Introduction
Cancer remains one of the major public health issues
worldwide and the second leading cause of mortality worldwide
(1). Numerous standard strategies
for cancer treatment have been used to overcome this health issue,
including chemotherapy, surgery and radiotherapy (2). Colorectal cancer (CRC) is a solid
tumor that generates a considerable health and economic burden, as
it ranks second in terms of mortality among other types of cancer
(3). Among all cancers in United
States in 2020, CRC is the most frequently diagnosed cancer
accounting for 9% of all cases in men and 8% for those in women
whereas, 9% of cancer deaths account for CRC in both sexes
separately (1,3). This type of tumor begins in the
colorectal tissues due to the accumulations of genetic or/and
epigenetic modifications. Furthermore, tumorgenicity of CRC may
develop through three defined molecular pathways: Chromosomal
instability, microsatellite instability (MSI), and cytosine
preceding guanine island methylator phenotype pathways (4). Several risk factors are associated
with the development of CRC including genetic factors, lifestyle
factors, sex, age and diet. Genetic factors mainly arise from the
presence of hereditary CRC syndromes including Lynch syndrome and
familial adenomatous polyposis. Mutations of oncogenes and tumor
suppressor genes could also increase the risk of CRC (4,5).
Moreover, red meat consumption, diet high in fat, low physical
activity, obesity and cigarette smoking are examples of diet and
lifestyle factors that have been linked with increase of CRC risk
(3,4). Over the past few decades, a notable
improvement in the treatment of CRC, whether in surgery,
radiotherapy or systemic chemotherapy, has been achieved (5). With progress in antineoplastic agent
identification and development over the past several decades,
>100 antineoplastic agents have been identified and approved by
the USA Food and Drug Administration (FDA) (6–8).
5-Fluorouracil (5-FU) is a member of the antimetabolite group
administered intravenously. As an analogue of pyrimidine, it is
incorporated into DNA and RNA, thus blocking its synthesis.
Capecitabine, a prodrug of 5-FU, is administered orally and
exhibits the same cellular mechanism as 5-FU (5,6).
Initially, capecitabine requires three-step enzymatic methods to
transform it to the antimetabolite 5-FU. The variation in the
presence of these metabolic enzymes between tumor and normal
tissues gives capecitabine a theoretical advantage over 5-FU in
terms of efficacy (5). The
objective of the current review is to provide a comprehensive
overview of 5-FU and capecitabine therapies regarding their
physical and chemical properties, metabolism, cellular mechanism,
safety and tolerability. In addition, the effects of 5-FU and
capecitabine treatments on the apoptotic mechanism and cell cycle
phases of certain CRC cell lines are discussed. Furthermore, the
three metabolizing enzymes required for the conversion of
capecitabine to 5-FU are explained, and the concept of a
chemotherapeutic approach combined with capecitabine treatment is
also presented.
Chemotherapeutic drugs
Depending on their mechanisms of action,
chemotherapeutic drugs can be broadly categorized into two basic
categories: Cytotoxic drugs and targeted drugs (8,9).
Cytotoxic drugs are defined as agents that can eliminate rapidly
dividing cancer cells by targeting components of the mitotic and/or
DNA replication pathways. Conversely, targeted drugs are agents
that can block the proliferation and spread of cancer cells by
interacting with molecular targets involved in pathways linked to
tumor growth, progression and metastasis (8,10).
Anticancer agents are classified as chemotherapy, immunotherapy or
hormonal therapy. Chemotherapy, as presented in Table I, involves a number of families
defined by their chemical structure and mechanism of action, and
categorized into the following eight groups: i) Alkylating agents,
such as nitrogen mustards and nitrosoureas; ii) antimetabolites,
including methotrexate, 5-FU, 6-mercaptopurine and hydroxyurea;
iii) antibiotics, such as doxorubicin and mitomycin C; iv) mitotic
inhibitors, including vincristine and vinblastine; v) topoisomerase
I inhibitors, such as irinotecan; vi) topoisomerase II inhibitors,
including etoposide; vii) platinum compounds, such as cisplatin;
and viii) others (transcription factor inhibitors) including
ecteinascidin (11,12).
 | Table I.Classical classification of
anticancer drugs. |
Table I.
Classical classification of
anticancer drugs.
| Anticancer
drug | Classification |
|---|
| Chemotherapy | Alkylators |
|
| Antibiotics |
|
|
Antimetabolites |
|
| Topoisomerase
inhibitors |
|
| Mitosis
inhibitors |
|
| Others (inhibitors
of transcription factors) |
| Hormonal
therapy | Steroids |
|
| Anti-estrogens |
|
| Anti-androgens |
|
| LH-RH
analogues |
|
| Anti-aromatase
agents |
| Immunotherapy | Interferon |
|
| Interleukin 2 |
|
| Vaccines |
Espinosa et al (12) proposed a method of anticancer drug
classification based on targets that could be located at the DNA,
RNA or protein levels. Drugs may be directed at different levels:
Tumor cells or other elements included in carcinogenesis, such as
the endothelium and extracellular matrix of the immune system or
host cells.
The present review focused on antimetabolite agents.
As aforementioned, the majority of anticancer drugs exert their
cytotoxicity either by inhibiting DNA synthesis or damaging the DNA
template (11). Antimetabolites, a
class of chemotherapeutic agents, are the oldest designed
anticancer drugs that target DNA and RNA molecules, and can be
considered the first generation of targeted drugs (13). Antimetabolites are defined as
substances that are structurally analogous to natural metabolites
and play a role in cellular metabolism, in which they compete with
or replace them, thus preventing or reducing their normal cellular
utilization (13,14). Antimetabolites are used to target
‘key’ enzymes in the de novo biosynthesis pathways of purine
(dATP and dGTP) and pyrimidine (UTP, dTTP and dCTP) nucleotides
(11).
Antimetabolite classes include folate analogs
(including aminopterin and methotrexate), pyrimidine analogs [such
as 5-fluoropyrimidines (5-FU, ftorafur and capecitabine),
gemcitabine and cytarabine] and purine analogs (including
thiopurines, 6-mercaptopurine, pentostatin and cladribine)
(11–13). According to the drug classification
of Espinosa et al (12),
antimetabolites can be grouped into drugs directed against tumor
DNA (sub-class: DNA-related proteins) and drugs directed against
tumor RNA. Antimetabolites can also be grouped into drugs directed
at protein-DNA complexes because they do not bind directly to DNA
molecules. Instead, they act by interfering with enzymes shared
during DNA synthesis. This group includes antifolates,
5-fluoropyrimidines (5-FU, ftorafur and capecitabine), raltitrexed,
cytarabine, gemcitabine and adenosine analogs (fludarabine,
pentostatin and cladribine). Conversely, antimetabolites classified
as drugs directed against tumor RNA include 5-fluoropyrimidines
(5-FU, ftorafur and capecitabine) and platinum compounds (12). Antimetabolite agents are widely used
to treat viral infections and multiple types of cancer. As cancer
cells have abnormal proliferation compared with normal cells,
blocking DNA and RNA synthesis is an efficient anticancer treatment
strategy (13). Antimetabolite
agents were first demonstrated to have clinical anticancer activity
~50 years ago (15). They are
currently used in chemotherapy combinations for the treatment of
different leukemias and solid tumors worldwide (16).
5-Fluoropyrimidine compounds, a class of
antimetabolite agents, are the result of a fluorine substitution at
a specific site of the pyrimidine ring; specifically, replacement
of a hydrogen atom at position 5 of the pyrimidine ring by a
similarly sized fluorine (F) atom (17). Naturally, to convert uracil (U)
nucleotides to thymine (T) nucleotides, the insertion of the methyl
group (−CH3) should take place at the 5-position of the
U ring instead of the hydrogen atom. During the development of
5-fluoropyrimidine compounds, the 5-position of the U ring, which
occupies the hydrogen atom, was selected as the specific site of
substitution with the F atom because it was considered to be more
efficient in inhibiting the subsequent conversion of the U
nucleotide to the T nucleotide, thus preventing DNA synthesis and
cellular proliferation (17).
5-Fluoropyrimidines are a class of anticancer drugs, specifically
antimetabolites (18). Clinically,
they are still used as a standard treatment regimen for solid
tumors, including breast cancer, CRC and cancers of the
aerodigestive tract (18,19). Examples of 5-fluoropyrimidine drugs
include 5-FU, ftorafur and capecitabine (12).
5-FU
5-FU was specifically created to resemble pyrimidine
bases (U and T) (11). It is a
fluorinated analog of the nitrogenous base U, and it was originally
synthesized in 1957 (20) and sold
under the brand name Adrucil® (21). It was the first pyrimidine analog
registered for cancer treatment (16). Approved by the USA FDA in 1962, 5-FU
is an antimetabolite commonly used as a chemotherapeutic agent,
with activity against several types of solid tumors, including head
and neck, breast, prostate, pancreas, liver, genitourinary,
gastrointestinal tract and ovary cancer, and CRC (20,22).
It has been the main chemotherapeutic agent for the treatment of
CRC for ~50 years (23,24), and has been used as a single agent
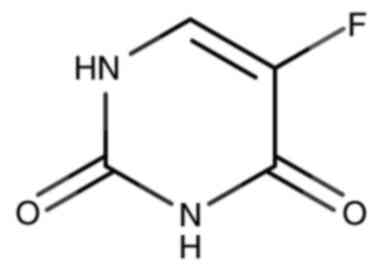
and in combination with other agents (24). In a 5-FU molecule, the hydrogen atom
at position 5 of the U base is replaced with a similarly sized F
atom (25–27), as shown in Fig. 1. This molecule was designed to
occupy the active sites of target enzymes, thereby blocking
metabolism in tumor cells (25).
The International Union of Pure and Applied Chemistry (IUPAC) name
for 5-FU is 5-fluoropyrimidine-2,4(1H,3H)-dione
(27). Various physical and
chemical properties of the 5-FU compound are listed in Table II.
| Table II.Physical and chemical properties of
5-flourouracil. |
Table II.
Physical and chemical properties of
5-flourouracil.
| Property name | Description | (Refs.) |
|---|
| Form
(Appearance) | Crystalline
powder | (28) |
| Color | White to nearly
white | (28) |
| Solubility | Less than 1 mg/ml
(at 66°F=18.89°C) | (29) |
| Molecular
weight | 130.08 g/mol | (30) |
| Molecular
formula |
C4H3FN2O2 | (26) |
| Formal charge | 0 | (30) |
| Hydrogen bond donor
count | 2 | (30) |
| Hydrogen bond
acceptor count | 3 | (30) |
| Rotatable bond
count | 0 | (30) |
| Number of
rings | 1 | (31,32) |
| Acid/Base
property | A weakly acidic
compound | (33) |
| pKa | 7.93 or 8.05 | (34,35) |
Metabolism of 5-FU
5-FU can enter efficiently a cell using a
facilitated transmembrane carrier system known as a nucleoside
transporter system, specifically the human equilibrative nucleoside
transporter (36), where it is then
transformed into several metabolites. This drug requires enzymatic
conversion into a nucleotide through ribosylation and
phosphorylation reactions to perform its cytostatic activity.
Moreover, conversion to nucleotides enhances intracellular
retention and further metabolism (37). The level of 5-FU available for
transformation into an active nucleotide is based on the extent to
which it is catabolized (22).
After penetration into a cell, 5-FU is metabolized via two routes;
either an anabolic or a catabolic route in competition with each
other (25). The anabolic route
(activation route) occurs through thymidine phosphorylase (TP),
which forms the active metabolites of 5-FU that are responsible for
cytotoxic activity. The catabolic route (inactivation route) occurs
through dihydropyrimidine dehydrogenase (DPD), which is responsible
for the inactivation, degradation, detoxification and subsequent
elimination of 5-FU from an organism (20,25).
The anabolic route, which can be complex,
demonstrates the mechanism of 5-FU cytotoxicity (20). Both cancerous and non-cancerous
cells metabolize 5-FU drugs intracellularly via three routes into
three different active metabolites that cause cell injury:
5-Fluoro-2′-deoxyuridine-5′-monophosphate (5-FdUMP);
5-fluoro-2′-deoxyuridine-5′-triphosphate (5-FdUTP) and
5-fluorouridine-5′-triphosphate (5-FUTP) (18,19,38),
as presented in Fig. 2.
