Introduction
Hepatocellular carcinoma (HCC) is the fourth most
common cause of cancer-associated mortality worldwide and is
associated with poor prognosis with overall 5-year survival <20%
(1,2). One key characteristic of HCC is its
high degree of heterogeneity, with distinct molecular alterations
in different subclones within each tumor as confirmed by recent
high-throughput sequencing analyses (3,4).
Adenosine deaminase acting on RNA-1 (ADAR1) is an
RNA editing enzyme that catalyzes the conversion of adenosine
residues to inosine in double-stranded RNAs (A-to-I RNA editing)
(5–7). ADAR1 is frequently amplified with
elevated activity in cancer types, consistent with the elevated
editing levels of the substrates (8,9).
Similar to somatic DNA mutations, most RNA mutations are passenger
mutations. However, A-to-I RNA editing at residue 367 of antizyme
inhibitor 1 (AZIN1) regulated by ADAR1 is observed in many primary
HCC samples and is linked with tumor initiation and development
(10). While other editing events
within coding or non-coding regions are also observed during
oncogenesis (11), AZIN1 is the
most common gene targeted by ADAR1 in the oncogenesis of numerous
types of cancers (10,12–15).
Extracellular vesicles (EVs) have been recognized as
functional modes of intercellular communication and have been shown
to be involved in physiological and disease-associated cellular
activities (6,16). There are two main subtypes of EVs,
microvesicles (typically 100 nm to 1 µm in diameter) and exosomes
(30–150 nm in diameter). EVs carry diverse cargo, including
miscellaneous proteins, lipids and nucleic acids, for intercellular
communication. Although EVs secreted by cancer cells are also
hypothesized to various signals to the neighboring environment, to
the best of our knowledge, the biological significance of cargo in
EVs secreted by cancer cells and its effects on recipient cells
have yet to be elucidated.
In the present study, it was hypothesized that EVs
secreted from HCC cells transmit ADAR1 protein to neighboring
cancer cells as well as to surrounding non-cancerous hepatocytes,
leading to intratumoral heterogeneity and tumor spread via RNA
editing. To determine if ADAR1 is transferred in EVs from HCC cells
to surrounding cells, highly sensitive split-luciferase technology,
a horizontal co-culture system and mutational detection by digital
PCR were used.
Materials and methods
Cells
Human HCC Huh7 cells were purchased from the
American Type Culture Collection (ATCC, Manassas, VA, USA).
SV40-mediated immortalized human normal hepatocyte Fa2N-4 cells
were purchased from Sekisui XenoTech. 293T cells were obtained from
ATCC and cultured in Dulbecco's Modified Eagle's Medium (DMEM)
supplemented with 10% fetal bovine serum (FUJIFILM Wako Pure
Chemical Corporation). Human iPS cells established from the
fibroblasts of a healthy donor (European/North African 32-year-old
male) were purchased from Takara Bio, Inc. Cells were maintained in
Cellartis DEF-CS 500 Culture System (Takara Bio, Inc.) according to
the manufacturer's instructions. All cells were incubated at 37°C
with 20% O2 and 5% CO2.
Differentiation of iPS into
hepatocyte-like cells
Human hepatocyte-like cells were induced from iPS
cells using a Cellartis Hepatocyte Differentiation System (Takara
Bio, Inc.) according to the manufacturer's instructions. Briefly,
iPS cells were differentiated into definitive endoderm (DE) cells
using 3 µM CHIR99021 (FUJIFILM Wako Pure Chemical Corporation) with
100 ng/ml Activin A (FUJIFILM Wako Pure Chemical Corporation) for 1
day, followed by 100 ng/ml Activin A for 2 days at 37°C in
RPMI-1640 medium with 0.0, 0.2 and 2.0% fetal bovine serum
(FUJIFILM Wako Pure Chemical Corporation). Then, the cells were
seeded onto 12-well plates at 90% confluency and differentiation
into hepatocyte-like cells was initiated by adding hepatocyte
progenitor medium for 7 days, followed by hepatocyte maturation
medium for 6 days (FUJIFILM Wako Pure Chemical Corporation). The
induced hepatocyte-like cells were maintained in hepatocyte
maintenance medium (FUJIFILM Wako Pure Chemical Corporation) for up
to 7 days at 37°C after induction. Hepatocyte-like cells were used
for experiments ~20 days after induction.
