involved in cell growth
and survival. Also, exogenous leptin modulates growth factor
expression and cell migration. In CICs isolated from gastric cancer
cells, leptin maintains stemness and EMT expression, and promotes
spheroid formation and invasion.
In the gastrointestinal system, leptin receptors are
expressed in the stomach and bowels, but leptin is produced only by
parietal cells and chief cells in the stomach (9,23).
Therefore, the stomach is unique in constitutively expressing both
leptin and leptin receptors, and is capable of transducing
autocrine leptin signals. Leptin is reportedly associated with more
aggressive gastric cancers, which are associated with poorer
survival (24,25). This suggests that obesity may be
associated with the progression of gastric cancer. The serum leptin
level, which is affected by adipose tissue mass, is elevated in
obese patients and associated with an increased risk of several
types of cancer (26–30). In the present study, the overweight
group had a significantly higher serum leptin level. In a study of
63 patients with gastric cancer, the serum leptin level was higher
in the high-than low-BMI group (20). In addition, the overweight group
was more likely to have advanced disease, and had poorer OS.
Therefore, obesity is associated with the prognosis of gastric
cancer in a manner involving the serum leptin level.
Leptin and leptin receptor (Ob-R) were predominantly
expressed in chief and parietal cells, but not in the surface
epithelium in normal gastric mucosa adjacent to cancer tissue
(31,32). Another study reported that the
expression level of both leptin and Ob-R tended to increase as the
depth of tumor invasion or TNM stage progressed (24). According to our previous study, the
expression of leptin was significantly associated with TNM stage
(33). Moreover, nodal and distant
metastasis was frequently detected in leptin-strong and Ob-R
positive tumors as compared with leptin-weak and Ob-R negative
tumors (24,25). The present results were consistent
with previous studies and suggested that leptin and Ob-R may
function as an autocrine growth factor during the development and
progression of gastric cancer. In the present study, the OS in the
overweight group tended to be poorer than in the normal group.
Therefore, there is a possibility that serum leptin level may have
relationship with OS. However, since the sample size of the present
study was small, it was not possible to perform subgroup analysis
by adjusting the TNM stage, which is a limitation to the present
study. Further larger cohort research with adjusting for TNM stage
should be conducted in the future.
Metastasis is a hallmark of malignancy and a leading
cause of cancer-related death (34). Cell migration and invasion are
crucial for tumor progression and metastasis. These processes
involve detachment of tumor cells from the primary site, migration
thereof through primary tissue and intravasation into the blood or
lymphatic systems, and invasion and proliferation in distant organs
(35). In the present study,
leptin induced cancer cell migration and invasion. Indeed, leptin
reportedly enhances gastric cancer cell migration by increasing
intercellular adhesion molecule-1 (ICAM-1) expression (36). In addition, leptin may exert
effects via the JAK-STAT and MEK pathways, likely by
phosphorylating AKT and ERK. The present study revealed that leptin
may activate the JAK-STAT and MEK pathways, which is consistent
with the previous findings that several signaling pathways
activated by leptin have been identified in gastric cancer, that
is, the JAK-STAT, ERK1/2 and MEK pathways, as well as the PI3K/AKT
pathways. A previous study revealed that leptin upregulated ICAM-1
expression in eosinophils by the combined activation of the MAPK
and JAK pathways (36–39). However, their changes were not
examined in the present study. Further experiments will be
conducted to test these possibilities. In vitro, leptin
induced gastric cancer cell migration via the Rho/ROCK pathway,
which regulates actin-myosin assembly and generates a traction
force (36,40).
The AKT signaling pathway seems to be implicated in
gastric cancer cell invasion. Membrane type 1-matrix
metalloproteinase (MT1-MMP) is important for tumor invasion and
MT1-MMP overexpression may be associated with gastric cancer cell
invasion (41,42). The present study indicated that
leptin affected the expression of MMP-9 protein and this effect was
likely to have association with the JAK-STAT and MEK signaling
pathways. Consistent with these results, previous studies reported
that leptin triggered the generation and surface localization of
MT1-MMP, likely in a manner involving the AKT pathway (43,44).
Similarly, leptin binding to the Ob-R activates several signaling
pathways. These findings suggested that leptin promotes cancer
migration and invasion by activating signaling pathways; blockade
of its action may have therapeutic potential.
Lymphatic metastasis of gastric cancer is an
important prognostic factor, and is included in the staging system
for gastric cancer. Lymphangiogenesis mediates cancer cell entry
into the lymphatic system. VEGF-C is important in
lymphangiogenesis, as is its receptor, which is primarily expressed
on the lymphatic vessel endothelium (45). VEGF-C is associated with lymphatic
metastasis in patients with early gastric cancer (46,47).
