Cancer is one of the leading cause of death
worldwide; according to estimates from the World Health
Organization, ~10 million cancer deaths occurred in 2020 (1). Cancer results from DNA aberrations
that cause the deregulation of pathways controlling cellular
processes involved in proliferation, cell survival, apoptosis and
DNA repair. Mutations in tumor suppressor genes and oncogenes are
responsible for cancer initiation, promotion and progression
(2). However, carcinogenesis cannot
be explained solely by genetic alterations, as it also involves
epigenetic processes. Epigenetic changes are defined as inheritable
modifications in gene expression that do not result from changes in
DNA sequences (3). Epigenetic
mechanisms were discovered as the knowledge regarding DNA structure
increased. DNA is packaged in the nucleus by histones into a
structure called chromatin. The basic unit of chromatin is the
nucleosome; each nucleosome comprises one histone octamer composed
of H3, H4, H2A and H2B subunits, with 147 bp of DNA coiling around
them. Chromatin is classified as either heterochromatin or
euchromatin. Heterochromatin is a region of DNA that is highly
condensed and transcriptionally inactive, whereas euchromatin is
less condensed and is actively transcribed (4). The chromatin structure creates a
barrier to DNA replication, damage repair or access of
transcription machinery to DNA. Chromatin is highly dynamic to
allow different regulators or transcriptional factors to access DNA
(5). Various epigenetic mechanisms
regulate the states of euchromatin or heterochromatin. Six
epigenetic mechanisms altering chromatin structure have been
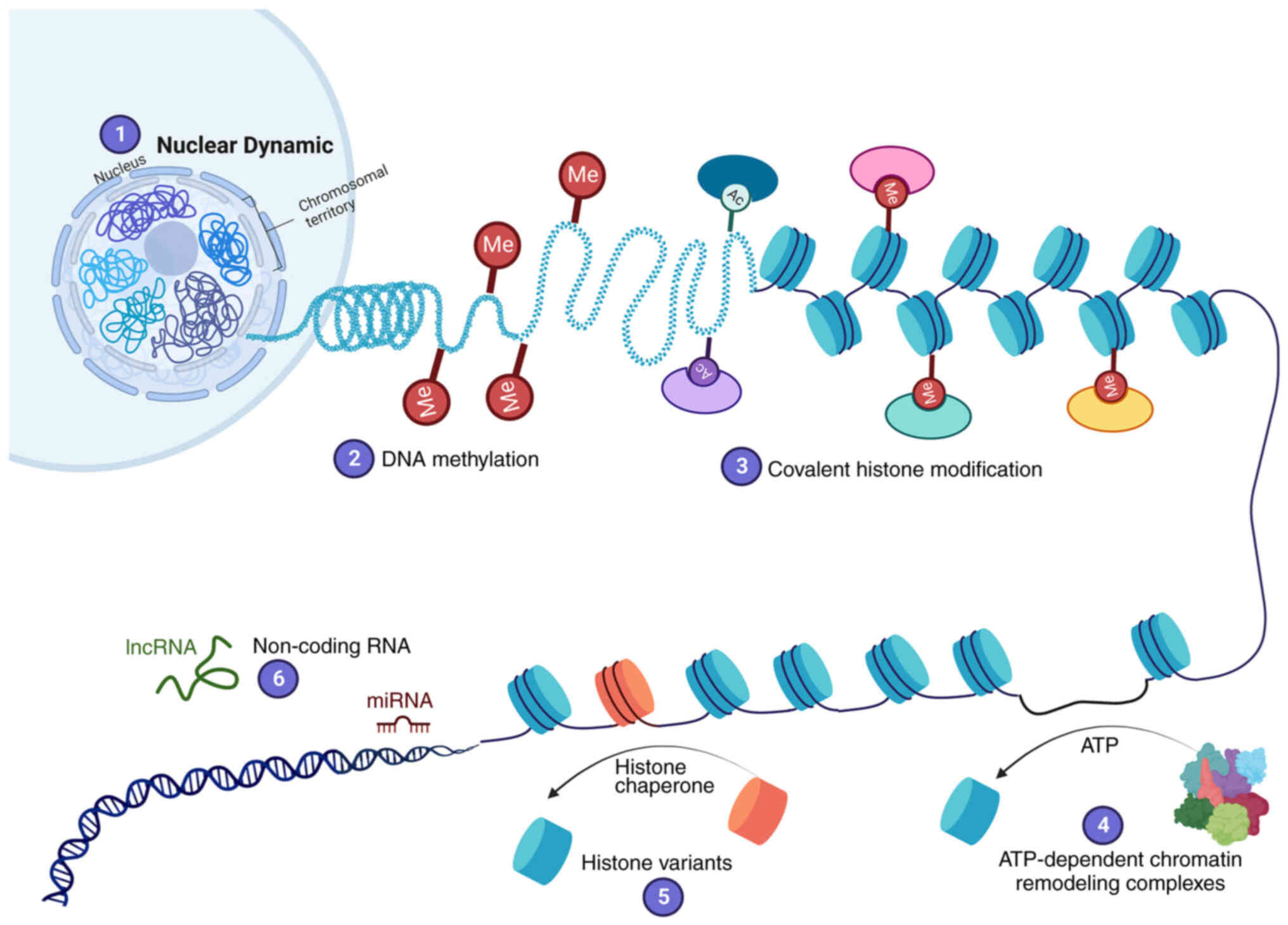
described (Fig. 1): i) Nuclear
dynamic; ii) DNA methylation; iii) covalent histone modification;
iv) ATP-dependent chromatin remodeling complexes; v) histone
variants; and vi) non-coding RNA (ncRNA), including microRNA
(miRNA/miR) and long ncRNA (lncRNA) (5–7).
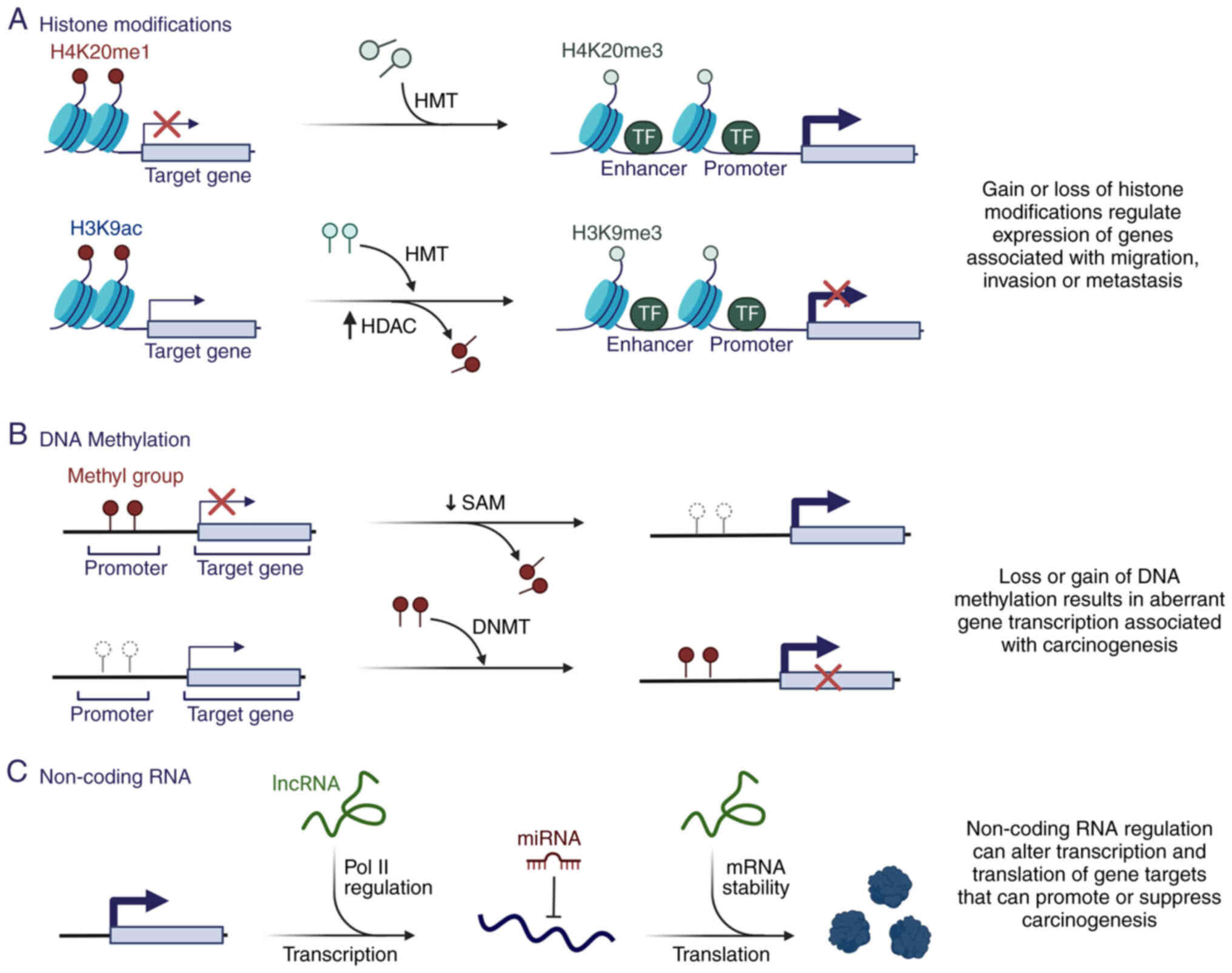
A number of studies have demonstrated that
dysregulation of epigenetic mechanisms is a critical contributor to
carcinogenesis (8–10) (Fig.
2), mainly due to regulation of DNA accessibility and the
compactness of chromatin structure, favoring the regulation of
different genes necessary for carcinogenesis (Fig. 2A and B).
Unlike genetic mutations, the cancer epigenomic
landscape can be reprogrammed. It represents one of the most
promising therapeutic targets for treatment and reversal of drug
resistance. Epigenetic alterations in cancer development and
progression may be the basis for individual variations in drug
response (11,12). The present review focuses on
epigenetic reprogramming during tumorigenesis and recent progress
in developing clinical strategies to target epigenetic codes.
DNA methylation is the most well-studied epigenetic
modification. It is a stable gene-silencing mechanism that
regulates gene expression and chromatin architecture through the
covalent addition of a methyl group to the C5 cytosines by DNA
methyltransferases (DNMTs), producing 5-methylcytosine (5mC)
(Fig. 1) (12). A total of five members of the DNMT
family have been identified in mammals: DNMT1, DNMT2, DNMT3A,
DNMT3B and DNMT3-like (DNMT3L) (12). However, only DNMT1, DNMT3A and
DNMT3B exhibit catalytic activity for DNA methylation (13). The de novo methyltransferases
DNMT3A and DNMT3B establish DNA methylation during embryonic
development (14,15), whereas DNMT1 is a housekeeping
methyltransferase that methylates preexisting hemi-methylated DNA
and preserves DNA methylation during DNA replication (14). DNMT3L has sequence homology with
DNMT3A/3B but has no catalytic domain; however, it is an essential
accessory protein for de novo methylation (16). Although the mechanism remains
unclear, DNMT3L modulates the activity of other DNMTs, including
DNMT3A (17) and DNMT2, which act
as RNA methyltransferases (18).
DNA methylation is reversible and is mediated by
three members of the ten-eleven translocation (TET) family: TET1,
TET2 and TET3 (19). TET enzymes
function in the demethylation of 5mC to 5-hydroxymethylcytosine
(5hmC), which is converted to unmethylated cytosine through
sequential oxidation to produce 5-formylcytosine (5fC) and
5-carboxylcytosine (5caC) intermediates (20). 5fC and 5caC are subsequently removed
by DNA glycosylases and the base excision repair machinery to
generate unmethylated cytosines at TET-targeted sites, or they are
passively demethylated by the loss of 5hmC during cell division
(20). Therefore, TET enzymes
regulate the chromatin structure and gene expression (21).
DNA methylation serves a fundamental role in
different processes, including embryonic development (22), X chromosome inactivation (23), genomic imprinting (24) and transcriptional silencing
(25). CpG islands are often
methylated in genome sequences rich in cytosine-guanine nucleotide
pairs in normal tissues (16). By
contrast, promoter regions are generally unmethylated, except for
the inactive X chromosome, silenced alleles of imprinted genes and
tissue-specific genes (21). DNA
methylation can also be detected in repetitive sequences, either in
tandem (satellite DNA), or short- or long-interspersed nuclear
elements; these modifications mainly help maintain genomic
integrity (26).
Approximately 60% of all gene promoters are enriched
in CpG islands; therefore, these genes are potentially
epigenetically regulated (26). In
cancer cells, alterations in DNA methylation are the first
epigenetic marker associated with cancer development (Fig. 2B) (27). Cancer cell genomes are
hypomethylated relative to normal counterparts, whereas a
paradoxical increment of site-specific CpG methylation is observed
in the gene promoter regions (28).
Loss of methylation in repetitive regions of the genome is the main
cause of DNA hypomethylation (29).
It is associated with genomic instability, changes in gene
imprinting and increased cancer risk (30). In addition, DNA methylation is
substantially more frequent than gene inactivation by genetic
mutations (31). It can induce gene
silencing directly or indirectly by inhibiting the binding of
specific transcription factors or by recruiting methyl-CpG-binding
domain proteins, respectively (27). The most crucial tumor-suppressor
gene silencing occurs through epigenetic mechanisms (32). This includes p14, p16/inhibitors of
CDK4 (33), retinoblastoma,
cyclin-dependent kinase inhibitor 2 and
methylguanine-DNA-methyltransferase, which favor uncontrolled
cellular proliferation (34). In
addition, alterations in DNA methylation patterns detected in
hematological malignancies are frequently associated with the
aberrant activity of enzymes regulating DNA methylation, such as
DNMT3A (35).
