Introduction
Oral squamous cell carcinoma (OSCC) is one of the
most common types of head and neck SCC (HNSCC), accounting for more
than half of HNSCC cases. OSCC originates in the squamous
epithelium of the dental area (1).
It is an aggressive neoplasia with a poor prognosis and a high
recurrence rate (2). Despite
progress in anti-cancer therapeutic research, the 5-year survival
rate for OSCC has remained at 30% when detected at advanced stages
(3). Notably, the mortality rate of
patients with OSCC increases up to 92% when recurrence is diagnosed
(4). Therefore, detailed pathogenic
mechanisms need to be identified.
For tumorigenesis, multiple critical steps are
required. First, cancer cells sustain autonomous proliferative
capacity that is maintained to avoid senescence that can be caused
by excessive cell proliferation (5). Second, cancer cells effectively resist
cell death by regulating the apoptotic machinery. Apoptosis
initiation can be negatively controlled by pro-survival members of
the Bcl-2 family. Third, carcinoma cells promote local invasion and
distant metastasis by activating epithelial-mesenchymal transition
and/or invasion-inducing signals. Additionally, cancer cells have
the potential for replicative immortality, angiogenesis, evading
growth suppressors, reprogramming cellular metabolism and
inhibiting eradication by the immune system (6). Therefore, key molecules involved in
these tumor hallmarks must be identified to develop anti-tumor
therapeutic strategies.
Forkhead transcriptional factor O1 (FoxO1) is a
critical gene for regulating cellular homeostasis. FoxO1 controls
the expression of target genes responsible for redox balance, cell
proliferation, inflammation, metabolism and apoptosis. FoxO1 has
been regarded as a putative tumor suppressor in numerous types of
neoplasms (7). Several in
vitro and in vivo studies have shown that FoxO1
suppresses cancer cell proliferation and induces apoptosis
(8). FoxO1 also exerts anti-tumor
effects by inhibiting invasion and angiogenesis (9,10).
Consistent with these previous findings, FoxO1 is downregulated,
mutated or inactivated in numerous cancers (11). However, it has also been
demonstrated that FoxO1 supports cancer progression; gain of
function mutations in FoxO1 have been reported in specific cancer
types (12). Furthermore, the
concurrent downregulation of FoxO1 and FoxO3 inhibits tumor growth
and metastasis (13). To the best
of our knowledge, however, the molecular functions of FoxO1 in OSCC
have not yet been investigated. Therefore, the present study aimed
to explore the potential intracellular roles of FoxO1 in OSCC.
Materials and methods
Plasmid construction
The lentiCRISPRv2 plasmid, a gift from Dr Feng Zhang
(Addgene, Inc.; cat. no. 52961) (14), was digested with BsmBI (New
England BioLabs, Inc.) according to the manufacturer's
instructions, followed by dephosphorylation and gel purification.
For genetic perturbation of FoxO1, a pair of oligos
(5′-CACCGGTTGCCCCACGCGTTGCGGC-3′ and
5′-AAACGCCGCAACGCGTGGGGCAACC-3′; Macrogen) were phosphorylated,
annealed at 60°C for 30 sec and ligated into the digested
lentiCRISPRv2 plasmid, which was called LC-FoxO1.
Cell culture and generation of
FoxO1-deficient cells
YD-9, YD-8, FaDu, and SNU1041 cells were purchased
from Korean Cell line bank. YD-9, YD-8, and SNU1041 cells were
maintained in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.), and FaDu cells were maintained in MEM (Cytiva), supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin-streptomycin in a humidified atmosphere
containing 5% CO2 at 37°C. To generate FoxO1-decifient
cells, the 0.5 µg of LC-FoxO1 construct was transfected into YD-9
cells via electroporation (pulse voltage: 1400 V, pulse width: 20
ms, pulse number: 2) using the Neon Transfection System
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. YD-9 cells transfected with
GFP-targeting plasmid were used as control cells. After 2 days of
transfection, the cells were selected with 2 µg/ml puromycin.
Puromycin-resistant control and FoxO1-deficient cells were
maintained in RPMI-1640 supplemented with 10% fetal bovine serum,
1% penicillin-streptomycin and 2 µg/ml puromycin. To assess the
effect of FoxO1 on YD-9 cell survival, control and FoxO1-deficient
YD-9 cells were treated with 10 ng/ml TNFα for 2 days.
Clinical samples
A total of seven (two males and 5 females)
randomized Korean patients diagnosed with oral lichen planus (OLP)
(age range: 47–62 years old) from April to July 2022 in the
Department of Oral Medicine, Jeonbuk National University Hospital
(Jeonju, Korea) were enrolled in the study. The inclusion criteria
were reticular/atrophic/erosive OLP verified according to the
clinicopathological criteria by World Health Organization (WHO)
(15), including visual validation
of whitish and erythematous mucosal lesions on the buccal mucosa
and confirmation using incisional biopsy. The following diseases
and systemic conditions were excluded, such as autoimmune diseases
including psoriasis and rheumatoid arthritis, other oral mucosal
infectious diseases including oral candidiasis and herpes virus
infection, and oral cancers. The present study was approved by the
Institutional Review Board of Jeonbuk National University Hospital
(approval no. CUH 2021-02-044-001), and written informed consent
was obtained from all the participants.
Protein preparation and immunoblot
analysis
YD-9 cells were directly disrupted in laemmli buffer
[60 mM Tris-HCl (pH 6.8), 2% (w/v) sodium dodecyl sulfate (SDS),
10% (v/v) glycerol and 0.02% (w/v) bromophenol blue], followed by
sonication and heat denaturation. Samples were separated on 12%
SDS-polyacrylamide gels, and proteins were transferred to a
polyvinylidene fluoride membrane. After being blocked with 5%
skimmed milk at room temperature for 30 min, the membranes were
incubated overnight at 4°C with the following primary antibodies
against phosphorylated (phospho) H3 at Ser 10 (1:1,000, #9701, Cell
Signaling Technology, Inc.), total H3 (1:50,000, ab1791, Abcam),
PCNA (1:1,000, BS1289, Bioworld Technology, Inc.), cyclin B (1:200,
sc-245, Santa Cruz Biotechnology, Inc.), ARF (1:1,000, PA1-127,
Thermo Fisher Scientific, Inc.), CDK5 (1:1,000, CSB-PA005067LA01HU,
Cusabio Technology, LLC), β actin (1:50,000, A5316, Sigma-Aldrich;
Merck KGaA), PRDX5 (peroxiredoxin 5) (1:1,000, A305-339A, Bethyl
Laboratories, Inc.), PRDX3 (1:1,000, A304-744A, Bethyl
Laboratories, Inc.), LC3 (1:1,000, NB100-2220, Novus Biologicals,
LLC), HIF1α (1:1,000, ab82832, Abcam), SQSTM1 (Sequestosome1)/p62
(1:1,000, cat. no. ab56416, Abcam), Beclin 1 (1:1,000, A302-566A,
Bethyl Laboratories, Inc.), survivin (1:1,000, #2808, Cell
Signaling Technology, Inc.), total JNK (1:1,000, #9252, Cell
Signaling Technology, Inc.), total ERK (1:1,000, #4695, Cell
Signaling Technology, Inc.), total NFκB (1:1,000, #8242, Cell
Signaling Technology, Inc.), phospho JNK (1:1,000, #4668, Cell
Signaling Technology, Inc.), phospho ERK (1:1,000, MA5-15174,
Thermo Fisher Scientific, Inc.), and phospho NFκB (1:1,000,
MA5-15160, Thermo Fisher Scientific, Inc.). The membranes were
incubated at room temperature for 1 h with secondary anti-rabbit
(1:5,000, ab205718, Abcam) and anti-mouse (1:10,000, A90-116P,
Bethyl Laboratories, Inc.) antibodies that were horseradish
peroxidase conjugated. Immunoreactive signals were detected using
the D-Plus™ ECL Femto system (ECL-FS200, Donginbiotech
Co., Ltd., Korea) and Fusion Solo S chemiluminescence imaging
system (Vilber). Densitometric analysis of western blot bands was
carried out using ImageJ software (National Institutes of
Health).