Knock-in of HiBiT peptide sequence
into the ADAR locus
The HiBiT peptide sequence was knocked into the 3′
terminus of the ADAR gene locus by CRISPR method. Briefly,
recombinant Cas9 protein (Integrated DNA Technologies, Inc.), guide
RNA [transactivating CRISPR RNA (tracrRNA) + CRISPR RNA (crRNA)];
5′-CACCCUAAUCCAUCUGUCACGUUUUAGAGCUAUGCU-3′) and donor
oligonucleotides
(5′-AAAAAGGCCTGAAGGATATGGGCTATGGGAACTGGATTAGCAAACCCCAGGAGGAAAAGAACTTTTATCTCTGCCCAGTAGTGAGCGGCTGGCGGCTGTTCAAGAGGGTGTGTCATACTAGGGTCTGAGAGAGGTAGGTCGTAGCATTCCTCAT-3′;
Integrated DNA Technologies, Inc.) were transfected into Huh7 cells
according to the manufacturer's instructions. Guide RNA sequences
were determined by using the ChopChop software
(chopchop.cbu.uib.no/). Cells were seeded by limiting dilution and
supernatants were subjected to HiBiT luciferase assay to determine
successfully knocked-in cells. After selecting the cells with
higher luciferase activity, genomic DNA was extracted for
genotyping to confirm successful knock-in.
Genomic DNA extraction
To confirm successful knock-in of the HiBiT peptide
sequence and examine AZIN1 mutation, cellular genomic DNA was
extracted using a QIAamp DNA Mini kit (Qiagen GmbH) according to
the manufacturer's instructions.
Genotyping PCR for confirmation of
successful knock-in
Genotyping PCR was performed using the following
primers (Eurofin): Forward (Fw), 5′-CAAGCCTCTGCCCTGACTTGC-3′ and
reverse (Rv), 5′-ATCCCCTGACCATGTGATGAG-3′. Thermocycling conditions
were as follows: Initial denature at 95°C for 2 min, denature at
95°C for 10 sec, annealing at 60°C, and extension at 72°C for 40
cycles, and final extension at 72°C for 5 min.
Western blotting
Western blotting was performed as described
previously (17). Briefly, lysate
samples were separated by SDS-PAGE on 10% polyacrylamide gels
followed by electrophoretic transfer onto PVDF membranes (GE
Healthcare). Following blocking with 5% dried milk, membranes were
probed with primary antibodies diluted in Immunoshot Reagent 1
(Cosmo Bio Co., Ltd.) overnight at 4°C. HRP-conjugated
corresponding secondary antibodies (GE Healthcare) were added.
Bound antibodies were detected using Immunostar LD reagent
(FUJIFILM Wako Pure Chemical Corporation). The following antibodies
were used: ADAR1 (cat. no. #81284), β-actin (cat. no. #5125; both
Cell Signaling Technology, Inc.) and Flag-tag (Medical &
Biological Laboratories Co., Ltd.).
Detection of ADAR1-HiBiT protein
Screening for the presence of HiBiT peptides in the
DMEM was performed using Nano Glo-Extracellular HiBiT detection
system (Promega Corporation) after removal of cellular debris,
according to the manufacturer's instructions. ADAR1-HiBiT within
cells or isolated exosomes by size exclusion method (performed by
MEIWAFORSIS Co.) was determined using the Nano Glo-Lytic HiBiT
detection system (Promega Corporation). Briefly, cells or exosomes
were lysed with ice-cold RIPA buffer (FUJIFILM Wako Pure Chemical
Corporation). Lysate was incubated with the provided recombinant
LgBiT protein and substrate. Luciferase activity by the NanoBit
produced by the coexistence of HiBiT peptide and LgBiT protein was
measured with Nano Glo-Extracellular Detection System (Promega
Corporation). A Nano Glo blotting system (Promega Corporation) was
used to detect HiBiT peptide by western blotting. Briefly, the
transferred membrane was incubated at room temperature for 2 h with
the provided recombinant LgBiT protein, followed by detection with
the substrate. Recombinant Halo-tagged HiBiT control protein
(Promega Corporation) was used as a positive control.
Detection of luciferase activity
The luciferase activity was detected by the GloMax
multi-detection system (Promega Corporation).
EV isolation and characterization
Huh7 cells were incubated with culture DMEM
containing exosome-depleted FBS (Thermo Fisher Scientific, Inc.)
for 48 h at 37°C. To isolate EVs from cell culture medium, qEV size
exclusion columns and an automated fraction collector (AFC; Izon
Science Ltd.) were used according to the manufacturer's
instructions. Briefly, the column was loaded into the AFC and
flushed with phosphate-buffered saline (PBS) before sample loading.