Alterations in the epigenome and deregulated
epigenetic mechanisms cannot entirely explain the process of
multistage carcinogenesis. However, epigenetic changes are
generally of a clonal nature, and occur in the early generation of
cancer cells; this initiates genetic instability resulting in the
acquisition of genetic mutations in tumor suppressor genes and the
activation of genetic mutations in oncogenes that facilitate cell
transformation (28). In addition,
epigenetic alterations can lead to the development of
drug-resistant phenotypes (Fig. 2).
Epigenetic inactivation can directly determine tumor
chemosensitivity and influence drug resistance and post-therapy
clinical outcomes (36–38). These findings and the reversible
nature of the underlying changes have made epigenetic alterations
potential rational targets for therapeutic approaches. Classes of
chromatin-modifying drugs have been approved by the US Food and
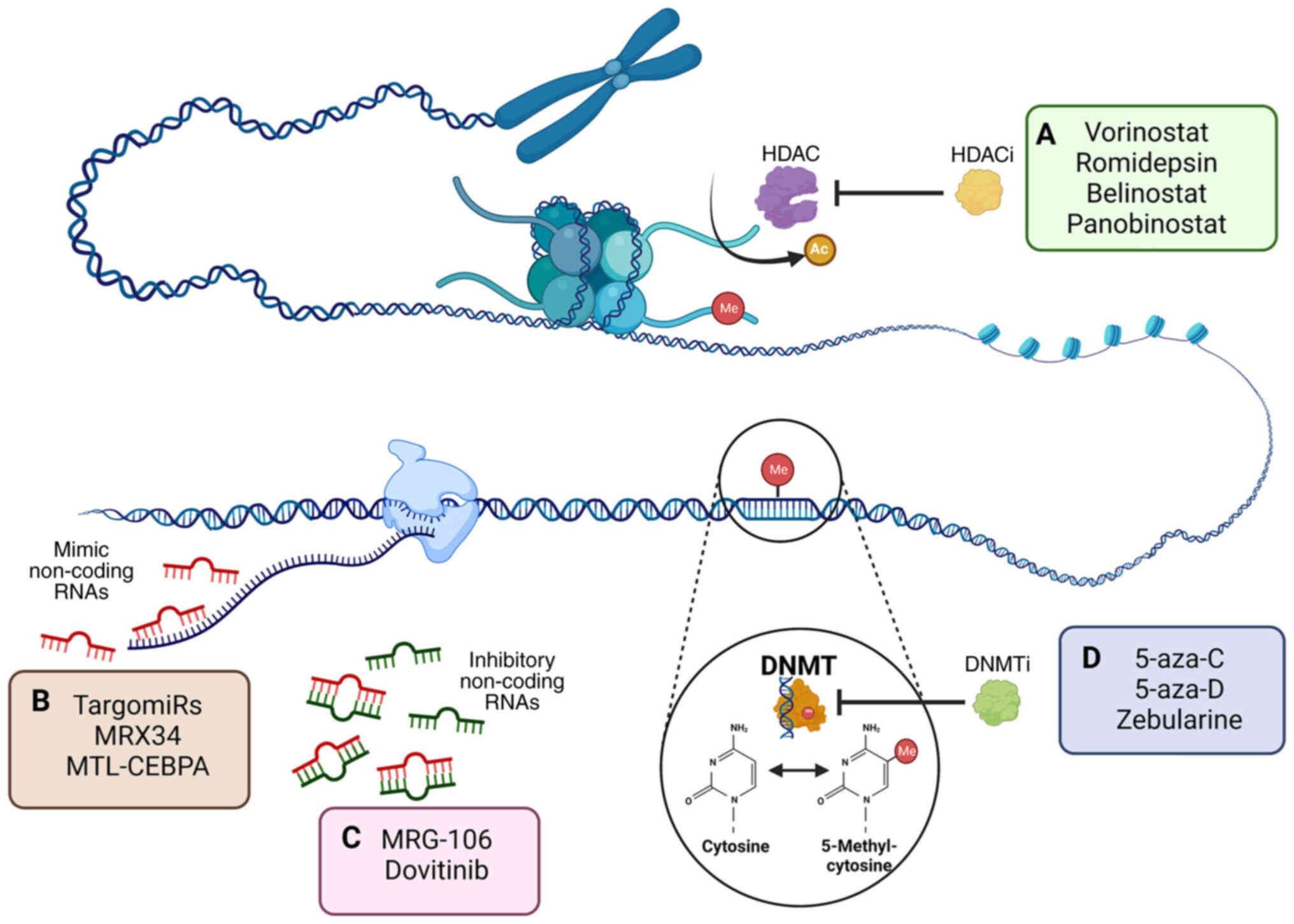
Drug Administration (FDA), including two main DNMT inhibitors
(DNMTi) and histone deacetylase (HDAC) inhibitors (HDACi; Fig. 3A) (39). Their application can reactivate
tumor suppressor genes and synergistically suppress the
proliferation of cancer cell lines in vitro and in
vivo (40,41). In addition, DNMTi are involved in
responses to radiotherapy and chemotherapy treatment, sensitizing
several chemo- and/or radiation-resistant cancer types in the
clinical setting (Fig. 3D)
(42,43). For example, treatment with
5-Aza-2′-deoxycytidine (5-aza-D) has been shown to be effective in
reversing cisplatin resistance in bladder cancer cells, which is
mainly associated with promoter demethylation of the HOXA9 gene
(44).
Drugs that inhibit DNA cytosine methylation are
divided into two groups: i) Nucleoside analogs, including
5-azacytidine (5-aza-C; Vidaza) and 5-aza-D (Dacogen® or
decitabine) and zebularine (Fig.
3A) (45); and ii)
non-nucleoside drugs, including procainamide, hydralazine,
epigallocatechin-3-gallate (EGCG), N-phthalyl-L-tryptophan and MG98
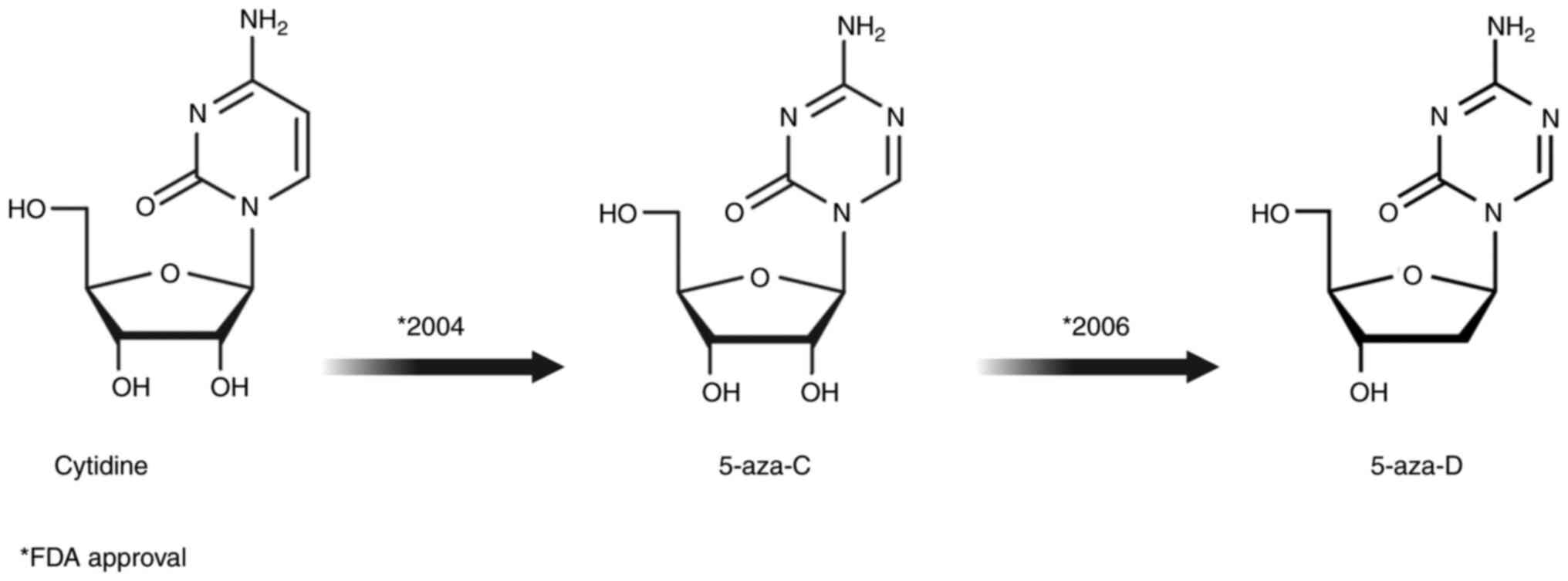
(43,46). Nucleoside analogs are characterized
by different modifications of the cytosine ring, such as
replacement of carbon with nitrogen at position 5 of the pyrimidine
ring (Fig. 4) (47). Cellular uptake of nucleotide analogs
is dependent on human equilibrative nucleoside transporter (hENT)-1
and hENT2 (48). Following
absorption, they are successively phosphorylated by intracellular
deoxycytidine kinase to produce the active tri-phosphorylated
metabolite 5-aza-2′-deoxycytidine 5′-triphosphate (49). This subsequently contributes to the
loss of methylation through its incorporation into newly
synthesized DNA and covalent bond formation with DNMTs (50). Thus, the enzyme remains bound to DNA
and its methyltransferase function is blocked (51). Furthermore, the covalent bond
compromises DNA functionality by triggering DNA damage signaling,
leading to the degradation of these enzymes and the passive
demethylation of the cellular genome during DNA replication
(45). 5-aza-C and 5-aza-D are the
most studied demethylating agents commonly used as chemotherapeutic
agents that produce numerous biological effects (Table I) (52,53).
5-aza-C was first synthesized in 1960 for use as a
classical cytostatic agent and was later shown to inhibit DNA
methylation in human cell lines (54–56).
It is the first epigenetic drug (epidrug) proposed for use in
cancer therapeutics, specifically in patients with acute
myelodysplastic syndrome (MDS). In May 2004, the US FDA approved it
as a first line treatment for MDS (57). It is also used to treat other
hematological malignancies, such as chronic myeloid leukemia
(58).
In preclinical studies, 5-aza-C inhibited the
antiapoptotic transcription factor NimA related kinase by
decreasing the phosphorylation of the upstream regulator IKKα/β,
which induces upregulation of the proapoptotic protein
phorbol-12-myristate-13-acetate-induced protein 1, resulting in
increased cell death (64,65). Furthermore, 5-aza-C has been
proposed to inhibit Wnt signaling, a pathway involved in oncogene
expression in acute myeloid leukemia (AML) and other cancer types,
such as colon cancer (66,67).
5-aza-D is a potent DNMTi included in the group of
analogs of the nucleoside cytidine. Because it contains a
deoxyribose, it can be incorporated into DNA (Fig. 4) (59,62),
unlike 5-aza-C, which predominantly incorporates into RNA (Table I). 5-aza-D covalently binds to DNA
during the S-phase of the cell cycle leading to rapid loss of
methylated cytosine (66). In 2006,
the FDA approved its use in MDS, and it is currently used to treat
chronic myelomonocytic leukemia and AML (68).
5-aza-D and 5-aza-C are used to treat various
hematological cancer types, including AML and MDS, because of their
ability to reactivate silenced tumor suppressor genes and DNA
damage-induced cell cycle arrest and apoptosis (76). Both drugs induce complete responses
and hematologic improvement; however, prolonged overall survival
has only been shown for 5-aza-C. Although the drugs only slightly
differ in their molecular structures, there are essential
differences in their mechanisms of action (Table I) (77). There are >135 epigenetics-related
clinical completed studies in different phases (49 in phase 1; 67
in phase 2; 22 in phase 3; and 1 in phase 4); Of which, 17 phase 3
interventional clinical trials using 5-aza-D and 5-aza-C in
combination with chemotherapy drugs or immunotherapy in cancer have
completed the recruitment stage (Table
II) (78).
In the early 1990s, second-generation nucleoside
analogs were synthesized, such as zebularine
[1-(β-D-ribofuranosyl)-2(1H)-pyrimidinone], a cytidine analog that
lacks the amino (N) group at position four of the pyrimidine ring
(79). It has high stability under
physiological conditions and inhibits DNA methylation through its
action on DNMTs and cytidine deaminase, an enzyme responsible for
resistance to nucleoside DNA methylation analogs (80). In addition, zebularine has low
cytotoxicity, which could translate into longer treatments with low
doses to maintain a demethylated state at an acidic or a neutral pH
(80). By comparison, 5-aza-C and
5-aza-D exhibit significant toxicity, low instability under
physiological conditions and short half-lives (59), which may complicate their use in
clinical settings.
Furthermore, a chemically diverse group of
non-nucleoside small-molecule compounds, such as hydralazine and
procainamide have been reported as DNMTi (81–83).
These drugs that are not incorporated into DNA or RNA but inhibit
DNMT activity alone or in combination with chemo/radiotherapy, have
shown promising results in various cancer types (82). Other small-molecule compounds, such
as guadecitabine, epigallocatechin-3-gallate (EGCG), RG-108 and
MG98, exhibit a DNA hypomethylating effect either through the
competitive inhibition or through the degradation of DNMT (46,84).
