Introduction
Hepatocellular carcinoma (HCC) is the fourth most
prevalent type of cancer worldwide and the sixth most frequent
malignancy (1). In 2020, 45% of the
year's new cases (910,000) and 47% of the year's fatalities
(830,000) occurred in China (2).
The data for GCC in China are still not encouraging, despite the
recent worldwide reduction in the incidence of this type of cancer.
The explanation for this may be related to a shift in the etiology;
an increasing number of HCC cases are being linked to faulty lipid
metabolism, which also causes non-alcoholic fatty liver disease
(NAFLD), non-alcoholic steatohepatitis and even HCC. As a result, a
new risk factor is replacing the classic theory of the evolution of
viral hepatitis, namely abnormal lipid metabolism.
The dysregulation of lipid droplets (LDs) may
interact with the endoplasmic reticulum (ER), mitochondria,
peroxisomes, vesicles and lysosomes under normal oxygen conditions
(3). In addition, the tumor
microenvironment may lead to dysregulation via factors, such
hyperinsulinemia caused by insulin resistance and increased
pro-inflammatory cytokine levels (4). Previous research has emphasized the
role of modifications in signaling pathways and enzyme metabolism
during the production of fatty acid synthesis (FAS) (5). However, available data on the
association between the ubiquitin regulatory X domain-containing
protein (UBX) family and endoplasmic reticulum-associated
degradation (ERAD) are limited. The UBX family, in contrast to
earlier research (6), increases
cellular stability, primarily through ERAD or increased
intranuclear translocation mechanisms, a process that even inhibits
FAS. Despite this apparent contradiction with the main line of
inquiry, there is still evidence to support the increased
expression of UBX family genes or proteins in HCC and their roles
as poor prognostic factors (7).
The UBX family, which consists of the 13 members
UBXN1, UBXN2A-2C, UBXN3A-3B, UBXN4 and UBXN6-11, has a structural
domain that is comparable to the N terminus of ubiquitin. The N
terminus of the receptor protein is identified and transported to
the proteasome, where it undergoes the standard protein degradation
process and finally degrades into smaller polypeptides and amino
acids (8). An essential organelle
for the synthesis, folding, secretion and recycling of proteins
across organelles is the ER. When elements such as Ca2+
levels, energy and nutrition are aberrant, abnormal protein folding
occurs in the ER, a process known as ER stress, which is required
to maintain ER homeostasis (9). As
a result, ERAD is a crucial process that eliminates unfolded or
misfolded proteins when ER stress arises in order to return the ER
to its normal state and function (10).
Of the UBX family, nine members, namely UBXN1,
UBXN2A-2C, UBXN3B, UBXN4, UBXN6, UBXN8 and UBXN10, are involved in
ERAD during lipid metabolism (Fig.
1). UBXN1 interacts with apoptosis protein inhibitors, blocks
and interferes with retinoic acid-inducible gene (RIG-I)-like
receptors and the nuclear factor (NF)-κB pathway, and is a key
player in cell signaling, endocytosis and DNA damage repair
(11). UBXN1 is a component of the
reverse translocation degradation complex (11) and is involved in ER stress-mediated
de-glycosylation (11). Trimers of
UBXN2A, 2B and 2C bind to cytoplasmic p97 and are involved in the
stability of the ER, the Golgi apparatus and the rearrangement of
the mitotic terminal (12). Based
on remodeling mechanisms, p97, an enzyme belonging to the adenosine
triphosphate (ATP) enzyme family, is connected to a number of
cellular processes and activities (13). p97 may alternatively be considered
as an ‘enzymatic dissociative activity’ protein (13). p97 has two different mechanisms for
protein degradation: One involves the p97-Ufd1-Npl4 complex, which
binds to various ubiquitinated ERAD substrates on the cytoplasmic
side of the ER membrane before being reverse transcribed and
transported to the proteasome for degradation (14); the other involves a protein
degradation pathway that connects the ER to p97 through a number of
cofactors, primarily members of the UBX family. In addition to
regulating the ERAD pathway to preserve ER function under
conditions of ER stress (15),
UBXN3B plays a crucial role in ERAD by limiting the activity of
phospholipases in LDs and preventing the degradation of LDs
(15). In order to attract
valosin-containing protein (VCP) to the ER and encourage ERAD,
UBXN4, an essential membrane protein of the ER, serves as a
platform (16). UBXN6 participates
in lysosomal degradation, may play a role in misfolded proteins in
ERAD, and may adversely affect ATP-driven VCP (17). One of the cofactors of p97, VCP, is
a component of the ATPase complex known as UBXN8, which binds to
p97 and promotes ERAD. VCP/p97 factor UBXN10, a VCP/p97 binding
protein necessary for cilia promotion, has a VCP/p97 substrate
specificity (18).