Reactive oxygen species (ROS)
detection
Intracellular ROS levels were determined using the
fluorogenic CellROX® Orange reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
CellROX reagent was added to cultured cells at a final
concentration of 5 µM for 30 min at 37°C. Nuclear DAPI staining was
conducted with NucBlue Live ReadyProbes Reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) for 5 min. Fluorescent microscopic images
were acquired using the EVOS FL Auto Imaging System (Thermo Fisher
Scientific, Inc.) (magnification, ×200).
Immunofluorescence
OLP tissues were fixed with 10% formalin at room
temperature for 24 h. Paraffin-embedded OLP tissue sections were
cut at 5–8 µm thickness and incubated at 55°C for 30 min before
staining. Two changes of xylene (10 min each) were used to dewax
the tissue sections and 100, 90, 80, 70 and 50% ethanol, and water
(10 min each) were used for hydration. Following deparaffinization
and hydration, the OLP tissue sections were incubated with blocking
serum (1% bovine serum albumin, 4% normal goat serum and 0.2%
Triton X-100 in PBS) (Vector Laboratories, Inc.) for 2 h at room
temperature and subsequently treated with primary antibody against
FoxO1 (1:200, ab39670, Abcam) overnight at 4°C. The next day,
sections were rinsed with PBS and incubated with Alexa Fluor® 555
(1:1,000, ab150078, Abcam) for 90 min at room temperature.
Fluorescent microscopic images were acquired using the EVOS FL Auto
Imaging System (Thermo Fisher Scientific, Inc.) (magnification,
×100).
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from YD-9 cells using a
FavorPrep™ Blood/Cultured Cell Total RNA Kit (Favorgen
Biotech Corporation), according to the manufacturer's instructions.
A total of 300 ng total RNA was treated with RNase-free DNase
(Sigma-Aldrich; Merck KGaA) for 15 min, followed by inactivation of
DNase with EDTA treatment and heating. Total RNA was
reverse-transcribed into cDNA using the First Strand cDNA Synthesis
kit (Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. RT-qPCR was performed on cDNA samples
with the Luna® Universal qPCR Master Mix (New England BioLabs,
Inc.) using the Mic qPCR Cycler (Bio Molecular Systems). The
thermocycling conditions were as follows: Initial denaturation at
95°C for 1 min was followed by cycles comprising denaturation at
95°C for 15 sec, annealing and extension at 60°C for 30 sec. The
relative mRNA expression was calculated via the 2−ΔΔCq
method (16). The sequences of the
forward and reverse primers are presented in Table I.
 | Table I.Forward and reverse primers used for
reverse transcription-quantitative PCR. |
Table I.
Forward and reverse primers used for
reverse transcription-quantitative PCR.
| Gene | Sequence,
5′→3′ |
|---|
| hRPL32 (F) |
GAAGTTCCTGGTCCACAACG |
| hRPL32 (R) |
GCGATCTCGGCACAGTAAG |
| hKRT76 (F) |
CCGCAGAGAATGAGTTTGTGGG |
| hKRT76 (R) |
CATAGAGGGTCCTCAGGAAGCT |
| hKRT80 (F) |
CTCAATGTGCGCATCCAGAAGC |
| hKRT80 (R) |
TTGGTCTTGGCATCCTGGAAGG |
| hKRT23 (F) |
GGTGACATCCACGAACTGAAGC |
| hKRT23 (R) |
AGCTTGCAGGAGTACCGAGACT |
| hKRT10 (F) |
CCTGCTTCAGATCGACAATGCC |
| hKRT10 (R) |
ATCTCCAGGTCAGCCTTGGTCA |
| hTNFα (F) |
AGCCCATGTTGTAGCAAACC |
| hTNFα (R) |
TGAGGTACAGGCCCTCTGAT |
| hCCL20 (F) |
TGCTGTACCAAGAGTTTGCTC |
| hCCL20 (R) |
CGCACACAGACAACTTTTTCTTT |
Colony formation assay
LC-GFP and LC-FoxO1 cells were seeded in 6-well
plates at a density of 500 cells/well and incubated at 37°C for 14
days. After incubation, the wells were washed twice with PBS, fixed
with 4% paraformaldehyde at room temperature for 15 min. Cells were
stained with 0.1% crystal violet at RT for 30 min and then lysed
with 1% SDS. Quantitative changes in clonogenicity was assessed at
OD570 nm by spectrometer.
Bioinformatics data analysis
Analysis of the FoxO1 Expression in the HNSCC was
performed using cBioPortal (cbioportal.org), a publicly available database for
tumor transcriptomics. Gene Expression Profiling Interactive
Analysis (gepia.cancer-pku.cn) was used to
compare FoxO1 gene expression among cancer types and tumor stages.
Transcriptome datasets from the National Center for Biotechnology
Information Gene Expression Omnibus (ncbi.nlm.nih.gov/geo/) were utilized to assess FoxO1
transcript levels in OSCC. G:Profiler (http://biit.cs.ut.ee/gprofiler/gost) was utilized to
perform gene ontology (GO) enrichment analysis for the biological
process on gene sets.
Transwell migration assay
A 24-well Transwell insert system with an 8.0 µm
pore size polycarbonate membrane was purchased from Corning, Inc.
10% FBS-containing RPMI was placed in the lower chambers to as a
chemoattractant. LC-GFP and LC-FoxO1 cells (5×104
cells/insert) in 300 µl serum-free RPMI medium were treated with
200 nM CoCl2 at 37°C for 12 h, seeded in the upper
chamber and allowed to migrate at 37°C for 24 h. The cells were
fixed with 4% paraformaldehyde at RT for 15 min and stained with
0.1% crystal violet at room temperature for 20 min. Non-migrated
cells were removed from the top of each insert using a cotton swab.