The serum VEGF-C level is associated with poor OS and
disease-specific survival, and thus has prognostic potential
(45). In the present study,
leptin induced VEGF-C secretion by gastric cancer cells via the
JAK-STAT pathway in a time-dependent manner. Ramucirumab, a
monoclonal antibody directed against VEGF receptor 2, is
recommended as second-line palliative chemotherapy by the Korean
Gastric Cancer Association (48).
By considering the leptin-VEGF-C relationship, antiangiogenetic
agents could be extended to adjuvant settings.
The EMT is an important step in metastasis (49,50).
The EMT is the process by which epithelial cells lose their
characteristic polarity and cell-cell adhesion ability, and acquire
migratory and invasive characteristics (51). E-cadherin is an important component
of adherent junctions and stabilizes cell-cell connections
(52). N-cadherin is related to
cell motility and migration. During the EMT, the E- to N-cadherin
switch is a hallmark of cancer progression (53). SNAIL is a major EMT transcription
factor, and is reportedly related to the downregulation of
E-cadherin in colon cancer (54).
CICs, a subgroup of cancer cells expressing EMT and stemness
markers within tumors, likely adapt and respond to environmental
stimuli (50). As in breast and
ovarian cancers, it was found that leptin induces the expression of
EMT markers (N-cadherin and Snail) and stemness markers (CD44) in
CICs isolated from gastric cancer (50,55).
In addition, leptin promoted spheroid formation and invasion of
Matrigel by CICs. Therefore, leptin promotes the maintenance of an
aggressive phenotype of gastric cancer. However, the flow
cytometric assay was not examined in the present study to prove
that leptin could maintain the stemness of gastric cancer cells,
and to detect the proportion of CD44+ cells or
CD133+ cells. Further experiments to elucidate more
markers will be required to confirm the results of the present
study.
There are several limitations to the present study.
The expression of leptin or its receptor in gastric cancer and
corresponding normal tissues, suggesting that leptin is produced
locally and functions in a paracrine or autocrine manner, was not
evaluated. In addition, further research to improve the reliability
of the results that leptin affects the JAK-STAT and MEK signaling
pathways is required in tissue or animals. Second, a retrospective
cohort study was conducted to evaluate the prognostic significance
of leptin and obesity, but certain clinicopathological
characteristics were imbalanced between the groups. Third, the
present study used a single-center design with small sample size
and was therefore prone to inherent bias. Although a relationship
was found between the serum leptin level and cancer progression,
further research with a larger cohort is required to validate the
association. Finally, the inhibition study was not conducted using
CICs and further research about the leptin-associated signaling
pathways in CICs is required.
In conclusion, a relationship between the serum
leptin level and obesity and gastric cancer progression was
identified, supporting an adverse effect of obesity in gastric
cancer. It was also demonstrated that leptin promotes gastric
cancer progression via the STAT and MEK signaling pathways, by
inducing the migration and invasion of gastric cancer cells.
Therefore, targeting leptin-associated signaling pathways could
have therapeutic potential for gastric cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by a research grant from St.
Vincent's Hospital, Catholic University of Korea (grant no.
SVHR201707).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KBP conducted investigation and wrote the original
draft. EYK conceptualized the present study and conducted
investigation. HC curated data, wrote, reviewed and edited the
manuscript. KBP and KHJ confirm the authenticity of all the raw
data. DJY conducted investigation. KHJ conceptualized and
supervised the study, performed investigation, wrote, reviewed and
edited the manuscript. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
The present study was approved (approval no.
VCM07BR016) by the Institutional Review Board of the College of
Medicine, Catholic University of Korea (Seoul, Republic of Korea)
and complied with its ethical standards. Written informed consent
was provided by all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
NCD Risk Factor Collaboration (NCD-RisC),
. Worldwide trends in body-mass index, underweight, overweight, and
obesity from 1975 to 2016: A pooled analysis of 2416
population-based measurement studies in 128·9 million children,
adolescents, and adults. Lancet. 390:2627–2642. 2017. View Article : Google Scholar
|
|
2
|
Lauby-Secretan B, Scoccianti C, Loomis D,
Grosse Y, Bianchini F and Straif K; International Agency for
Research on Cancer Handbook Working Group, : Body fatness and
cancer-viewpoint of the IARC working group. N Engl J Med.