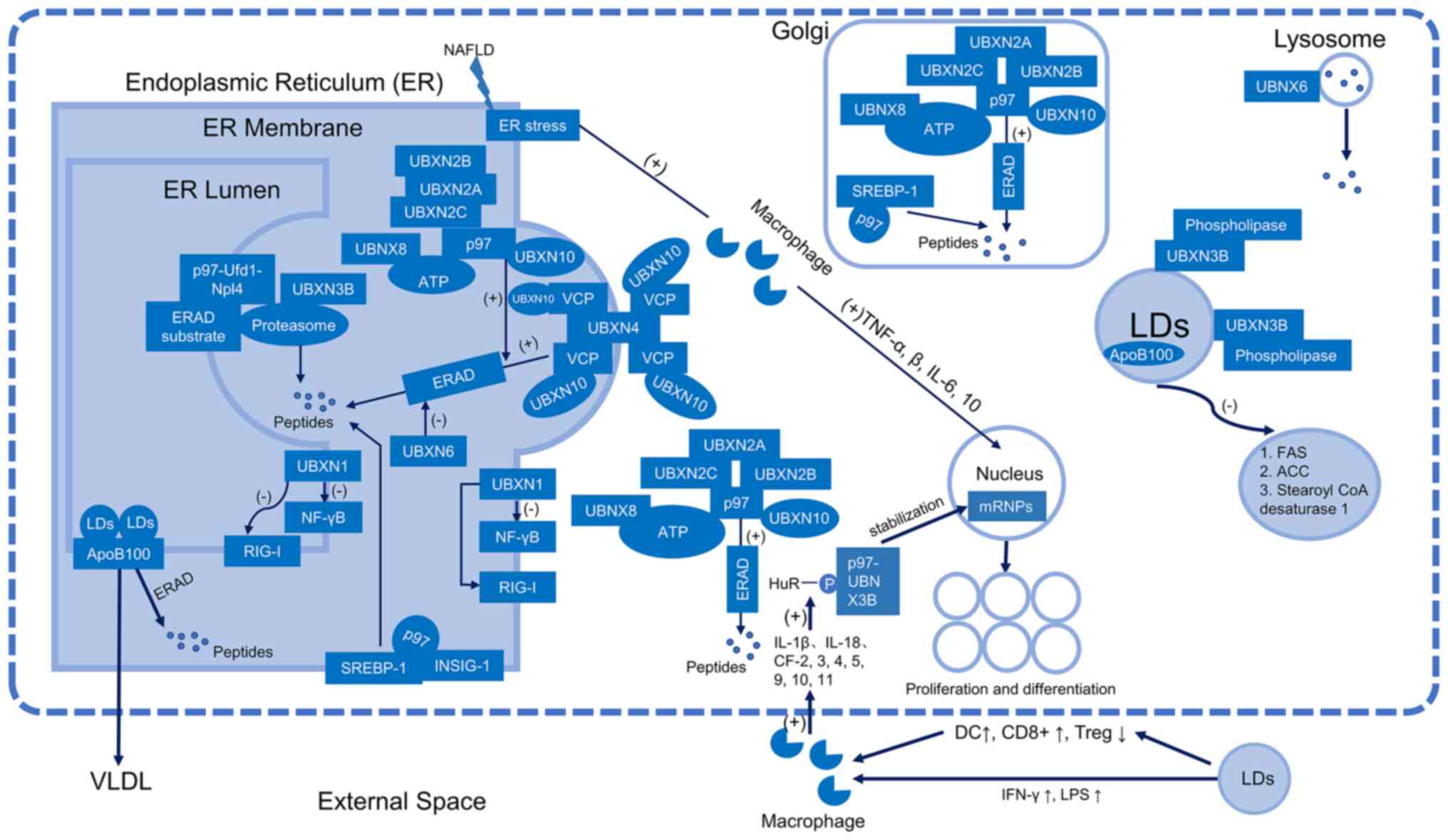 | Figure 1.Schematic representation of the ER
and LD model with the UBXN family and the lipolysis of LDs. ER,
endoplasmic reticulum; LDs, lipid droplets; VLDL, very low-density
lipoprotein; Apo, apolipoprotein; ERAD, endoplasmic
reticulum-associated degradation; UBXN, ubiquitin regulatory X
domain-containing protein; NF-κB, nuclear factor-κB; SREBP-1,
sterol regulatory element-binding protein 1; INSIG-1,
insulin-inducible gene 1; RIG-1, retinoic acid-inducible; ATP,
adenosine triphosphate; NAFLD, non-alcoholic fatty liver disease;
mRNPs, mRNA-protein complexes; FAS, fatty acid synthase; ACC,
acetyl CoA carboxylase; LPS, lipopolysaccharide; DC, dendritic
cell; IFN, interferon; IL, interleukin; CF, chemokine factor; VCP,
valosin-containing protein. |
In the UBX family, UBXN3B has a specific and crucial
function in cancer cells when LD production and enhanced ERAD occur
simultaneously. Other members, on the other hand, perform unique
tasks relating to cellular activity, apoptosis, innate immunity,
Golgi apparatus and lysosomes. As a result, the main aim of the
present review was to describe the mechanisms through which UBXN3B
contributes to the development of HCC.
Transcriptional regulation of UBXN3B
UBXN3B maintains ER stability
UBXN3B is a hairpin-like structural protein that
resembles a hairpin and is comprised of hydrophobic amino acid
residues that are introduced into the ER cytoplasm. UBXD8 migrates
from the ER to the surface of fatty acid-rich LDs in fatty
acid-deficient cells (19), and it
has a UBX structural domain that interacts with p97 (13), which is necessary for ERAD (8). By repressing the transcriptional
activity and target of NAFLD, UBXD8 inhibits the transcriptional
activity of the liver X receptor (LXR), a key regulator of the
enterohepatic cycle. It also inhibits the transcriptional activity
of the sterol regulatory element-binding protein 1 (SREBP-1), and
it stimulates the transcription of genes encoding proteins
necessary for FAS (20). FAS,
acetyl coenzyme A carboxylase, and stearoyl coenzyme A desaturase-1
are all directly activated by the LXR (20). The first and rate-limiting steps of
FAS, which are carried out by these three enzymes, control how
rapidly monounsaturated fatty acids are produced. The transcription
factor termed SREBP-1, which is found in the ER, stimulates the
expression of all the genes necessary for fatty acid metabolism
(21). Its cytoplasmic N-terminal
structural domain is connected to the insulin-inducible gene 1
(INSIG-1) and is located there (22). Without fatty acids, UBXD8 binds to
INSIG-1 and induces the rapid proteasomal degradation of the
receptor protein by attracting p97 to the protein (23). SREBP-1 is moved from the ER to the
Golgi without INSIG-1, where it is broken by two Golgi-localized
proteases (24). This cleavage
allows SREBP-1 to enter the nucleus and activate all FAS-required
genes, as it frees the protein's N-terminal structural domain from
the membrane (21).
By identifying and destroying the ubiquitin-like
structural domains of lipid-binding proteins in the ER, ERAD
guarantees the structural normalcy of the ER. Hepatocytes release
apolipoprotein (Apo) B-100, a glycoprotein with a molecular weight
>500 kDa. Several phases in the lipoprotein transport mechanism
control the production of this protein. When there are more lipids
in the ER lumen, Apo B-100 may develop. Lipid-carrying Apo B-100 is
ubiquitinated by ERAD, which causes the proteasome to degrade
lipid-poor Apo B-100. On the other hand, ubiquitinated Apo B-100
builds up in the LDs when the proteasome is blocked, and it has
been proposed that this may act as a platform for Apo B-100
breakdown (25). It should be noted
that this is only conjecture. According to another study, Apo B-100
predominantly lipidates and builds up in LDs (25). As lipidation only occurs in the ER
lumen, lipidated Apo B-100 is moved to the cytoplasmic side near
the LDs. Thus, the buildup of lipidated Apo B-100 aids in the
development of specific Apo B-100 structures linked to ER-LD. It
should be noted that UBX family members are ubiquitinated proteins
that the ubiquitination-proteasome system degrades under
‘non-essential’ conditions (26).
In other words, ubiquitinated proteins may be deubiquitinated and
retrieved in a hypothesized non-degradation mechanism rather than
necessarily being subject to destruction (26). According to this hypothesis, damaged
proteins either accumulate or continue to degrade in response to
environmental changes and ERAD only functions under specified
circumstances. An essential protein involved in ER stability is the
p97-UBXN3B complex.
UBXN3B maintains fatty acid and
triglyceride homeostasis
The p97-UBXN3B complex regulates the production of
triglycerides and breaks down ubiquitinated proteins in the ER.
p97-UBXN3B controls triglyceride metabolism, in addition to serving
as a sensor of long-chain unsaturated fatty acids (15,23,24).