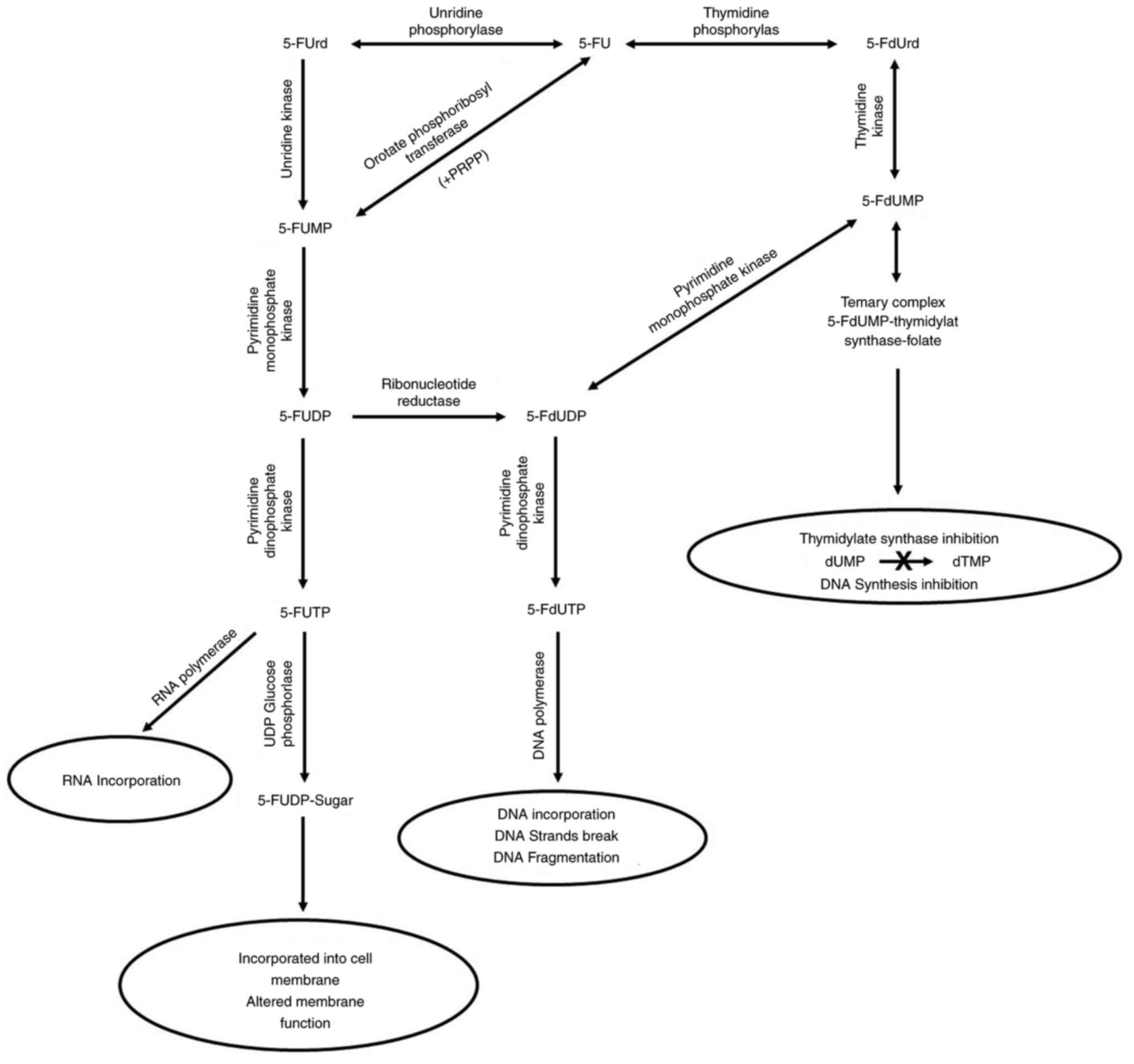 | Figure 2.Intracellular anabolic route of 5-FU.
This drug is converted via three routes into three different
cytotoxic metabolites: 5-FdUmp, 5-FdUTP and 5-FUTP. Thereafter, the
cytotoxic effects of 5-FU can occur through the incorporation of
these cytotoxic metabolites into DNA, RNA and the cell membrane.
These mechanisms inhibit DNA and RNA synthesis, and alter the cell
membrane function. 5-FU, 5-fluorouracil; 5-FUrd, 5-fluorouridine;
5-FUMP, 5-fluorouridine monophosphate; 5-FUDP,
5-fluorouridine-5-diphosphate; 5-FdUDP,
5-fluoro-2′-deoxyuridine-5′-diphosphate; 5-FUTP,
5-fluorouridine-5′-triphosphate; 5-FdUTP,
5-fluoro-2′-deoxyuridine-5′-triphosphate; 5-FUDP-Sugar,
5-FU-nucleotide sugar; 5-FdUrd, 5-fluoro-2′-deoxyuridine; 5-FdUmp,
5-fluoro-2′-deoxyuridine-5′-monophosphate; dTMP, deoxythymidine
monophosphate. |
The first route, which is the conversion of 5-FU
into 5-fluoro-2′-deoxyuridine (5-FdUrd), is performed through the
TP enzyme, followed by the subsequent phosphorylation of 5-FdUrd
through the thymidine kinase enzyme, which results in the formation
of the cytotoxic nucleotide 5-FdUMP (18,22).
In the presence of a cofactor, reduced folate 5-FdUMP, which is a
powerful inhibitor of the thymidylate synthase (TS) enzyme, binds
to TS to form a covalent complex. This binding blocks the
production of thymidylate, which is a necessary precursor of deoxy
thymidine triphosphate (dTTP), which is essential for DNA synthesis
(19,22,38),
thus inhibiting cell division (19). In addition, the clinical response to
5-FU treatment correlates with the level of TS inhibition of
5-FdUMP in patient tumors (38).
The second route, which is the formation of
5-fluorouridine-5′-monophosphate (5-FUMP) from 5-FU, is carried out
either through direct conversion by orotate phosphoribosyl
transferase in the presence of 5′-phosphoribosyl-1-pyrophosphate as
a co-substrate or through intermediate conversion into a
ribonucleoside 5-fluorouridine by uridine and subsequent conversion
into 5-FUMP by uridine kinase. The formed 5-FUMP can undergo two
steps of phosphorylation to form 5-fluorouridine-5-diphosphate
(5-FUDP) and 5-FUTP through pyrimidine monophosphate kinase and
pyrimidine diphosphate kinase (PDPK), respectively (25,39).
Nuclear transcriptional enzymes can mistakenly incorporate 5-FUTP
instead of uridine triphosphate during the synthesis of RNA
molecules (19,25). The previous mistake can overlap with
RNA processing (post-transcriptional process) and protein synthesis
(translation) (19). Moreover,
5-FUTP can be conjugated to sugars through UDP glucose
phosphorylase, producing 5-FU-nucleotide sugars (5-FUDP-sugars)
that are incorporated into the cell membrane, thereby altering the
membrane function (25).
In the third route, both 5-FUDP and 5-FdUMP can be
converted into 5-fluoro-2′-deoxyuridine-5′-diphosphate, which is
phosphorylated by PDPK to 5-FdUTP. 5-FdUTP acts as a substrate for
DNA polymerases, which are enzymes responsible for DNA replication,
and can therefore be incorporated into a DNA molecule, leading to
the production of single-strand breaks and DNA fragmentation (which
inhibits DNA replication) (11,25,40,41).
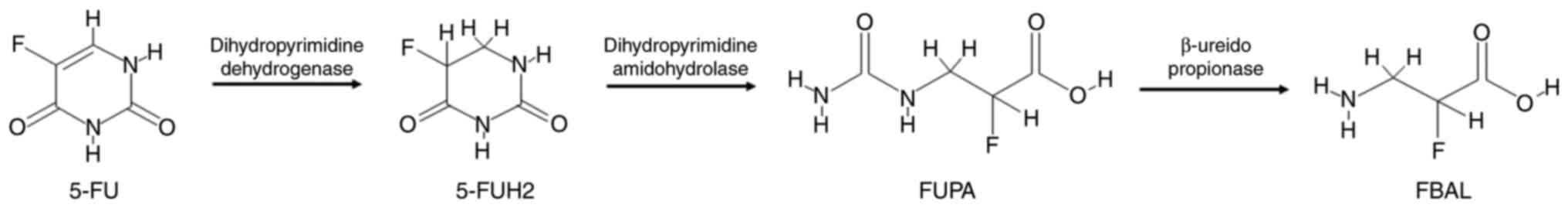
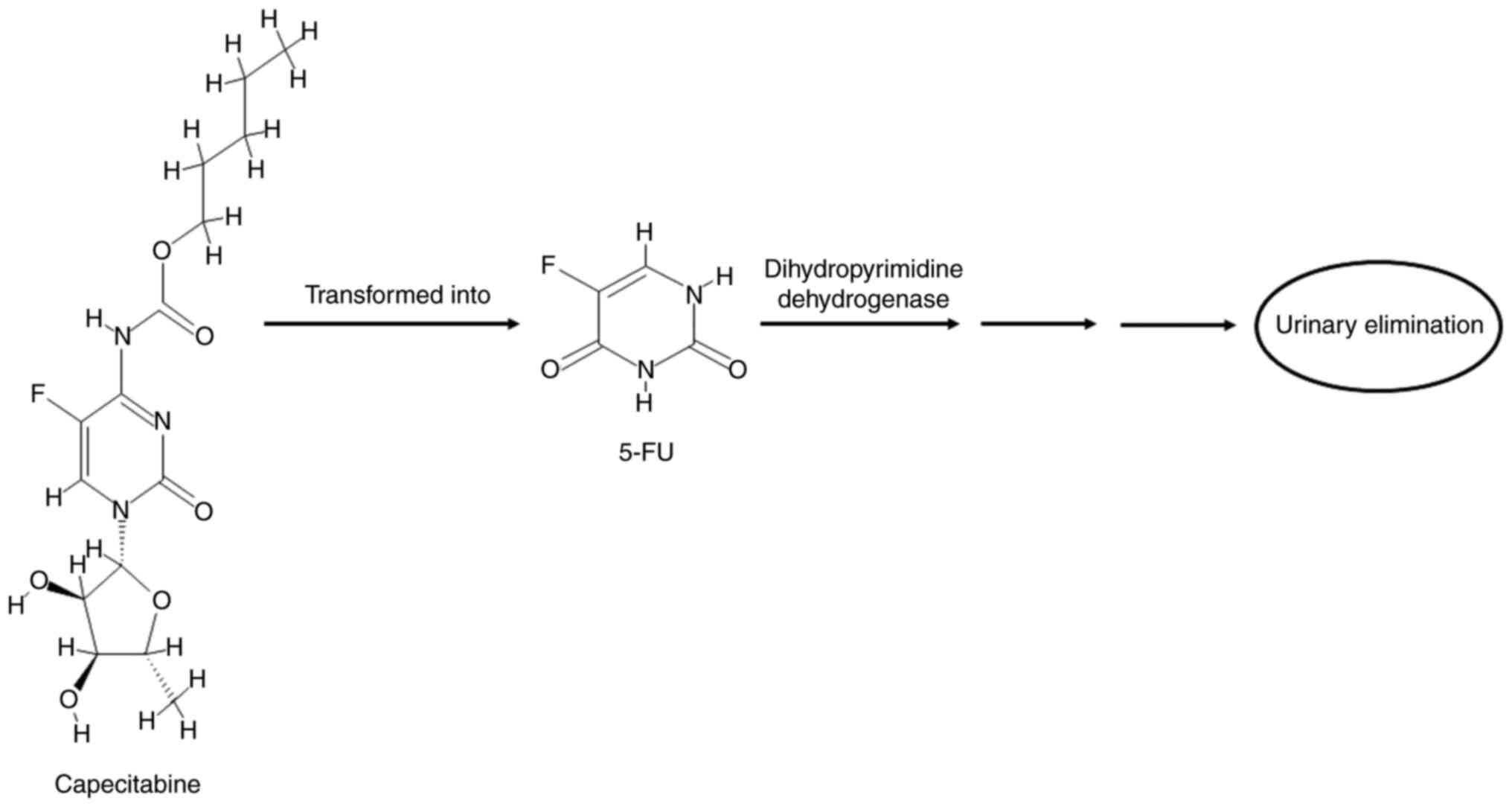
The degradation of 5-FU (the catabolic route) is
mediated by the DPD enzyme, resulting in the reduction of 5-FU to
the markedly less toxic 5-fluoro-5,6-dihydrouracil compound
(20,25,41),
which then undergoes two cleavage reactions. In the first reaction,
the dihydropyrimidinase enzyme cleaves the pyrimidine to produce
α-fluoro-β-ureidopropionic acid (FUPA) (19,41).
In the second step, the β-ureido-propionase enzyme cleaves FUPA to
produce α-fluoro-β-alanine, which is the major catabolite of 5-FU
and is cleared by urinary elimination (19,42).