375:794–798. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guilford P, Hopkins J, Harraway J, McLeod
M, McLeod N, Harawira P, Taite H, Scoular R, Miller A and Reeve AE:
E-cadherin germline mutations in familial gastric cancer. Nature.
392:402–405. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hirabayashi M, Inoue M, Sawada N, Saito E,
Abe SK, Hidaka A, Iwasaki M, Yamaji T, Shimazu T, Shibuya K, et al:
Effect of body-mass index on the risk of gastric cancer: A
population-based cohort study in A Japanese population. Cancer
Epidemiol. 63:1016222019. View Article : Google Scholar
|
|
5
|
Turati F, Tramacere I, La Vecchia C and
Negri E: A meta-analysis of body mass index and esophageal and
gastric cardia adenocarcinoma. Ann Oncol. 24:609–617. 2013.
View Article : Google Scholar
|
|
6
|
Renehan AG, Zwahlen M and Egger M:
Adiposity and cancer risk: New mechanistic insights from
epidemiology. Nat Rev Cancer. 15:484–498. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rubinstein MM, Brown KA and Iyengar NM:
Targeting obesity-related dysfunction in hormonally driven cancers.
Br J Cancer. 125:495–509. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Masuzaki H, Ogawa Y, Sagawa N, Hosoda K,
Matsumoto T, Mise H, Nishimura H, Yoshimasa Y, Tanaka I, Mori T and
Nakao K: Nonadipose tissue production of leptin: Leptin as a novel
placenta-derived hormone in humans. Nat Med. 3:1029–1033. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bado A, Levasseur S, Attoub S, Kermorgant
S, Laigneau JP, Bortoluzzi MN, Moizo L, Lehy T, Guerre-Millo M, Le
Marchand-Brustel Y and Lewin MJ: The stomach is a source of leptin.
Nature. 394:790–793. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kolaczynski JW, Considine RV, Ohannesian
J, Marco C, Opentanova I, Nyce MR, Myint M and Caro JF: Responses
of leptin to short-term fasting and refeeding in humans: A link
with ketogenesis but not ketones themselves. Diabetes.
45:1511–1515. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Surmacz E: Obesity hormone leptin: A new
target in breast cancer? Breast Cancer Res. 9:3012007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tartaglia LA: The leptin receptor. J Biol
Chem. 272:6093–6096. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saxena NK, Sharma D, Ding X, Lin S, Marra
F, Merlin D and Anania FA: Concomitant activation of the JAK/STAT,
PI3K/AKT, and ERK signaling is involved in leptin-mediated
promotion of invasion and migration of hepatocellular carcinoma
cells. Cancer Res. 67:2497–2507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Carino C, Olawaiye AB, Cherfils S,
Serikawa T, Lynch MP, Rueda BR and Gonzalez RR: Leptin regulation
of proangiogenic molecules in benign and cancerous endometrial
cells. Int J Cancer. 123:2782–2790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Donato J Jr, Frazão R and Elias CF: The
PI3K signaling pathway mediates the biological effects of leptin.
Arq Bras Endocrinol Metabol. 54:591–602. 2010. View Article : Google Scholar
|
|
16
|
Crean-Tate KK and Reizes O: Leptin
regulation of cancer stem cells in breast and gynecologic cancer.
Endocrinology. 159:3069–3080. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cao R, Brakenhielm E, Wahlestedt C,
Thyberg J and Cao Y: Leptin induces vascular permeability and
synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc
Natl Acad Sci USA. 98:6390–6395. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barone I, Catalano S, Gelsomino L, Marsico
S, Giordano C, Panza S, Bonofiglio D, Bossi G, Covington KR, Fuqua
SA and Andò S: Leptin mediates tumor-stromal interactions that
promote the invasive growth of breast cancer cells. Cancer Res.
72:1416–1427. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee KN, Choi HS, Yang SY, Park HK, Lee YY,
Lee OY, Yoon BC, Hahm JS and Paik SS: The role of leptin in gastric
cancer: Clinicopathologic features and molecular mechanisms.
Biochem Biophys Res Commun. 446:822–829. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tas F, Karabulut S, Erturk K and
Duranyildiz D: Clinical significance of serum leptin level in
patients with gastric cancer. Eur Cytokine Netw. 29:52–58. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Information Committee of the Korean
Gastric Cancer Association, . Korean gastric cancer association-led
nationwide survey on surgically treated gastric cancers in 2019. J
Gastric Cancer. 21:221–235. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mix H, Widjaja A, Jandl O, Cornberg M,
Kaul A, Göke M, Beil W, Kuske M, Brabant G, Manns MP and Wagner S:
Expression of leptin and leptin receptor isoforms in the human
stomach. Gut. 47:481–486. 2000. View Article : Google Scholar
|
|
24
|
Ishikawa M, Kitayama J and Nagawa H:
Expression pattern of leptin and leptin receptor (OB-R) in human
gastric cancer. World J Gastroenterol. 12:5517–5522. 2006.