In addition, the inhibition of histone acetylation and generation
of reactive oxygen species are effects associated with the
treatment with biologically active non-nucleoside compounds
(85,86).
In eukaryotic cells, 145–147 bp DNA is wrapped
around histone octamers, comprising a core of histone dimers 2A,
H2B, H3 and H4, to form nucleosomes, the basic units of chromatin.
Histone tails extending from the core of the nucleosome can be
post-translationally modified by methylation, acetylation,
phosphorylation, SUMOylation and ubiquitination (Fig. 1) (87). Histone modifications serve essential
roles in various cellular processes, including DNA repair,
replication, stemness and changes in cell state (88). Aberrant histone post-translation
modifications occur during the initiation and progression of cancer
(89).
Two families of antagonistic enzymes control histone
acetylation: Histone acetyltransferases (HATs), which acetylate
lysine residues at the N-terminus of histone proteins, and HDACs,
which remove acetyl groups from histones. Histone acetylation
reduces attraction between negative and positive charges of DNA and
histone tails, respectively (90,91).
This structural change favors the decompaction of chromatin,
allowing access to the gene sequence for its transcription. The
altered activity of HATs observed in several types of cancer,
including esophageal, lung and liver cancer, has been associated
with the aberrant activation of genes whose expression is necessary
for maintaining tumor growth (92–94).
Chemical modifications to histones form the basis of histone codes
and may be associated with gene expression or silencing (88). Based on their sequence similarities
to yeast HDACs, the 18 identified human HDACs are divided into four
classes: i) Class I isoenzyme HDACs, which include HDAC1, HDAC2,
HDAC3 and HDAC8; ii) class II HDACs, which are homologous to yeast
protein lysine deacetylase and are further divided into class IIa
(including HDAC4, HDAC5, HDAC7 and HDAC9) and class IIb (HDAC6 and
HDAC10) subgroups; iii) class III HDACs are similar to yeast
sirtuins (SIRTs), and include SIRT1-SIRT7; and iv) class IV HDACs,
which comprise only HDAC11 (95).
SIRTs are NAD+-dependent enzymes, whereas the other
three classes are zinc cation (Zn2+)-dependent HDACs
(96).
HDACs serve a role in primary biological functions,
such as transcription, metastasis (97), autophagy, cell cycle (98), DNA damage repair, angiogenesis
(99), stress responses (100) and senescence (101), through histone deacetylation and
through non-histone proteins. However, aberrant expression of HDACs
can contribute to carcinogenesis by altering gene expression
through the deacetylation of different histone residues (Table III) (84). In addition, HDACs can deacetylate
non-histone proteins involved in apoptosis, differentiation, cell
proliferation control and transcription (102). HDAC upregulation in solid and
hematologic tumors promotes tumor suppressor gene silencing or
alters gene expression of oncogenic pathways (99). Furthermore, HDAC upregulation is
useful for the diagnosis and prognosis of gastric and breast cancer
(103).
Histone methylation is an essential
post-transcriptional modification generally occurring at the lysine
and arginine residues of histones H3 and H4 through the addition of
methyl groups (104). Mono-, di-
or trimethylated states may occur in both lysine and arginine,
which subsequently affects transcriptional regulation (88). Histone methylation is mediated by
HMTs, of which there are six major classes of histone lysine
methyltransferase complexes (KMT1-KMT6) (97,105).
The identification of lysine demethylases revealed the
reversibility of lysine methylation (106). H3K4-trimethylation (me3), H3K36me3
and H3K79me3 are associated with transcriptional activation,
whereas methylation of H3K9me3, H4K20me3 and H3K27me3 is associated
with a repressive chromatin state. The interaction and level of
methylation of these histone marks serve an important role in
transcriptional regulation (103).
Furthermore, mutations detected in different cancer types (bladder,
melanoma and lung cancer) have been associated with altered
activity of methylating enzymes (107). Mutations in enhancer of zeste
homolog 2 (EZH2) are found in follicular and diffuse large B-cell
lymphomas, gliomas and bone cancers affecting H3K27 (108). The HMT SET domain containing 1A is
upregulated in breast cancer, favoring the increase in H3K4me3 in
genes associated with migration and metastasis (109). In addition, H3K36me3 is altered in
various cancers, such as chondroblastoma, and colon, and head and
neck cancer (110,111). These alterations in histone
modifications and methylation enzymes contribute to changes in gene
expression that may promote carcinogenesis. Therefore, HMT
inhibitors have been developed for use as antineoplastic drugs.
Clinical trials of protein arginine N-methyltransferase 1, EZH2 and
lysine-specific histone demethylase 1 inhibitors have been
summarized by Yang et al (112).
The increasing understanding of the reversibility of
epigenetic processes has led to the identification of drugs that
inhibit HDAC activation and prevent tumor development. Four HDACi
affecting different cellular processes are FDA-approved (105,113), including vorinostat, romidepsin,
belinostat and panobinostat (Fig.
3A; Table IV). Additionally,
interventional clinical trials using HDACi in combination with
therapeutic drugs for cancer are summarized in Table V.
Vorinostat (also known as suberoylanilide hydroxamic
acid) is a class I- and class II HDACi structurally belonging to
the hydroxamate group; it does not inhibit class III HDACs
(114). Vorinostat binds to zinc
in the catalytic site of HDAC, which blocks the access of target
proteins and inhibits its HDAC activity (115). Therefore, this HDACi can modify
expression of proteins through structural changes on chromatin, but
also by modifying the activity of non-histones proteins (p53, EKLF,
E2F1, HIF-1 and GATA) (115,116). Clinical studies have demonstrated
that vorinostat treatment reduced solid tumors when administered
alone or in combination with alkylating agents, proteasome
inhibitors, anthracyclines and antiangiogenic and/or antimetabolite
agents (78,113,117,118).
Panobinostat is a pan-HDACi belonging to the
hydroxamate family. Some clinical studies have tested its use alone
or in combination with other drugs to particularly treat solid
tumors, such as sarcoma, breast, lung, renal, prostate, pancreas,
thyroid and malignant brain cancer, including hematological cancer
types such as Hodgkin and non-Hodgkin lymphoma; however, the
mechanism of action is uncharacterized (78,127).
In addition to DNA methylation and histone
modifications, ncRNAs also serve a key role in epigenetic control
(Fig. 1). ncRNAs are divided into
two main groups: Housekeeping and regulatory ncRNA. Housekeeping
ncRNAs include transfer RNAs, ribosomal RNAs, small nuclear RNAs
(snRNAs) and small nucleolar RNAs (snoRNAs), which enable the flow
of genetic information from DNA to proteins (128,129). snRNAs and snoRNAs participate in
the modulation of alternative splicing, which drives mRNA
maturation (130). In some
instances, these splicing-devoted ncRNAs have epigenetic regulatory
functions, as is the case with snRNAs U1 and 7SK, which modulate
transcription (130). U1 is
associated with the general transcription initiation factor
transcription factor IIH (131),
and 7SK interacts with the chromatin factor and transcriptional
regulator high mobility group AT-hook 1 (132).
By contrast, regulatory ncRNAs are
non-constitutively expressed RNAs that serve roles in epigenetic
control by modulating chromatin structure, RNA polymerase activity
and mRNA stability. Regulatory ncRNAs are divided according to
length: Small [small non-coding RNA (sncRNA); ≤200 nucleotides] and
long [long non-coding RNA (lncRNA); >200 nucleotides] (128). The best-understood sncRNAs are
miRNAs/miRs and small interfering RNAs (siRNAs). Other small ncRNAs
include trans-acting siRNAs, small-scan siRNAs, repeated-associated
siRNAs and piwi-interacting RNAs (piRNAs) (133,134). Classification of lncRNAs is even
more complex, and efforts have been made to obtain more
comprehensive annotations. Thus, one classification system for
lncRNAs involves four subdivisions: i) Long intergenic non-coding
RNAs (lincRNAs) and intronic lncRNAs; ii) sense and antisense
lncRNAs; iii) cis- or trans-acting lncRNAs; and iv) transcriptional
or post-transcriptional lncRNAs (135). Notably, the function of several
lncRNAs remains unknown; therefore, they are denoted as
non-functional RNAs and considered as transcriptional noise
(136). Circular RNAs (circRNAs)
can be consider as lncRNAs in terms of size (mean length ~50 nt);
however, due to their structure, some authors consider them as a
third group of ncRNAs (137). They
comprise between one and five exons and flanking introns, but
differ from the previously mentioned lncRNAs since they are
single-stranded RNA that form a loop by covalently joining their 5′
and 3′ ends (135).
siRNAs and miRNAs are the most understood sncRNAs.
They are double-stranded RNAs (dsRNAs) whose genesis and function
partially overlap. Both can promote RNA interference (RNAi), a
post-translational regulatory mechanism that provokes gene
silencing through mRNA degradation (138). The genesis of siRNAs and miRNAs
relies on Dicer, an RNase III enzyme that cleaves dsRNAs (139). A capped and polyadenylated RNA
with a stem-loop structure is cleaved by Drosha to form genetically
coded precursor miRNAs (pre-miRNAs) (140). miRNA genes are transcribed in the
cell nucleus by RNA polymerase II giving rise to primary miRNAs
(pri-miRNA) (141). Pre-miRNAs and
dsRNAs are processed by Dicer in the cytoplasm and interact with
the RNA-induced silencing complex (RISC) (142). The siRNA or miRNA sequence guides
RISC to the target mRNA (139).
After binding to its target by base pairing, argonaute 2
endonuclease (a part of RISC) cleaves the mRNA, causing gene
silencing (138,143).
There are essential differences between siRNAs and
miRNAs: siRNAs are molecules 21–23 nucleotides long that can
modulate more than one mRNA because they are partially
complementary to their targets; whereas miRNAs are 19–25
nucleotides long and highly specific to one mRNA sequence, but they
can potentially regulate more than one target gene (138). The partial complementarity of
siRNAs with the gene sequence suggests that inhibitory activity is
not solely due to the direct degradation of target mRNA by the RISC
complex but can also increase the mRNA decay through the rapid
deadenylation of target transcripts (144,145). In addition, siRNAs can recruit
chromatin-targeted RNAi silencing components to heterochromatin
(146).
piRNAs are likely miRNAs that are genetically coded
in the piRNA cluster of genes and are highly expressed in gonadal
tissue to regulate transposable elements in the germline (147). Precursor piRNAs (pre-piRNAs) are
transcribed by RNA polymerase II. Once the newly transcribed RNA
leaves the nucleus, pre-piRNAs interact with the germline-specific
subclade or argonaute proteins known as PIWI proteins (148). Transgenerational inheritance of
piRNAs, such as maternal inheritance through oocytes, alters the
progeny phenotype, suggesting that it may contribute to diverse
cellular processes over generations (149).
There is strong evidence showing the association
between miRNAs and cancer development (150–153). miRNA expression is commonly
dysregulated in various tumors and deletion of miRNA genes is also
observed in other oncogenic genes (Fig.
2C). For example, miR-15a and miR-16-1 are commonly deleted in
B-cell chronic lymphocytic leukemia (154). These miRNAs are tumor suppressors
that inhibit the expression of the antiapoptotic factor Bcl-1
(155).
By contrast, miRNAs with oncogenic potential are
known oncogenes. The best-studied oncomiR is the miR-17-92 cluster
(known as oncomiR-1); it is part of chromosome 13 and includes
miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92a-1, which
are often dysregulated in hematopoietic and solid cancers (156). Transcriptional regulation of
miRNAs is also common in cancer. For instance, transcription
factors and the tumor suppressor protein p53 are the most commonly
mutated genes in several human cancer types (e.g., prostate, ovary,
skin, lung and breast cancer), and thus, a high number of
dysregulated miRNAs are associated with carcinogenesis (157,158). Notably, the highly mutagenic
landscape seen in the p53 gene gives rise to different variants,
each with a particular behavior, with specific miRNA targets
(159). Notably, several
oncoviruses express proteins that inactivate p53 and
consequently alter host miRNAs (160,161).
The therapeutic potential of miRNAs is delayed
through problems associated with the toxicity of the administration
methods and biological factors (clearance, non-specific
distribution and degradation in circulation) that may affect the
efficacy of the molecules. In addition to side effects unwanted
related mainly with the possible activation immune response and
off-target silencing of RNAs that play essential roles in cell
mechanisms. Synthetic RNA molecules improve biostability and reduce
toxicity through chemical modifications, such as 20-fluoro-35 and
20-O-methyl, indicating that they may solve some of the problems
associated with miRNAs (161).
lncRNAs are the most abundant RNA, with
16,000-100,000 known human lncRNA genes (162). They share similarities with mRNAs
because they exhibit transcriptional regulation, and a considerable
number of them are 3′ polyadenylated and 5′ capped and undergo
lncRNA splicing (159).
lncRNAs modulate gene transcription and epigenetics
in the cell nucleus and regulate mRNA stability, protein
translation and competing endogenous RNA (ceRNA) in the cytoplasm
(163). These RNAs have various
mechanisms, such as transmitting transcriptional signals,
functioning as decoys, scaffolds, RNA enhancers or coding for short
peptides (163,164).
Signal lncRNAs regulate transcription in response
to several stimuli such as inflammatory response, metabolic changes
and oxidative stress (163). Their
transcription occurs at specific times and locations, thus serving
as molecular signals. For example, X-chromosome inactivation occurs
during female development (165).