Neither of these metabolites has antiproliferative effects
(20,25), as shown in Fig. 3. DPD is the initial rate-limiting
enzyme of 5-FU catabolism (i.e., the first enzyme in the catabolic
cycle of 5-FU), and it is abundant in the liver, intestinal mucosa,
pancreas, lungs, kidneys and peripheral blood (22,23).
~90% of 5-FU drugs are catabolized by DPD, while only 10% are
excreted without a change in urine (22). Due to the role of DPD in the
detoxification of 5-FU, deficiency of this enzyme in patients can
result in severe and even lethal 5-FU toxicity (18,20).
5-FU cytotoxicity is caused by interference with RNA
and DNA synthesis (22,25). Specifically, it interferes with RNA
synthesis through the inclusion of a drug metabolite (5-FUTP) in
replicating RNA molecules, thus impeding RNA processing and
subsequent protein synthesis (inhibition of RNA synthesis and
function) (19,22,40).
It also interferes with DNA synthesis by inhibiting the TS enzyme,
resulting in the depletion of the thymidine nitrogenous base, which
is necessary for DNA synthesis (19,20,43).
Moreover, the cellular damage resulting from 5-FU induces three
modes of cell proliferation modulation: i) Accumulation or loss of
S-phase cells; ii) blocking of the G2/M phase; iii) and
arrest of the G1/S phase (22). The cause of the accumulation or loss
of S-phase cells is the blocking of the de novo synthesis of
T nucleotides, which leads to a lack of T in tumor cells, resulting
in the inability of cells to create DNA molecules, which accumulate
at the beginning of the S-phase (44). Another mechanism that could be
partly responsible for the cytotoxic effect of 5-FU was reported,
which involves the alteration of membrane function after the
treatment of cells with 5-FU. This alteration is associated with
the formation of 5-FUDP-sugars after 5-FUTP conjugation to sugars
and their incorporation into the cell membrane (25). The extent to which any of these
mechanisms predominate in human cancer is unknown, and varies
depending on the cancer type, route of administration and dose of
drug. It has been suggested that longer exposure to low doses of
5-FU [as intravenous (IV) treatment] results in a TS-inhibited
mechanism (DNA damage), which mediates the process of cell death,
whereas the bolus route of 5-FU results in an RNA-mediated process
of cell death (RNA damage) (18,25).
The present review used DrugBank 5.1.8 (https://www.drugbank.com) to identify the direct
cellular targets (referred as DTs) of 5-FU. DrugBank is a
comprehensive online database that provides specific drug data and
information on drug actions and targets. 5-FU was output as DB00544
in DrugBank 5.1.8, and various DTs of 5-FU that involved DNA, RNA,
TS, TP, DPD, and multidrug resistance-associated proteins 4 and 5
were selected (32), as
demonstrated in Table III.
| Table III.Identification of certain direct
cellular targets of 5-FU in humans using DrugBank 5.1.8. |
Table III.
Identification of certain direct
cellular targets of 5-FU in humans using DrugBank 5.1.8.
| Drug name | DrugBank Identity
(DB-ID) | Target name | Type of target | Gene name | Uniprot ID |
|---|
| 5-FU | DB00544 | DNA | Nucleotide | - | - |
|
|
| RNA | Nucleotide | - | - |
|
|
| Thymidylate
synthase | Protein | TYMS | P04818 |
|
|
| Thymidine
phosphorylase | Protein | TYMP | P19971 |
|
|
| Dihydropyrimidine
dehydrogenase [NADP (+)] | Protein | DPYD | Q12882 |
|
|
| Multidrug
resistance-associated protein 4 | Protein | ABCC4 | O15439 |
|
|
| Multidrug
resistance-associated protein 5 | Protein | ABCC5 | O15440 |
The SwissADME (http://www.swissadme.ch/) and DrugBank 5.1.8 databases
were used to represent certain pharmacokinetic properties. 5-FU was
found to be readily (highly) absorbed from the human intestine and
did not cross the blood-brain barrier (BBB) (32,45).
Therefore, it could not cause toxicity to the central nervous
system (CNS).
Effect of 5-FU treatment on the
apoptotic pathway and cell cycle phases of CRC cell lines
Apoptosis is the mechanism of programmed cell death
that normally occurs during development and aging. It is a
homeostatic process that maintains cell populations in tissues and
a defense mechanism when cells are damaged. There are two main
apoptotic pathways, namely the extrinsic and intrinsic pathways, in
addition to a third new pathway, which is the perforin/granzyme
pathway. The extrinsic pathway is called the death receptor
pathway, and is triggered by the binding of a death ligand to death
receptors, such as Fas and tumor necrosis factor receptors, which
are expressed on the cell surface. The intrinsic pathway is called
the mitochondrial-dependent pathway, and is initiated by
mitochondria. The perforin/granzyme pathway involves
perforin/granzyme-dependent cell killing and T-cell-mediated
cytotoxicity. Each pathway has a cascade of molecular events and
activates its own initiator caspases (8–10),
which in turn activates the executioner caspase 3 (46,47).
The cell cycle is a complicated process involved in cell
proliferation and development, regulation of DNA repair mechanisms,
tissue hyperplasia, and diseases such as cancer (48). Cell cycle arrest has been considered
a target for cancer therapy, in which cancer cell proliferation and
metastasis can be prevented by specific cell cycle regulation
(49). Cyclin-dependent kinases
(CDKs) are regulatory proteins that act in specific pathways to
determine whether the cell cycle may remain arrested between stages
or proceed (50). The cell cycle
transition is an ordered, regulated process that includes multiple
checkpoints, which assess extracellular growth signals, cell size
and DNA integrity (51). Cell
division has three stages: i) The interphase (G0,
G1, S and G2 phases); ii) mitotic phase (M
phase); and iii) cytokinesis. The interphase is the first and
longest stage of the cell cycle, and consists of four steps: i) Gap
0 phase (G0); ii) growth phase 1 (G1); iii)
synthesis phase (S); and iv) growth phase 2 (G2).
Overall, these steps take 12–24 h in mammalian tissues, in which
the cell increases in size and produces proteins that synthesize
RNA (52). The G0 phase
is the arresting time when the cell stops dividing and leaves the
cycle, while the G1 phase is when the cell increases in
size and starts producing RNA and synthesizing proteins. This step
is important to ensure that everything is prepared for DNA
synthesis (S phase) (51). The S
phase is where DNA replication begins, and it aims to produce new
daughter cells that have two identical chromosomes. Accuracy is
important in preventing any genetic abnormalities that can lead to
disease or cell death (52). In the
G2 phase, the cells proliferate and produce new
proteins. The checkpoint of this phase determines whether the cell
is ready to enter a new stage (mitotic M phase) and divide
(51). The mitosis phase, which is
the second stage, is shorter than the interphase, in which the
chromosomes split between the two daughter cells. In the middle of
this phase there is a checkpoint similar to the G1 and
G2 phases, which ensures the cell is ready to complete
the division (52). Cytokinesis,
the final stage of the cell cycle, is initiated during the late
phase of mitosis, during which the two daughter cells are
completely separated (51,53).
DNA-damaging chemotherapeutic agents, including
antimetabolites, generally trigger cell death in tumor cells by
inducing apoptosis. The extent to which cells are susceptible to
apoptosis depends on the status of the genes (e.g., tumor
suppressor gene p53) that regulate the critical components of the
cell death mechanism (54,55). These antimetabolites, including 5-FU
and the prodrug capecitabine, are considered to stimulate the
intrinsic apoptotic pathway (mitochondria-dependent pathway)
through caspase-8 and/or caspase-3 activation. Moreover, they cause
cell cycle arrest in the S phase (56).
The following section discusses the effects of 5-FU
treatment on apoptosis and the changes in the cell cycle phases of
CRC cell lines reported by several studies. The human colon cancer
cell line Caco-2 was treated with 5-FU (2 and 5 µM) and underwent
the intrinsic apoptotic mechanism through the activation of caspase
9. This treatment significantly increased the intracellular
reactive oxygen species (ROS) level, which caused oxidative stress
apoptotic pathways in cancer cells (57). The percentage of the human colon
cancer cell line HCT-116 in the G1 phase of the cell
cycle decreased after 5-FU treatment (10 µg/l), whereas that in the
G2/M phase increased. No significant apoptotic effect of
5-FU chemotherapy was observed in the same cancer cells (58). 5-FU treatment was added to
TP53 5-FU-resistant cell lines (ContinB and ContinD)
generated from the HCT-116 human colon cancer cell line to
determine its effects on cell cycle and apoptosis. The results
revealed that 5-FU treatment caused S phase arrest, accumulation in
the G2/M phase, upregulation of cell cycle regulation
(CDKN1A) and apoptosis induction in both parental and
resistant cell lines with variable levels of response (high or
low). Additionally, it resulted in the upregulation of
p53-target genes in the DNA damage response and
apoptosis-regulatory pathways (Fas) in the same cells (59). Analysis of the SW-620 colorectal
adenocarcinoma cell line treated with the half-maximal inhibitory
concentration of 5-FU indicated that 5-FU therapy resulted in
apoptosis and increased percentage of cells in the S phase of the
cell cycle (60). The cell cycle
alterations in human colon cancer cell lines (HCT-116 and
metastatic LoVo) treated with 5-FU were evaluated as follows: In
HCT-116 cells, 5-FU induced significant arrest in the S phase,
while LoVo cells accumulated in the G2/M phase. Afrin
et al (61) identified the
molecular mechanisms involved in cell cycle arrest using 5-FU
treatment. Notably, the mRNA levels of CDK2 and CDK4 and cell cycle
regulatory proteins (cyclin D1 and cyclin E) decreased in HCT-116
cells, while the LoVo cells did not demonstrate any alteration in
cyclin E, CDK2 or CDK4 mRNA levels after 5-FU treatment, but cyclin
D1 was suppressed. In both cell lines, the percentage of cells
undergoing apoptosis significantly increased after 5-FU treatment.
The apoptotic effects were confirmed by examining the mRNA
expression of extrinsic and intrinsic apoptotic markers. In both
cell lines, caspase 3, p53 and cleaved poly (ADP-ribose) polymerase
levels increased after 5-FU treatment. The expression of the
intrinsic apoptotic markers Bax/Bcl-2, cytochrome c and
caspase 9 was increased in both cell lines. Furthermore, the
expression of the extrinsic apoptotic markers FasL and caspase 8
increased after 5-FU treatment in both cell lines (61).
Summary of the safety, tolerability
and disadvantages of 5-FU treatment
Oral administration of 5-FU is not clinically useful
due to the large-scale metabolism by the DPD enzyme in the mucosa
of the gastrointestinal tract and liver, which leads to extremely
variable bioavailability (19).
Therefore, this drug must be administered intravenously because of
its rapid clearance, degradation and variable gastrointestinal
absorption (20,22). This route is complicated by hospital
visits and the risk of infection with IV administration (20). Moreover, this regimen is
non-curative, and its effect on survival is unclear (23). Although 5-FU-based chemotherapy is
the most widely used treatment for solid gastrointestinal tumors,
it has various disadvantages, including toxicity, lack of
selectivity (poor tissue specificity), lack of effectiveness and
development of resistance (22).
The systemic toxicities of this chemotherapeutic agent are
neutropenia, stomatitis and diarrhea, which usually occur due to
non-selective cytotoxicity (20).
The principal side effects of continuous infusion of 5-FU were
hand-foot syndrome (HFS), diarrhea, nausea, vomiting and mucositis
(42).
The resistance of tumor cells to 5-FU chemotherapy
is associated with the TS enzyme, which is one of the most
important mechanisms of 5-FU resistance. Carrillo et al
(22) summarized the proposed
mechanisms of 5-FU resistance, including TS induction, decreased
accumulation of activated metabolites, gene amplification and
mutation, aberrant enzyme kinetics, imbalanced folate pools,
imbalanced metabolite accumulation, imbalanced ATP: dTTP ratio,
pharmacokinetic resistance, availability of the compound in the
tumor, drug distribution, increased drug elimination, high DPD
enzyme activity, increased transporter activity, ATP-binding
cassette proteins and multidrug resistance proteins.
Overcoming 5-FU complications
The side effects of 5-FU treatment need to be
overcome by i) improving selectivity toward cancer cells; ii)
enhancing cell absorption and metabolic stability; iii) increasing
specific tumor cell toxicity; and iv) increasing its
bioavailability. Overcoming the development of 5-FU resistance,
which is highly required due to the resistance of tumor cells to
conventional chemotherapy, plays a key role in the fight against
cancer. To fulfil these objectives, several oral fluoropyrimidines
have been developed to achieve the need for more convenient
therapy, and to enhance the safety and efficacy profile (22,40).