Fluorescent microscopic images were acquired using the EVOS FL Auto
Imaging System (Thermo Fisher Scientific, Inc.; magnification,
×100).
Wound healing assay
The migration ability of YD-9 cells was measured
using wound healing assay. YD-9 cells were suspended in serum-free
RPMI and stained with 5 µl/ml
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate)
37°C for 30 min. Cells were washed with PBS and seeded in 12-well
plates at a density of 1×105 cells/well. When cells grew
to ~100% confluency in a monolayer, a scratch was made using a
1,000 µl pipette tip. After removing cell debris by washing three
times with PBS, the cells were incubated with RPMI containing 10%
FBS at 37°C. Images were acquired at the 0, 6, 12 and 24 h
post-scratching. Fluorescent microscopic images were acquired using
the EVOS FL Auto Imaging System (Thermo Fisher Scientific, Inc.;
magnification, ×100).
Statistical analysis
Unpaired two-tailed Student's t test was used for
experiments comparing two groups. One-way ANOVA followed by post
hoc Bonferroni's correction was used to assess >2 groups. All
data are expressed as the mean ± SEM of ≥3 independent experimental
repeats. GraphPad Prism software (version 9; GraphPad Software,
Inc.; Dotmatics) was used for all statistical analyses. P<0.05
was considered to indicate a statistically significant
difference.
Results
Analysis of FoxO1 expression in
premalignant and malignant lesions
To explore the potential roles of FoxO1 in the
pathogenesis of OSCC, expression of FoxO1 was assessed. Previously,
Shi et al (17) performed
transcriptome profiling with clinical samples and demonstrated that
FoxO1 mRNA levels are significantly increased in both OLP, a
premalignant lesion, and OSCC. The present immunofluorescence
staining of normal and OLP samples validated that FoxO1 protein
expression was significantly upregulated in both reticular and
ulcerative forms of OLP compared with normal tissue (Fig. 1A).
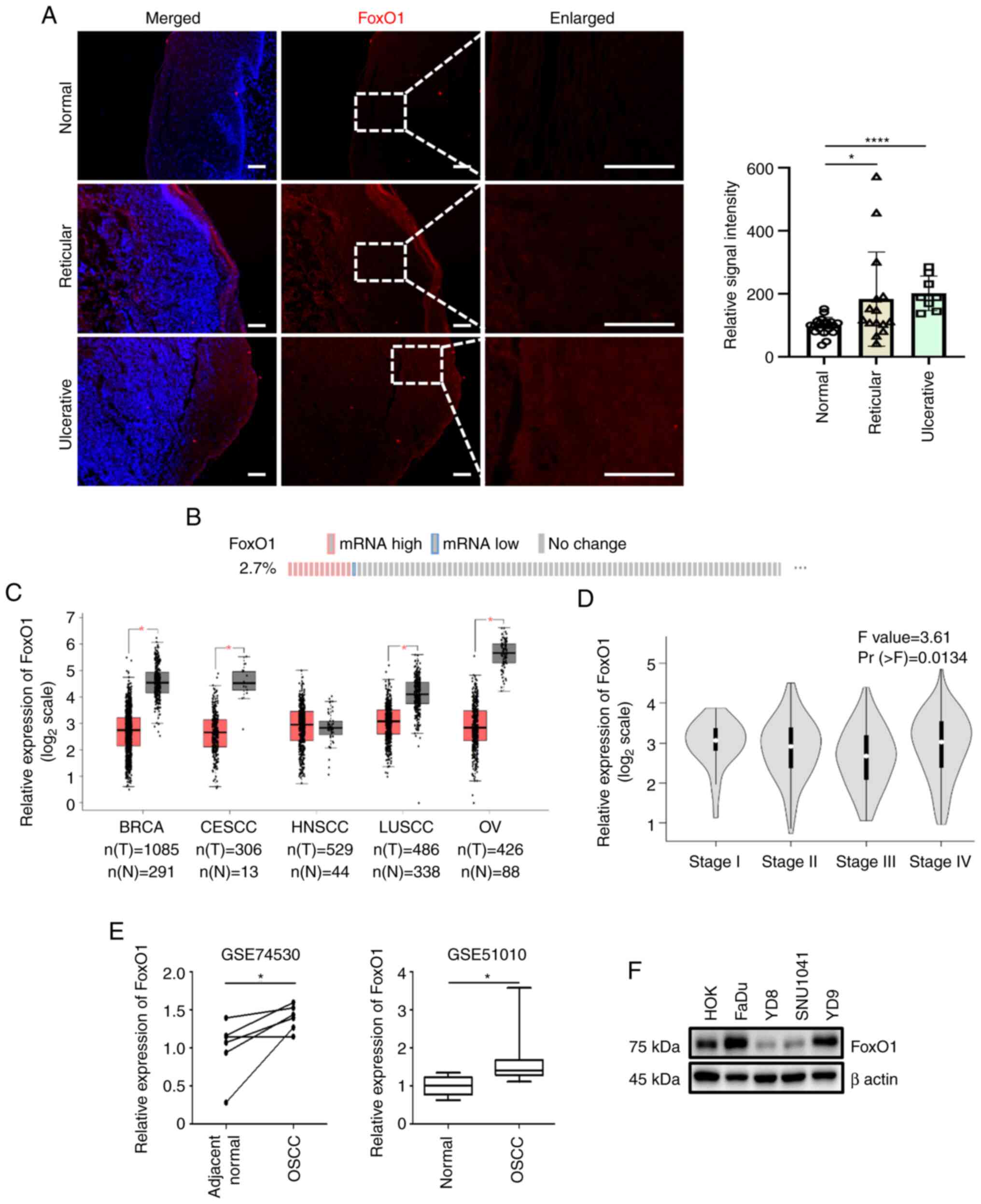 | Figure 1.FoxO1 expression profile. (A) Normal
and reticular- and ulcerative-type oral lichen planus samples were
immunostained with FoxO1 antibody (red). Nuclear DAPI staining is
shown in blue. Scale bar, 50 µm. Relative FoxO1 fluorescence
intensities are shown. (B) Alteration of FoxO1 expression in HNSCC.
The number of patients with HNSCC with upregulated (red) or
downregulated (blue) expression of FoxO1 is presented. Data were
derived from cBioPortal (cbioportal.org). (C) Expression levels of FoxO1 in
cancer were evaluated via Gene Expression Profiling Interactive
Analysis. (D) FoxO1 expression based on pathological HNSCC stage.
(E) Comparison of FoxO1 mRNA expression between N and OSCC cells.