Unsaturated fatty acids enhance the purification and polymerization
of UBXN3B (15) when it is
cultivated in vitro; in cells without lipids, UBXN3B
prevents triglyceride production, since this process requires the
attachment and the conversion of fatty acids (27). Fatty acids in cells are linked to
phospholipids and do not participate in triglyceride production
when triglyceride synthesis is terminated (15). Excess long-chain unsaturated fatty
acids have the ability to polymerize UBXN3B and interfere with its
ability to perform its functions, resulting in unaltered
triglyceride production (15). By
blocking the rate-limiting enzyme of triglyceride production, the
recruitment of p97 from the ER to the LD surface by UBXN3B
increases the size of LDs and prevents triglyceride hydrolysis to
fatty acids (15). While saturated
fatty acids are unable to interact with UBXN3B, they promote the
conversion of extra unsaturated fatty acids into triglycerides for
storage in the LDs and prevent breakdown by attaching to
phospholipases and blocking their activity (15). Since UBXN3B promotes triglyceride
accumulation in the LDs, while inhibiting triglyceride production
and binding to phospholipids in the ER, this protein constantly
cycles between the two tissues to carry out its various roles
(19).
Other mechanisms
Coenzyme 3-hydroxy-3-methylglutaryl coenzyme A
reductase (HMGCR) is a necessary protein for the ER membrane, and
p97 and ERAD are required for its destruction (26). The degradation of HMGCR induced by
sterols is prevented by the silencing of UBXN3B (28). In hepatocytes, UBXN3B is a poor
predictor of HMGCR degradation. It can thus be hypothesized that
UBXN3B knockdown promotes the production of cholesterol. However,
in hepatocytes with higher HMGCR levels induced by sterol
depletion, there were no changes in messenger ribonucleic acid
(mRNA) or protein expression levels, indicating that
sterol-dependent UBXN3B expression did not promote HMGCR breakdown
(28). Type I interferon and
immunological inflammatory reactions are promoted by the
stimulatory interferon genes (STING) (29). The UBX protein family activates and
controls the biological activities of the interferon-stimulated
response element (ISRE) (30). High
amounts of UBXN3B do not, however, substantially promote ISRE,
indicating that UBXN3B and STING-dependent signaling pathways only
have a positive connection. Additionally, STING is ubiquitinated,
dimerized and is maintained in a phosphorylated state by UBXN3B,
which causes SRING to be destroyed by binding to p97 and the other
E3 ubiquitin ligases (8). Although
the aforementioned HMGCR degradation, ISRE activation and STING
stimulation do not appear to directly interact with UBXN3B,
numerous specific mechanisms of these pathways still need to be
addressed by more extensive research.
Mechanisms of progression from NAFLD to
HCC
LD metabolic disorders are associated with a number
of metabolic illnesses, including obesity, fatty liver, diabetes
and cardiovascular disease (31).
In clinical practice, blood concentrations of total cholesterol,
high-density lipoprotein (HDL), low-density lipoprotein (LDL),
triglycerides and free fatty acids (FFA) are used to determine the
lipid status of a patient. Diseases with high levels of LDs enhance
the risk of tumorigenesis in viral infections (32).
The liver serves as the hub for lipid metabolism
(33). Issues regarding systemic
glucose and NAFLD are tightly related (34). Although excess hepatic lipid levels
have been established to be an independent risk factor and are
strongly associated with the development of HCC, the mechanisms
through which this contributes to fatty acid metabolism remain
unclear (35).
By interfering with signaling pathways and cytokines
via a variety of methods, lipid dysregulation may either directly
or indirectly contribute to the development of cancers (e.g., lipid
regulation, spontaneous synthesis of lipids, ER stress, increased
inflammatory cytokines and immune cells).
Abnormal lipid regulation
Enzymes expressed by a variety of key
transcriptional proteins control normal lipid metabolism (36). Triglycerides do not specifically
harm or destroy cells (8). When the
accumulation of triglycerides sensitizes cells to damage, it
disrupts signaling pathways and gene function, leading to the
dysfunction of lipid-related factors (37).
De novo biosynthesis
The proliferation, invasion and metastasis of cancer
cells, including those in hepatocellular, breast, renal, colorectal
and prostate malignancies, are mediated by high levels of LDs
(38). The unique function of the
liver is to both use and resynthesize FFAs, which are released by
other organs. External lipid absorption is the primary mechanism
through which the enhanced metabolism required for lipolysis as a
means of cell survival is accomplished. Instead, during the process
of cellular energy acquisition, the citric acid cycle, ATP citrate
lyase, and acetyl coenzyme A carboxylase may transform the pyruvate
generated by glycolysis into oxaloacetate and acetyl coenzyme in
the mitochondria (39). Saturated
fatty acids may be created from acetyl coenzyme A via binding and
conversion mechanisms (40).
Stearoyl coenzyme A desaturase converts them to monounsaturated
fatty acids (39,40). In this particular de novo
biosynthesis procedure, the key building blocks of prostaglandin
and membrane production are unsaturated fatty acids. By triggering
autophagy, promoting cell membrane renewal, affecting intracellular
signaling and gene transcription, and boosting energy generation,
the unsaturated fatty acids are crucial for cell survival. Energy
is produced in this process by lipid ‘starvation’, which combines
extracellular resources with lipid de novo production
(41). ATP citrate lyase and acetyl
coenzyme A carboxylase are abundantly expressed in NAFLD after FAS
has developed, increasing FAS, an early sign of NAFLD and fibrosis
(42).
ER stress
ER stress may be observed in patients with NAFLD
(43). The reduction of ER stress
improves NAFLD (43). Even in the
absence of carcinogenic therapy, spontaneous fatty liver disease
may result in HCC, indicating that ER stress is sufficient to
convert NAFLD to HCC. Mechanistically, increased levels of ER
stress cause macrophages to produce increased levels of tumor
necrosis factor (TNF), which promotes cell growth, anti-apoptotic
activity and eventually, tumor development (13). These findings suggest that when
NAFLD progresses to HCC, ER stress may initiate malignant
transformation. The section that follows provides a more in-depth
discussion of what is connected to ER stress.
Inflammatory factors associated with
NAFLD
TNF-α and interleukin (IL)-6 are two inflammatory
factors that are linked to NAFLD (44). Additionally, NAFLD induces the
production of anti-inflammatory cytokines, such as IL-10 or IL-1
receptor antagonists, which prevent NF-κB from being activated and
prevent the release of chemokines, TNF-α and IL-6 (44,45).
TNF-α expression is elevated in individuals with NAFLD, cirrhosis
and HCC, which results in the release of additional cytokines and
chemokines (45,46). TNF-α gene polymorphisms also have a
greater propensity to cirrhosis and NAFLD (45,46).
IL-6 is a significant inducer of C-reactive protein
and hepatocyte production (47) and
may play a role in NAFLD. The direct pathogenic factors are IL-6
and TNF-α. In addition to IL-6 and TNF-α, hepatocyte injury, the
disruption of signaling cascades and functional protein loss are
also induced by IL-10, IL-1 inhibitors and growth factors, which in
turn result in aberrant lipid metabolism. In response to
stress-induced intracellular changes, a surge in inflammatory
substances alters LD proteins and signaling pathways, which in turn
causes cancer cell conversion.