View Article : Google Scholar
|
|
25
|
Zhao X, Huang K, Zhu Z, Chen S and Hu R:
Correlation between expression of leptin and clinicopathological
features and prognosis in patients with gastric cancer. J
Gastroenterol Hepatol. 22:1317–1321. 2007. View Article : Google Scholar
|
|
26
|
Stattin P, Lukanova A, Biessy C, Söderberg
S, Palmqvist R, Kaaks R, Olsson T and Jellum E: Obesity and colon
cancer: Does leptin provide a link? Int J Cancer. 109:149–152.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Garofalo C, Koda M, Cascio S, Sulkowska M,
Kanczuga-Koda L, Golaszewska J, Russo A, Sulkowski S and Surmacz E:
Increased expression of leptin and the leptin receptor as a marker
of breast cancer progression: Possible role of obesity-related
stimuli. Clin Cancer Res. 12:1447–1453. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yoon YS, Kwon AR, Lee YK and Oh SW:
Circulating adipokines and risk of obesity related cancers: A
systematic review and meta-analysis. Obes Res Clin Pract.
13:329–339. 2019. View Article : Google Scholar
|
|
29
|
Thomas T, Burguera B, Melton LJ III,
Atkinson EJ, O'Fallon WM, Riggs BL and Khosla S: Relationship of
serum leptin levels with body composition and sex steroid and
insulin levels in men and women. Metabolism. 49:1278–1284. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mantzoros CS: The role of leptin in human
obesity and disease: A review of current evidence. Ann Intern Med.
130:671–680. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Azuma T, Suto H, Ito Y, Ohtani M, Dojo M,
Kuriyama M and Kato T: Gastric leptin and Helicobacter pylori
infection. Gut. 49:324–329. 2001. View Article : Google Scholar
|
|
32
|
Schneider R, Bornstein SR, Chrousos GP,
Boxberger S, Ehninger G and Breidert M: Leptin mediates a
proliferative response in human gastric mucosa cells with
functional receptor. Horm Metab Res. 33:1–6. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim JH, Jung H, Jun KH, Kim SK, Chin HM,
Jung JH, Kim W, Jeon HM, Park CH, Park SM, et al: Correlation
between the serum leptin level and the expression of leptin in
stomach cancer patients. J Korean Gastric Cancer Assoc. 8:176–181.
2008. View Article : Google Scholar
|
|
34
|
Seyfried TN and Huysentruyt LC: On the
origin of cancer metastasis. Crit Rev Oncog. 18:43–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lambert AW, Pattabiraman DR and Weinberg
RA: Emerging biological principles of metastasis. Cell.
168:670–691. 2017. View Article : Google Scholar
|
|
36
|
Dong Z, Fu S, Xu X, Yang Y, Du L, Li W,
Kan S, Li Z, Zhang X, Wang L, et al: Leptin-mediated regulation of
ICAM-1 is Rho/ROCK dependent and enhances gastric cancer cell
migration. Br J Cancer. 110:1801–1810. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ji BC, Hsiao YP, Tsai CH, Chang SJ, Hsu
SC, Liu HC, Huang YP, Lien JC and Chung JG: Cantharidin impairs
cell migration and invasion of A375.S2 human melanoma cells by
suppressing MMP-2 and −9 through PI3K/NF-κB signaling pathways.
Anticancer Res. 35:729–738. 2015.PubMed/NCBI
|
|
38
|
Mavrommati I, Cisse O, Falasca M and
Maffucci T: Novel roles for class II Phosphoinositide 3-Kinase C2β
in signalling pathways involved in prostate cancer cell invasion.
Sci Rep. 6:232772016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wong CK, Cheung PF and Lam CW:
Leptin-mediated cytokine release and migration of eosinophils:
Implications for immunopathophysiology of allergic inflammation.