These antimetabolites were designed to have a novel mechanism of
action, bypass drug resistance, or be used in combination to
enhance the effects of other drugs (2). Oral fluoropyrimidines are classified
into three groups: 5-FU prodrugs, 5-FU combined with a DPD
inhibitor, and 5-FU prodrugs combined with a DPD inhibitor
(40). Efforts to identify more
efficient chemotherapeutic agents for CRC have resulted in oral
formulations and prodrugs of 5-FU with equivalent activity
(23). Oral prodrugs of 5-FU were
developed to reduce the problems of IV administration through oral
dosing, and to ameliorate safety and efficacy by selectively
delivering the active drug to the target cancer cell, thereby
avoiding healthy tissue (20). In
addition, the oral route may simulate continuous infusion schedules
without the inconvenience, morbidity or cost associated with the IV
route (23).
A prodrug is a pharmacologically inactive compound,
that is, the precursor of a drug, which is converted into an active
agent by a metabolic biotransformation process (25). These prodrugs are activated by
biological mediators only when they enter specific tissues or
cells. Moreover, these derivatives are created by modifying 5-FU,
for example by coupling with peptides, amino acids, phospholipids
and polymers (22). Among 5-FU
prodrugs, capecitabine has been demonstrated to have clinical
efficacy with low toxicity compared with 5-FU (19), as well as increased metabolic
stability and selectivity toward tumor tissue (22).
Capecitabine
Capecitabine, a fluorouracil prodrug, is the only
novel, universally approved and orally home-administered drug
(19). Its brand name is
Xeloda® (25,62). It was synthesized in the 1990s
(25) and approved by the USA FDA
in 2005 (20,24) for use as a first-line therapy in
patients with metastatic CRC when single-agent fluoropyrimidine
therapy is preferred (20). This
oral-formulation prodrug is a nucleoside metabolic inhibitor used
for the treatment of CRC in three settings: i) Monotherapy; ii)
adjuvant treatment; iii) and combination therapy with other agents
for metastatic or advanced disease (22,24).
This drug has been studied in large-scale clinical trials of
several solid tumors, including CRC, breast cancer and gastric
cancer (19).
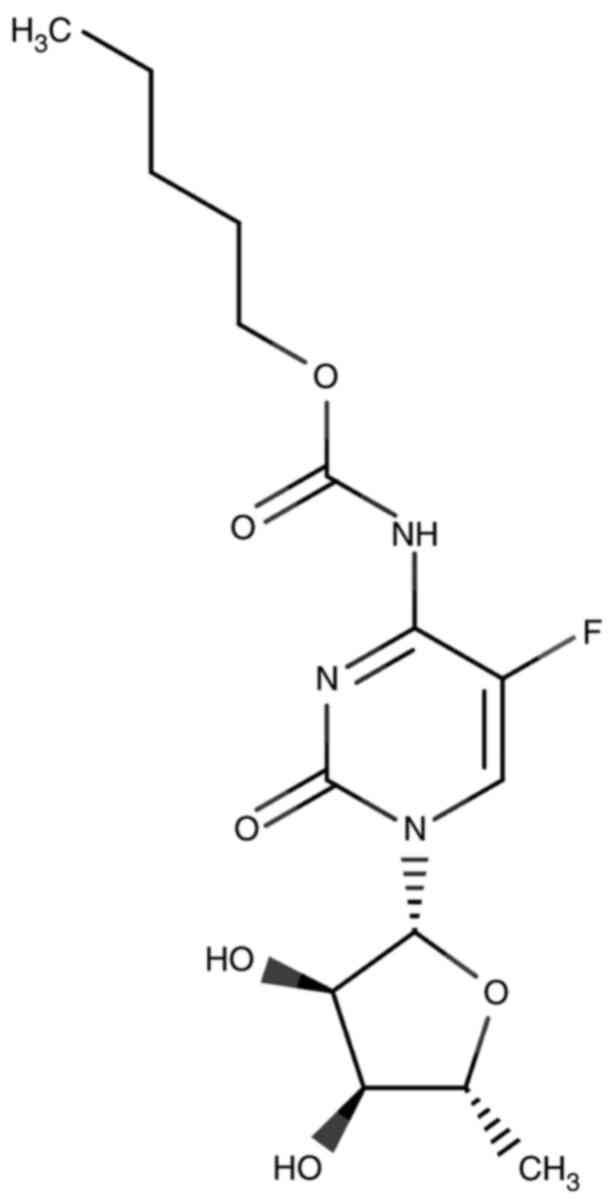
Capecitabine, as shown in Fig. 4, is a carbamate ester that is a
cytidine nucleoside. A cytosine base is attached to a ribose sugar
ring. The hydrogen atom at position 5 is replaced by a F atom, in
which the amino group (NH2) attached to position 4 is
converted to its N-(pentyloxy) carbonyl derivative. It is also a
carbamate ester (derived from carbamic acid, an organofluorine
compound and a member of the cytidine class) (63,64).
Moreover, it is a prodrug of fluoropyrimidine carbamate (20,65)
and a carbamate derivative of 5′-deoxy-5-fluorouridine (5′-DFUR)
(20). It is stable for ≥9 months
in tablet form (20,66). The IUPAC name for capecitabine is
5′-deoxy-5-fluoro-N-[(pentyloxy) carbonyl] cytidine
(64). The physical and chemical
properties of capecitabine are listed in Table IV.
| Table IV.Physical and chemical properties of
capecitabine. |
Table IV.
Physical and chemical properties of
capecitabine.
| Property name | Description | (Refs.) |
|---|
| Form
(Appearance) | Crystalline
powder | (67) |
| Color | White to
off-white | (67) |
| Solubility | In water 26 mg/ml
at 20°C | (68) |
| Molecular
weight | 359.35 g/mol | (30) |
| Molecular
formula |
C15H22FN3O6 | (63) |
| Formal charge | 0 | (30) |
| Rotatable bonds
count | 8 | (45) |
| Hydrogen bond donor
count | 3 | (30) |
| Hydrogen bond
acceptor count | 8 | (45) |
| Number of
rings | 2 | (31,32) |
| Acid/Base
property | An extremely weak
basic compound | (63) |
| pKa | 1.9 | (63) |
Compared with 5-FU therapy, the developed
capecitabine is an alternative to 5-FU therapy and offers multiple
advantages over the original fluorinated analog of uracil 5-FU,
including reduced toxicity, convenience, tolerability, efficient
agents and ease of administration (19,69).
Patients can be free from hospital visits that do not require
continuous infusion (unlike 5-FU) (18,19),
which facilitates the adherence of patients to the treatment
regimen and provides an improved clinical outcome (22). Therefore, the development of orally
administered 5-FU that prevents its degradation in the
gastrointestinal mucosa and other tissues, is expected to make the
oral prodrug of 5-FU more bioavailable and increase the systemic
exposure to the drug (23).
Capecitabine has ~100% bioavailability in adults (20,40).
The oral fluoropyrimidine carbamate mimics the serum concentration
levels of 5-FU continuous infusion (24,69).
Capecitabine is a tumor-selective cytotoxic agent that is
selectively activated by the TP enzyme (42). This drug was designed as a prodrug
to form 5-FU preferentially in situ or at the tumor site
(69).
Metabolism of capecitabine
Capecitabine is a relatively non-cytotoxic drug
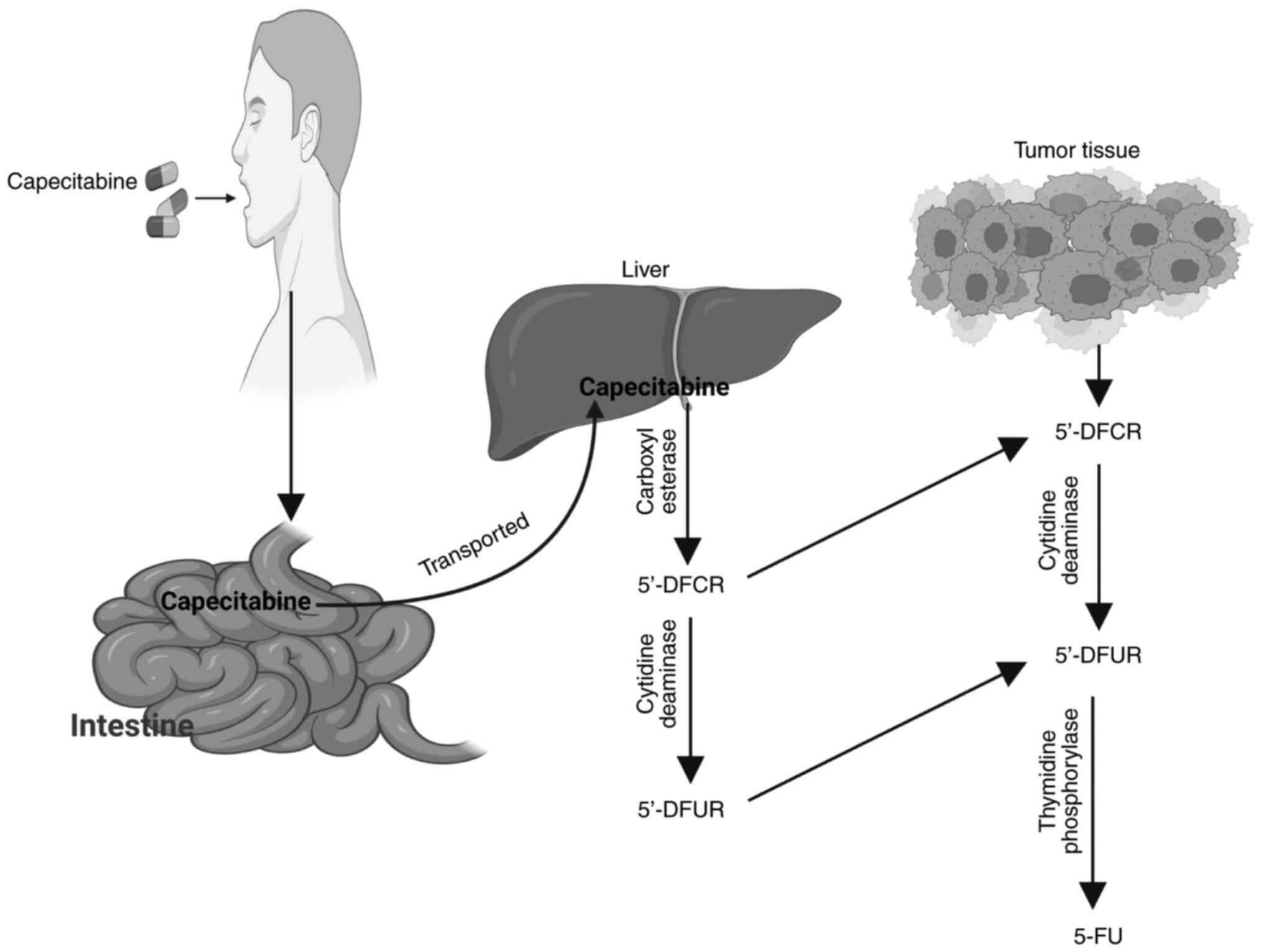
in vivo and in vitro compared with 5-FU (19). Following oral administration
(activation route), capecitabine was readily absorbed as an intact
molecule in the prodrug form through the intestinal wall, and was
then extensively metabolized enzymatically to its active compound
(5-FU) through three metabolic activation steps (Fig. 5) (19,20,24).
Once capecitabine was readily absorbed through the gastrointestinal
tract, hepatic carboxylesterase (CES) hydrolyzed the majority of
the compound to 5′-deoxy-5-fluorocytidine (5′-DFCR) (19,20,42).
Cytidine deaminase (CDA), a ubiquitous enzyme detected in high
concentrations in most tissues, including liver, plasma and tumor
tissues, converted 5′-DFCR to 5′-DFUR (19,20,42).
Finally, the TP enzyme hydrolyzed 5′-DFUR to the active drug 5-FU
(19,20,42).
This final metabolic step was considered to occur preferentially in
tumor tissues because TP was overexpressed at consistently higher
concentrations in various human solid tumors compared with normal
adjacent tissues (19,20,24,42,69),
and played a critical role in tumor growth, angiogenesis, invasion
and metastasis (69). To evaluate
this hypothesis, Schüller et al (70) conducted a study and observed that
5-FU was present at a concentration level 3.2-fold higher in CRC
tissues than in adjacent healthy tissues. Moreover, the mean
tissue/plasma 5-FU concentration ratios exceeded 20 for CRC, while
they were 8–10 for other tissues. In brief, capecitabine reached
higher levels of 5-FU in tumor tissues than in plasma or other
tissues (71). This theoretically
allows for the targeted intratumoral (in situ) release of
5-FU, thus enhancing the selective activation of capecitabine, and
demonstrating improved tolerability of the drug and less systemic
toxicity compared with IV 5-FU (19,20).