Macrophage function
Numerous immune cells are found in the liver, where
they are affected by portal blood and endure a special tolerant
environment where they may react to foreign pathogens, but avoid
innocuous antigens caused by dietary antigens and microbial
products (48,49). An increased inflammatory activity is
associated with the lipid burden in macrophages (50). M1- and M2-macrophages may arise as a
result of macrophage involvement, depending on the changed
microenvironment. While the latter has an anti-inflammatory and
immunomodulatory function, the former causes the generation of
inflammatory cytokines (51).
M1-macrophages are known to play a role in NAFLD,
with the bacterial endotoxin, lipopolysaccharide, and interferon-γ
helping to activate M1-macrophages and increase the production of
pro-inflammatory cytokines and chemokines, reactive oxygen species
and nitric oxide. According to research, individuals with NAFLD
have higher hepatic levels of certain M1-specific cytokines and
chemokines, such as IL-1β, IL-18, and chemokines 2–5 and 9–11
(52). The increased activation of
cytokines and chemokines, in addition to lipids, oxidation
products, or chemicals produced following hepatocyte damage that
may directly contribute to liver injury, can aggravate
M1-transformation (53). Changes in
the M1/M2 phenotype of macrophages may be influenced by the
environment. Lipid-rich dendritic cells (DCs) struggle to digest
antigens in the presence of tumors (54). Similar to this, under conditions of
steatosis, DCs are recruited into hepatocytes and are maintained at
high levels, despite the fact that their function is activated.
Conversely, the depletion of DCs leads to an increase in liver
inflammation, a decrease in the numbers of Treg, an increase in
CD8+ T-cell function, an increase in immune effector
cell activity and the production of pro-inflammatory cytokines, an
increase in hepatocyte apoptosis, and ultimately, in the
acceleration of liver fibrosis (55). However, another study found higher
levels of natural killer (NK)p46+ cells in NAFLD, which
trigger local and invading macrophages to differentiate into M1 and
stop fibrosis from inducing M2-cells (56). M2-macrophages, however, have been
linked to HCC brought on by NAFLD (57). The influence of the macroenvironment
on macrophage activity and its involvement in the development of
NAFLD-promoted HCC warrant further investigation.
Role of UBXN3B in HCC
ER stress
Increasing attention has been paid to the role that
ER stress plays in the growth, metastasis, angiogenesis and even
treatment resistance of HCC cells (58). Proteotoxic ER stress refers to
disruptions in protein folding in the ER, which triggers unfolded
protein responses (UPRs). UPR activation by ER stress is often
observed as an adaptive mechanism to preserve in vivo
protein homeostasis. In terms of stability to mRNA and ERAD
substrates and modified signaling pathways, the present review also
discusses the mechanisms through which the UBXN3B protein reacts to
ER stress.
Stability of mRNA
The biogenesis and metabolic functions of mRNAs are
connected to a number of different proteins. Pre-mRNA processing,
nuclear export, translation, localization and mRNA decay processes
are the key factors influencing the remodeling events that the
resultant mRNA-protein complexes (mRNPs) go through (59). The most extensively studied effect
on mRNPs is the influence of ATP-dependent RNA helicases, through
which mRNAs are modified to facilitate the ‘metabolism’ of mRNPs
(58). The UBX family plays a
significant role in preserving their stability, since they rely on
ubiquitination signals; HuR, a dominant binding protein that binds
multiple AU-rich regions, is one of these mRNA stabilizers and
performs a crucial stabilizing function as a cytokine and
transcription factor under conditions of cellular stress (58). HuR is often overexpressed and serves
as a representation of the very dynamic alterations that occur
during the recombination and dissociation of mRNPs (60). Researchers have investigated how
phosphorylated HuR regulates protein abundance by influencing the
location and stability of organelles (61). That is, phosphorylated HuR maintains
mRNA stability, while transporting proteins to the nucleus or
degrading proteins in the ER to regulate protein abundance in the
cytoplasm. Through a non-degradative ubiquitination signaling
mechanism that disrupts the metabolism of mRNPs, the p97-UBXD8
complex affects HuR. In fact, HuR only interacts with the p97-UBXD8
complex and not p97 or UBXD8 alone (7). The primary characteristic of the
p97-UBXD8 complex, among the ubiquitination signals of the
non-degradation route, is that the ubiquitinated HuR is not
degraded by the ubiquitin-proteasome system (62). The protein may instead be
deubiquitinated and regenerated via a process known as a
non-degradation route. According to a previous study (63), the p97-UBXD8 complex is involved in
HuR-mRNA modification during the stress response. When cancer
develops, the stabilizing factors, the p97-UBXD8 complex and the
phosphorylated HuR transition, are activated to serve their
stabilizing roles after organelle function and structure have been
compromised, mostly by transitory proliferation and metabolic
abnormalities. The UBX family, particularly the p97-UBXD8 complex,
is one of these modifications that not only improves cellular
stability, but also controls it through autoregulation.
Stabilization of ERAD substrates
Based on the heterogeneity of the N-terminal
structural domains of the UBX family, wherein only five of the 13
proteins in this family possess N-terminal AAA-enriched structures
in mammals (UBXN1, UBXN2C, UBXN3A, UBXN3B and UBXN7), the UBX
proteins have been split into two groups (8,11,64,).
Park et al (64) reported
that these five proteins fold abnormally to the ER, as ERAD
substrates. The expression of all five proteins was shown to be
upregulated in cells treated with cyclooxygenase, according to
RT-PCR data (64). Among these
proteins, UBXN2C, UBXN1 and UBXN3B are ‘immediate’ responders to
endogenous stress, whereas UBXN3A and UBXN7 are ‘late’ responders.
This indicates that whereas UBXN3A and UBXN7 are affected by other
variables and do not reflect the ER stress in a timely and
efficient manner, UBXN2C, UBXN1 and UBXN3B directly reflect
increased ER stress.
Of note, UBXN2C and UBXN3B have higher expression
levels than UBXN1 across all ERAD substrates, but UBXN1 has lower
expression levels (11). Additional
research revealed that UBXN1 has no affinity for certain ERAD
substrates. Stable α-T-cell antigen receptor (α-TCR)-expressing
cells under conditions of stress exhibit higher levels of UBXN2C
and UBXN3B and lower levels of UBXN1. When UBXN2C or UBXN3B are
overexpressed, the degradation of α-TCR occurs more rapidly; UBXN1
has the reverse effect. The degradation of α-TCR is caused by the
overexpression of UBXN1. ERAD is one of the key elements in
overcoming ER stress by activating the UPR (11). These findings imply that the
expression levels of these five genes are altered by both ER stress
and the overexpression of ERAD substrates (64). Although these five proteins function
as the primary proteins for proteasomal degradation, the expression
of several genes affects the outcomes when a certain role is
played. This paradox can be explained by the fact that, on the one
hand, UPRs build up in the ER and that, under conditions of ER
stress, the overexpression of ERAD substrates can partially reduce
this buildup; on the other hand, an increase in misfolded proteins
in the cell and an increase in their demand can decrease the rate
of proteasomal degradation of ERAD substrates. When UBXN2C and
UBXN3B levels are high in a significant number of misfolded
proteins, particularly in cells expressing α-TCR, and when UBXN1 is
downregulated to further promote degradation, this may explain the
enhanced participation of UBXN2C and UBXN3B in the feedback loop
during ER stress.