Eur J Immunol. 37:2337–2348. 2007. View Article : Google Scholar
|
|
40
|
Ghasemi A, Saeidi J, Azimi-Nejad M and
Hashemy SI: Leptin-induced signaling pathways in cancer cell
migration and invasion. Cell Oncol (Dordr). 42:243–260. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mimori K, Fukagawa T, Kosaka Y, Ishikawa
K, Iwatsuki M, Yokobori T, Hirasaki S, Takatsuno Y, Sakashita H,
Ishii H, et al: A large-scale study of MT1-MMP as a marker for
isolated tumor cells in peripheral blood and bone marrow in gastric
cancer cases. Ann Surg Oncol. 15:2934–2942. 2008. View Article : Google Scholar
|
|
42
|
Poincloux R, Lizárraga F and Chavrier P:
Matrix invasion by tumour cells: A focus on MT1-MMP trafficking to
invadopodia. J Cell Sci. 122:3015–3024. 2009. View Article : Google Scholar
|
|
43
|
Dong Z, Xu X, Du L, Yang Y, Cheng H, Zhang
X, Li Z, Wang L, Li J, Liu H, et al: Leptin-mediated regulation of
MT1-MMP localization is KIF1B dependent and enhances gastric cancer
cell invasion. Carcinogenesis. 34:974–983. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jin G, Peng L, Zhang J, Qu L and Shou C:
Cancer and embryo expression protein 65 promotes cancer cell growth
and metastasis. Oncol Lett. 9:1772–1778. 2015. View Article : Google Scholar
|
|
45
|
Petrillo A, Laterza MM, Tirino G, Pompella
L, Pappalardo A, Ventriglia J, Savastano B, Auricchio A, Orditura
M, Ciardiello F, et al: Increased circulating levels of vascular
endothelial growth factor C can predict outcome in resectable
gastric cancer patients. J Gastrointest Oncol. 10:314–323. 2019.
View Article : Google Scholar
|
|
46
|
Onogawa S, Kitadai Y, Amioka T, Kodama M,
Cho S, Kuroda T, Ochiumi T, Kimura S, Kuwai T, Tanaka S and Chayama
K: Expression of vascular endothelial growth factor (VEGF)-C and
VEGF-D in early gastric carcinoma: Correlation with
clinicopathological parameters. Cancer Lett. 226:85–90. 2005.
View Article : Google Scholar
|
|
47
|
Arigami T, Natsugoe S, Uenosono Y,
Yanagita S, Ehi K, Arima H, Mataki Y, Nakajo A, Ishigami S and
Aikou T: Vascular endothelial growth factor-C and -D expression
correlates with lymph node micrometastasis in pN0 early gastric
cancer. J Surg Oncol. 99:148–153. 2009. View Article : Google Scholar
|
|
48
|
Guideline Committee of the Korean Gastric
Cancer Association (KGCA), . Development Working Group & Review
Panel: Korean practice guideline for gastric cancer 2018: An
evidence-based, multi-disciplinary approach. J Gastric Cancer.
19:1–48. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Voulgari A and Pintzas A:
Epithelial-mesenchymal transition in cancer metastasis: Mechanisms,
markers and strategies to overcome drug resistance in the clinic.
Biochim Biophys Acta. 1796:75–90. 2009.
|
|
50
|
Kato S, Abarzua-Catalan L, Trigo C,
Delpiano A, Sanhueza C, García K, Ibañez C, Hormazábal K, Diaz D,
Brañes J, et al: Leptin stimulates migration and invasion and
maintains cancer stem-like properties in ovarian cancer cells: An
explanation for poor outcomes in obese women. Oncotarget.
6:21100–21119. 2015. View Article : Google Scholar
|
|
51
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Frixen UH, Behrens J, Sachs M, Eberle G,
Voss B, Warda A, Löchner D and Birchmeier W: E-cadherin-mediated
cell-cell adhesion prevents invasiveness of human carcinoma cells.
J Cell Biol. 113:173–185. 1991. View Article : Google Scholar
|
|
53
|
Wheelock MJ, Shintani Y, Maeda M, Fukumoto
Y and Johnson KR: Cadherin switching. J Cell Sci. 121:727–735.
2008. View Article : Google Scholar
|
|
54
|
Peña C, García JM, Silva J, García V,
Rodríguez R, Alonso I, Millán I, Salas C, de Herreros AG, Muñoz A
and Bonilla F: E-cadherin and vitamin D receptor regulation by
SNAIL and ZEB1 in colon cancer: Clinicopathological correlations.
Hum Mol Genet. 14:3361–3370. 2005. View Article : Google Scholar
|
|
55
|
Zheng Q, Banaszak L, Fracci S, Basali D,
Dunlap SM, Hursting SD, Rich JN, Hjlemeland AB, Vasanji A, Berger
NA, et al: Leptin receptor maintains cancer stem-like properties in
triple negative breast cancer cells. Endocr Relat Cancer.
20:797–808. 2013. View Article : Google Scholar : PubMed/NCBI
|