The lncRNA Xist is transcribed from the X chromosome that
will be inactivated, and the sole expression of Xist
indicates active silencing at the genomic locus. Signal lncRNAs can
be classified as decoys or scaffolds (166), and a single lncRNA can have more
than one mode of action. For instance, lincRNA-p21 has
essential functions in cancer development by modulating the
transcription of p21 downstream of p53 activity
(167). lincRNA-p21
expression is a marker of DNA damage and apoptosis (168). However, it is also a scaffold
molecule for the transcriptional complex assembled over the
p21 promoter by recruiting heterogeneous nuclear
ribonucleoprotein (hnRNP)-K, which mediates p53 binding to
such promoters (169).
Furthermore, lincRNA-p21 interacts with MDM2 proto-oncogene
(MDM2) to release p53 from repression (166).
Decoy lncRNAs trap transcriptional regulatory
factors by presenting decoy binding sites, thus lowering the
availability of elements needed for mRNA transcription (170). Scaffold lncRNAs allow the
formation of multiple complexes that either enhance or repress
transcription (164). For
instance, paraspeckles are mammalian-specific protein-rich nuclear
organelles assembled on top of the lncRNA NEAT1. These organelles
can trap RNAs and nucleoplasmic proteins, thereby reducing their
availability (171). Gastric
cancer lncRNA serves as a modular scaffold for the histone-modified
components WD repeat-containing protein 5 and lysine
acetyltransferase 2A in gastric cancer. It guides these components
to target genes to specify histone modification patterns (172). The guiding function of lncRNAs
helps direct ribonucleoproteins to target genes, and enhancer
lncRNAs influence the three-dimensional organization of DNA
(164).
It has been demonstrated that some ncRNAs possess
open reading frames encoding peptides (173). pri-miRNAs may encode proteins if
transported to the cytosol without processing (174). circRNAs can also be translated
into protein through the internal ribosome entry site (IRES)
(175). Several lncRNAs have been
identified in a number cancer types. For instance, lncRNA
HOXB-AS3 encodes a conserved 53-amino-acid-long peptide that
interacts with hnRNPA1 to repress glucose metabolism reprogramming,
thus suppressing colon cancer growth (176). Notably, the lncRNA peptidome has
been established for colorectal adenocarcinoma (177). Five universally expressed lncRNA
peptides show greater expression in tumors than normal tissues,
suggesting that they are promising biomarkers for this type of
cancer (154). Furthermore, the
peptide products of these lncRNAs are more stable than RNA
molecules in circulation; therefore, patient plasma can be a
reliable sample for diagnosis (178).
circRNAs are ncRNAs that are similar in length to
lncRNAs but they lack 5′ and 3′ modifications and are covalently
linked, forming a closed circular structure (129). These circular structures are
usually formed between the 5′ and 3′ spliced sites of exons,
although intron circularization also exists (179). Intronic circRNAs (ciRNAs) are
distinguished from exonic circRNAs (180). Exonic circRNAs are transported to
the cytoplasm by ATP-dependent RNA helicases, whereas ciRNAs remain
in the nucleus and modulate transcription (129).
Cytoplasmic circRNAs act as miRNA sponges that
regulate downstream targets or bind proteins (181). For instance, numerous circRNAs are
sponges of miR-7, a crucial tumor suppressor in several cancer
types. ciRS-7 expression is upregulated in colorectal and gastric
cancer, and blocks the suppressive function of miR-7 (181). ciRS-7 contains ~70 conserved miR-7
target sites, and downregulation of this miRNA promotes
retinoblastoma and astrocytoma development (182). circZFR binds to different miRNAs
and exerts opposing functions; it inhibits gastric cancer
proliferation through PTEN upregulation by targeting the
miR-130a/107 (183). Conversely,
circFZR promotes hepatocellular carcinoma proliferation by
modulating miR-1261/4302/3619 (184).
In addition, circRNAs interact with RNA-binding
proteins, which regulate RNA transcription, splicing, stability,
localization and translation (185). The interaction of circRNAs with
proteins acts as either a decoy or a scaffold (186). For instance, circ-DNMT1,
originating from the splicing of DNMT1, is upregulated in breast
cancer and contributes to cell proliferation (187). It interacts with p53 and
ribonucleoprotein hnRNPD and mediates the nuclear translocation of
both proteins (187). The binding
of circ-DNMT1 to hnRNPD stabilizes DNMT1 mRNA and increases
its translation (188).
circ-forkhead box O3 (FOXO3) expression serves a determining role
in the control of cell cycle progression. The formation of a
ternary complex with CDK2 and p21 proteins inhibits the
G1/S transition, mainly by blocking the interaction of
CDK2 with cyclin E (189).
circ-FOXO3 expression is dysregulated in tumor cells (breast and
renal cancer), while, under oxidative stress conditions, its
upregulation is associated with cell cycle arrest and apoptosis
(190). The interaction of
Circ-FOXO3 with MDM2 facilitates p53 ubiquitination in the cytosol,
which reduces FOXO3 degradation by MDM2 and upregulates the
expression of the p53 upregulated modulator of apoptosis
gene (187).
Some circRNAs can be translated into peptides due
to IRESs or N6-methyladenosine modifications (184). For example, circ-FBXW7 encodes a
21 kDa protein that inhibits cancer cell proliferation and reduces
c-Myc half-life; its expression is positively associated with the
overall survival of patients with glioblastoma (191).
RNA-based therapy is an expanding drug category
with rapid success found through the development of an mRNA-based
vaccine against severe acute respiratory syndrome coronavirus-2 to
tackle the coronavirus disease 2019 pandemic (192). ncRNA-based therapies are aimed at
modulating the transcriptional or post-transcriptional expression
of endogenous mRNAs to control the expression of specific proteins
involved in cancer development (193). Most ncRNA therapies are based on
miRNAs because they potentially target several genes
simultaneously, which is useful in multigenetic diseases (137). However, this multi-target action
may also have off-target drawbacks, and thus, siRNAs are preferred
for target identification and drug discovery (138). Similarly, lncRNAs may be used to
directly target or modulate other molecules, such as miRNAs, with
various molecules reaching the clinical trial phase (194). To the best of our knowledge, there
are no current preclinical reports using circRNAs alone as targets
or therapeutic vectors for cancer treatment; however, this will
likely change in the future (195).
Targeting ncRNAs in cancer serves one of two aims:
To restore tumor suppressor activity or to inhibit oncogene
expression (Fig. 3C). The ncRNA may
directly related with activation of genes controlling growth and
differentiation cellular, or promote the transformation through
indirect inhibition of genes of downstream pathways (196). In the second case, ncRNAs modulate
gene expression through RNAi or through transcriptional gene
silencing, which can result in long-term stable, inheritable
epigenetic changes (197). This
second mechanism is more commonly described for lncRNAs and RNAi
than for sncRNAs, although both types of ncRNAs can exert each mode
of action (144).
Replacement therapy involves delivering mimicking
molecules, synthetic RNA (particularly sncRNA), to restore
downregulated targets (Fig. 3B)
(198). Exogenous inhibitory RNAs
mimic tiers in the RNAi pathway; synthetic RNAs can be delivered to
cells in three ways. The first is artificial miRNAs (amiRNAs),
which are equivalent to endogenous pri-miRNAs. amiRNAs are
double-stranded stems flanked by a single-stranded basal segment
and an apical loop containing a recombinant pri-miRNA scaffold and
siRNA insert (199). These
synthetic RNAs enter the cell nucleus, undergo Drosha processing
and are exported to the cytoplasm to be loaded into the Dicer
complex. Therapeutically, amiRNAs can exert a long-lasting effect
due to their stable expression, which implies treatment regimens
with few doses, thus reducing the risk of toxicity. In addition,
they do not saturate miRNA machinery (198). Second, vector-based short hairpin
RNAs are the most widely used molecules for silencing in mammalian
cells. They mimic pre-miRNAs, which are processed double-stranded
stem-loop molecules that undergo Dicer processing. Third, synthetic
siRNAs are molecules of dsRNA that can be directly processed by
Dicer in a similar manner to miRNAs (198,199). This last group includes small
activating RNAs (saRNAs), which are dsRNAs that specifically target
and activate promoters. saRNAs are processed by argonaute proteins,
which translocate as a complex to the nucleus, bind to the promoter
to open the chromatin, and recruit positive transcriptional
regulators to the complex (200).
Tables VI and VII present examples of ncRNAs associated
with cancer development and therapeutic approaches applied in
clinical trials.
Inhibition therapy depletes oncogenic ncRNA
expression. Antisense oligonucleotides (ASOs) are 18–30 bp in
length and bind to target mRNAs and promote their degradation by
RNAse H, which recognizes the DNA:RNA hybrid and cleaves the RNA in
the complex (201). Alternatively,
ASOs can bind to miRNAs [anti-miRNA oligonucleotide (AMOs) also
known as antagomirs] (202). ASOs
and AMOs are subjected to nuclease degradation and weakly bind to
proteins; thus, their stability and uptake must be improved by
specific chemical modifications (201). One such modification is locked
nucleic acids (LNAs), a methylene linkage connecting the 2′ and 4′
carbons of the RNA backbone ribose (203). BlockmiRs are another class of ASOs
that specifically target the miRNA binding site in mRNAs, thus
blocking miRNA binding (204).
Aptamers are a class of synthetic DNA or RNA
oligonucleotides that target proteins. They recognize the tertiary
or quaternary structure of proteins rather than the linear
sequence, as is the case for ASOs; thus, aptamers are also known as
chemical antibodies (205).
Gapmers are ASOs containing a central DNA flanked by modified
oligonucleotides. These molecules are considered powerful options
for targeting nuclear lncRNAs due to the high level of RNAse H
activity observed during application (206).
Small molecules can also inhibit ncRNAs. These
molecules target either RNA-interacting molecules or ncRNAs. For
example, bisphenol A and diethylstilbestrol can modify chromatin
structure and gene activation via the increased expression of the
lncRNA HOTAIR (207). Their
advantage over oligonucleotides is that they are easily delivered,
soluble, bioavailable, stable and can be designed on a
structure-based approach. In addition, some small-molecule
inhibitors approved by the FDA can manipulate ncRNAs; this would
shorten the time taken to translate experiments to clinical therapy
(208).
Endogenously expressed lncRNAs and circRNAs
function as ceRNAs by sequestering miRNAs (209). This inherent ability of lncRNAs to
block the activity of miRNAs was exploited to engineer decoys, also
referred to as target mimics or sponges (177). Both lncRNAs and artificial sponges
harbor short sequences that are homologous to miRNA binding sites
on their endogenous targets (210). Therefore, decoys sequester miRNAs
and prevent their binding to targets (211). Engineered sponges typically
contain 10 binding sites separated by a few nucleotides (212). Sponges with imperfect binding
sites provoking a bulge are more effective in attracting miRNAs and
are more stable (213). Tough
decoys are 60-bp-long harpin sponges with an internal bulge
containing two miRNA sites (214).
The CRISPR-CRISPR associated protein 9
(CRISPR-Cas9) toolbox enables the manipulation of genes based on
RNA-guided DNA recognition (193).
A single-guide RNA is used to mediate DNA endonuclease Cas-9 to
introduce gaps into the DNA; thus, CRISPR-Cas9 is designed to
remove DNA fragments from the genome (215). The addition of donor DNA with the
CRISPR-Cas9 system may cause DNA breakage through homology-directed
repair, which uses and introduces the donor DNA into the genome.
Although several studies have confirmed the potential therapeutic
value of CRISPR-Cas9, there are still concerns regarding off-target
effects and ethical concerns raised by this technique in germline
cells (216–219). Tables VIII and IX present examples of ncRNA-inhibitory
therapies and clinical trials for cancer, respectively.
Although carcinogenesis has a complex and
multifactorial origin, alterations in epigenetic processes are
considered among the first genomic aberrations that occur during
cancer development and are closely related to its progression. The
dynamic and reversible nature of epigenetic modifications has
allowed the use of epidrug agents that improve cancer treatment,
including reducing chemoresistance. Although the use of epidrugs in
combination with chemotherapeutic agents may have clinical
benefits, several issues must be considered. The reversible nature
of methylation patterns persists after drug treatment, and
re-methylation and silencing are major unresolved issues. The
toxicity and instability of 5-aza-C and 5-aza-D may complicate
their use in clinical settings under physiological conditions
(59,62). Furthermore, the lack of specificity
of the methylating agent may result in the activation of normally
silenced genes and contribute to tumorigenesis (62); this poses a significant obstacle in
epigenetic therapy.
ncRNAs also represent an essential epigenetic
mechanism. Thousands of ncRNAs have been identified and their roles
in carcinogenesis are defined. This enables their clinical
application through the development of ncRNA-based diagnostic kits
for the early detection of cancer and for anticancer therapy.
However, this field is not as successful as epidrugs because there
is still a large gap between the identification of functional
ncRNAs and their clinical applications. In addition,
delivery-associated toxicity, poor transfection efficiency,
systemic clearance, non-specific biodistribution, degradation in
circulation and rapid renal clearance, have delayed the therapeutic
potential of ncRNAs (220,221).
The use of epidrugs promises to be an effective
tool to help overcome drug resistance in some patients with cancer,
particularly for hematological cancer types. However,
multidisciplinary work is needed to optimize drug engineering and
to design clinical trials to assess the safety and scope of these
drugs in cancer therapy for solid tumors.
Not applicable.
This work was supported by the National Council on Science and
Technology (CONACYT), Instituto Nacional de Cancerología, Mexico
(grant no. CF-2019-263979).
Not applicable.