Clinical evidence to support this were observed in a study of
patients with CRC (19).
As presented in Fig.
6, after the transformation of capecitabine into the active
form 5-FU, it is degraded (inactivation route) into fewer toxic
metabolites. This is mediated by the DPD enzyme, followed by
urinary elimination, as aforementioned. Briefly, when capecitabine
is absorbed unchanged (inactive form) through the intestinal wall
(20), it is selectively activated
in tumors by the TP enzyme into its only active metabolite, 5-FU
(20,22,65).
Once 5-FU is activated, it exerts cytotoxic effects.
The present review used DrugBank 5.1.8 to identify
the DTs of capecitabine. Capecitabine was output as DB01101 from
DrugBank 5.1.8, and the DTs of capecitabine, including DNA, RNA,
TS, liver CES 1, CDA, TP and DPD were selected (32) (Table
V). When the SwissADME and DrugBank 5.1.8 databases were used
to represent pharmacokinetic properties, it was revealed that
capecitabine was readily absorbed from the intestine and did not
cross the BBB (32,45), thus not causing toxicity to the CNS,
similar to its parental 5-FU.
| Table V.Identification of selected direct
cellular targets of capecitabine in humans using DrugBank
5.1.8. |
Table V.
Identification of selected direct
cellular targets of capecitabine in humans using DrugBank
5.1.8.
| Drug name | DrugBank Identity
(DB-ID) | Target name | Type of target | Gene name | Uniprot ID |
|---|
| Capecitabine | DB01101 | DNA | Nucleotide | - | - |
|
|
| RNA | Nucleotide | - | - |
|
|
| Thymidylate
synthase | Protein | TYMS | P04818 |
|
|
| Liver
carboxylesterase 1 | Protein | CES1 | P23141 |
|
|
| Cytidine
deaminase | Protein | CDA | P32320 |
|
|
| Thymidine
phosphorylase | Protein | TYMP | P19971 |
|
|
| Dihydropyrimidine
dehydrogenase [NADP (+)] | Protein | DPYD | Q12882 |
Effect of capecitabine treatment on
the apoptotic pathway and the cell cycle phases of CRC cell
lines
When the human colon cancer cell line Caco-2 was
exposed to capecitabine treatment (2 µM), the intrinsic apoptotic
mechanism was induced through the activation of caspase-9. This
drug induced the apoptotic-oxidative stress pathway by increasing
intracellular ROS levels (57). A
previous study reported that capecitabine treatment of the human
colon cancer cell line HCT-15 caused DNA condensation and triggered
apoptosis in a dose-dependent manner. Additionally, capecitabine (5
µM) increased the population of HCT-15 cells in the
G0/G1 phase, decreased the population of
cells in S phase and caused a significant increase in the
production of ROS compared with untreated cells (72). Capecitabine (300 µM) was evaluated
on human CRC cell lines (LS174T and TP-transfected LS174T-c2) to
identify the role of the Fas system in the apoptotic mechanism of
these cell lines. Early and late apoptosis were induced by
capecitabine in TP-transfected LS174T-c2 cells, while late
apoptosis was induced in LS174T cells. This sensitivity to
capecitabine treatment was accompanied by a strong overexpression
of the CD95-Fas receptor (apoptosis receptor) on the cell surface
of the treated cells (73).
Capecitabine was applied as a single agent to the human colon
cancer cell lines HCT-116, HT-29 and Caco-2, and it significantly
stimulated apoptosis in HT-29 and Caco-2 cells, but no apoptotic
effect was observed in HCT-116 cells (74). In Caco-2 cells, 100 µM capecitabine
significantly increased the number of apoptotic cells compared with
the control group (75).
Clinical outcomes from using
capecitabine therapy over 5-FU therapy
In patients with metastatic CRC, the clinical
outcomes of capecitabine as monotherapy were evaluated in
comparison to IV 5-FU/leucovorin (LV) combination regimen, and the
results revealed a higher response rate (RR) than 5-FU/LV, and an
equivalency to 5-FU/LV in terms of overall and progression-free
survival (19). Although the
beneficial outcome of capecitabine in the increasing of RR was
determined, the lack of differences in other variables indicated
that the advantages of such combination in improving outcomes were
similar to those of treatment with capecitabine as a single
agent.
Summary of the safety and tolerability
of capecitabine treatment
Since capecitabine is not a cytotoxic drug
(42), its toxicity profile
reflects that of its major active metabolite 5-FU (19,42).
This prodrug has a well-established safety profile and can be
provided safely to patients with advanced age, and renal and
hepatic dysfunction (40). However,
capecitabine should be administered with caution in these critical
cases. Moreover, this oral therapy demonstrated a favorable
tolerability profile (76). The
most reported adverse effects associated with capecitabine therapy
were HFS, diarrhea (19,40,42),
nausea, vomiting, stomatitis (19,40)
and hyperbilirubinemia (20).
Previous adverse effects, including diarrhea, stomatitis and
nausea, occurred significantly less frequently with continuous
infusion of 5-FU (77). These toxic
effects of capecitabine are considered secondary to 5-FU
phosphorylation in the gastrointestinal tract (40). Myelosuppression with low incidence,
weakness, fatigue, abdominal pain (20), myocardial infarction, angina and
anemia (77) have also been
reported as adverse effects of capecitabine therapy (20). Capecitabine treatment is less
expensive than other chemotherapies for the treatment of toxic side
effects (65). HFS may be supported
with the use of pyridoxine (vitamin B6), while nausea and vomiting
are easily controlled with antiemetics (19). However, a previous study reported
that combined treatment of pyridoxine with capecitabine was not
effective against HFS (78).
Metabolizing enzymes required for the
activation of capecitabine to 5-FU
Capecitabine prodrug (inactive form) can be
converted into 5-FU (active form) using three ordered metabolic
activation steps, which involve CESs, CDA and TP (20,24).
CESs
Mammalian CESs are key enzymes belonging to the
superfamily of serine hydrolase enzymes (79–83).
Three CES isoenzymes are detected in the human body: CES1, CES2 and
CES3. They are classic xenobiotic-metabolizing enzymes that play
critical roles in the metabolism of a wide variety of endogenous
esters, ester-containing drugs and environmental toxicants
(83). Two human carboxylesterase
genes (CES1 and CES2) code for the mature 533-amino
acid enzyme (84). Structurally,
CESs are members of the superfamily of α/β-fold proteins, which
comprises alternate α-helices and β-sheets joined together by loops
of different lengths (85). The
three-dimensional structure of CES1 consists of a central catalytic
domain, an αβ domain and an adjacent regulatory domain that
contains a low-affinity surface ligand-binding Z-site (86,87).
As their name implies (serine hydrolases), these enzymes are
characterized by the presence of the amino acid serine at the
enzyme active site (83). CESs can
be found as monomers, trimers or hexamers (83), with a molecular weight of 60 kDa
(80,83). The majority of mammalian CESs are
intracellular proteins located within the lumen of the endoplasmic
reticulum in numerous tissues (79,81,82,83).
The elementary physiological function of CES appears to be
xenobiotic metabolism (83). CES
enzymes exhibit broad substrate specificity, which efficiently
catalyzes the ester cleavage of a large number of endogenous and
xenobiotic substrates with ester, thioester, carbamate and amide
bonds into the corresponding carboxylic acid, alcohols, thiols and
amines (80,83,88).
Therefore, they play major roles in the endobiotic metabolism and
activation and/or detoxification of xenobiotics (83). Furthermore, CESs are considered
classic xenobiotic-metabolic enzymes responsible for the
biotransformation of a wide range of ester-containing drugs,
prodrugs and environmental toxins. CESs can be readily hydrolyzed
by numerous clinical drugs with ester moieties, such as anticancer
prodrugs (irinotecan and capecitabine) (83,88).
Typically, both CES1 and CES2 are highly expressed in the epithelia
of most metabolic organs, including liver, kidney and intestine,
indicating that these two isoenzymes play protective roles against
xenobiotics (82,83,89).
Notably, the expression levels of CES1 and CES2 in tumor tissues
and cancer cell lines are markedly different from those in normal
healthy tissues and cells. For example, the human colon carcinoma
cell line Caco-2 mainly expresses CES1, whereas the expression
level of CES1 in the normal human intestine is markedly low
(89). CES2 is overexpressed in
various cancer cell lines (90).
Both CES1A1 and CES2 isoenzymes are expressed in colon
adenocarcinoma (91).
CDA
CDA is an ubiquitous enzyme that is a member of the
cytidine and deoxycytidylate deaminase families (92). Human CDA is involved in the
pyrimidine salvage pathway, which metabolizes numerous cytidine
analogs used as prodrugs in chemotherapy (93). The human CDA gene codes a
146-aminoacid protein that relies on zinc (Zn2+)
binding. Human CDA is a 52-kDa homotetrameric enzyme with a
molecular weight of 15 kDa of each subunit, with all four subunits
requiring an essential zinc atom at their active site, which plays
a crucial role in its catalytic activity (92–96).
CDA is a cytoplasmic protein that may also be located in the
nucleus (92). Alternatively, CDA
may be referred to as cytidine amino hydrolase, in which the
molecular role of CDA is the hydrolytic deamination function, which
plays a major role in the recycling of free pyrimidines (92). CDA is an enzyme of the pyrimidine
salvage pathway that catalyzes the biotransformation of cytidines
and deoxycytidines into uridines and deoxyuridines, respectively,
by hydrolyzing the amine moiety into ketones with the release of
free ammonia (92,93,96).
The pyrimidine salvage pathway contributes to two different aims:
i) The recycling of pyrimidines for the synthesis of other
nucleotides, which will be integrated into the synthesis of DNA and
RNA molecules; and ii) the catabolism of pyrimidines to ensure a
constant source of carbon and nitrogen, leading to the formation of
β-alanine. Of note, this hydrolase enzyme plays a vital role in
both pathways (92). The clinical
relevance of human CDA is its ability to deaminate its natural
substrate and several chemotherapeutic agents, including prodrugs
(93). CDA is commonly expressed in
the liver, spleen and bone marrow, and moderately expressed in
other tissues, including kidney, lung, large intestinal mucosa and
colon mucosa (92,93,97).
This enzyme is mainly located in liver and tumor tissues (98). A previous study reported that
pancreatic cancer tissues mainly expressed high levels of CDA in
epithelial tumor cells, not in the stroma (99). CDA overexpression facilitated the
metabolism of synthetic cytidine analogs for integration into DNA
molecules, leading to cell cycle arrest and cell death (92). In healthy normal cells, the
metabolites of these synthetic cytidine analogs are not inserted
into the DNA molecule due to the presence of cytidine monophosphate
kinase 1 enzyme, which is involved in nucleoside recycling and
metabolic pathways, and acts as a barrier to protect the genome.
These observations provide a therapeutic opportunity to treat
CDA-overexpressing tumors (92).
TP
TP is a key enzyme in nucleoside metabolism and
plays a critical role in the pyrimidine salvage pathway (100). Human TP is a tumor-associated
angiogenic growth factor, the sole endothelial mitogen (101), and a member of the pyrimidine
nucleoside phosphorylase family (101,102). TP is also known as
platelet-derived endothelial cell growth factor (PD-ECGF) (100,103). The human TP gene encodes a
protein of 482 amino acids with a molecular weight of 51 kDa. Human
TP is a homodimeric protein composed of two identical subunits.