Data were extracted from the National Center for Biotechnology
Information Gene Expression Omnibus profile database. (F)
Immunoblot analysis of FoxO1 protein. β actin was used as a loading
control. BRCA, breast invasive carcinoma; CESCC, cervical squamous
cell carcinoma and endocervical adenocarcinoma; LUSCC, lung
squamous cell carcinoma; OV, ovarian serous cystadenocarcinoma;
FoxO1, forkhead transcriptional factor O1; HNSCC, head and neck
squamous cell carcinoma; OSCC, oral squamous cell carcinoma; T,
tumor; N, normal. *P<0.05, ****P<0.0001. |
To analyze FoxO1 expression levels in OSCC, publicly
available gene expression datasets were used. According to the data
from The Cancer Genome Atlas and cBioPortal (18,19),
abnormal FoxO1 expression was detected in 2.7% of HNSCC cases.
FoxO1 upregulation was more common than FoxO1 downregulation
(Fig. 1B). Gene Expression
Profiling Interactive Analysis (gepia.cancer-pku.cn/) was used to
compare FoxO1 gene expression in cancer types. By contrast to other
cancer types in which FoxO1 was downregulated, FoxO1 expression was
not altered in HNSCC (Fig. 1C)
(20). The expression of FoxO1
varied during tumor progression (Fig.
1D). FoxO1 transcript levels were assessed in two oral cancer
datasets (accession nos. GSE74530 and GSE51010) from the National
Center for Biotechnology Information Gene Expression Omnibus
profile database. Although the clinical sample number was
insufficient to draw a general conclusion, FoxO1 was significantly
upregulated in OSCC (Fig. 1E).
FoxO1 protein levels were assessed in four HNSCC
(including OSCC) cell lines. Compared with normal human oral
keratinocytes, FoxO1 protein was upregulated in FaDu and YD-9 cells
but downregulated in YD-8 and SNU1041 cells (Fig. 1F), suggesting that FoxO1 expression
was increased in a subpopulation of patients with OSCC. To
determine the molecular function of FoxO1 in OSCC, YD-9 was
selected as a cell model. YD-9 is a human papillomavirus
DNA-negative OSCC acquired from buccal mucosa where premalignant
OLP frequently occurs. As FoxO1 is overexpressed in both OLP and
YD-9 cells, the present study explored the pathophysiological roles
of FoxO1 in OLP and OSCC. Compared with the other HNSCC three cell
lines that contain the mutant form of p53, YD-9 cells express
wild-type p53. Therefore, molecular function of FoxO1 could be
determined excluding the effect of the p53 mutation (21).
FoxO1 inhibits proliferation in YD-9
human OSCC cells
To determine the disrupted intracellular biological
processes in OSCC, Gene Ontology (GO) enrichment analysis of
transcriptome data (accession no. GSE70665) was performed (22–24).
As a result, 1,890 genes (687 up- and 1,203 downregulated) were
differentially expressed in OSCC compared with normal samples.
Upregulated genes were highly enriched in ‘keratinization’ and
‘cell division’ (Fig. S1).
To determine whether FoxO1 was involved in the
keratinization process, FoxO1 expression was knocked out in YD-9
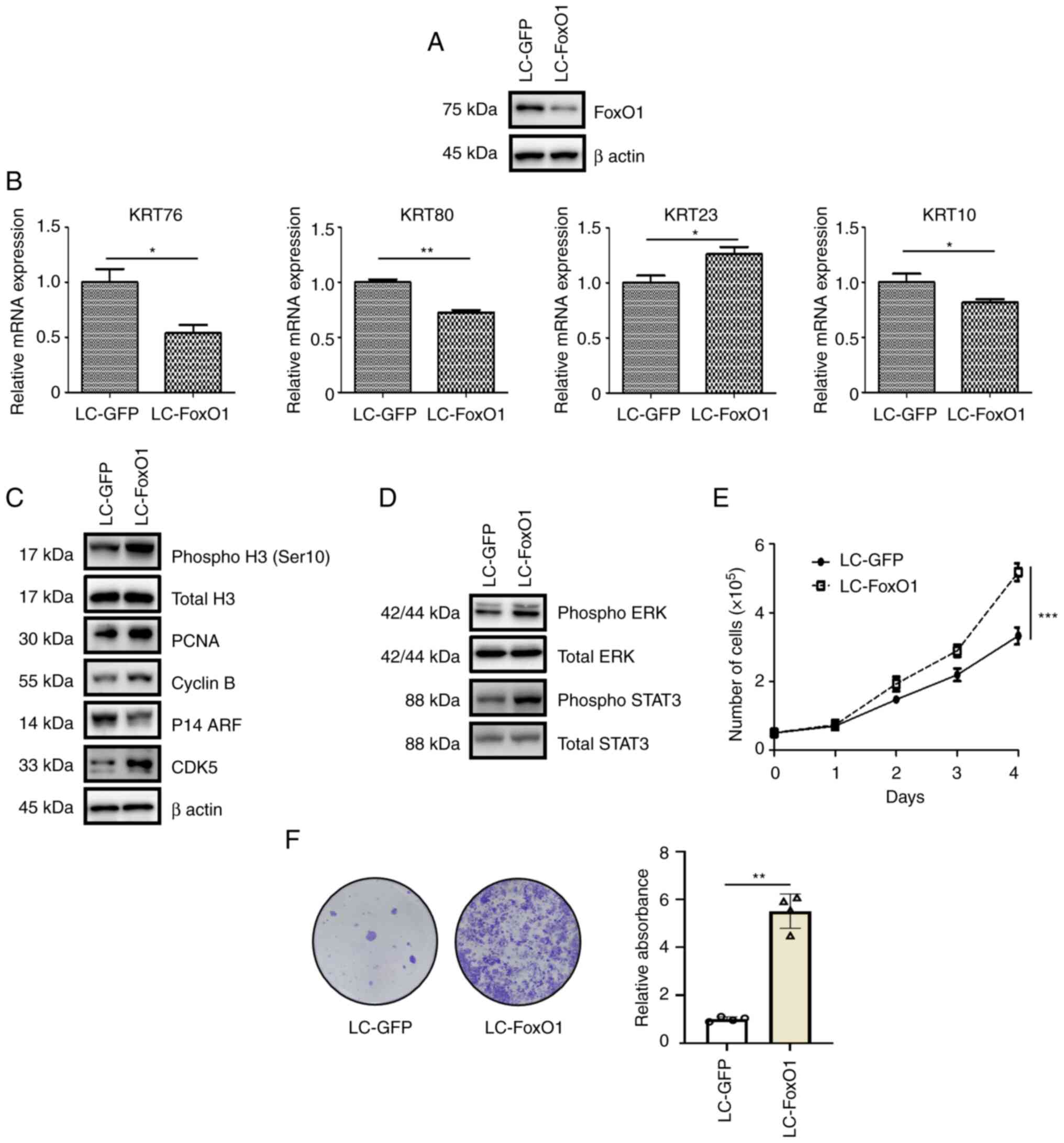
cells using Crispr/Cas9 (LC-FoxO1; Fig.