LJCM, EVU, CS, ML, EDLCH and ACP performed the
bibliographic review and wrote and critically revised the
manuscript. ACP conceived and directed the manuscript. Data
authentication is not applicable. All authors read and approved the
final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mantovani F, Collavin L and Del Sal G:
Mutant p53 as a guardian of the cancer cell. Cell Death Differ.
26:199–212. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deans C and Maggert KA: What do you mean,
‘epigenetic’? Genetics. 199:887–896. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bashyam MD, Animireddy S, Bala P, Naz A
and George SA: The Yin and Yang of cancer genes. Gene. 704:121–133.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen P, Li W and Li G: Structures and
functions of chromatin fibers. Annu Rev Biophys. 50:95–116. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ferreira HJ and Esteller M: Non-coding
RNAs, epigenetics, and cancer: Tying it all together. Cancer
Metastasis Rev. 37:55–73. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang L, Lu Q and Chang C: Epigenetics in
health and disease. Adv Exp Med Biol. 1253:3–55. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu Y, Chan YT, Tan HY, Li S, Wang N and
Feng Y: Epigenetic regulation in human cancer: The potential role
of epi-drug in cancer therapy. Mol Cancer. 19:792020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ding X, He M, Chan AWH, Song QX, Sze SC,
Chen H, Man MKH, Man K, Chan SL, Lai PBS, et al: Genomic and
epigenomic features of primary and recurrent hepatocellular
carcinomas. Gastroenterology. 157:1630–1645.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Malouf GG, Taube JH, Lu Y, Roysarkar T,
Panjarian S, Estecio MR, Jelinek J, Yamazaki J, Raynal NJ, Long H,
et al: Architecture of epigenetic reprogramming following
Twist1-mediated epithelial-mesenchymal transition. Genome Biol.
14:R1442013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miranda Furtado CL, Dos Santos Luciano MC,
Silva Santos RD, Furtado GP, Moraes MO and Pessoa C: Epidrugs:
targeting epigenetic marks in cancer treatment. Epigenetics.
14:1164–1176. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lyko F: The DNA methyltransferase family:
a versatile toolkit for epigenetic regulation. Nat Rev Genet.
19:81–92. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ren W, Gao L and Song J: Structural basis
of DNMT1 and DNMT3A-mediated DNA methylation. Genes (Basel).
9:6202018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen Z and Zhang Y: Role of mammalian DNA
methyltransferases in development. Annu Rev Biochem. 89:135–158.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Smith ZD and Meissner A: DNA methylation:
Roles in mammalian development. Nat Rev Genet. 14:204–220. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang ZM, Lu R, Wang P, Yu Y, Chen D, Gao
L, Liu S, Ji D, Rothbart SB, Wang Y, et al: Structural basis for
DNMT3A-mediated de novo DNA methylation. Nature. 554:387–391. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Veland N, Lu Y, Hardikar S, Gaddis S, Zeng
Y, Liu B, Estecio MR, Takata Y, Lin K, Tomida MW, et al: DNMT3L
facilitates DNA methylation partly by maintaining DNMT3A stability
in mouse embryonic stem cells. Nucleic Acids Res. 47:152–167. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Loaeza-Loaeza J, Beltran AS and
Hernández-Sotelo D: DNMTs and impact of CpG content, transcription
factors, consensus motifs, lncRNAs, and histone marks on DNA
methylation. Genes (Basel). 11:13362020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Onodera A, González-Avalos E, Lio CWJ,
Georges RO, Bellacosa A, Nakayama T and Rao A: Roles of TET and TDG
in DNA demethylation in proliferating and non-proliferating immune
cells. Genome Biol. 22:1862021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rasmussen KD and Helin K: Role of TET
enzymes in DNA methylation, development, and cancer. Genes Dev.
30:733–750. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sabino JC, de Almeida MR, Abreu PL,
Ferreira AM, Caldas P, Domingues MM, Santos NC, Azzalin CM, Grosso
AR and de Almeida SF: Epigenetic reprogramming by TET enzymes
impacts co-transcriptional R-loops. Elife. 11:e694762022.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li C, Fan Y, Li G, Xu X, Duan J, Li R,
Kang X, Ma X, Chen X, Ke Y, et al: DNA methylation reprogramming of
functional elements during mammalian embryonic development. Cell
Discov. 4:412018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dossin F, Pinheiro I, Żylicz JJ, Roensch
J, Collombet S, Le Saux A, Chelmicki T, Attia M, Kapoor V, Zhan Y,
et al: SPEN integrates transcriptional and epigenetic control of
X-inactivation. Nature. 578:455–460. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tucci V, Isles AR, Kelsey G and
Ferguson-Smith AC; Erice Imprinting Group, : Genomic imprinting and
physiological processes in mammals. Cell. 176:952–965. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang M, Wu J, Zhong W, Zhao Z and He W:
DNA-methylation-induced silencing of DIO3OS drives non-small cell
lung cancer progression via activating hnRNPK-MYC-CDC25A axis. Mol
Ther Oncolytics. 23:205–219. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kennedy EM, Goehring GN, Nichols MH,
Robins C, Mehta D, Klengel T, Eskin E, Smith AK and Conneely KN: An
integrated-omics analysis of the epigenetic landscape of gene
expression in human blood cells. BMC Genomics. 19:4762018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kanwal R, Gupta K and Gupta S: Cancer
epigenetics: An introduction. Methods Mol Biol. 1238:3–25. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Klutstein M, Nejman D, Greenfield R and
Cedar H: DNA methylation in cancer and aging. Cancer Res.
76:3446–3450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sheaffer KL, Elliott EN and Kaestner KH:
DNA hypomethylation contributes to genomic instability and
intestinal cancer initiation. Cancer Prev Res (Phila). 9:534–546.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hatziapostolou M and Iliopoulos D:
Epigenetic aberrations during oncogenesis. Cell Mol Life Sci.
68:1681–1702. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guerrero-Preston R, Michailidi C,
Marchionni L, Pickering CR, Frederick MJ, Myers JN,
Yegnasubramanian S, Hadar T, Noordhuis MG, Zizkova V, et al: Key
tumor suppressor genes inactivated by ‘greater promoter’
methylation and somatic mutations in head and neck cancer.
Epigenetics. 9:1031–1046. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Faam B, Ghaffari MA, Khorsandi L, Ghadiri
AA, Totonchi M, Amouzegar A, Fanaei SA, Azizi F, Shahbazian HB and
Hashemi Tabar M: RAP1GAP functions as a tumor suppressor gene and
is regulated by DNA methylation in differentiated thyroid cancer.
Cytogenet Genome Res. 161:227–235. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chantre-Justino M, Gonçalves da Veiga
Pires I, Cardoso Figueiredo M, Dos Santos Moreira A, Alves G and
Faria Ornellas MH: Genetic and methylation status of CDKN2A
(p14ARF/p16INK4A) and TP53 genes in recurrent
respiratory papillomatosis. Hum Pathol. 119:94–104. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Han B, Yang X, Zhang P, Zhang Y, Tu Y, He
Z, Li Y, Yuan J, Dong Y, Hosseini DK, et al: DNA methylation
biomarkers for nasopharyngeal carcinoma. PLoS One. 15:e02305242020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hoang NM and Rui L: DNA methyltransferases
in hematological malignancies. J Genet Genomics. 47:361–372. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Steele N, Finn P, Brown R and Plumb JA:
Combined inhibition of DNA methylation and histone acetylation
enhances gene re-expression and drug sensitivity in vivo. Br J
Cancer. 100:758–763. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bao Y, Oguz G, Lee WC, Lee PL, Ghosh K, Li
J, Wang P, Lobie PE, Ehmsen S, Ditzel HJ, et al: EZH2-mediated PP2A
inactivation confers resistance to HER2-targeted breast cancer
therapy. Nat Commun. 11:58782020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vijayaraghavalu S and Labhasetwar V:
Nanogel-mediated delivery of a cocktail of epigenetic drugs plus
doxorubicin overcomes drug resistance in breast cancer cells. Drug
Deliv Transl Res. 8:1289–1299. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou Z, Li HQ and Liu F: DNA
methyltransferase inhibitors and their therapeutic potential. Curr
Top Med Chem. 18:2448–2457. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kedhari Sundaram M, Hussain A, Haque S,
Raina R and Afroze N: Quercetin modifies 5′CpG promoter methylation
and reactivates various tumor suppressor genes by modulating
epigenetic marks in human cervical cancer cells. J Cell Biochem.
120:18357–18369. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sanaei M, Kavoosi F and Behjoo H: Effect
of valproic acid and zebularine on SOCS-1 and SOCS-3 gene
expression in colon carcinoma SW48 cell line. Exp Oncol.
42:183–187. 2020.PubMed/NCBI
|
|
42
|
Capdevila J, Arqués O, Hernández Mora JR,
Matito J, Caratù G, Mancuso FM, Landolfi S, Barriuso J,
Jimenez-Fonseca P, Lopez Lopez C, et al: Epigenetic EGFR gene
repression confers sensitivity to therapeutic BRAFV600E blockade in
colon neuroendocrine carcinomas. Clin Cancer Res. 26:902–909. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Marques-Magalhães Â, Graça I, Henrique R
and Jerónimo C: Targeting DNA methyltranferases in urological
tumors. Front Pharmacol. 9:3662018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xylinas E, Hassler MR, Zhuang D,
Krzywinski M, Erdem Z, Robinson BD, Elemento O, Clozel T and
Shariat SF: An epigenomic approach to improving response to
neoadjuvant cisplatin chemotherapy in bladder cancer. Biomolecules.
6:372016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Stresemann C and Lyko F: Modes of action
of the DNA methyltransferase inhibitors azacytidine and decitabine.
Int J Cancer. 123:8–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chakrawarti L, Agrawal R, Dang S, Gupta S
and Gabrani R: Therapeutic effects of EGCG: A patent review. Expert
Opin Ther Pat. 26:907–916. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Beisler JA: Isolation, characterization,
and properties of a labile hydrolysis product of the antitumor
nucleoside, 5-azacytidine. J Med Chem. 21:204–208. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rahman MF, Raj R and Govindarajan R:
Identification of structural and molecular features involved in the
transport of 3′-Deoxy-nucleoside analogs by human equilibrative
nucleoside transporter 3. Drug Metab Dispos. 46:600–609. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Momparler RL and Derse D: Kinetics of
phosphorylation of 5-aza-2′-deoxyycytidine by deoxycytidine kinase.
Biochem Pharmacol. 28:1443–1444. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Santi DV, Garrett CE and Barr PJ: On the
mechanism of inhibition of DNA-cytosine methyltransferases by
cytosine analogs. Cell. 33:9–10. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Seelan RS, Mukhopadhyay P, Pisano MM and
Greene RM: Effects of 5-Aza-2′-deoxycytidine (decitabine) on gene
expression. Drug Metab Rev. 50:193–207. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zheng Z, Li L, Liu X, Wang D, Tu B, Wang
L, Wang H and Zhu WG: 5-Aza-2′-deoxycytidine reactivates gene
expression via degradation of pRb pocket proteins. FASEB J.
26:449–459. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sorm F, Pískala A, Cihák A and Veselý J:
5-Azacytidine, a new, highly effective cancerostatic. Experientia.
20:202–203. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sorm F and Vesely J: The activity of a new
antimetabolite, 5-azacytidine, against lymphoid leukaemia in AK
mice. Neoplasma. 11:123–130. 1964.PubMed/NCBI
|
|
55
|
Case DC Jr: 5-azacytidine in refractory
acute leukemia. Oncology. 39:218–221. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tanaka K, Appella E and Jay G:
Developmental activation of the H-2K gene is correlated with an
increase in DNA methylation. Cell. 35:457–465. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Vogler WR, Miller DS and Keller JW:
5-Azacytidine (NSC 102816): A new drug for the treatment of
myeloblastic leukemia. Blood. 48:331–337. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kaminskas E, Farrell A, Abraham S, Baird
A, Hsieh LS, Lee SL, Leighton JK, Patel H, Rahman A, Sridhara R, et
al: Approval summary: Azacitidine for treatment of myelodysplastic
syndrome subtypes. Clin Cancer Res. 11:3604–3608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ganesan A, Arimondo PB, Rots MG, Jeronimo
C and Berdasco M: The timeline of epigenetic drug discovery: From
reality to dreams. Clin Epigenetics. 11:1742019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Duchmann M and Itzykson R: Clinical update
on hypomethylating agents. Int J Hematol. 110:161–169. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Schaefer M, Hagemann S, Hanna K and Lyko
F: Azacytidine inhibits RNA methylation at DNMT2 target sites in
human cancer cell lines. Cancer Res. 69:8127–8132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hollenbach PW, Nguyen AN, Brady H,
Williams M, Ning Y, Richard N, Krushel L, Aukerman SL, Heise C and
MacBeth KJ: A comparison of azacitidine and decitabine activities
in acute myeloid leukemia cell lines. PLoS One. 5:e90012010.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Venturelli S, Berger A, Weiland T, Essmann
F, Waibel M, Nuebling T, Häcker S, Schenk M, Schulze-Osthoff K,
Salih HR, et al: Differential induction of apoptosis and senescence
by the DNA methyltransferase inhibitors 5-azacytidine and
5-aza-2′-deoxycytidine in solid tumor cells. Mol Cancer Ther.
12:2226–2236. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jin S, Cojocari D, Purkal JJ, Popovic R,
Talaty NN, Xiao Y, Solomon LR, Boghaert ER, Leverson JD and
Phillips DC: 5-Azacitidine induces NOXA to prime AML cells for
venetoclax-mediated apoptosis. Clin Cancer Res. 26:3371–3383. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fabre C, Grosjean J, Tailler M, Boehrer S,
Adès L, Perfettini JL, de Botton S, Fenaux P and Kroemer G: A novel
effect of DNA methyltransferase and histone deacetylase inhibitors:
NFkappaB inhibition in malignant myeloblasts. Cell Cycle.