Altered signaling pathways
Under typical circumstances, the three primary
transmembrane sensors inositolase-1α, protein kinase RNA-like ER
kinase (PERK) and activated transcription factor-6α (ATF-6α) are
all active in the lumen. Apoptosis may be caused if ER homeostasis
is not recovered.
The inositolase-1α pathway is a transmembrane
protein that is present in the UPRs and has kinase and ribonucleic
acid endonuclease as its cytoplasmic structural domains (65). In a healthy state, inositolase-1
binds to immunoglobulins and is inactive. In response to stress,
inositolase-1 is released, dimerized and activated, resulting in
conformational alterations. As an alternative, unfolded proteins
may bind directly to the structural domain of inositolase-1,
causing conformational alterations and activation (66). The transcription factor known as the
X-box binding protein, which is produced as a result of activated
inositolase-1α, improves the capacity of the protein to fold and
the function of ERAD in the ER. When ER stress is unrecoverable,
the function of inositolase-1 is interrupted, which causes mRNA
that is linked to it to degrade or to be recruited by TNF, which
then stimulates cellular and mitochondrial death via apoptotic
signaling (67). As a result, the
inositolase-1 pathway reacts to ER stress by either boosting
apoptotic pathways or upregulating ERAD, both of which include the
UBX protein family. By inhibiting and interfering with the
RIG-I-like receptors and the NF-κB pathway, UBXN1 is a member of
the complex necessary to engage in the de-glycosylation and
proteasome-mediated destruction of misfolded proteins under ER
stress via reverse translocation (11). Similar to inositolase-1, PERK is a
transmembrane protein that is inactive in a physiological setting
(68). Dimerization and tetrameric
pattern-mediated autophosphorylation are required for its
activation (69). When PERK is
activated, it may also phosphorylate translation initiation factors
(70), which causes ERAD to reduce
the amount of protein (71). This
pathway also causes cell death by upregulating apoptosis-related
genes (71). ATF-6α is translocated
to the Golgi apparatus when the ER is stressed, despite the fact
that it is primarily engaged in the control of ER plasmalogens
(72).
Abnormal lipid metabolism
As a type of proteotoxic ER stress, the changed
protein folding burden in the ER (73) interferes with aberrant protein
folding, misfolded, unfolded or altered folding density. UPRs are
also known as lipotoxic ER stress, as they may be directly
triggered by toxic lipids in addition to being reliant on the
buildup of misfolded proteins. Both forms of stress are induced by
the involvement of the ER in the folding and transport of proteins,
which is connected to lipid production and transport and activates
UPRs to bring about homeostasis.
Genes associated with FAS, such as ATP acetyl
coenzyme A carboxylase, which causes the conversion of citric acid
into acetyl coenzyme A, malonyl coenzyme A, and fatty acids, are
often overexpressed or upregulated in HCC (74). Through the examination of gene
expression, a number of studies have investigated the causes and
purposes of HCC development (6–8,11,14–18). As has already been
established, as UBXN3B levels increase, so do the amounts of
lipidated Apo B-100 in LDs and ubiquitinated Apo B-100 in ER. Both
ubiquitinated and lipidated Apo B-100 have the ability to speed up
proteasomal breakdown, while promoting lipid transport. The
associated metabolism between LD and ER is regulated by UBXN3B.
Changes in the microenvironment
NK T-cells (NKT cells) are innate and adaptive
immune cells with a variety of immunomodulatory functions (75). Changes in the metabolic profile of
tumor lipids may also modify the type of lipid antigens, which may
affect the immunomodulatory activity of NKT cells, as they
predominantly detect lipid antigens (76). In a mouse model of NAFLD, increased
lipids were shown to cause NKT cell death, which reduced the amount
of hepatic NKT cells (77). Type I
NKT cells are activated by a lipid surplus, and this leads to a
more potent pro-inflammatory cytokine environment. However, despite
the higher hepatic lipid content in HCC, another study found no
significant difference in the number of NKT cells (78). These contradictory results suggest
that further more in-depth experiments are required to examine the
effects of lipid changes on NKT cells in NAFLD and HCC.
Additionally, processes including aberrant lipid
metabolism and Golgi expansion control the proliferation of cells,
such as myeloid-derived suppressor cells, CD8+ T-cells,
DCs and tumor-associated macrophages, which are all implicated in
the growth of tumors (79). The
expression of the UBX protein and the development of HCC may be
affected by mitochondrial defects, changes in lipid signaling
molecules and pathways, fatty acid biosynthetic pathways,
lipidomics, other genetic mutations, chronic viral infections,
cholesterol efflux factors, ER autophagy, and post-translational
modifications of proteins. The mechanisms through which the UBX
protein family affects ER stress, lipid metabolism and
microenvironmental changes remain unknown.
Conclusions and future perspectives
As a member of the UBX family, UBXN3B is primarily
involved in the ER stress mechanism known as ERAD. It performs a
variety of roles in LDs and ERs under various clinical conditions
and develops into a new HCC biomarker. In order to maintain fatty
acid and triglyceride homeostasis, maintain intracellular signaling
pathways, and normalize cytokines, UBXN3B participates in ER
stability, maintains fatty acid and triglyceride homeostasis, and
has an impact on cholesterol production. UBXN3B is also involved in
the progression of ER stress, the dysregulation of lipid
metabolism, the driving of inflammatory factors and the immune
microenvironment. However, a number of physiological processes of
UBXN3B remain unknown and require support and clarification by
further fundamental experimental and clinical studies.
Acknowledgements
Not applicable.
Funding
The present study was supported by a Peking University
International Hospital Research Grant (no. YN2022QN12).
Availability of data and materials
Not applicable.