2A). mRNA levels of keratin (KRT) genes were up-(KRT23) or
downregulated (KRT76, KRT80 and KRT10) following FoxO1 silencing
(Fig. 2B). It was previously
reported that certain KRT genes are involved in tumor growth. While
KRT23 promotes cancer proliferation (25), KRT76 and KRT10 suppress cancer
growth (26,27), raising the possibility that FoxO1
could regulate cell proliferation. FoxO1 reduction increased the
levels of the cell proliferation markers phospho H3 (Ser10) and
PCNA (Fig. 2C). In addition, FoxO1
depletion led to upregulation of cell cycle-promoting factors
Cyclin B and CDK5 and downregulation of the ARF tumor suppressor
(Fig. 2C). In line with a previous
report that ERK and STAT3 signaling are key for OSCC proliferation
(28), FoxO1 deficiency increased
the ERK and STAT3 phosphorylation (Fig.
2D). The enhancement of cell proliferation by FoxO1 silencing
was validated by time-dependent changes in the total live cell
counts (Fig. 2E). Furthermore,
FoxO1 deficiency increased clonogenicity (Fig. 2F). Collectively, these data
suggested that FoxO1 served an anti-proliferative function in OSCC
pathogenesis.
FoxO1 suppresses migration in YD-9
human OSCC cells
As simultaneous activation of ERK and STAT3
signaling is key for the migration/invasion and proliferation of
cancer cells (29), it was
investigated with FoxO1 was implicated in the migration of YD-9
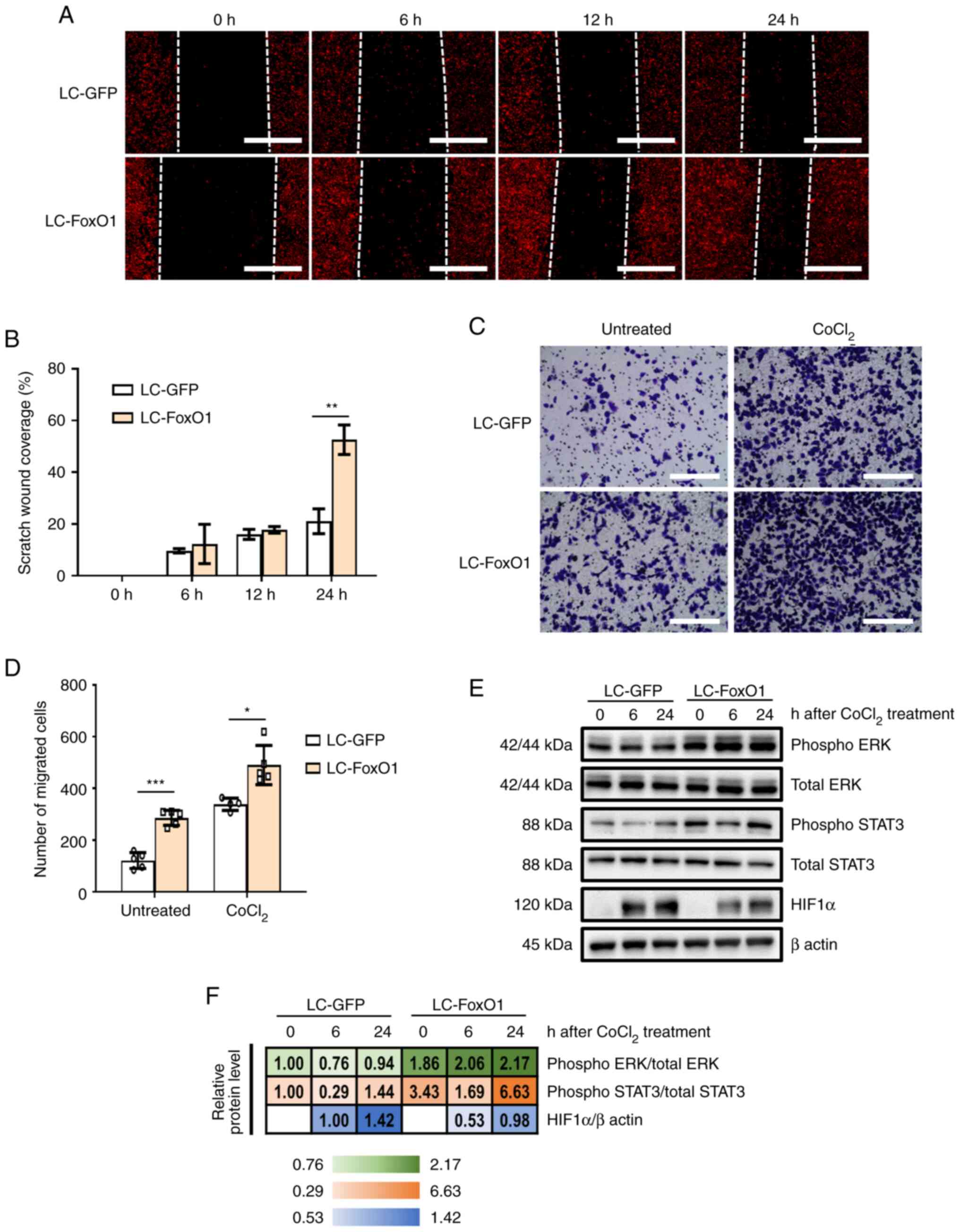
cells. In vitro wound healing assay demonstrated that
FoxO1-disrupted cells showed higher migration capacity than control
cells (Fig. 3A and B). Transwell
migration assay showed that FoxO1-deficient cells had higher
migration capacity than control cells. When cell migration was
stimulated with CoCl2, LC-FoxO1 cells exhibited stronger
migration potential than control cells (Fig. 3C and D). Consistently, increased
phosphorylation of ERK and STAT3 following FoxO1 silencing was
maintained when cells were treated with CoCl2 (Fig. 3E and F). These data suggest that
FoxO1 served an inhibitory function in the migration of OSCC
cells.
FoxO1 silencing decreases oxidative
stress and apoptosis in YD-9 cells
FoxO1 is implicated in the oxidative stress response
(30). In addition, oxidative
stress is involved in numerous disorders, including OLP and OSCC
(31). To assess the effect of
FoxO1 depletion on intracellular oxidative stress, ROS levels were
determined by CellROX staining in control and FoxO1-deficient
cells. Intracellular ROS levels significantly decreased following
FoxO1 silencing (Fig. 4A).