Aliquots of 150 µl culture medium were loaded into the column and
flushed through with PBS. The AFC was programmed to elute the first
~1 ml fraction as discarded void volume and collect the following
three 0.2 ml fractions as the EV-containing fractions. Acquired EVs
were characterized by number and size distribution by nano tracking
analysis (NanoSight LM10; Malvern Instruments Ltd.). Isolated EVs
were visualized by electron microscopy by the Hanaichi
UltraStructure Research Institute (Okazaki, Japan). To prevent
aggregation, visualization was performed the day after acquisition
of EVs.
Exosome labeling
Fluorescent exosome labeling was performed using an
ExoSparkler Exosome Labeling kit (Dojindo Laboratories, Inc.)
according to the manufacturer's instructions. Briefly,
~1×1010 exosomes were mixed at room temperature with the
provided dye for 30 min and the free dye was removed by filtration.
Labeled exosomes were recovered in 50 µl PBS. Labeled exosomes were
added to DMEM at 37°C for 24 h. After washing with PBS,
intracellular fluorescence was examined using an Olympus DP72
fluorescent microscope (with objective lenses at 20×) digital
camera system with Olympus DP2-TWAIN software (Olympus
Corporation).
Plasmids
PCR-amplified LgBit sequences with the flag tag
sequence at the 3′ terminus were subcloned into the pCDH-puromycin
vector (System Biosciences, LLC) at the NotI site. The
primers were as follows: Fw,
5′-ATCGGATCCGCGGCCGCGTCTTCACACTCGAAGATTTCGTTG-3′ and Rv,
5′-AGATCCTTCGCGGCCGCTCACTTGTCATCGTCATCCTTGTAGTCGCTGTTGATGGTTACTCGGAACAG-3′.
A C-terminal flag-tagged ADAR1 p110 expression plasmid was
constructed by inserting PCR-amplified ADAR1 cDNA using the
Halo-tagged ADAR1 plasmid obtained from the Kazusa open reading
frame clone collection (Promega Corporation) as a template, into
the pCDH-puromycin vector at the NotI site with flag tag sequences.
The following primers were used: Fw,
5′-CATAGAAGATTCTAGAACCATGGATTACAAGGATGA-3′ and Rv,
5′-AGATCCTTCGCGGCCGCTCATGGGCAGAGATAAAAGTTCTT-3′.
Transfection and lentivirus
transduction
To generate stably expressing polyclonal cells,
Lentivirus Packaging System (System Biosciences, LLC) was used
according to the manufacturer's protocol. Briefly, 1 µg LgBiT-flag
overexpression vector and 5 µg pPACKH1 packaging plasmid mix
(System Biosciences, LLC) for generating 3rd generation
lentiviruses, were transfected into 293T cells using Effectene
Transfection Reagent (Qiagen GmbH) at 37°C. After 24 h, collected
culture medium was mixed with 1/5 volume of PEG-it Reagent (System
Biosciences, LLC) overnight at 4°C to concentrate the lentiviruses.
The centrifuged pellet collected at 1,250 g at 4°C for 30 min was
resuspended in 1X PBS and aliquots were stored at −80°C and used
within 6 months. Lentiviruses were added to Fa2N4 and Huh7 cells
with Polybrene Reagent (Santa Cruz Biotechnology, Inc.) at 37°C and
puromycin selection (2–6 µg/ml) was started after 48 h.
Interactive horizontal co-culture
Instead of a traditional vertical co-culture system,
such as Transwell, a horizontal co-culture system (Interactive
Co-Culture Plate, ICCP; Ginreilab, Inc.) was used to examine
cell-to-cell communication (18).
Cells were seeded at 80% confluency into two culture vessels
separately at 37°C and DMEM was added to a level below the
connection port. On the following day, medium was added to bring
the two vessels into a horizontal co-culture state, followed by
incubation for 48 h. Vessels were separated by filters with pore
sizes of 0.60 or 0.03 µm.