Authors' contributions
ZG and JL drafted, read and approved the final
manuscript, and conceived and designed the study. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
UBXN3B
|
ubiquitin regulatory X
domain-containing proteins 3B
|
|
ER
|
endoplasmic reticulum
|
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
ERAD
|
endoplasmic reticulum-associated
degradation
|
|
NASH
|
non-alcoholic steatohepatitis
|
|
LDs
|
lipid droplets
|
|
FAS
|
fatty acid synthase
|
|
RIG-I
|
retinoic acid-inducible
|
|
NF-κB
|
nuclear factor-κB
|
|
ATP
|
adenosine triphosphate
|
|
VCP
|
valosin-containing protein
|
|
LXRα
|
liver X receptor α
|
|
SREBP-1
|
sterol regulatory element-binding
protein 1
|
|
INSIG-1
|
insulin-inducible gene 1
|
|
HMGCR
|
3-hydroxy-3-methylglutaryl coenzyme A
reductase
|
|
mRNA
|
messenger ribonucleic acid
|
|
STING
|
stimulator of interferon genes
|
|
IFN-I
|
type I interferon
|
|
ISRE
|
interferon-stimulated response
element
|
|
HDL
|
high-density lipoprotein
|
|
LDL
|
low-density lipoprotein
|
|
FFAs
|
free fatty acids
|
|
TNF
|
tumor necrosis factor
|
|
IL
|
interleukin
|
|
DCs
|
dendritic cells
|
|
NKT cells
|
natural killer T-cells
|
|
UPR
|
unfolded protein response
|
|
mRNPs
|
mRNA-protein complexes
|
|
AREs
|
AU-rich elements
|
|
α-TCR
|
α-T-cell antigen receptor
|
|
PERK
|
protein kinase RNA like ER kinase
|
|
ATF-6α
|
activated transcription factor-6α
|
References
|
1
|
Rawla P, Sunkara T, Muralidharan P and Raj
JP: Update in global trends and aetiology of hepatocellular
carcinoma. Contemp Oncol (Pozn). 22:141–150. 2018.PubMed/NCBI
|
|
2
|
World Health Organization, . GLOBOCAN 2020
Graph production. http://gco.iarc.fr/todaySeptember. 2021
|
|
3
|
Blanchette-Mackie EJ, Dwyer NK, Barber T,
Coxey RA, Takeda T, Rondinone CM, Theodorakis JL, Greenberg AS and
Londos C: Perilipin is located on the surface layer of
intracellular lipid droplets in adipocytes. J Lipid Res.
36:1211–1226. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shalapour S, Lin XJ, Bastian IN, Brain J,
Burt AD, Aksenov AA, Vrbanac AF, Li W, Perkins A, Matsutani T, et
al: Inflammation-induced IgA+ cells dismantle anti-liver cancer
immunity. Nature. 551:340–345. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Muir K, Hazim A, He Y, Peyressatre M, Kim
DY, Song X and Beretta L: Proteomic and lipidomic signatures of
lipid metabolism in NASH-associated hepatocellular carcinoma.
Cancer Res. 73:4722–4731. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou HL, Geng C, Luo G and Lou H: The
p97-UBXD8 complex destabilizes mRNA by promoting release of
ubiquitinated HuR from mRNP. Genes Dev. 27:1046–1058. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kloppsteck P, Ewens CA, Förster A, Zhang X
and Freemont PS: Regulation of p97 in the ubiquitin-proteasome
system by the UBX protein-family. Biochim Biophys Acta.
1823:125–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rezvani K: UBXD proteins: A family of
proteins with diverse functions in cancer. Int J Mol Sci.
17:17242016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liao Z, Luo R, Li G, Song Y, Zhan S, Zhao
K, Hua W, Zhang Y, Wu X and Yang C: Exosomes from mesenchymal stem
cells modulate endoplasmic reticulum stress to protect against
nucleus pulposus cell death and ameliorate intervertebral disc
degeneration in vivo. Theranostics. 9:4084–4100. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakatsukasa K, Huyer G, Michaelis S and
Brodsky JL: Dissecting the ER-associated degradation of a misfolded
polytopic membrane protein. Cell. 132:101–112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mukkavalli S, Klickstein JA, Ortiz B, Juo
P and Raman M: The p97-UBXN1 complex regulates aggresome formation.
J Cell Sci. 134:jcs2542012021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Uchiyama K, Totsukawa G, Puhka M, Kaneko
Y, Jokitalo E, Dreveny I, Beuron F, Zhang X, Freemont P and Kondo
H: p37 is a p97 adaptssswor required for Golgi and ER biogenesis in
interphase and at the end of mitosis. Dev Cell. 11:803–816. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Her NG, Toth JI, Ma CT, Wei Y,
Motamedchaboki K, Sergienko E and Petroski MD: p97 composition
changes caused by allosteric inhibition are suppressed by an
on-target mechanism that increases the enzyme's ATPase activity.
Cell Chem Biol. 23:517–528. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ye Y, Meyer HH and Rapoport TA: The AAA
ATPase Cdc48/p97 and its partners transport proteins from the ER
into the cytosol. Nature. 414:652–656. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olzmann JA, Richter CM and Kopito RR:
Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated
lipid droplet turnover. Proc Natl Acad Sci USA. 110:1345–1350.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang J, Yin C, Doong H, Fang S, Peterhoff
C, Nixon RA and Monteiro MJ: Characterization of erasin (UBXD2): A
new ER protein that promotes ER-associated protein degradation. J
Cell Sci. 119:4011–4024. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nagahama M, Ohnishi M, Kawate Y, Matsui T,
Miyake H, Yuasa K, Tani K, Tagaya M and Tsuji A: UBXD1 is a
VCP-interacting protein that is involved in ER-associated
degradation. Biochem Biophys Res Commun. 382:303–308. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Raman M, Sergeev M, Garnaas M, Lydeard JR,
Huttlin EL, Goessling W, Shah JV and Harper JW: Systematic
proteomics of the VCP-UBXD adaptor network identifies a role for
UBXN10 in regulating ciliogenesis. Nat Cell Biol. 17:1356–1369.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schrul B and Kopito RR: Peroxin-dependent
targeting of a lipid-droplet-destined membrane protein to ER
subdomains. Nat Cell Biol. 18:740–751. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pawar A, Botolin D, Mangelsdorf DJ and
Jump DB: The role of liver X receptor-alpha in the fatty acid
regulation of hepatic gene expression. J Biol Chem.
278:40736–40743. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Horton JD, Shah NA, Warrington JA,
Anderson NN, Park SW, Brown MS and Goldstein JL: Combined analysis
of oligonucleotide microarray data from transgenic and knockout
mice identifies direct SREBP target genes. Proc Natl Acad Sci USA.
100:12027–12032. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gong Yi, Lee JN, Lee PC, Goldstein JL,
Brown MS and Ye J: Sterol-regulated ubiquitination and degradation
of Insig-1 creates a convergent mechanism for feedback control of
cholesterol synthesis and uptake. Cell Metab. 3:15–24. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee JN, Zhang X, Feramisco JD, Gong Y and
Ye J: Unsaturated fatty acids inhibit proteasomal degradation of
Insig-1 at a postubiquitination step. J Biol Chem. 283:33772–33783.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nohturfft A, Yabe D, Goldstein JL, Brown
MS and Espenshade PJ: Regulated step in cholesterol feedback
localized to budding of SCAP from ER membranes. Cell. 102:315–323.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohsaki Y, Cheng J, Suzuki M, Fujita A and
Fujimoto T: Lipid droplets are arrested in the ER membrane by tight
binding of lipidated apolipoprotein B-100. J Cell Sci.