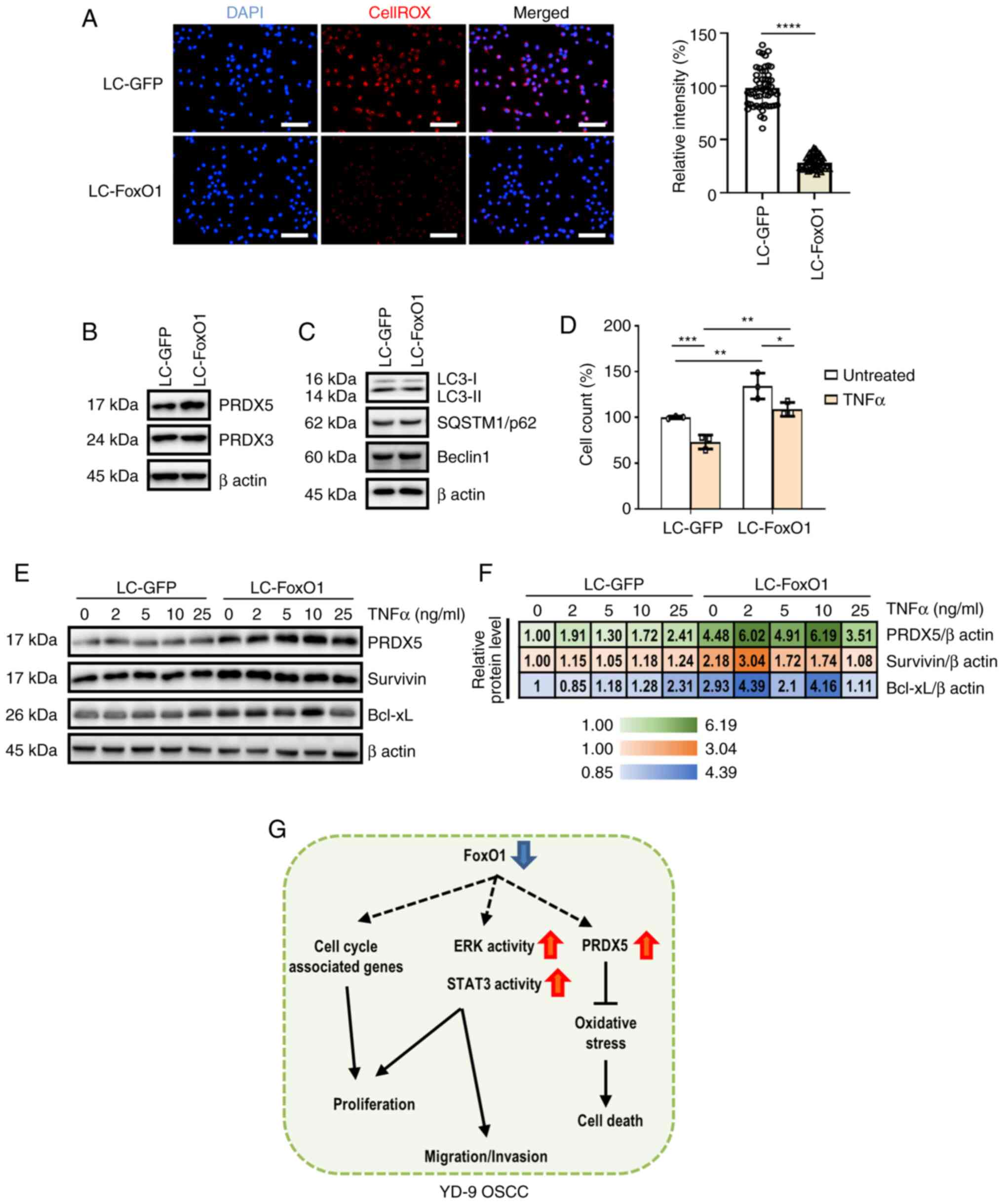 | Figure 4.FoxO1 deficiency suppresses
intracellular oxidative stress and death in YD-9 cells. (A) Control
and FoxO1-deficient cells were subjected to CellROX staining.
Nuclear DAPI signal is shown in blue. Scale bar, 100 µm. The
relative CellROX signal intensities are shown. Immunoblot analysis
of proteins involved in (B) reactive oxygen species scavenging and
(C) autophagy response. (D) Relative cell numbers at 2 days after
cell seeding in the presence or absence of 10 ng/ml TNFα treatment.
(E) Immunoblot analysis of PRDX5, survivin, and Bcl-xL in control
and FoxO1-deficient cells following treatment with TNFα for 2 days.
β actin was used as a loading control. (F) Quantification of
immunoblot analysis. (G) Proposed schematic of the anti-tumor
properties of FoxO1 in YD-9 cells. FoxO1, forkhead transcriptional
factor O1; PRDX5, peroxiredoxin 5; SQSTM1, sequestosome 1; OSCC,
oral squamous cell carcinoma. *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. |
Peroxiredoxins (PRDXs) are key factors contributing
to ROS scavenging. PRDX3 and PRDX5 mRNA expression is
regulated by FoxO family members (32,33).
Although the PRDX3 protein expression was not changed by FoxO1
deficiency, PRDX5 was notably upregulated by FoxO1 disruption,
which could partly explain the decreased ROS levels in the LC-FoxO1
cells (Fig. 4B). Considering the
interplay between autophagy and oxidative stress (34), autophagic flux following FoxO1
silencing was assessed. However, we could not observe any signature
of autophagy activation/suppression by FoxO1 depletion in YD-9
cells, which was determined by the unchanged LC3 level (Fig. 4C).
As inflammation promotes OSCC pathogenesis, the
effect of FoxO1 on the intracellular inflammatory response was
assessed. As expected, the phosphorylation of JNK and NFκB was
increased by TNFα stimulation. However, none of JNK and NFκB
pathways seemed to be affected by FoxO1 perturbation (Fig. S2A). Consistently, mRNA levels of
the pro-inflammatory genes TNFα and CC Motif Chemokine Ligand 20
were clearly induced by TNFα but those were unchanged by FoxO1
perturbation (Fig. S2B),
suggesting that FoxO1 exerted a negligible effect on the acute
inflammatory response in YD-9 cells.
A strong intracellular inflammatory status
contributes to tumor cell death (35). YD-9 cells were treated with TNFα for
2 days before measurement of cell survival. Exposure to TNFα
significantly decreased YD-9 cell survival. However, FoxO1
deficiency effectively suppressed OSCC cell death (Fig. 4D). Consistent with this data,
Survivin and Bcl-xL protein levels remained high in FoxO1-deficient
cells (Fig. 4E and F), again
indicating the anti-tumor role of FoxO1 in YD-9 OSCC (Fig. 4G).
Discussion
Unlike other tumor types where FoxO1 is
downregulated, in the present study, FoxO1 transcript abundance was
unchanged or increased in some populations of OSCC. Expression
enhancement of specific genes during pathogenesis may indicate that
these genes promote progression of disease. Alternatively, cells
could operate defensive mechanisms by upregulating specific genes
that have suppressive roles in the pathogenesis of disease. The
present results support the latter by showing that FoxO1 had
suppressive roles in proliferation and migration but promoted cell
death in OSCC.