Each subunit consists of a large α/β domain that contains a
phosphate-binding site and a small α-helical domain that comprises
a thymidine-binding site (100,102,104,105). TP is located intracellularly
(102) in the cytosol and nucleus
(106). In the cytoplasm, it
presents enzymatic activity, while in the nucleus, it adjusts the
pyrimidine nucleoside pool for DNA synthesis (100). TP is abundant in macrophages,
platelets, stromal cells and cancer cells (107). The enzyme plays a dual role in the
body by serving a metabolic function and a vital role in
angiogenesis (100). The metabolic
function of TP is to drive the pyrimidine nucleoside salvage
pathway as a key enzyme (100). In
the presence of inorganic phosphate (Pi), TP catalyzes
the reversible conversion of thymidine to T and
2-deoxy-α-D-ribose-1-phosphate (2DDRP) (100,102,103), and the phosphorolysis of
deoxyuridine to U and 2DDRP (100). The latter is further degraded to
2-D-deoxyribose (102). This
reaction occurs through the cleavage of the glycosidic bond of
pyrimidine 2-deoxynucleotides, most likely through an SN2-like
transition state involving the nucleobase 2′-deoxyribose and
Pi (101). Moreover, TP
has deoxyribosyl transferase activity, which involves the transfer
of a deoxyribosyl moiety from a pyrimidine nucleoside to another
pyrimidine base, resulting in a new pyrimidine nucleoside (102). This enzyme maintains a sufficient
pool of pyrimidine nucleotides available for DNA repair and
replication processes (100,101). In addition to its metabolic
function, TP plays a key role in angiogenesis because it is
identical to the angiogenic factor PD-ECGF (100). The TP enzyme utilizes thymidine
and uridine nucleoside as substrates due to the overlapping
substrate specificity for both the TP and uridine phosphorylase
enzymes, as reported in a previous study on patients with colon
cancer (100,108). In cancer, TP plays a complex role
in cancer progression through its role in angiogenesis and
determines the response to tumor treatments (100). TP catalyzes the conversion of the
oral fluoropyrimidine prodrug capecitabine to the active form of
5-FU, which interferes with DNA synthesis (100,101). Furthermore, this enzyme is
considered a predictive marker of the response to fluoropyrimidine
(100). TP is expressed by various
cells (102) and is frequently
co-expressed with the vital angiogenic factor vascular endothelial
growth factor (109). A previous
study has shown that TP is highly expressed in a wide range of
solid tumors compared with normal healthy tissues (103). Moreover, this enzyme is
overexpressed in lung, gastric, breast, colorectal, bladder,
cervical, esophageal cancer and oral squamous carcinoma (100). Overexpression of TP in cell
cultures and xenograft models has been revealed to increase the
sensitivity to 5-FU anticancer agents (100).
Opinion on capecitabine combined
chemotherapy
Capecitabine represents a major advance in cancer
therapeutics and has demonstrated promising anticancer activity as
monotherapy or in combination with other chemotherapeutic drugs in
colorectal, breast, pancreatic and head and neck cancer (40). Specific drug combinations in cancer
treatment may provide additional positive effects that may be
absent in a single drug, such as enhancement of tumor therapy
efficiency (110). Therefore, the
purpose of multidrug regimens (combined therapy) that usually
involve drugs referred to different classes is to increase
efficacy, decrease toxicity, bypass drug resistance and increase
survival, at least when classical chemotherapy is concerned
(12,13,111).
One of the most widely studied chemotherapies in neoadjuvant and
adjuvant settings is capecitabine treatment (111). Neoadjuvant and adjuvant
chemotherapy combined with capecitabine has been used clinically,
resulting in a significant improvement in patient survival and
safety in different tumors, including CRC and breast cancer
(111,112). An example of such combination
therapy is XELOX (CAP + oxaliplatin) combined with camrelizumab
(immune checkpoint inhibitor) plus bevacizumab or regorafenib
(anti-angiogenic agents), which increases the rate of responses in
patients with mCRC with microsatellite stability (113).
Drug resistance to 5-FU-based chemotherapy is one
of the greatest challenges in CRC management. It can be acquired or
intrinsic during treatment, and is considered to occur in ~50% of
patients with metastatic CRC. Therefore, one of the emerging fields
of precision medicine is determining the biological mechanisms
associated with 5-FU-based chemotherapy response. This approach is
expected to have a significant role in identifying patients likely
to benefit from 5-FU-based chemotherapy in the future. Therefore,
resistance to treatment may be predicted or overcame (114).
Factors that affect the response to
capecitabine therapy
Chemoresistance to capecitabine may be due to
various conditions that are relevant to the molecular features
(genetic or/and epigenetic) and metabolic characteristic of
patients with CRC, including: i) High MSI CRC, which is a CRC tumor
with high MSI that arises from a deficiency in mismatch repair
(MMR) genes (115); ii)
single-nucleotide polymorphisms (SNPs), which are genetic variants
of genes that are included in capecitabine activation steps
(CES, CDA and TYMP), as well as the TYMS and
DPYD genes, which are included in 5-FU metabolism (116); iii) TP53 mutation (117); and iv) epigenetic alterations,
including DNA methylation, histone modifications and abnormal
changes of non-coding RNAs as regulators of the gene expression
(118).
The majority of genetic (such as MSI and
TP53 mutation) and epigenetic modifications were mainly
involved in the molecular pathogenicity of CRC, and these altered
mechanisms accounted for a significant proportion of the molecular
classification in CRC cases (4).
Multiple cellular and molecular changes have been
reported to play crucial roles in the lack of CRC response to
capecitabine treatment (119).
These changes can be classified into four categories: i)
Intracellular factors; ii) extracellular factors; iii) cell surface
factors; and iv) cell-phenotype state. These factors can affect the
response to capecitabine drug or/and its parental 5-FU.
Changes in intracellular factors
Various intracellular factors alter the response to
therapy. Mutation of tumor-suppressor genes including TP53
(117) and APC mutations
were reported to lower the sensitivity to therapy (119). Changes in drug metabolism enzymes
could affect the response to treatment as aforementioned (SNPs in
CES, CDA, TYMP and DPYD genes) (116). Moreover, changes in drug targets
such as SNPs in the TYMS gene cause a worse clinical outcome
(116). Irregularities of
non-coding RNA including microRNA-520g participation in resistance
to 5-FU therapy (120). In
addition, changes in DNA repairing mechanisms may affect the
response to the treatment, which involves three different
mechanisms, including: i) Changes in nucleotide excision repair
mechanism as SNPs in the ERCC1 and ERCC2 genes
(121); ii) defects in the MMR
mechanism, which are accompanied by deficient MMR members in CRC,
which account for developing a MSI phenotype in CRC tumors
(115); and iii) changes in the
base excision repair (BER) mechanism, which is accompanied by the
high expression of BER proteins, and is correlated with more
aggressive tumor characteristics and poor clinical outcomes in CRC
cases (122). Alterations in the
first two aforementioned mechanisms lead to resistance of
capecitabine and 5-FU therapies, and reduced the overall survival
of patients with mCRC (119). In
addition, aberrant survival signaling pathways in CRC that involved
hyperactivation of the Wnt/β-catenin, Notch and NF-κB signaling
pathways could further enhance chemoresistance, cell proliferation
and survival (119). The final
variable is metabolic reprogramming such as the Warburg effect
(119,123) which is associated with tumor
aggressiveness and poor clinical outcome in CRC (123).
Changes in cell surface factors
The first variable of cell surface factors is
changes in drug export pumps, such as SNPs in the ABCC4 gene
that affect multidrug resistance protein 4 function and lead to a
decrease in intracellular drug concentration and therefore a lower
clinical response (124). The
second variable is the aberrant activation of the survival
signaling pathway, which is represented by hyperactivation of the
Wnt/β-catenin and Notch signaling pathways in CRC, which are
relevant to resistance (119).
Notably, the signal cascades of these pathways begin with the
stimulation of their cell surface receptors, and then are
transmitted to the intracellular space and maybe to the nucleus,
such as the Wnt/β-catenin signaling pathway.
Changes in extracellular factors
The extracellular factors are known as the
adaptation to the tumor microenvironment, which can affect the
response to such therapy, and involve hypoxic condition, gut
microbiota, impairment of pro- and anti-inflammatory cytokines, and
the extracellular matrix surrounding CRC cells (119).
Cell-phenotype state
The first cellular phenotype is the
epithelial-to-mesenchymal transition (EMT), which contributes to
enhance the cell migratory capacity and thus promotes tumor
metastasis. The EMT process is also associated with the malignancy
of CRC such as drug resistance (125). The second one is the quiescent
state of cancer stem cells (CSCs), which increases the chance of
CSCs to become resistant. Moreover, as the majority of conventional
chemotherapeutic drugs target proliferating cancer cells, a number
of CSCs often survive and promote cancer relapse (119).
Conclusion
Chemotherapeutic agents include several drugs with
different mechanisms of action, including antimetabolites that
target DNA and RNA molecules. Although 5-FU, an antimetabolite
drug, is the main treatment for several solid tumors, including CRC
and breast cancer, IV 5-FU is associated with certain health
issues, such as systemic toxicity and development of resistance.
Capecitabine is a prodrug of 5-FU that was developed to eliminate
the local (GI) toxicity of 5-FU. Capecitabine has been demonstrated
to be effective in the treatment of adenocarcinoma of colorectal
and breast origin. Moreover, the expression of three metabolizing
activation enzymes (CES, CDA and TP) that catalyze the conversion
of the capecitabine prodrug into 5-FU in CRC tissues indicates that
capecitabine is a preferred, effective and targeted therapy for the
treatment of CRC. The cellular mechanism of action of capecitabine
is the same as that of its parental 5-FU when it is activated and
enters cancer cells. Blocking DNA and RNA synthesis is the
predominant cellular mechanism of action of 5-FU and capecitabine.
Moreover, they alter the cell cycle phases and cause apoptosis in
several CRC cell lines. Capecitabine is a promising anticancer
agent, either as a monotherapy or in combination with other drugs.
Combined chemotherapy with capecitabine plays a significant role in
efficacy, resistance and survival. The chemoresistance to
capecitabine treatment could be according to changes that are
categorized as intracellular factors, extracellular factors and
cell surface factors, or cell-phenotype state. Future research on
CRC should expand the study of the effect of capecitabine with
novel and safe approved FDA drugs that target cellular pathways
other than those that target nucleic acids, such as the
β-adrenergic pathway (also called the stress pathway), which are
implicated in carcinogenesis, tumor progression and metastasis.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
SA and HA designed the present review, collected
information, and wrote the manuscript. AA and PP wrote and reviewed
the manuscript. All authors read and approved the final version of
the manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Seigel RL, Miller K and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar
|
|
2
|
Blecher E, Chaney-Graves K, DeSantis C,
Edwards B, Ferlay J, Forman D, Grey N, Harford J, Kramer J, McMikel
A and McNeal B: Global cancer facts and figures. American Cancer
Society; Atlanta, GA, USA: 2011
|
|
3
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alzahrani SM, Al Doghaither HA and
Al-Ghafari AB: General insight into cancer: An overview of
colorectal cancer (review). Mol Clin Oncol. 15:2712021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Centelles JJ: General aspects of
colorectal cancer. ISRN Oncol. 2012:1392682012.PubMed/NCBI
|
|
6
|
Blagosklonny MV: Analysis of FDA approved
anticancer drugs reveals the future of cancer therapy. Cell Cycle.
3:1033–1040. 2004. View Article : Google Scholar
|
|
7
|
Kinch MS: An analysis of FDA-approved
drugs for oncology. Drug Discov Today. 19:1831–1835. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun J, Wei Q, Zhou Y, Wang J, Liu Q and Xu
H: A systematic analysis of FDA-approved anticancer drugs. BMC Syst
Biol. 11 (Suppl 5):S872017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Winkler GC, Barle EL, Galati G and Kluwe
WM: Functional differentiation of cytotoxic cancer drugs and
targeted cancer therapeutics. Regul Toxicol Pharmacol. 70:46–53.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tseng HH and He B: Molecular markers as
therapeutic targets in lung cancer. Chin J Cancer. 32:59–62. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kinsella AR, Smith D and Pickard M:
Resistance to chemotherapeutic antimetabolites: A function of
salvage pathway involvement and cellular response to DNA damage. Br
J Cancer. 75:935–945. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Espinosa E, Zamora P, Feliu J and Barón
MG: Classification of anticancer drugs-a new system based on
therapeutic targets. Cancer Treat Rev. 29:515–523. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peters GJ: Novel developments in the use
of antimetabolites. Nucleosides Nucleotides Nucleic Acids.
33:358–374. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peters GJ and Jansen G: Antimetabolites.
Souhami RL, Tannock I, Hohenberger P and Horiot JC: ‘Oxford
Textbook of Oncology’. Oxford University Press; pp. 663–713.
2001
|
|
15
|
Kaye SB: New antimetabolites in cancer
chemotherapy and their clinical impact. Br J Cancer. 78 (Suppl
3):S1–S7. 1998. View Article : Google Scholar
|
|
16
|
Peters GJ, Van der Wilt CL, Van Moorsel
CJ, Kroep JR, Bergman AM and Ackland SP: Basis for effective
combination cancer chemotherapy with antimetabolites. Pharmacol
Ther. 87:227–253. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pizzorno G, Diasio RB and Cheng YC:
Pyrimidine analogs. In Holland-Frei Cancer Medicine. 6th edition.