RNA extraction and reverse
transcription-quantitative (RT-q)PCR
RNA was extracted using Isogen II (Nippon Gene Co.,
Ltd.) according to the manufacturer's protocol. RT-qPCR was
performed as described previously (17). All values were normalized to β-actin
mRNA expression. Relative expression was calculated using the
ΔΔCq method as follows:
ΔΔCq=ΔCqsample-ΔCqβ-actin
(19). The primers were as follows:
Albumin (Alb) Fw, 5′-GCACAGTTACCTTGGTGAACAG-3′ and Rv,
5′-ATGGAAGGTGAATGTTTTCAGCA-3′; Cyp3A4 Fw,
5′-AAGTCGCCTCGAAGATACACA-3′ and Rv, 5′-AAGGAGAGAACACTGCTCGTG-3′;
SOX2 Fw, 5′-GGCAGCTACAGCATGATGATGCAGGAGC-3′ and Rv
5′-ATTTCTGCTCCGGCTCTATG-3′ and β-actin Fw
5′-TCCCTGGAGAAGAGCTACGA-3′ and Rv 5′-AGCACTGTGTTGGCGTACAG-3′.
Detection of AZIN1 mutation by droplet
digital PCR (ddPCR)
To detect A-to-I RNA editing at residue 367 of AZIN1
(10), a custom TaqMan probe was
designed and purchased from Bio-Rad Laboratories, Inc. ddPCR was
conducted using the QX100 Droplet Digital PCR system (Bio-Rad
Laboratories, Inc.) as described previously (20). Briefly, 3.3 µl template cDNA with
20X primer and TaqMan probe set were partitioned into ~20,000
droplets by the QX100 Droplet Generator for amplification. The
cycling conditions were 95°C for 10 min, followed by 50 cycles of
95°C for 15 sec and 60°C for 1 min with a final 10 min incubation
at 98°C. The droplets were read automatically by the QX10 droplet
reader. The data were analyzed with QuantaSoft analysis software
(ver. 1.3.2.0; Bio-Rad Laboratories, Inc.).
Prognosis of HCC according to ADAR1
expression
The data of Kaplan-Meier analysis of HCC cases with
high and low ADAR1 protein expression levels were obtained from the
Pathology Atlas in the Human Protein Atlas Database (21).
Clinical samples
Surgically resected HCC samples were obtained
sequentially from April 2018 to March 2020 at the Department of
Surgery, Dokkyo Medical University (Tochigi, Japan). Cancerous and
surrounding non-cancerous tissue from 30 cases (age, 53–82 years
old; sex, 22 male and 8 female cases; cases with poor tissue
quality were excluded) were subjected to immunohistochemistry to
examine ADAR1 expression and intracellular localization. Data from
the surrounding non-cancerous tissue in two cases and tumor tissues
in one case were not tested due to lack of availability and
necrosis, respectively. Written informed consent was obtained from
all patients and the study protocols were approved by the Ethics
Committee of Dokkyo Medical University (approval no. 28110).
Immunohistochemistry
Immunohistochemistry was performed as described
previously (22). Briefly, resected
liver specimens were fixed in 10% v/v formalin at room temperature
for 12 h, cut into blocks and embedded in paraffin by Sept. Sapie
Co., Ltd.). The blocks were cut into sections 4-µm thick and
stained with hematoxylin and eosin at room temperature for 1 min or
used for immunohistochemical analyses. The sections were subjected
to dewaxing, heat-induced epitope retrieval with citrate buffer,
antibody incubation and counterstaining on a BOND Max automated
immunostainer (Leica Microsystems GmbH). Anti-ADAR1 antibody (cat.
no. HPA003890) was obtained from Atlas Antibodies.
Statistical analysis
All statistical analysis was performed using
GraphPad PRISM9 (GraphPad Software, Inc.; Dotmatics). Differences
between groups were analyzed using Welch's t test or one-way ANOVA
followed by Tukey's post hoc test. Data are presented as mean ± SD
of three or four experiments. P<0.05 was considered to indicate
a statistically significant difference.