In line with the present study, FoxO1 is regarded as
a tumor suppressor. FoxO1 is highly expressed in non-Hodgkin
lymphoma and constitutive activation of FoxO1 inhibits
proliferation and promotes cell cycle arrest/apoptosis (36). In prostate cancer, the reduction of
FoxO1 activity and expression increases cancer cell proliferation
(37). FoxO1 deficiency decreases
cell death in response to energy stress (38). On the other hand, however, studies
have demonstrated that FoxO1 has oncogenic properties: The
cooperation of FoxO1 with β-catenin mediates hematopoiesis and
induces malignant transformation of hematopoietic stem cells into
leukemia cells (39). In ovarian
cancer, cell viability and migration are disrupted by FoxO1
knockdown (40). Collectively,
FoxO1 shows complicated molecular functions in cancer and thorough
studies must be conducted into intra- and extracellular factors
that affect these roles.
Given the importance of ERK and STAT3 signaling in
tumor progression (28), the
present study focused on their hyperactivation under FoxO1
silencing in this study. HIF1α protein levels were decreased in
FoxO1-deficient YD-9 cells upon CoCl2 treatment.
Considering the tumor-promoting roles of HIF1α, decreased HIF1α in
FoxO1-deficient cells with high proliferative and migratory
capacity may appear contradictory. Although the present study did
not assess HIF1α mRNA levels in the absence of FoxO1, FoxO1 may act
as a transcription factor for Hif1α. Vasquez et al (41) demonstrated that HIF1α is a direct
transcriptional target of FoxO1 and that HIF1α mRNA levels are
decreased in human endometrial stromal cells when FoxO1 is
silenced. In addition, intracellular redox status has a strong
influence on HIF1α protein levels. Gao et al (42) demonstrated that N-Acetyl Cysteine
significantly decreases HIF1α protein levels in human P493 B cells
by lowering oxidative stress. Notably, the ROS scavenging protein
PRDX5 has been shown to inhibit HIF1α stabilization and HIF
activity (43). According to the
present results, FoxO1 depletion led to the upregulation of PRDX5
protein levels and downregulation of oxidative stress, subsequently
contributing to a decrease in HIF1α protein levels. Therefore,
dysregulated ERK and STAT3 activation may override the effect of
decreased HIF1α levels in FoxO1-deficient YD-9 cells.
Although the present study determined the molecular
function of FoxO1 in YD-9 OSCC cells, the study has limitations.
First, it did not determine why FoxO1 expression is elevated in a
subset of oral cancer cell lines and patients with OSCC. Second,
the roles of FoxO1 in other oral cancer cells infected with human
papillomavirus or mutated p53 were not investigated. Given tumor
heterogeneity, it is important to explore the molecular functions
of FoxO1 in different types of tumor. Third, the detailed mechanism
of FoxO1 knockdown-induced activation of ERK and STAT3 was not
determined. Understanding the crosstalk between molecules and
signaling pathways is key in tumor biology.
Based on the present results, further studies need
to be performed. First, the molecular mechanisms of malignant
transformation from OLP to OSCC need to be documented. OLP and OSCC
share certain molecular features and cellular phenotypes. To
explore the link between OLP and OSCC and to determine whether
FoxO1 is implicated in disease progression is important for
development of therapeutic strategies. In addition, it should be
tested whether FoxO1 overexpression or activation suppresses or
reverses the pathogenesis of OSCC. Several therapeutic options have
been developed for the treatment of OSCC (44). Nonetheless, complete recovery from
OSCC is not easy. Although further evaluations are required, the
present results suggest FoxO1 activation as a potential therapeutic
strategy against OSCC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Research
Foundation of Korea funded by the Korean government (grant nos.
2017R1A5A2015391, 2022R1C1C1006181 and 2020R1C1C1006757) and the
Basic Science Research Program through the National Research
Foundation of Korea funded by the Ministry of Education (grant no.
2019R1I1A2A01062430).
Availability of data and materials
The datasets and materials used in the current study
are available from the corresponding author on reasonable
request.
Authors' contributions
KHL, JSB and DYK conceived the study. YGK, CES, KAC,
KHL, JSB and DYK wrote the manuscript. YGK, CES, KAC, SML, TJK, HJK
and JHC performed the experiments. YGK, CES, KAC, SML, TJK, HJK,
JHC, JCS, WJ, SJ, KHL, JSB and DYK designed the methodology. YGK,
CES, KAC, KHL, JSB and DYK collected data. WJ, SJ, JCS and KHL
provided resources. JSB and DYK obtained funding. YGK and DYK
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of Jeonbuk National University Hospital (approval no.
CUH 2021-02-044-001), and written informed consent was obtained
from all the participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rivera C: Essentials of oral cancer. Int J
Clin Exp Pathol. 8:11884–11894. 2015.PubMed/NCBI
|
|
2
|
Weckx A, Riekert M, Grandoch A, Schick V,
Zöller JE and Kreppel M: Time to recurrence and patient survival in
recurrent oral squamous cell carcinoma. Oral Oncol. 94:8–13. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Omar E: Current concepts and future of
noninvasive procedures for diagnosing oral squamous cell
carcinoma-a systematic review. Head Face Med. 11:62015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Safi AF, Kauke M, Grandoch A, Nickenig HJ,
Zöller JE and Kreppel M: Analysis of clinicopathological risk
factors for locoregional recurrence of oral squamous cell
carcinoma-retrospective analysis of 517 patients. J
Craniomaxillofac Surg. 45:1749–1753. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Collado M and Serrano M: Senescence in
tumours: Evidence from mice and humans. Nat Rev Cancer. 10:51–57.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yadav RK, Chauhan AS, Zhuang L and Gan B:
FoxO transcription factors in cancer metabolism. Semin Cancer Biol.
50:65–76. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chae YC, Kim JY, Park JW, Kim KB, Oh H,
Lee KH and Seo SB: FOXO1 degradation via G9a-mediated methylation
promotes cell proliferation in colon cancer. Nucleic Acids Res.
47:1692–1705. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dong T, Zhang Y, Chen Y, Liu P, An T,
Zhang J, Yang H, Zhu W and Yang X: FOXO1 inhibits the invasion and
metastasis of hepatocellular carcinoma by reversing ZEB2-induced
epithelial-mesenchymal transition. Oncotarget. 8:1703–1713. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Furuyama T, Kitayama K, Shimoda Y, Ogawa
M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K,
Nakayama K, et al: Abnormal angiogenesis in Foxo1 (Fkhr)-deficient
mice. J Biol Chem. 279:34741–34749. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cancer Genome Atlas Research Network, .