BC Decker; 2003, Available from:. https://www.ncbi.nlm.nih.gov/books/NBK13287/
|
|
18
|
Thorn CF, Marsh S, Carrillo MW, McLeod HL,
Klein TE and Altman RB: PharmGKB summary: Fluoropyrimidine
pathways. Pharmacogenet Genomics. 21:237–242. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saif MW: Targeting cancers in the
gastrointestinal tract: Role of capecitabine. Onco Targets Ther.
2:29–41. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Walko CM and Lindley C: Capecitabine: A
review. Clin Ther. 27:23–44. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fluorouracil: Uses, Interactions,
Mechanism of Action | DrugBank Online. (2022). Retrieved.
4–May;2022.from. https://go.drugbank.com/drugs/DB00544
|
|
22
|
Carrillo E, Navarro SA, Ramírez A, García
MÁ, Griñán-Lisón C, Perán M and Marchal JA: 5-Fluorouracil
derivatives: A patent review (2012–2014). Expert Opin Ther Pat.
25:1131–1144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Adjei AA: A review of the pharmacology and
clinical activity of new chemotherapy agents for the treatment of
colorectal cancer. Br J Clin Pharmacol. 48:265–277. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hirsch BR and Zafar SY: Capecitabine in
the management of colorectal cancer. Cancer Manag Res. 3:79–89.
2011.PubMed/NCBI
|
|
25
|
Malet-Martino M and Martino R: Clinical
studies of three oral prodrugs of 5-fluorouracil (capecitabine,
UFT, S-1): A review. Oncologist. 7:288–323. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
DrugBank Online, . 5-Fluorouracil.
https://go.drugbank.com/structures/DB00544/image.svgRetrieved.
September 29–2022.
|
|
27
|
EMBL's European Bioinformatics Institute
(EMBL-EBI), . 5-fluorouracil (CHEBI:46345). http://www.ebi.ac.uk/chebi/searchId.do?chebiId=CHEBI:46345Retrieved.
May 4–2022.
|
|
28
|
National Center for Biotechnology
Information, . FLUOROURACIL. https://pubchem.ncbi.nlm.nih.gov/source/hsdb/3228Retrieved.
May 4–2022.
|
|
29
|
CAMEO Chemicals, . FLUOROURACIL.
https://cameochemicals.noaa.gov/chemical/5005Retrieved.
May 4–2022.
|
|
30
|
PubChem, . https://pubchem.ncbi.nlm.nih.govRetrieved. May
4–2022.
|
|
31
|
ChemAxon, . Calculators and Predictors.
https://chemaxon.com/products/calculators-and-predictors#topology_analysisRetrieved.
4–May;2022.
|
|
32
|
DrugBank Online, . DrugBank Release
Version 5.1.8. https://go.drugbank.com/releases/latestRetrieved. May
4–2022.
|
|
33
|
Wielińska J, Nowacki A and Liberek B:
5-Fluorouracil-complete insight into its neutral and ionised forms.
Molecules. 24:36832019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Privat EJ and Sowers LC: A proposed
mechanism for the mutagenicity of 5-formyluracil. Mutat Res.
354:151–156. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Şanli N, Şanli S and Alsancak G:
Determination of dissociation constants of folinic acid
(leucovorin), 5-fluorouracil, and irinotecan in hydro-organic media
by a spectrophotometric method. J Chem Eng Data. 55:2695–2699.
2010. View Article : Google Scholar
|
|
36
|
Phua LC, Mal M, Koh PK, Cheah PY, Chan EC
and Ho HK: Investigating the role of nucleoside transporters in the
resistance of colorectal cancer to 5-fluorouracil therapy. Cancer
Chemother Pharmacol. 71:817–823. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Álvarez P, Marchal JA, Boulaiz H, Carrillo
E, Vélez C, Rodríguez-Serrano F, Melguizo C, Prados J, Madeddu R
and Aranega A: 5-Fluorouracil derivatives: A patent review. Expert
Opin Ther Pat. 22:107–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gustavsson B, Carlsson G, Machover D,
Petrelli N, Roth A, Schmoll HJ, Tveit KM and Gibson F: A review of
the evolution of systemic chemotherapy in the management of
colorectal cancer. Clin Colorectal Cancer. 14:1–0. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Piedbois P, Buyse M, Blijham G, Glimelius
B, Herrmann RB, Valone F, Carlson R, Machiavelli M, Delfino C, Abad
A and Petrelli N: Meta-analysis of randomized trials testing the
biochemical modulation of fluorouracil by methotrexate in
metastatic colorectal cancer. In Database of Abstracts of Reviews
of Effects (DARE): Quality-assessed Reviews [Internet]. Centre for
Reviews and Dissemination (UK); 1994, Available from:. https://www.ncbi.nlm.nih.gov/books/NBK66225/
|
|
40
|
Mikhail SE, Sun JF and Marshall JL: Safety
of capecitabine: A review. Expert Opin Drug Saf. 9:831–841. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Van der Jeught K, Xu HC, Li YJ, Lu XB and
Ji G: Drug resistance and new therapies in colorectal cancer. World
J Gastroenterol. 24:3834–3848. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Di Costanzo F, Sdrobolini A and Gasperoni
S: Capecitabine, a new oral fluoropyrimidine for the treatment of
colorectal cancer. Crit Rev Oncol Hematol. 35:101–108. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mohammadian M, Zeynali S, Azarbaijani AF,
Ansari MH and Kheradmand F: Cytotoxic effects of the
newly-developed chemotherapeutic agents 17-AAG in combination with
oxaliplatin and capecitabine in colorectal cancer cell lines. Res
Pharm Sci. 12:517–525. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sharma R, Adam E and Schumacher U: The
action of 5-fluorouracil on human HT29 colon cancer cells grown in
SCID mice: Mitosis, apoptosis and cell differentiation. Br J
Cancer. 76:1011–1016. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Swiss Institute of Bioinformatics, .
SwissADME. http://www.swissadme.ch/index.phpRetrieved. May
4–2022.
|
|
46
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
D'Arcy MS: Cell death: A review of the
major forms of apoptosis, necrosis and autophagy. Cell Biol Int.
43:582–592. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Schafer KA: The cell cycle: A review. Vet
Pathol. 35:461–478. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dickson MA and Schwartz GK: Development of
cell-cycle inhibitors for cancer therapy. Curr Oncol. 16:36–43.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nigg EA: Cyclin-dependent protein kinases:
Key regulators of the eukaryotic cell cycle. Bioessays. 17:471–480.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Park MT and Lee SJ: Cell cycle and cancer.
J Biochem Mol Biol. 36:60–65. 2003.PubMed/NCBI
|
|
52
|
Alberts B, Johnson A, Lewis J, Raff M,
Roberts K and Walter P: Molecular biology of the cell. 4th edition.
New York: Garland Science; 2002, Available from:. https://www.ncbi.nlm.nih.gov/books/NBK21054/
|
|
53
|
Sagona AP and Stenmark H: Cytokinesis and
cancer. FEBS Lett. 584:2652–2661. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Green DR, McGahon A and Martin SJ:
Regulation of apoptosis by oncogenes. J Cell Biochem. 60:33–38.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Brown JM and Wouters BG: Apoptosis, p53,
and tumor cell sensitivity to anticancer agents. Cancer Res.
59:1391–1399. 1999.PubMed/NCBI
|
|
56
|
Tiwari M: Antimetabolites: Established
cancer therapy. J Cancer Res Ther. 8:510–519. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Övey İS and Güler Y: Apoptotic efficiency
of capecitabine and 5-fluorouracil on human cancer cells through
TRPV1 channels. NISCAIR-CSIR. pp64–72. 2020.http://nopr.niscair.res.in/handle/123456789/54047
|
|
58
|
Shi H, Jiang J, Ji J, Shi M, Cai Q, Chen
X, Yu Y, Liu B, Zhu Z and Zhang J: Anti-angiogenesis participates
in antitumor effects of metronomic capecitabine on colon cancer.
Cancer Lett. 349:128–135. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
De Angelis PM, Svendsrud DH, Kravik KL and
Stokke T: Cellular response to 5-fluorouracil (5-FU) in
5-FU-resistant colon cancer cell lines during treatment and
recovery. Mol Cancer. 5:202006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gao L, Shen L, Yu M, Ni J, Dong X, Zhou Y
and Wu S: Colon cancer cells treated with 5-fluorouracil exhibit
changes in polylactosamine-type N-glycans. Mol Med Rep.
9:1697–1702. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Afrin S, Giampieri F, Cianciosi D,
Alvarez-Suarez JM, Bullon B, Amici A, Quiles JL, Forbes-Hernández
TY and Battino M: Strawberry tree honey in combination with
5-fluorouracil enhances chemosensitivity in human colon
adenocarcinoma cells. Food Chem Toxicol. 156:1124842021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
DrugBank Online, . Capecitabine: Uses,
Interactions, Mechanism of Action. https://go.drugbank.com/drugs/DB01101Retrieved. May
4–2022.
|
|
63
|
DrugBank Online, . Capecitabine.
https://go.drugbank.com/structures/DB01101/image.svgRetrieved.
September 29–2022.
|
|
64
|
Team, E. Capecitabine (CHEBI:31348),
2022b, . Retrieved. 4–May;2022.from. http://www.ebi.ac.uk/chebi/searchId.do?chebiId=CHEBI:31348
|
|
65
|
Twelves C, Boyer M, Findlay M, Cassidy J,
Weitzel C, Barker C, Osterwalder B, Jamieson C and Hieke K; Xeloda
Colorectal Cancer Study Group, : Capecitabine (Xeloda) improves
medical resource use compared with 5-fluorouracil plus leucovorin
in a phase III trial conducted in patients with advanced colorectal
carcinoma. Eur J Cancer. 37:597–604. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Budman DR, Meropol NJ, Reigner B, Creaven
PJ, Lichtman SM, Berghorn E, Behr J, Gordon RJ, Osterwalder B and
Griffin T: Preliminary studies of a novel oral fluoropyrimidine
carbamate: Capecitabine. J Clin Oncol. 16:1795–1802. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
National Center for Biotechnology
Information, . CAPECITABINE. https://pubchem.ncbi.nlm.nih.gov/source/hsdb/7656Retrieved.
May 4–2022.
|
|
68
|
O'Neil MJ: The merck index-an encyclopedia
of chemicals, drugs and biologicals. Merck and Co. Inc.; Whitehouse
Station, NJ: pp. pp17232006
|
|
69
|
Loo WT, Chow LW, Suzuki T, Ono K, Ishida
T, Hirakawa H, Ohuchi N and Sasano H: Expression of thymidine
phosphorylase and dihydropyrimidine dehydrogenase in human breast
carcinoma cells and tissues. Anticancer Res. 29:2525–2530.
2009.PubMed/NCBI
|
|
70
|
Schüller J, Cassidy J, Dumont E, Roos B,
Durston S, Banken L, Utoh M, Mori K, Weidekamm E and Reigner B:
Preferential activation of capecitabine in tumor following oral
administration to colorectal cancer patients. Cancer Chemother
Pharmacol. 45:291–297. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Brito RA, Medgyesy D, Zukowski TH, Royce
ME, Ravandi-Kashani F, Hoff PM and Pazdur R: Fluoropyrimidines: A
critical evaluation. Oncology. 57 (Suppl 1):S2–S8. 1999. View Article : Google Scholar
|
|
72
|
Li M, Zhang N and Li M: Capecitabine
treatment of HCT-15 colon cancer cells induces apoptosis via
mitochondrial pathway. Trop J Pharm Res. 16:1529–1536. 2017.
View Article : Google Scholar
|
|
73
|
Ciccolini J, Fina F, Bezulier K,
Giacometti S, Roussel M, Evrard A, Cuq P, Romain S, Martin PM and
Aubert C: Transmission of apoptosis in human colorectal tumor cells
exposed to capecitabine, Xeloda, is mediated via Fas. Mol Cancer
Ther. 1:923–927. 2002.PubMed/NCBI
|
|
74
|
Prasad S, Yadav VR, Sung B, Reuter S,
Kannappan R, Deorukhkar A, Diagaradjane P, Wei C,
Baladandayuthapani V, Krishnan S, et al: Ursolic acid inhibits
growth and metastasis of human colorectal cancer in an orthotopic
nude mouse model by targeting multiple cell signaling pathways:
Chemosensitization with capecitabine. Clin Cancer Res.