Results
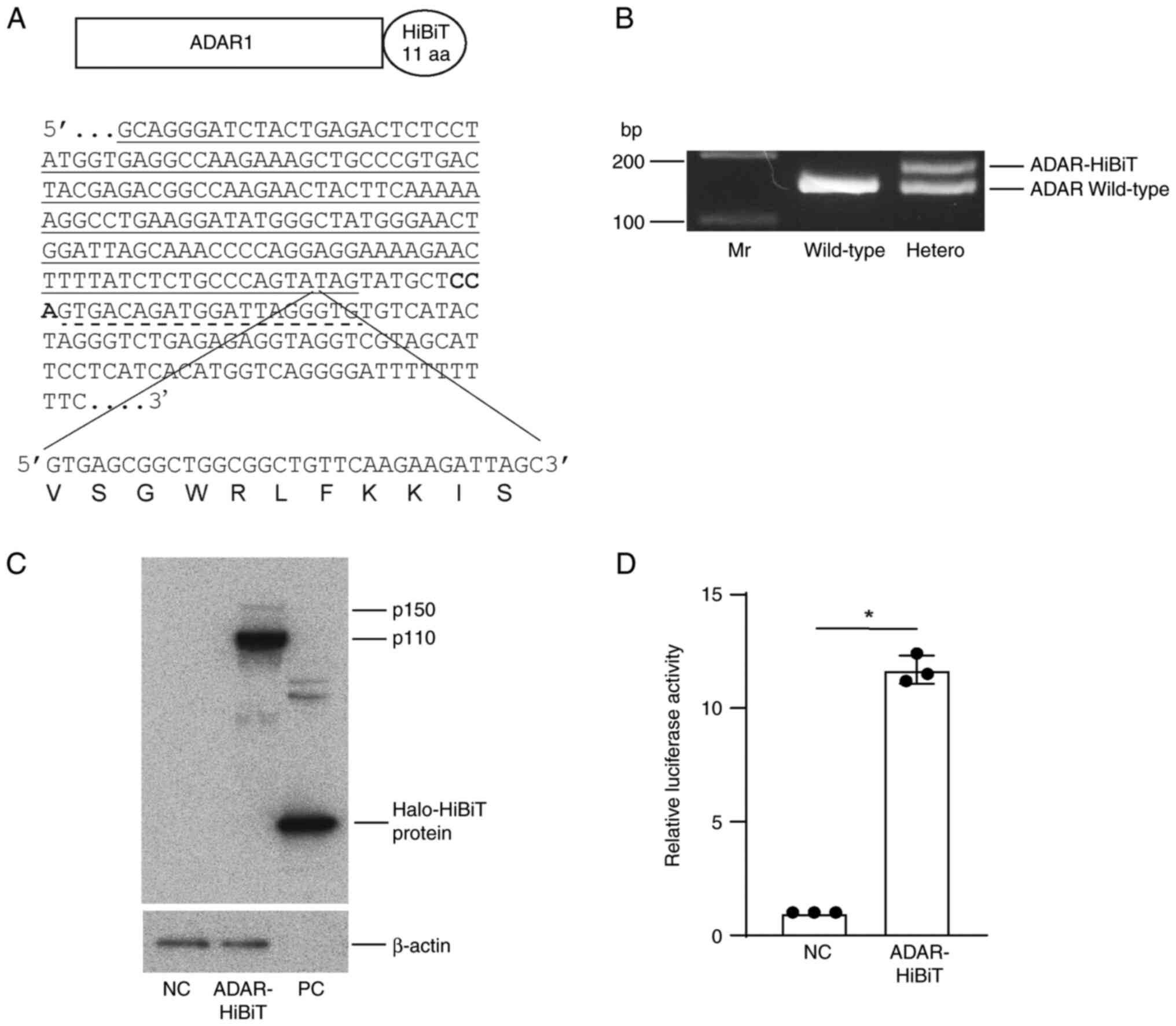
Establishment of HCC cells with HiBiT
peptide-conjugated ADAR1 protein
To monitor ADAR1 protein with high sensitivity, a
sequence corresponding to the 11-amino acid HiBiT peptide at the
C-terminus of the ADAR1 locus was knocked into the human HCC cell
line Huh7 by gene editing (Fig.
1A). Following limiting dilution and expansion, cells
heterozygous for the knocked-in sequence were successfully isolated
(Fig. 1B). While both ADAR1 p150
and p110 isoforms were detected by western blotting, p110 was the
main isoform of ADAR1 in Huh7 cells (Fig. 1C). Cellular lysates with ADAR1-HiBiT
protein expression showed significantly higher luciferase activity
when recombinant LgBiT protein was added, suggesting that HiBiT
peptides and LgBiT protein efficiently formed a complete luciferase
enzyme (Fig. 1D).