7:2139–2145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Carbajo-García MC, Corachán A,
Segura-Benitez M, Monleón J, Escrig J, Faus A, Pellicer A, Cervelló
I and Ferrero H: 5-aza-2′-deoxycitidine inhibits cell
proliferation, extracellular matrix formation and Wnt/β-catenin
pathway in human uterine leiomyomas. Reprod Biol Endocrinol.
19:1062021. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Linnekamp JF, Kandimalla R, Fessler E, de
Jong JH, Rodermond HM, van Bochove GGW, The FO, Punt CJA, Bemelman
WA, van de Ven AWH, et al: Pre-operative decitabine in colon cancer
patients: Analyses on WNT target methylation and expression.
Cancers (Basel). 13:23572021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Santini V: How I treat MDS after
hypomethylating agent failure. Blood. 133:521–529. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Mabaera R, Greene MR, Richardson CA,
Conine SJ, Kozul CD and Lowrey CH: Neither DNA hypomethylation nor
changes in the kinetics of erythroid differentiation explain
5-azacytidine's ability to induce human fetal hemoglobin. Blood.
111:411–420. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Susanto JM, Colvin EK, Pinese M, Chang DK,
Pajic M, Mawson A, Caldon CE, Musgrove EA, Henshall SM, Sutherland
RL, et al: The epigenetic agents suberoylanilide hydroxamic acid
and 5-AZA-2′ deoxycytidine decrease cell proliferation, induce cell
death and delay the growth of MiaPaCa2 pancreatic cancer cells in
vivo. Int J Oncol. 46:2223–2230. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Evans IC, Barnes JL, Garner IM, Pearce DR,
Maher TM, Shiwen X, Renzoni EA, Wells AU, Denton CP, Laurent GJ, et
al: Epigenetic regulation of cyclooxygenase-2 by methylation of
c8orf4 in pulmonary fibrosis. Clin Sci (Lond). 130:575–586. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Allis CD and Jenuwein T: The molecular
hallmarks of epigenetic control. Nat Rev Genet. 17:487–500. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Meng CF, Zhu XJ, Peng G and Dai DQ:
Promoter histone H3 lysine 9 di-methylation is associated with DNA
methylation and aberrant expression of p16 in gastric cancer cells.
Oncol Rep. 22:1221–1227. 2009.PubMed/NCBI
|
|
74
|
Lee SH, Kim J, Kim WH and Lee YM: Hypoxic
silencing of tumor suppressor RUNX3 by histone modification in
gastric cancer cells. Oncogene. 28:184–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Nguyen CT, Weisenberger DJ, Velicescu M,
Gonzales FA, Lin JCY, Liang G and Jones PA: Histone H3-lysine 9
methylation is associated with aberrant gene silencing in cancer
cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer
Res. 62:6456–6461. 2002.PubMed/NCBI
|
|
76
|
Griffiths EA and Gore SD: Epigenetic
therapies in MDS and AML. Adv Exp Med Biol. 754:253–283. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Nguyen AN, Hollenbach PW, Richard N,
Luna-Moran A, Brady H, Heise C and MacBeth KJ: Azacitidine and
decitabine have different mechanisms of action in non-small cell
lung cancer cell lines. Lung Cancer (Auckl). 1:119–140.
2010.PubMed/NCBI
|
|
78
|
Home-ClinicalTrials.govNovember 22–2022https://clinicaltrials.gov/
|
|
79
|
Hurd PJ, Whitmarsh AJ, Baldwin GS, Kelly
SM, Waltho JP, Price NC, Connolly BA and Hornby DP: Mechanism-based
inhibition of C5-cytosine DNA methyltransferases by 2-H
pyrimidinone. J Mol Biol. 286:389–401. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yoo CB, Cheng JC and Jones PA: Zebularine:
A new drug for epigenetic therapy. Biochem Soc Trans. 32:910–912.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ferguson LR, Tatham AL, Lin Z and Denny
WA: Epigenetic regulation of gene expression as an anticancer drug
target. Curr Cancer Drug Targets. 11:199–212. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Dueñas-Gonzalez A, Coronel J, Cetina L,
González-Fierro A, Chavez-Blanco A and Taja-Chayeb L:
Hydralazine-valproate: A repositioned drug combination for the
epigenetic therapy of cancer. Expert Opin Drug Metab Toxicol.
10:1433–1444. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Singh N, Dueñas-González A, Lyko F and
Medina-Franco JL: Molecular modeling and molecular dynamics studies
of hydralazine with human DNA methyltransferase 1. ChemMedChem.
4:792–799. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Daher-Reyes GS, Merchan BM and Yee KWL:
Guadecitabine (SGI-110): An investigational drug for the treatment
of myelodysplastic syndrome and acute myeloid leukemia. Expert Opin
Investig Drugs. 28:835–849. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Salvador LA and Luesch H: Discovery and
mechanism of natural products as modulators of histone acetylation.
Curr Drug Targets. 13:1029–1047. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhang Y, Wang X, Han L, Zhou Y and Sun S:
Green tea polyphenol EGCG reverse cisplatin resistance of A549/DDP
cell line through candidate genes demethylation. Biomed
Pharmacother. 69:285–290. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Alaskhar Alhamwe B, Khalaila R, Wolf J,
von Bülow V, Harb H, Alhamdan F, Hii CS, Prescott SL, Ferrante A,
Renz H, et al: Histone modifications and their role in epigenetics
of atopy and allergic diseases. Allergy Asthma Clin Immunol.
14:392018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lawrence M, Daujat S and Schneider R:
Lateral thinking: How histone modifications regulate gene
expression. Trends Genet. 32:42–56. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Lakshmaiah KC, Jacob LA, Aparna S,
Lokanatha D and Saldanha SC: Epigenetic therapy of cancer with
histone deacetylase inhibitors. J Cancer Res Ther. 10:469–478.
2014.PubMed/NCBI
|
|
90
|
Albaugh BN, Arnold KM and Denu JM:
KAT(ching) metabolism by the tail: Insight into the links between
lysine acetyltransferases and metabolism. Chembiochem. 12:290–298.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yang J, Song C and Zhan X: The role of
protein acetylation in carcinogenesis and targeted drug discovery.
Front Endocrinol (Lausanne). 13:9723122022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Cohen I, Poręba E, Kamieniarz K and
Schneider R: Histone modifiers in cancer: Friends or foes? Genes
Cancer. 2:631–647. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Fan P, Zhao J, Meng Z, Wu H, Wang B, Wu H
and Jin X: Overexpressed histone acetyltransferase 1 regulates
cancer immunity by increasing programmed death-ligand 1 expression
in pancreatic cancer. J Exp Clin Cancer Res. 38:472019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Sun XJ, Man N, Tan Y, Nimer SD and Wang L:
The role of histone acetyltransferases in normal and malignant
hematopoiesis. Front Oncol. 5:1082015. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wang P, Wang Z and Liu J: Role of HDACs in
normal and malignant hematopoiesis. Mol Cancer. 19:52020.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Carraway HE, Malkaram SA, Cen Y, Shatnawi
A, Fan J, Ali HEA, Abd Elmageed ZY, Buttolph T, Denvir J, Primerano
DA and Fandy TE: Activation of SIRT6 by DNA hypomethylating agents
and clinical consequences on combination therapy in leukemia. Sci
Rep. 10:103252020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hu XT, Xing W, Zhao RS, Tan Y, Wu XF, Ao
LQ, Li Z, Yao MW, Yuan M, Guo W, et al: HDAC2 inhibits EMT-mediated
cancer metastasis by downregulating the long noncoding RNA H19 in
colorectal cancer. J Exp Clin Cancer Res. 39:2702020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Körholz K, Ridinger J, Krunic D, Najafi S,
Gerloff XF, Frese K, Meder B, Peterziel H, Vega-Rubin-de-Celis S,
Witt O and Oehme I: Broad-spectrum HDAC inhibitors promote
autophagy through FOXO transcription factors in neuroblastoma.
Cells. 10:10012021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Chun P: Histone deacetylase inhibitors in
hematological malignancies and solid tumors. Arch Pharm Res.
38:933–949. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Jęśko H, Wencel P, Strosznajder RP and
Strosznajder JB: Sirtuins and their roles in brain aging and
neurodegenerative disorders. Neurochem Res. 42:876–890. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Willis-Martinez D, Richards HW, Timchenko
NA and Medrano EE: Role of HDAC1 in senescence, aging, and cancer.
Exp Gerontol. 45:279–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Glozak MA and Seto E: Histone deacetylases
and cancer. Oncogene. 26:5420–5432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Audia JE and Campbell RM: Histone
modifications and cancer. Cold Spring Harb Perspect Biol.
8:a0195212016. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bergamin E, Sarvan S, Malette J, Eram MS,
Yeung S, Mongeon V, Joshi M, Brunzelle JS, Michaels SD, Blais A, et
al: Molecular basis for the methylation specificity of ATXR5 for
histone H3. Nucleic Acids Res. 45:6375–6387. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Mohan M, Herz HM and Shilatifard A:
SnapShot: Histone lysine methylase complexes. Cell. 149:498–498.e1.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
From the American Association of
Neurological Surgeons (AANS), American Society of Neuroradiology
(ASNR), Cardiovascular and Interventional Radiology Society of
Europe (CIRSE), Canadian Interventional Radiology Association
(CIRA), Congress of Neurological Surgeons (CNS), European Society
of Minimally Invasive Neurological Therapy (ESMINT), European
Society of Neuroradiology (ESNR), European Stroke Organization
(ESO), Society for Cardiovascular Angiography and Interventions
(SCAI), Society of Interventional Radiology (SIR), ; et al:
Multisociety consensus quality improvement revised consensus
statement for endovascular therapy of acute ischemic stroke. Int J
Stroke. 13:612–632. 2018.PubMed/NCBI
|
|
107
|
McGrath J and Trojer P: Targeting histone
lysine methylation in cancer. Pharmacol Ther. 150:1–22. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Wan YCE, Liu J and Chan KM: Histone H3
mutations in cancer. Curr Pharmacol Rep. 4:292–300. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Salz T, Deng C, Pampo C, Siemann D, Qiu Y,
Brown K and Huang S: Histone methyltransferase hSETD1A is a novel
regulator of metastasis in breast cancer. Mol Cancer Res.
13:461–469. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Behjati S, Tarpey PS, Presneau N, Scheipl
S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et
al: Distinct H3F3A and H3F3B driver mutations define
chondroblastoma and giant cell tumor of bone. Nat Genet.
45:1479–1482. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Papillon-Cavanagh S, Lu C, Gayden T,
Mikael LG, Bechet D, Karamboulas C, Ailles L, Karamchandani J,
Marchione DM, Garcia BA, et al: Impaired H3K36 methylation defines
a subset of head and neck squamous cell carcinomas. Nat Genet.
49:180–185. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Yang T, Yang Y and Wang Y: Predictive
biomarkers and potential drug combinations of epi-drugs in cancer
therapy. Clin Epigenetics. 13:1132021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Bates SE: Epigenetic therapies for cancer.
N Engl J Med. 383:650–663. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Yaseen A, Chen S, Hock S, Rosato R, Dent
P, Dai Y and Grant S: Resveratrol sensitizes acute myelogenous
leukemia cells to histone deacetylase inhibitors through reactive
oxygen species-mediated activation of the extrinsic apoptotic
pathway. Mol Pharmacol. 82:1030–1041. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Bubna AK: Vorinostat-an overview. Indian J
Dermatol. 60:4192015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Richon VM: Cancer biology: Mechanism of
antitumour action of vorinostat (suberoylanilide hydroxamic acid),
a novel histone deacetylase inhibitor. Br J Cancer. 95 (Suppl
1):S2–S6. 2006. View Article : Google Scholar
|
|
117
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Siegel D, Hussein M, Belani C, Robert F,
Galanis E, Richon VM, Garcia-Vargas J, Sanz-Rodriguez C and Rizvi
S: Vorinostat in solid and hematologic malignancies. J Hematol
Oncol. 2:312009. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Nakajima H, Kim YB, Terano H, Yoshida M
and Horinouchi S: FR901228, a potent antitumor antibiotic, is a
novel histone deacetylase inhibitor. Exp Cell Res. 241:126–133.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
VanderMolen KM, McCulloch W, Pearce CJ and
Oberlies NH: Romidepsin (Istodax, NSC 630176, FR901228, FK228,
depsipeptide): A natural product recently approved for cutaneous
T-cell lymphoma. J Antibiot (Tokyo). 64:525–531. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Marshall JL, Rizvi N, Kauh J, Dahut W,
Figuera M, Kang MH, Figg WD, Wainer I, Chaissang C, Li MZ and
Hawkins MJ: A phase I trial of depsipeptide (FR901228) in patients
with advanced cancer. J Exp Ther Oncol. 2:325–332. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Petrich A and Nabhan C: Use of class I
histone deacetylase inhibitor romidepsin in combination regimens.