18:4942–4953. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Namvaran A, Fazeli M, Farajnia S, Hamidian
G and Rezazadeh H: Apoptosis and caspase 3 pathway role on
anti-proliferative effects of scrophulariaoxy sepala methanolic
extract on caco-2 cells. Drug Res (Stuttg). 67:547–552. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Loo WT, Sasano H and Chow LW: Evaluation
of therapeutic efficacy of capecitabine on human breast carcinoma
tissues and cell lines in vitro. Biomed Pharmacother. 61:553–557.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Nazari-Vanani R, Karimian K, Azarpira N
and Heli H: Capecitabine-loaded nanoniosomes and evaluation of
anticancer efficacy. Artif Cells Nanomed Biotechnol. 47:420–426.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Kang YK, Lee SS, Yoon DH, Lee SY, Chun YJ,
Kim MS, Ryu MH, Chang HM, Lee JL and Kim TW: Pyridoxine is not
effective to prevent hand-foot syndrome associated with
capecitabine therapy: Results of a randomized, double-blind,
placebo-controlled study. J Clin Oncol. 28:3824–3829. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Satoh T and Hosokawa M: The mammalian
carboxylesterases: From molecules to functions. Annu Rev Pharmacol
Toxicol. 38:257–288. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sanghani SP, Quinney SK, Fredenburg TB,
Sun Z, Davis WI, Murry DJ, Cummings OW, Seitz DE and Bosron WF:
Carboxylesterases expressed in human colon tumor tissue and their
role in CPT-11 hydrolysis. Clin Cancer Res. 9:4983–4991.
2003.PubMed/NCBI
|
|
81
|
Satoh T and Hosokawa M: Structure,
function and regulation of carboxylesterases. Chem Biol Interact.
162:195–211. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Sanghani SP, Sanghani PC, Schiel MA and
Bosron WF: Human carboxylesterases: An update on CES1, CES2 and
CES3. Protein Pept Lett. 16:1207–1214. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wang D, Zou L, Jin Q, Hou J, Ge G and Yang
L: Human carboxylesterases: A comprehensive review. Acta Pharm Sin
B. 8:699–712. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Pindel EV, Kedishvili NY, Abraham TL,
Brzezinski MR, Zhang J, Dean RA and Bosron WF: Purification and
cloning of a broad substrate specificity human liver
carboxylesterase that catalyzes the hydrolysis of cocaine and
heroin. J Biol Chem. 272:14769–14775. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Oakeshott JG, Claudianos C, Russell RJ and
Robin GC: Carboxyl/cholinesterases: A case study of the evolution
of a successful multigene family. Bioessays. 21:1031–1042. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kim KK, Song HK, Shin DH, Hwang KY, Choe
S, Yoo OJ and Suh SW: Crystal structure of carboxylesterase from
Pseudomonas fluorescens, an alpha/beta hydrolase with broad
substrate specificity. Structure. 5:1571–1584. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Fleming CD, Edwards CC, Kirby SD, Maxwell
DM, Potter PM, Cerasoli DM and Redinbo MR: Crystal structures of
human carboxylesterase 1 in covalent complexes with the chemical
warfare agents soman and tabun. Biochemistry. 46:5063–5071. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hosokawa M: Structure and catalytic
properties of carboxylesterase isozymes involved in metabolic
activation of prodrugs. Molecules. 13:412–431. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Taketani M, Shii M, Ohura K, Ninomiya S
and Imai T: Carboxylesterase in the liver and small intestine of
experimental animals and human. Life Sci. 81:924–932. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yano H, Kayukawa S, Iida S, Nakagawa C,
Oguri T, Sanda T, Ding J, Mori F, Ito A, Ri M, et al:
Overexpression of carboxylesterase-2 results in enhanced efficacy
of topoisomerase I inhibitor, irinotecan (CPT-11), for multiple
myeloma. Cancer Sci. 99:2309–2314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Xie M, Yang D, Liu L, Xue B and Yan B:
Human and rodent carboxylesterases: Immunorelatedness, overlapping
substrate specificity, differential sensitivity to serine enzyme
inhibitors, and tumor-related expression. Drug Metab Dispos.
30:541–547. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Frances A and Cordelier P: The emerging
role of cytidine deaminase in human diseases: A new opportunity for
therapy? Mol Ther. 28:357–366. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Micozzi D, Carpi FM, Pucciarelli S,
Polzonetti V, Polidori P, Vilar S, Williams B, Costanzi S and
Vincenzetti S: Human cytidine deaminase: A biochemical
characterization of its naturally occurring variants. Int J Biol
Macromol. 63:64–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Vincenzetti S, Quadrini B, Mariani P, De
Sanctis G, Cammertoni N, Polzonetti V, Pucciarelli S, Natalini P
and Vita A: Modulation of human cytidine deaminase by specific
aminoacids involved in the intersubunit interactions. Proteins.
70:144–156. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Micozzi D, Pucciarelli S, Carpi FM,
Costanzi S, De Sanctis G, Polzonetti V, Natalini P, Santarelli IF,
Vita A and Vincenzetti S: Role of tyrosine 33 residue for the
stabilization of the tetrameric structure of human cytidine
deaminase. Int J Biol Macromol. 47:471–482. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Vincenzetti S, Pucciarelli S, Carpi FM,
Micozzi D, Polzonetti V, Natalini P, Santarelli I, Polidori P and
Vita A: Site directed mutagenesis as a tool to understand the
catalytic mechanism of human cytidine deaminase. Protein Pept Lett.
20:538–549. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ho DH: Distribution of kinase and
deaminase of 1-beta-D-arabinofuranosylcytosine in tissues of man
and mouse. Cancer Res. 33:2816–2820. 1973.PubMed/NCBI
|
|
98
|
Ishikawa T, Sawada N, Sekiguchi F, Fukase
Y and Ishitsuka H: Xeloda™ (capecitabine), a new oral
fluoropyrimidine carbamate with an improved efficacy profile over
other fluoropyrimidines. Proc Am Soc Clin Oncol. 16:226a1997.
|
|
99
|
Hessmann E, Patzak MS, Klein L, Chen N,
Kari V, Ramu I, Bapiro TE, Frese KK, Gopinathan A, Richards FM, et
al: Fibroblast drug scavenging increases intratumoural gemcitabine
accumulation in murine pancreas cancer. Gut. 67:497–507. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Elamin YY, Rafee S, Osman N, O Byrne KJ
and Gately K: Thymidine phosphorylase in cancer; enemy or friend?
Cancer Microenviron. 9:33–43. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Mitsiki E, Papageorgiou AC, Iyer S,
Thiyagarajan N, Prior SH, Sleep D, Finnis C and Acharya KR:
Structures of native human thymidine phosphorylase and in complex
with 5-iodouracil. Biochem Biophys Res Commun. 386:666–670. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li W and Yue H: Thymidine phosphorylase: A
potential new target for treating cardiovascular disease. Trends
Cardiovasc Med. 28:157–171. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Bijnsdorp IV, Capriotti F, Kruyt FA,
Losekoot N, Fukushima M, Griffioen AW, Thijssen VL and Peters GJ:
Thymidine phosphorylase in cancer cells stimulates human
endothelial cell migration and invasion by the secretion of
angiogenic factors. Br J Cancer. 104:1185–1192. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ishikawa F, Miyazono K, Hellman U, Drexler
H, Wernstedt C, Hagiwara K, Usuki K, Takaku F, Risau W and Heldin
CH: Identification of angiogenic activity and the cloning and
expression of platelet-derived endothelial cell growth factor.
Nature. 338:557–562. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Bronckaers A, Gago F, Balzarini J and
Liekens S: The dual role of thymidine phosphorylase in cancer
development and chemotherapy. Med Res Rev. 29:903–953. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kamatani N, Jinnah HA, Hennekam RC and van
Kuilenburg AB: Purine and pyrimidine metabolism. In Emery and
Rimoin's Principles and Practice of Medical Genetics. Academic
Press. 1–38. 2013. View Article : Google Scholar
|
|
107
|
Harris AL and Generali D: Inhibitors of
tumor angiogenesis. Cancer Drug Design and Discovery. 275–306.
2008.
|
|
108
|
Temmink OH, de Bruin M, Turksma AW, Cricca
S, Laan AC and Peters GJ: Activity and substrate specificity of
pyrimidine phosphorylases and their role in fluoropyrimidine
sensitivity in colon cancer cell lines. Int J Biochem Cell Biol.
39:565–575. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Sivridis E, Giatromanolaki A, Anastasiadis
P, Georgiou L, Gatter KC, Harris AL, Bicknell R and Koukourakis MI;
Tumour Angiogenesis Research Group, : Angiogenic co-operation of
VEGF and stromal cell TP in endometrial carcinomas. J Pathol.
196:416–422. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Xiao X, Wang T, Li L, Zhu Z, Zhang W, Cui
G and Li W: Co-delivery of cisplatin(IV) and capecitabine as an
effective and non-toxic cancer treatment. Front Pharmacol.
10:1102019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Huo X, Li J, Zhao F, Ren D, Ahmad R, Yuan
X, Du F and Zhao J: The role of capecitabine-based neoadjuvant and
adjuvant chemotherapy in early-stage triple-negative breast cancer:
A systematic review and meta-analysis. BMC Cancer. 21:782021.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Voegeli M and Wicki A: Neoadjuvant,
adjuvant and palliative systemic therapy of colorectal cancer. Ther
Umsch. 75:622–626. 2018.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zhou H, Wang Y, Lin Y, Cai W, Li X and He
X: Preliminary efficacy and safety of camrelizumab in combination
with XELOX plus bevacizumab or regorafenib in patients with
metastatic colorectal cancer: A retrospective study. Front Oncol.
11:7744452021. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sabeti Aghabozorgi A, Moradi Sarabi M,
Jafarzadeh-Esfehani R, Koochakkhani S, Hassanzadeh M, Kavousipour S
and Eftekhar E: Molecular determinants of response to
5-fluorouracil-based chemotherapy in colorectal cancer: The
undisputable role of micro-ribonucleic acids. World J Gastrointest
Oncol. 12:942–956. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Oneda E and Zaniboni A: Adjuvant treatment
of colon cancer with microsatellite instability-the state of the
art. Crit Rev Oncol Hematol. 169:1035372022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Cura Y, Pérez-Ramírez C, Sánchez-Martín A,
Membrive-Jimenez C, Valverde-Merino MI, González-Flores E and
Morales AJ: Influence of single-nucleotide polymorphisms on
clinical outcomes of capecitabine-based chemotherapy in colorectal
cancer patients: A systematic review. Cancers (Basel). 15:18212023.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Michel M, Kaps L, Maderer A, Galle PR and
Moehler M: The role of p53 dysfunction in colorectal cancer and its
implication for therapy. Cancers (Basel). 13:22962021. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Jung G, Hernández-Illán E, Moreira L,
Balaguer F and Goel A: Epigenetics of colorectal cancer: Biomarker
and therapeutic potential. Nat Rev Gastroenterol Hepatol.
17:111–130. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Marin JJG, Macias RIR, Monte MJ, Herraez
E, Peleteiro-Vigil A, Blas BS, Sanchon-Sanchez P, Temprano AG,
Espinosa-Escudero RA, Lozano E, et al: Cellular mechanisms
accounting for the refractoriness of colorectal carcinoma to
pharmacological treatment. Cancers (Basel). 12:26052020. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zhang Y, Geng L, Talmon G and Wang J:
MicroRNA-520g confers drug resistance by regulating p21 expression
in colorectal cancer. J Biol Chem. 290:6215–6225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Boige V, Mollevi C, Gourgou S, Azria D,
Seitz JF, Vincent M, Bigot L, Juzyna B, Miran I, Gerard JP and
Laurent-Puig P: Impact of single-nucleotide polymorphisms in DNA
repair pathway genes on response to chemoradiotherapy in rectal
cancer patients: Results from ACCORD-12/PRODIGE-2 phase III trial.
Int J Cancer. 145:3163–3172. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Leguisamo NM, Gloria HC, Kalil AN, Martins
TV, Azambuja DB, Meira LB and Saffi J: Base excision repair
imbalance in colorectal cancer has prognostic value and modulates
response to chemotherapy. Oncotarget. 8:54199–54214. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Brown RE, Short SP and Williams CS:
Colorectal cancer and metabolism. Curr Colorectal Cancer Rep.
14:226–241. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Chen Q, Meng F, Wang L, Mao Y, Zhou H, Hua
D, Zhang H and Wang W: A polymorphism in ABCC4 is related to
efficacy of 5-FU/capecitabine-based chemotherapy in colorectal
cancer patients. Sci Rep. 7:70592017. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Cao H, Xu E, Liu H, Wan L and Lai M:
Epithelial-mesenchymal transition in colorectal cancer metastasis:
A system review. Pathol Res Pract. 211:557–569. 2015. View Article : Google Scholar : PubMed/NCBI
|