121:2415–2422. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sasako T, Ohsugi M, Kubota N, Itoh S,
Okazaki Y, Terai A, Kubota T, Yamashita S, Nakatsukasa K, Kamura T,
et al: Hepatic Sdf2l1 controls feeding-induced ER stress and
regulates metabolism. Nat Commun. 10:9472019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yen CL, Monetti M, Burri BJ and Farese RV
Jr: The triacylglycerol synthesis enzyme DGAT1 also catalyzes the
synthesis of diacylglycerols, waxes, and retinyl esters. J Lipid
Res. 46:1502–1511. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Loregger A, Raaben M, Tan J, Scheij S,
Moeton M, van den Berg M, Gelberg-Etel H, Stickel E, Roitelman J,
Brummelkamp T and Zelcer N: Haploid mammalian genetic screen
identifies UBXD8 as a key determinant of HMGCR degradation and
cholesterol biosynthesis. Arterioscler Thromb Vasc Biol.
37:2064–2074. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ishikawa H, Ma Z and Barber GN: STING
regulates intracellular DNA-mediated, type I interferon-dependent
innate immunity. Nature. 461:788–792. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang L, Wang L, Ketkar H, Ma J, Yang G,
Cui S, Geng T, Mordue DG, Fujimoto T, Cheng G, et al: UBXN3B
positively regulates STING-mediated antiviral immune responses. Nat
Commun. 9:23292018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Preuss C, Jelenik T, Bódis K, Müssig K,
Burkart V, Szendroedi J, Roden M and Markgraf DF: A new targeted
lipidomics approach reveals lipid droplets in liver, muscle and
heart as a repository for diacylglycerol and ceramide species in
non-alcoholic fatty liver. Cells. 8:2772019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tian Y, Yang B, Qiu W, Hao Y, Zhang Z,
Yang B, Li N, Cheng S, Lin Z, Rui YC, et al: ER-residential Nogo-B
accelerates NAFLD-associated HCC mediated by metabolic
reprogramming of oxLDL lipophagy. Nat Commun. 10:33912019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Trefts E, Gannon M and Wasserman DH: The
liver. Curr Biol. 27:R1147–R1151. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lonardo A, Ballestri S, Marchesini G,
Angulo P and Loria P: Nonalcoholic fatty liver disease: A precursor
of the metabolic syndrome. Dig Liver Dis. 47:181–190. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guri Y, Colombi M, Dazert E, Hindupur SK,
Roszik J, Moes S, Jenoe P, Heim MH, Riezman I, Riezman H and Hall
MN: mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell.
32:807–823.e12. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang X, Fan M and Huang W: Pleiotropic
roles of FXR in liver and colorectal cancers. Mol Cell Endocrinol.
543:1115432022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang S, Koteish A, Lin H, Huang J, Roskams
T, Dawson V and Diehl AM: Oval cells compensate for damage and
replicative senescence of mature hepatocytes in mice with fatty
liver disease. Hepatology. 39:403–411. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang T, Zhang Y, Liu J, Ma Y, Ye Q, Yan X
and Ding L: MicroRNA-377-3p inhibits hepatocellular carcinoma
growth and metastasis through negative regulation of CPT1C-mediated
fatty acid oxidation. Cancer Metab. 10:22022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bauer DE, Hatzivassiliou G, Zhao F,
Andreadis C and Thompson CB: ATP citrate lyase is an important
component of cell growth and transformation. Oncogene.
24:6314–6322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Litwack G: Chapter 9-lipids. Litwack G:
Human biochemistry Boston: Academic Press; pp. 199–255. 2018
|
|
41
|
Medes G, Thomas A and Wernhouse S:
Metabolism of neoplastic tissue. IV. A study of lipid synthesis in
neoplastic tissue slices in vitro. Cancer Res. 13:27–29.
1953.PubMed/NCBI
|
|
42
|
Fullerton MD, Galic S, Marcinko K, Sikkema
S, Pulinilkunnil T, Chen ZP, O'Neill HM, Ford RJ, Palanivel R,
O'Brien M, et al: Single phosphorylation sites in Acc1 and Acc2
regulate lipid homeostasis and the insulin-sensitizing effects of
metformin. Nat Med. 19:1649–1654. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lake AD, Novak P, Hardwick RN,
Flores-Keown B, Zhao F, Klimecki WT and Cherrington NJ: The
adaptive endoplasmic reticulum stress response to lipotoxicity in
progressive human nonalcoholic fatty liver disease. Toxicol Sci.
137:26–35. 2014.Oyadomari S, Harding HP, Zhang Y, Oyadomari M and
Ron D: Dephosphorylation of translation initiation factor 2alpha
enhances glucose tolerance and attenuates hepatosteatosis in mice.
Cell Metab. 7:520–532. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pinto Lde F, Compri CM, Fornari JV,
Bartchewsky W, Cintra DE, Trevisan M, Carvalho Pde O, Ribeiro ML,
Velloso LA, Saad MJ, et al: The immunosuppressant drug,
thalidomide, improves hepatic alterations induced by a high-fat
diet in mice. Liver Int. 30:603–610. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Crespo J, Cayón A, Fernández-Gil P,
Hernández-Guerra M, Mayorga M, Domínguez-Díez A,
Fernández-Escalante JC and Pons-Romero F: Gene expression of tumor
necrosis factor alpha and TNF-receptors, p55 and p75, in
nonalcoholic steatohepatitis patients. Hepatology. 34:1158–1163.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhou YJ, Li YY, Nie YQ, Yang H, Zhan Q,
Huang J, Shi SL, Lai XB and Huang HL: Influence of polygenetic
polymorphisms on the susceptibility to non-alcoholic fatty liver
disease of Chinese people. J Gastroenterol Hepatol. 25:772–777.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kramer F, Torzewski J, Kamenz J, Veit K,
Hombach V, Dedio J and Ivashchenko Y: Interleukin-1beta stimulates
acute phase response and C-reactive protein synthesis by inducing
an NFkappaB- and C/EBPbeta-dependent autocrine interleukin-6 loop.
Mol Immunol. 45:2678–2689. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Banroques J, Doère M, Dreyfus M, Linder P
and Tanner NK: Motif III in superfamily 2 ‘helicases’ helps convert
the binding energy of ATP into a high-affinity RNA binding site in
the yeast DEAD-box protein Ded1. J Mol Biol. 396:949–966. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Catalá M, Antón A and Portolés MT:
Characterization of the simultaneous binding of Escherichia coli
endotoxin to Kupffer and endothelial liver cells by flow cytometry.
Cytometry. 36:123–130. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Luo W, Xu Q, Wang Q, Wu H and Hua J:
Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2
polarization in the development of non-alcoholic fatty liver
disease. Sci Rep. 7:446122017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu PS, Wang H, Li X, Chao T, Teav T,
Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M, et al:
α-ketoglutarate orchestrates macrophage activation through
metabolic and epigenetic reprogramming. Nat Immunol. 18:985–994.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bertola A, Bonnafous S, Anty R, Patouraux
S, Saint-Paul MC, Iannelli A, Gugenheim J, Barr J, Mato JM, Le
Marchand-Brustel Y, et al: Hepatic expression patterns of
inflammatory and immune response genes associated with obesity and
NASH in morbidly obese patients. PLoS One. 5:e135772010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Maina V, Sutti S, Locatelli I, Vidali M,
Mombello C, Bozzola C and Albano E: Bias in macrophage activation
pattern influences non-alcoholic steatohepatitis (NASH) in mice.