Leuk Lymphoma. 57:1755–1765. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
O'Connor OA, Horwitz S, Masszi T, Van Hoof
A, Brown P, Doorduijn J, Hess G, Jurczak W, Knoblauch P, Chawla S,
et al: Belinostat in patients with relapsed or refractory
peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF
(CLN-19) study. J Clin Oncol. 33:2492–2499. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Poole RM: Belinostat: First global
approval. Drugs. 74:1543–1554. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Autin P, Blanquart C and Fradin D:
Epigenetic drugs for cancer and microRNAs: A focus on histone
deacetylase inhibitors. Cancers (Basel). 11:15302019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Spratlin JL, Pitts TM, Kulikowski GN,
Morelli MP, Tentler JJ, Serkova NJ and Eckhardt SG: Synergistic
activity of histone deacetylase and proteasome inhibition against
pancreatic and hepatocellular cancer cell lines. Anticancer Res.
31:1093–1103. 2011.PubMed/NCBI
|
|
127
|
Lee MJ, Kim YS, Kummar S, Giaccone G and
Trepel JB: Histone deacetylase inhibitors in cancer therapy. Curr
Opin Oncol. 20:639–649. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Gibney ER and Nolan CM: Epigenetics and
gene expression. Heredity (Edinb). 105:4–13. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
He J, Xie Q, Xu H, Li J and Li Y: Circular
RNAs and cancer. Cancer Lett. 396:138–144. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Khanna A and Stamm S: Regulation of
alternative splicing by short non-coding nuclear RNAs. RNA Biol.
7:480–485. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Kwek KY, Murphy S, Furger A, Thomas B,
O'Gorman W, Kimura H, Proudfoot NJ and Akoulitchev A: U1 snRNA
associates with TFIIH and regulates transcriptional initiation. Nat
Struct Mol Biol. 9:800–805. 2002.PubMed/NCBI
|
|
132
|
Eilebrecht S, Brysbaert G, Wegert T,
Urlaub H, Benecke BJ and Benecke A: 7SK small nuclear RNA directly
affects HMGA1 function in transcription regulation. Nucleic Acids
Res. 39:2057–2072. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Szymanski M, Erdmann VA and Barciszewski
J: Noncoding RNAs database (ncRNAdb). Nucleic Acids Res.
35:(Database Issue). D162–D164. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Chu CY and Rana TM: Small RNAs: Regulators
and guardians of the genome. J Cell Physiol. 213:412–419. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Abi A, Farahani N, Molavi G and Gheibi
Hayat SM: Circular RNAs: Epigenetic regulators in cancerous and
noncancerous skin diseases. Cancer Gene Ther. 27:280–293. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Quinn JJ and Chang HY: Unique features of
long non-coding RNA biogenesis and function. Nat Rev Genet.
17:47–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Ding X, Zhang S, Li X, Feng C, Huang Q,
Wang S, Wang S, Xia W, Yang F, Yin R, et al: Profiling expression
of coding genes, long noncoding RNA, and circular RNA in lung
adenocarcinoma by ribosomal RNA-depleted RNA sequencing. FEBS Open
Bio. 8:544–555. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Lam JKW, Chow MYT, Zhang Y and Leung SWS:
siRNA versus miRNA as therapeutics for gene silencing. Mol Ther
Nucleic Acids. 4:e2522015. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Meister G and Tuschl T: Mechanisms of gene
silencing by double-stranded RNA. Nature. 431:343–349. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Kim VN: MicroRNA biogenesis: Coordinated
cropping and dicing. Nat Rev Mol Cell Biol. 6:376–385. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek
SH and Kim VN: MicroRNA genes are transcribed by RNA polymerase II.
EMBO J. 23:4051–4060. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Tomari Y and Zamore PD: Perspective:
Machines for RNAi. Genes Dev. 19:517–529. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Paturi S and Deshmukh MV: A glimpse of
‘dicer biology’ through the structural and functional perspective.
Front Mol Biosci. 8:6436572021. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Alagia A, Jorge AF, Aviñó A, Cova TFGG,
Crehuet R, Grijalvo S, Pais AACC and Eritja R: Exploring
PAZ/3′-overhang interaction to improve siRNA specificity. A
combined experimental and modeling study. Chem Sci. 9:2074–2086.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Wu L, Fan J and Belasco JG: MicroRNAs
direct rapid deadenylation of mRNA. Proc Natl Acad Sci USA.
103:4034–4039. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Chen L, Dahlstrom JE, Lee SH and Rangasamy
D: Naturally occurring endo-siRNA silences LINE-1 retrotransposons
in human cells through DNA methylation. Epigenetics. 7:758–771.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Guo B, Li D, Du L and Zhu X: piRNAs:
Biogenesis and their potential roles in cancer. Cancer Metastasis
Rev. 39:567–575. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Sparmann A: piRNAs-guardians of the
germline. Nat Res. 2019.
|
|
149
|
Iwasaki YW, Siomi MC and Siomi H:
PIWI-interacting RNA: Its biogenesis and functions. Annu Rev
Biochem. 84:405–433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Caramuta S, Egyházi S, Rodolfo M, Witten
D, Hansson J, Larsson C and Lui WO: MicroRNA expression profiles
associated with mutational status and survival in malignant
melanoma. J Invest Dermatol. 130:2062–2070. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Chi J, Ballabio E, Chen XH, Kušec R,
Taylor S, Hay D, Tramonti D, Saunders NJ, Littlewood T, Pezzella F,
et al: MicroRNA expression in multiple myeloma is associated with
genetic subtype, isotype and survival. Biol Direct. 6:232011.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Garzon R, Garofalo M, Martelli MP,
Briesewitz R, Wang L, Fernandez-Cymering C, Volinia S, Liu CG,
Schnittger S, Haferlach T, et al: Distinctive microRNA signature of
acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin.
Proc Natl Acad Sci USA. 105:3945–3950. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Iorio MV, Ferracin M, Liu CG, Veronese A,
Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M,
et al: MicroRNA gene expression deregulation in human breast
cancer. Cancer Res. 65:7065–7070. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Calin GA, Cimmino A, Fabbri M, Ferracin M,
Wojcik SE, Shimizu M, Taccioli C, Zanesi N, Garzon R, Aqeilan RI,
et al: MiR-15a and miR-16-1 cluster functions in human leukemia.
Proc Natl Acad Sci USA. 105:5166–5171. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Cimmino A, Calin GA, Fabbri M, Iorio MV,
Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et
al: miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Mogilyansky E and Rigoutsos I: The
miR-17/92 cluster: A comprehensive update on its genomics,
genetics, functions and increasingly important and numerous roles
in health and disease. Cell Death Differ. 20:1603–1614. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: Lessons from recent developments in the IARC TP53
database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Liao JM, Cao B, Zhou X and Lu H: New
insights into p53 functions through its target microRNAs. J Mol
Cell Biol. 6:206–213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Madrigal T, Hernández-Monge J, Herrera LA,
González-De la Rosa CH, Domínguez-Gómez G, Candelaria M,
Luna-Maldonado F, Calderón González KG and Díaz-Chávez J:
Regulation of miRNAs expression by mutant p53 gain of function in
cancer. Front Cell Dev Biol. 9:6957232021. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Tornesello ML, Annunziata C, Tornesello
AL, Buonaguro L and Buonaguro FM: Human oncoviruses and p53 tumor
suppressor pathway deregulation at the origin of human cancers.
Cancers (Basel). 10:2132018. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Hassler MR, Turanov AA, Alterman JF,
Haraszti RA, Coles AH, Osborn MF, Echeverria D, Nikan M, Salomon
WE, Roux L, et al: Comparison of partially and fully
chemically-modified siRNA in conjugate-mediated delivery in vivo.
Nucleic Acids Res. 46:2185–2196. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Statello L, Guo CJ, Chen LL and Huarte M:
Gene regulation by long non-coding RNAs and its biological
functions. Nat Rev Mol Cell Biol. 22:96–118. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Gao N, Li Y, Li J, Gao Z, Yang Z, Li Y,
Liu H and Fan T: Long non-coding RNAs: The regulatory mechanisms,
research strategies, and future directions in cancers. Front Oncol.
10:5988172020. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Fang Y and Fullwood MJ: Roles, functions,
and mechanisms of long non-coding RNAs in cancer. Genomics
Proteomics Bioinformatics. 14:42–54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Mathy NW and Chen XM: Long non-coding RNAs
(lncRNAs) and their transcriptional control of inflammatory
responses. J Biol Chem. 292:12375–12382. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Amirinejad R, Rezaei M and
Shirvani-Farsani Z: An update on long intergenic noncoding RNA p21:
A regulatory molecule with various significant functions in cancer.
Cell Biosci. 10:822020. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Hall JR, Messenger ZJ, Tam HW, Phillips
SL, Recio L and Smart RC: Long noncoding RNA lincRNA-p21 is the
major mediator of UVB-induced and p53-dependent apoptosis in
keratinocytes. Cell Death Dis. 6:e17002015. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Huarte M, Guttman M, Feldser D, Garber M,
Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M,
et al: A large intergenic noncoding RNA induced by p53 mediates
global gene repression in the p53 response. Cell. 142:409–419.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Dai X, Kaushik AC and Zhang J: The
emerging role of major regulatory RNAs in cancer control. Front
Oncol. 9:9202019. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Fox AH, Nakagawa S, Hirose T and Bond CS:
Paraspeckles: Where long noncoding RNA meets phase separation.
Trends Biochem Sci. 43:124–135. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Sun TT, He J, Liang Q, Ren LL, Yan TT, Yu
TC, Tang JY, Bao YJ, Hu Y, Lin Y, et al: LncRNA GClnc1 promotes
gastric carcinogenesis and may Act as a modular scaffold of WDR5
and KAT2A complexes to specify the histone modification pattern.
Cancer Discov. 6:784–801. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Wang J, Zhu S, Meng N, He Y, Lu R and Yan
GR: ncRNA-encoded peptides or proteins and cancer. Mol Ther.
27:1718–1725. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Xing J, Liu H, Jiang W and Wang L:
LncRNA-encoded peptide: Functions and predicting methods. Front
Oncol. 10:6222942021. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Kong S, Tao M, Shen X and Ju S:
Translatable circRNAs and lncRNAs: Driving mechanisms and functions
of their translation products. Cancer Lett. 483:59–65. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Huang JZ, Chen M, Chen D, Gao XC, Zhu S,
Huang H, Hu M, Zhu H and Yan GR: A peptide encoded by a putative
lncRNA HOXB-AS3 suppresses colon cancer growth. Mol Cell.
68:171–184.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Chen Y, Long W, Yang L, Zhao Y, Wu X, Li
M, Du F, Chen Y, Yang Z, Wen Q, et al: Functional peptides encoded
by long non-coding RNAs in gastrointestinal cancer. Front Oncol.
11:7773742021. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Chakraborty S, Andrieux G, Hasan AMM,
Ahmed M, Hosen MI, Rahman T, Hossain MA and Boerries M: Harnessing
the tissue and plasma lncRNA-peptidome to discover peptide-based
cancer biomarkers. Sci Rep. 9:123222019. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Welden JR and Stamm S: Pre-mRNA structures
forming circular RNAs. Biochim Biophys Acta Gene Regul Mech.
1862:1944102019. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Eger N, Schoppe L, Schuster S, Laufs U and
Boeckel JN: Circular RNA splicing. Xiao J: Circular RNAs. Advances
in Experimental Medicine and Biology. 1087. Springer; Singapore:
pp. 41–52. 2018, View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Nisar S, Bhat AA, Singh M, Karedath T,
Rizwan A, Hashem S, Bagga P, Reddy R, Jamal F, Uddin S, et al:
Insights into the role of CircRNAs: Biogenesis, characterization,
functional, and clinical impact in human malignancies. Front Cell
Dev Biol. 9:6172812021. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Zhao J, Tao Y, Zhou Y, Qin N, Chen C, Tian
D and Xu L: MicroRNA-7: A promising new target in cancer therapy.
Cancer Cell Int. 15:1032015. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Liu T, Liu S, Xu Y, Shu R, Wang F, Chen C,
Zeng Y and Luo H: Circular RNA-ZFR inhibited cell proliferation and
promoted apoptosis in gastric cancer by sponging miR-130a/miR-107
and modulating PTEN. Cancer Res Treat. 50:1396–1417. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Yu CY and Kuo HC: The emerging roles and
functions of circular RNAs and their generation. J Biomed Sci.
26:292019. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Jiang MP, Xu WX, Hou JC, Xu Q, Wang DD and
Tang JH: The emerging role of the interactions between circular
RNAs and RNA-binding proteins in common human cancers. J Cancer.
12:5206–5219. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Huang A, Zheng H, Wu Z, Chen M and Huang
Y: Circular RNA-protein interactions: Functions, mechanisms, and
identification. Theranostics. 10:3503–3517. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Du WW, Yang W, Li X, Awan FM, Yang Z, Fang
L, Lyu J, Li F, Peng C, Krylov SN, et al: A circular RNA circ-DNMT1
enhances breast cancer progression by activating autophagy.
Oncogene. 37:5829–5842. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Chen S, Thorne RF, Zhang XD, Wu M and Liu
L: Non-coding RNAs, guardians of the p53 galaxy. Semin Cancer Biol.
75:72–83. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Du WW, Yang W, Liu E, Yang Z, Dhaliwal P
and Yang BB: Foxo3 circular RNA retards cell cycle progression via
forming ternary complexes with p21 and CDK2. Nucleic Acids Res.
44:2846–2858. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Wang C, Tao W, Ni S and Chen Q: Circular
RNA circ-Foxo3 induced cell apoptosis in urothelial carcinoma via
interaction with miR-191-5p. Onco Targets Ther. 12:8085–8094. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Yang Y, Gao X, Zhang M, Yan S, Sun C, Xiao
F, Huang N, Yang X, Zhao K, Zhou H, et al: Novel role of FBXW7
circular RNA in repressing glioma tumorigenesis. J Natl Cancer
Inst. 110:304–315. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Park JW, Lagniton PNP, Liu Y and Xu RH:
mRNA vaccines for COVID-19: What, why and how. Int J Biol Sci.