The molecular taxonomy of primary prostate cancer. Cell.
163:1011–1025. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Trinh DL, Scott DW, Morin RD, Mendez-Lago
M, An J, Jones SJ, Mungall AJ, Zhao Y, Schein J, Steidl C, et al:
Analysis of FOXO1 mutations in diffuse large B-cell lymphoma.
Blood. 121:3666–3674. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hornsveld M, Smits LMM, Meerlo M, van
Amersfoort M, Groot Koerkamp MJA, van Leenen D, Kloet DEA, Holstege
FCP, Derksen PWB, Burgering BMT and Dansen TB: FOXO transcription
factors both suppress and support breast cancer progression. Cancer
Res. 78:2356–2369. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sanjana NE, Shalem O and Zhang F: Improved
vectors and genome-wide libraries for CRISPR screening. Nat
Methods. 11:783–784. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van der Meij EH and van der Waal I: Lack
of clinicopathologic correlation in the diagnosis of oral lichen
planus based on the presently available diagnostic criteria and
suggestions for modifications. J Oral Pathol Med. 32:507–512. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi W, Yang J, Li S, Shan X, Liu X, Hua H,
Zhao C, Feng Z, Cai Z, Zhang L and Zhou D: Potential involvement of
miR-375 in the premalignant progression of oral squamous cell
carcinoma mediated via transcription factor KLF5. Oncotarget.
6:40172–40185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45((W1)):
W98–W102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee EJ, Kim J, Lee SA, Kim EJ, Chun YC,
Ryu MH and Yook JI: Characterization of newly established oral
cancer cell lines derived from six squamous cell carcinoma and two
mucoepidermoid carcinoma cells. Exp Mol Med. 37:379–390. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gene Ontology Consortium: The gene
ontology resource: Enriching a GOld mine. Nucleic Acids Res.
49(D1): D325–D334. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mi H, Muruganujan A, Ebert D, Huang X and
Thomas PD: PANTHER version 14: More genomes, a new PANTHER GO-slim
and improvements in enrichment analysis tools. Nucleic Acids Res.
47(D1): D419–D426. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Birkenkamp-Demtröder K, Hahn SA, Mansilla
F, Thorsen K, Maghnouj A, Christensen R, Øster B and Ørntoft TF:
Keratin23 (KRT23) knockdown decreases proliferation and affects the
DNA damage response of colon cancer cells. PLoS One. 8:e735932013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sequeira I and Watt FM: The role of
keratins in modulating carcinogenesis via communication with cells
of the immune system. Cell Stress. 3:136–138. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu H, Wang K, Liu W and Hao Q: PTEN
overexpression improves cisplatin-resistance of human ovarian
cancer cells through upregulating KRT10 expression. Biochem Biophys
Res Commun. 444:141–146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gkouveris I, Nikitakis N, Karanikou M,
Rassidakis G and Sklavounou A: Erk1/2 activation and modulation of
STAT3 signaling in oral cancer. Oncol Rep. 32:2175–2182. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Saxena NK, Sharma D, Ding X, Lin S, Marra
F, Merlin D and Anania FA: Concomitant activation of the JAK/STAT,
PI3K/AKT, and ERK signaling is involved in leptin-mediated
promotion of invasion and migration of hepatocellular carcinoma
cells. Cancer Res. 67:2497–2507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ponugoti B, Xu F, Zhang C, Tian C, Pacios
S and Graves DT: FOXO1 promotes wound healing through the
up-regulation of TGF-β1 and prevention of oxidative stress. J Cell
Biol. 203:327–343. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kesarwala AH, Krishna MC and Mitchell JB:
Oxidative stress in oral diseases. Oral Dis. 22:9–18. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Olmos Y, Sánchez-Gómez FJ, Wild B,
García-Quintans N, Cabezudo S, Lamas S and Monsalve M: SirT1
regulation of antioxidant genes is dependent on the formation of a
FoxO3a/PGC-1α complex. Antioxid Redox Signal. 19:1507–1521. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Q, Sztukowska M, Ojo A, Scott DA,
Wang H and Lamont RJ: FOXO responses to Porphyromonas gingivalis in
epithelial cells. Cell Microbiol. 17:1605–1617. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: The clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y
and Li Y: Inflammation and tumor progression: Signaling pathways
and targeted intervention. Signal Transduct Target Ther. 6:2632021.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xie L, Ushmorov A, Leithäuser F, Guan H,
Steidl C, Färbinger J, Pelzer C, Vogel MJ, Maier HJ, Gascoyne RD,
et al: FOXO1 is a tumor suppressor in classical Hodgkin lymphoma.
Blood. 119:3503–3511. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Duan X, Kong Z, Liu Y, Zeng Z, Li S, Wu W,
Ji W, Yang B, Zhao Z and Zeng G: β-Arrestin2 contributes to cell
viability and proliferation via the down-regulation of FOXO1 in
castration-resistant prostate cancer. J Cell Physiol.
230:2371–2381. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lin A, Yao J, Zhuang L, Wang D, Han J, Lam
EW and Gan B: The FoxO-BNIP3 axis exerts a unique regulation of
mTORC1 and cell survival under energy stress. Oncogene.
33:3183–3194. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kode A, Mosialou I, Manavalan SJ, Rathinam
CV, Friedman RA, Teruya-Feldstein J, Bhagat G, Berman E and
Kousteni S: FoxO1-dependent induction of acute myeloid leukemia by
osteoblasts in mice. Leukemia. 30:1–13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Han GH, Chay DB, Nam S, Cho H, Chung JY
and Kim JH: Prognostic implications of forkhead box protein O1
(FOXO1) and paired box 3 (PAX3) in epithelial ovarian cancer. BMC
Cancer. 19:12022019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vasquez YM, Mazur EC, Li X, Kommagani R,
Jiang L, Chen R, Lanz RB, Kovanci E, Gibbons WE and DeMayo FJ:
FOXO1 is required for binding of PR on IRF4, novel transcriptional
regulator of endometrial stromal decidualization. Mol Endocrinol.
29:421–433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gao P, Zhang H, Dinavahi R, Li F, Xiang Y,
Raman V, Bhujwalla ZM, Felsher DW, Cheng L, Pevsner J, et al:
HIF-dependent antitumorigenic effect of antioxidants in vivo.
Cancer Cell. 12:230–238. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sabharwal SS, Waypa GB, Marks JD and
Schumacker PT: Peroxiredoxin-5 targeted to the mitochondrial
intermembrane space attenuates hypoxia-induced reactive oxygen
species signalling. Biochem J. 456:337–346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dwivedi R, Pandey R, Chandra S and
Mehrotra D: Dendritic cell-based immunotherapy: A potential player
in oral cancer therapeutics. Immunotherapy. 15:457–469. 2023.
View Article : Google Scholar : PubMed/NCBI
|