Clin Sci (Lond). 122:545–553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Herber DL, Cao W, Nefedova Y, Novitskiy
SV, Nagaraj S, Tyurin VA, Corzo A, Cho HI, Celis E, Lennox B, et
al: Lipid accumulation and dendritic cell dysfunction in cancer.
Nat Med. 16:880–886. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Henning JR, Graffeo CS, Rehman A, Fallon
NC, Zambirinis CP, Ochi A, Barilla R, Jamal M, Deutsch M, Greco S,
et al: Dendritic cells limit fibroinflammatory injury in
nonalcoholic steatohepatitis in mice. Hepatology. 58:589–602. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tosello-Trampont AC, Krueger P, Narayanan
S, Landes SG, Leitinger N and Hahn YS: NKp46(+) natural killer
cells attenuate metabolism-induced hepatic fibrosis by regulating
macrophage activation in mice. Hepatology. 63:799–812. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ambade A, Satishchandran A, Saha B,
Gyongyosi B, Lowe P, Kodys K, Catalano D and Szabo G:
Hepatocellular carcinoma is accelerated by NASH involving M2
macrophage polarization mediated by hif-1αinduced IL-10.
Oncoimmunology. 5:e12215572016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang W, Furneaux H, Cheng H, Caldwell MC,
Hutter D, Liu Y, Holbrook N and Gorospe M: HuR regulates p21 mRNA
stabilization by UV light. Mol Cell Biol. 20:760–769. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gerber AP, Herschlag D and Brown PO:
Extensive association of functionally and cytotopically related
mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol.
2:E792004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Clement SL, Scheckel C, Stoecklin G and
Lykke-Andersen J: Phosphorylation of tristetraprolin by MK2 impairs
AU-rich element mRNA decay by preventing deadenylase recruitment.
Mol Cell Biol. 31:256–266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lafarga V, Cuadrado A, Lopez de Silanes I,
Bengoechea R, Fernandez-Capetillo O and Nebreda AR: p38
Mitogen-activated protein kinase- and HuR-dependent stabilization
of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol Cell Biol.
9:4341–4351. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Meerang M, Ritz D, Paliwal S, Garajova Z,
Bosshard M, Mailand N, Janscak P, Hübscher U, Meyer H and Ramadan
K: The ubiquitin-selective segregase VCP/p97 orchestrates the
response to DNA double-strand breaks. Nat Cell Biol. 13:1376–1382.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kim HH, Abdelmohsen K, Lal A, Pullmann R
Jr, Yang X, Galban S, Srikantan S, Martindale JL, Blethrow J,
Shokat KM and Gorospe M: Nuclear HuR accumulation through
phosphorylation by Cdk1. Genes Dev. 22:1804–1815. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Park ES, Yoo YJ and Elangovan M: The
opposite role of two UBA-UBX containing proteins, p47 and SAKS1 in
the degradation of a single ERAD substrate, α-TCR. Mol Cell
Biochem. 425:37–45. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sepulveda D, Rojas-Rivera D, Rodríguez DA,
Groenendyk J, Köhler A, Lebeaupin C, Ito S, Urra H, Carreras-Sureda
A, Hazari Y, et al: Interactome screening identifies the ER luminal
chaperone Hsp47 as a regulator of the unfolded protein response
transducer IRE1α. Mol Cell. 69:238–252.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Karagöz GE, Acosta-Alvear D, Nguyen HT,
Lee CP, Chu F and Walter P: An unfolded protein-induced
conformational switch activates mammalian IRE1. Elife.
6:e307002017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bertolotti A, Zhang Y, Hendershot LM,
Harding HP and Ron D: Dynamic interaction of BiP and ER stress
transducers in the unfolded-protein response. Nat Cell Biol.
2:326–332. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Carrara M, Prischi F, Nowak PR and Ali MM:
Crystal structures reveal transient PERK luminal domain
tetramerization in endoplasmic reticulum stress signaling. EMBO J.
34:1589–1600. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Harding HP, Zhang Y, Zeng H, Novoa I, Lu
PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al: An
integrated stress response regulates amino acid metabolism and
resistance to oxidative stress. Mol Cell. 11:619–633. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Blais JD, Filipenko V, Bi M, Harding HP,
Ron D, Koumenis C, Wouters BG and Bell JC: Activating transcription
factor 4 is translationally regulated by hypoxic stress. Mol Cell
Biol. 24:7469–7482. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Adachi Y, Yamamoto K, Okada T, Yoshida H,
Harada A and Mori K: ATF6 is a transcription factor specializing in
the regulation of quality control proteins in the endoplasmic
reticulum. Cell Struct Funct. 33:75–89. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Schindler AJ and Schekman R: In vitro
reconstitution of ER-stress induced ATF6 transport in COPII
vesicles. Proc Natl Acad Sci USA. 106:17775–17780. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lally JSV, Ghoshal S, DePeralta DK, Moaven
O, Wei L, Masia R, Erstad DJ, Fujiwara N, Leong V, Houde VP, et al:
Inhibition of acetyl-CoA carboxylase by phosphorylation or the
inhibitor ND-654 suppresses lipogenesis and hepatocellular
carcinoma. Cell Metab. 29:174–182.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhu H, Zhang Q and Chen G: CXCR6
deficiency ameliorates ischemia-reperfusion injury by reducing the
recruitment and cytokine production of hepatic NKT cells in a mouse
model of non-alcoholic fatty liver disease. Int Immunopharmacol.
72:224–234. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhou ZJ, Xin HY, Li J, Hu ZQ, Luo CB and
Zhou SL: Intratumoral plasmacytoid dendritic cells as a poor
prognostic factor for hepatocellular carcinoma following curative
resection. Cancer Immunol Immunother. 68:1223–1233. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Tang T, Sui Y, Lian M, Li Z and Hua J:
Pro-inflammatory activated Kupffer cells by lipids induce hepatic
NKT cells deficiency through activation-induced cell death. PLoS
One. 8:e819492013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wu L, Parekh VV, Gabriel CL, Bracy DP,
Marks-Shulman PA, Tamboli RA, Kim S, Mendez-Fernandez YV, Besra GS,
Lomenick JP, et al: Activation of invariant natural killer T cells
by lipid excess promotes tissue inflammation, insulin resistance,
and hepatic steatosis in obese mice. Proc Natl Acad Sci USA.
109:E1143–E1152. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ben Haij N, Planès R, Leghmari K, Serrero
M, Delobel P, Izopet J, BenMohamed L and Bahraoui E: HIV-1 Tat
protein induces production of proinflammatory cytokines by human
dendritic cells and monocytes/macrophages through engagement of
TLR4-MD2-CD14 complex and activation of NF-κB pathway. PLoS One.
10:e01294252015. View Article : Google Scholar : PubMed/NCBI
|