17:1446–1460. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Damase TR, Sukhovershin R, Boada C,
Taraballi F, Pettigrew RI and Cooke JP: The limitless future of RNA
therapeutics. Front Bioeng Biotechnol. 9:6281372021. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Qian Y, Shi L and Luo Z: Long non-coding
RNAs in cancer: Implications for diagnosis, prognosis, and therapy.
Front Med (Lausanne). 7:6123932020. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Kristensen LS, Hansen TB, Venø MT and
Kjems J: Circular RNAs in cancer: Opportunities and challenges in
the field. Oncogene. 37:555–565. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Ling H: Non-coding RNAs: Therapeutic
strategies and delivery systems. Adv Exp Med Biol. 937:229–237.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Weinberg MS and Morris KV: Transcriptional
gene silencing in humans. Nucleic Acids Res. 44:6505–6517. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Kotowska-Zimmer A, Pewinska M and
Olejniczak M: Artificial miRNAs as therapeutic tools: Challenges
and opportunities. Wiley Interdiscip Rev RNA. 12:e16402021.
View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Turner AMW and Morris KV: Controlling
transcription with noncoding RNAs in mammalian cells.
Biotechniques. 48:9–16. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Yoon S and Rossi JJ: Therapeutic potential
of small activating RNAs (saRNAs) in human cancers. Curr Pharm
Biotechnol. 19:604–610. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Scoles DR, Minikel EV and Pulst SM:
Antisense oligonucleotides: A primer. Neurol Genet. 5:e3232019.
View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Raghavendra P and Pullaiah T: RNA-based
applications in diagnostic and therapeutics for cancer. Advances in
Cell and Molecular Diagnostics. Elsevier; pp. 33–55. 2018,
View Article : Google Scholar
|
|
203
|
Ratti M, Lampis A, Ghidini M, Salati M,
Mirchev MB, Valeri N and Hahne JC: MicroRNAs (miRNAs) and long
non-coding RNAs (lncRNAs) as new tools for cancer therapy: First
steps from bench to bedside. Targ Oncol. 15:261–278. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Vicentini C, Galuppini F, Corbo V and
Fassan M: Current role of non-coding RNAs in the clinical setting.
Noncoding RNA Res. 4:82–85. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Giudice V, Mensitieri F, Izzo V,
Filippelli A and Selleri C: Aptamers and antisense oligonucleotides
for diagnosis and treatment of hematological diseases. Int J Mol
Sci. 21:32522020. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Maruyama R and Yokota T: Knocking down
long noncoding RNAs using antisense oligonucleotide gapmers.
Methods Mol Biol. 2176:49–56. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Bhan A, Hussain I, Ansari KI, Bobzean SAM,
Perrotti LI and Mandal SS: Bisphenol-A and diethylstilbestrol
exposure induces the expression of breast cancer associated long
noncoding RNA HOTAIR in vitro and in vivo. J Steroid Biochem Mol
Biol. 141:160–170. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Simonson B and Das S: MicroRNA
therapeutics: The next magic bullet? Mini Rev Med Chem. 15:467–474.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Zhong Y, Du Y, Yang X, Mo Y, Fan C, Xiong
F, Ren D, Ye X, Li C, Wang Y, et al: Circular RNAs function as
ceRNAs to regulate and control human cancer progression. Mol
Cancer. 17:792018. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Ebert MS, Neilson JR and Sharp PA:
MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian
cells. Nat Methods. 4:721–726. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Banks IR, Zhang Y, Wiggins BE, Heck GR and
Ivashuta S: RNA decoys: An emerging component of plant regulatory
networks? Plant Signal Behav. 7:1188–1193. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Ebert MS and Sharp PA: MicroRNA sponges:
Progress and possibilities. RNA. 16:2043–2050. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Bernardo BC, Ooi JY, Lin RC and McMullen
JR: miRNA therapeutics: A new class of drugs with potential
therapeutic applications in the heart. Future Med Chem.
7:1771–1792. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Yoo J, Hajjar R and Jeong D: Generation of
efficient miRNA inhibitors using tough decoy constructs. Ishikawa
K: Cardiac Gene Therapy. Methods in Molecular Biology. 1521. Humana
Press; New York, NY: pp. 41–53. 2017, View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Feng R, Patil S, Zhao X, Miao Z and Qian
A: RNA therapeutics-research and clinical advancements. Front Mol
Biosci. 8:7107382021. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Aimo A, Castiglione V, Rapezzi C, Franzini
M, Panichella G, Vergaro G, Gillmore J, Fontana M, Passino C and
Emdin M: RNA-targeting and gene editing therapies for transthyretin
amyloidosis. Nat Rev Cardiol. 19:655–667. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Dever DP, Bak RO, Reinisch A, Camarena J,
Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB,
Mantri S, et al: CRISPR/Cas9 β-globin gene targeting in human
haematopoietic stem cells. Nature. 539:384–389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Long C, Amoasii L, Mireault AA, McAnally
JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby
R and Olson EN: Postnatal genome editing partially restores
dystrophin expression in a mouse model of muscular dystrophy.
Science. 351:400–403. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Yin H, Xue W, Chen S, Bogorad RL,
Benedetti E, Grompe M, Koteliansky V, Sharp PA, Jacks T and
Anderson DG: Genome editing with Cas9 in adult mice corrects a
disease mutation and phenotype. Nat Biotechnol. 32:551–553. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Lee SWL, Paoletti C, Campisi M, Osaki T,
Adriani G, Kamm RD, Mattu C and Chiono V: MicroRNA delivery through
nanoparticles. J Control Release. 313:80–95. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Kawakami S and Hashida M: Targeted
delivery systems of small interfering RNA by systemic
administration. Drug Metab Pharmacokinet. 22:142–151. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Soengas MS, Capodieci P, Polsky D, Mora J,
Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL,
Lazebnik YA, et al: Inactivation of the apoptosis effector Apaf-1
in malignant melanoma. Nature. 409:207–211. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Mann BS, Johnson JR, Cohen MH, Justice R
and Pazdur R: FDA approval summary: Vorinostat for treatment of
advanced primary cutaneous T-cell lymphoma. Oncologist.
12:1247–1252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Grant C, Rahman F, Piekarz R, Peer C, Frye
R, Robey RW, Gardner ER, Figg WD and Bates SE: Romidepsin: A new
therapy for cutaneous T-cell lymphoma and a potential therapy for
solid tumors. Expert Rev Anticancer Ther. 10:997–1008. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Moore D: Panobinostat (Farydak): A novel
option for the treatment of relapsed or relapsed and refractory
multiple myeloma. P T. 41:296–300. 2016.PubMed/NCBI
|
|
226
|
Gilles ME and Slack FJ: Let-7 microRNA as
a potential therapeutic target with implications for immunotherapy.
Expert Opin Ther Targets. 22:929–939. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Segal M, Biscans A, Gilles ME,
Anastasiadou E, De Luca R, Lim J, Khvorova A and Slack FJ:
Hydrophobically modified let-7b miRNA enhances biodistribution to
NSCLC and downregulates HMGA2 in vivo. Mol Ther Nucleic Acids.
19:267–277. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Wu L, Wang Q, Yao J, Jiang H, Xiao C and
Wu F: MicroRNA let-7g and let-7i inhibit hepatoma cell growth
concurrently via downregulation of the anti-apoptotic protein
B-cell lymphoma-extra large. Oncol Lett. 9:213–218. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Pan X, Wang G and Wang B: MicroRNA-1182
and let-7a exert synergistic inhibition on invasion, migration and
autophagy of cholangiocarcinoma cells through down-regulation of
NUAK1. Cancer Cell Int. 21:1612021. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Li M, Dou J, Pan M, Xu H and Xu Z:
MicroRNA-7 agomir is a potential bioactive material for breast
cancer therapy by inhibiting breast cancer stem cell
tumorigenicity. Mater Express. 11:824–831. 2021. View Article : Google Scholar
|
|
231
|
Hong DS, Kang YK, Borad M, Sachdev J,
Ejadi S, Lim HY, Brenner AJ, Park K, Lee JL, Kim TY, et al: Phase 1
study of MRX34, a liposomal miR-34a mimic, in patients with
advanced solid tumours. Br J Cancer. 122:1630–1637. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
232
|
van Zandwijk N, Pavlakis N, Kao SC, Linton
A, Boyer MJ, Clarke S, Huynh Y, Chrzanowska A, Fulham MJ, Bailey
DL, et al: Safety and activity of microRNA-loaded minicells in
patients with recurrent malignant pleural mesothelioma: A
first-in-man, phase 1, open-label, dose-escalation study. Lancet
Oncol. 18:1386–1396. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Winata P, Williams M, McGowan E, Nassif N,
van Zandwijk N and Reid G: The analysis of novel microRNA mimic
sequences in cancer cells reveals lack of specificity in stem-loop
RT-qPCR-based microRNA detection. BMC Res Notes. 10:6002017.
View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Filippova EA, Fridman MV, Burdennyy AM,
Loginov VI, Pronina IV, Lukina SS, Dmitriev AA and Braga EA: Long
Noncoding RNA GAS5 in breast cancer: Epigenetic mechanisms and
biological functions. Int J Mol Sci. 22:68102021. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Williams GT and Pickard MR: Long
non-coding RNAs: New opportunities and old challenges in cancer
therapy. Transl Cancer Res. 5 (Suppl 3):S564–S565. 2016. View Article : Google Scholar
|
|
236
|
Liu W, Zhan J, Zhong R, Li R, Sheng X, Xu
M, Lu Z and Zhang S: Upregulation of long noncoding RNA_GAS5
suppresses cell proliferation and metastasis in laryngeal cancer
via regulating PI3K/AKT/mTOR signaling pathway. Technol Cancer Res
Treat. 20:15330338219900742021. View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Smaldone MC and Davies BJ: BC-819, a
plasmid comprising the H19 gene regulatory sequences and diphtheria
toxin A, for the potential targeted therapy of cancers. Curr Opin
Mol Ther. 12:607–616. 2010.PubMed/NCBI
|
|
238
|
Zhao X, Reebye V, Hitchen P, Fan J, Jiang
H, Sætrom P, Rossi J, Habib NA and Huang KW: Mechanisms involved in
the activation of C/EBPα by small activating RNA in hepatocellular
carcinoma. Oncogene. 38:3446–3457. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
239
|
Reebye V, Sætrom P, Mintz PJ, Huang KW,
Swiderski P, Peng L, Liu C, Liu X, Lindkaer-Jensen S, Zacharoulis
D, et al: Novel RNA oligonucleotide improves liver function and
inhibits liver carcinogenesis in vivo. Hepatology. 59:216–227.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Seto AG, Beatty X, Lynch JM, Hermreck M,
Tetzlaff M, Duvic M and Jackson AL: Cobomarsen, an oligonucleotide
inhibitor of miR-155, co-ordinately regulates multiple survival
pathways to reduce cellular proliferation and survival in cutaneous
T-cell lymphoma. Br J Haematol. 183:428–444. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Ijaz M, Wang F, Shahbaz M, Jiang W, Fathy
AH and Nesa EU: The role of Grb2 in cancer and peptides as Grb2
antagonists. Protein Pept Lett. 24:1084–1095. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
242
|
Ohanian M, Tari Ashizawa A, Garcia-Manero
G, Pemmaraju N, Kadia T, Jabbour E, Ravandi F, Borthakur G,
Andreeff M, Konopleva M, et al: Liposomal Grb2 antisense
oligodeoxynucleotide (BP1001) in patients with refractory or
relapsed haematological malignancies: A single-centre, open-label,
dose-escalation, phase 1/1b trial. Lancet Haematol. 5:e136–e146.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Golan T, Khvalevsky EZ, Hubert A, Gabai
RM, Hen N, Segal A, Domb A, Harari G, David EB, Raskin S, et al:
RNAi therapy targeting KRAS in combination with chemotherapy for
locally advanced pancreatic cancer patients. Oncotarget.
6:24560–24570. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Timar J and Kashofer K: Molecular
epidemiology and diagnostics of KRAS mutations in human cancer.
Cancer Metastasis Rev. 39:1029–1038. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
245
|
Zhang P, Liu X, Abegg D, Tanaka T, Tong Y,
Benhamou RI, Baisden J, Crynen G, Meyer SM, Cameron MD, et al:
Repro-gramming of protein-targeted small-molecule medicines to RNA
by ribonuclease recruitment. J Am Chem Soc. 143:13044–13055. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Shortridge MD, Chaubey B, Zhang HJ,
Pavelitz T, Olsen GL, Calin GA and Varani G: Drug-like small
molecules that inhibit expression of the oncogenic microRNA-21.
bioRxiv. doi:. https://doi.org/10.1101/2022.04.30.490150
|
|
247
|
Donlic A, Morgan BS, Xu JL, Liu A, Roble C
Jr and Hargrove AE: Discovery of small molecule ligands for MALAT1
by tuning an RNA-binding scaffold. Angew Chem Int Ed Engl.
57:13242–13247. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
248
|
Goyal B, Yadav SRM, Awasthee N, Gupta S,
Kunnumakkara AB and Gupta SC: Diagnostic, prognostic, and
therapeutic significance of long non-coding RNA MALAT1 in cancer.
Biochim Biophys Acta Rev Cancer. 1875:1885022021. View Article : Google Scholar : PubMed/NCBI
|