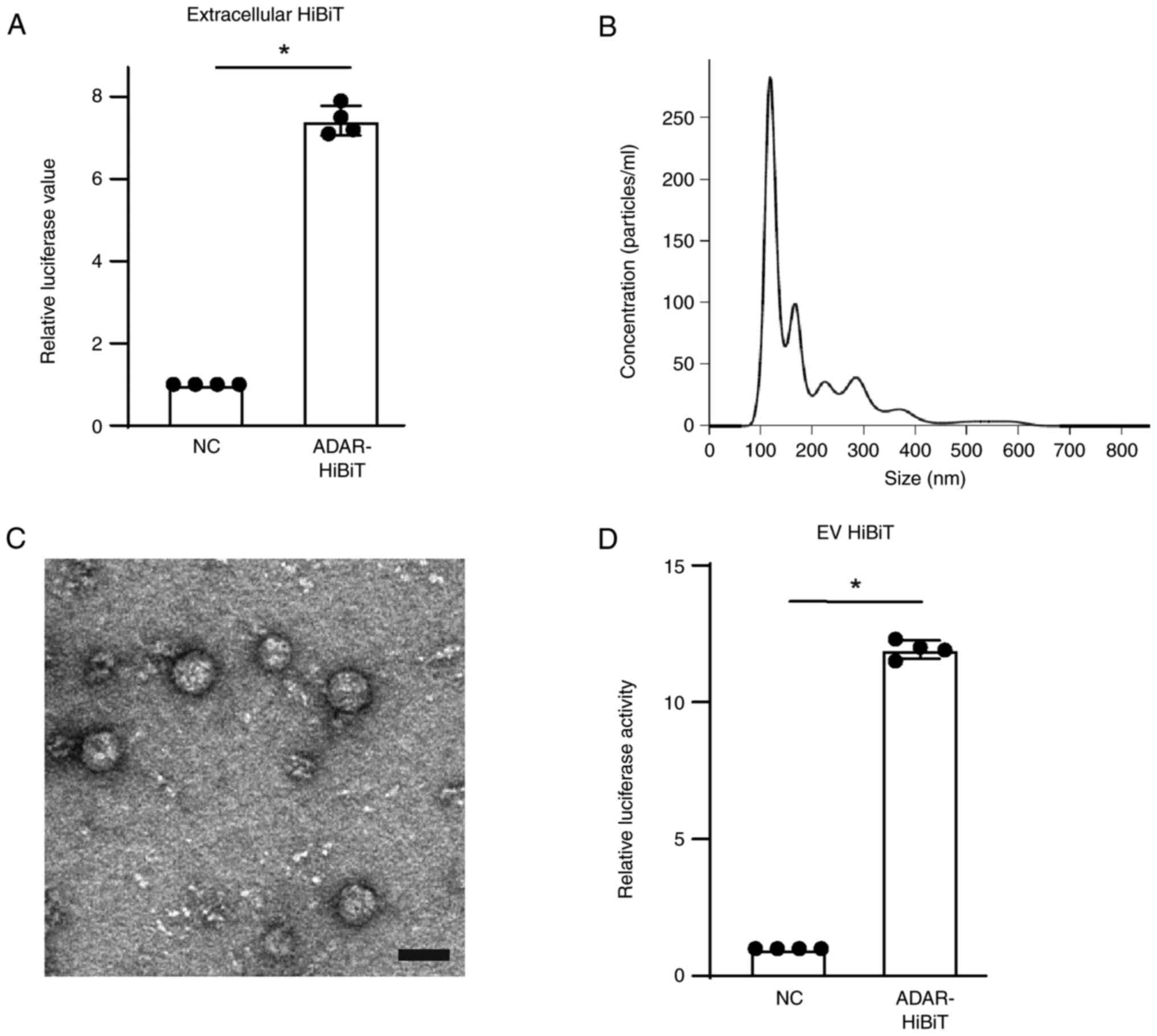
ADAR1 protein is included in EVs
To determine whether ADAR1 protein is released into
the extracellular environment, ADAR1-HiBiT in cell culture medium
was examined by mixing with recombinant LgBiT protein following
removal of cell debris. The luciferase activity was significantly
higher when using culture media of cells expressing ADAR1-HiBiT
(Fig. 2A), suggesting that ADAR1
protein was released from cells into the supernatant. As EVs carry
cellular components and are released from the cells (16), it was hypothesized that ADAR1 may be
included in EVs and actively released. EVs were isolated from cell
culture medium by size exclusion chromatography using qEV columns.
The size distribution of the EVs peaked at ~120 nm (Fig. 2B), corresponding to the size of
exosomes (6), and the concentration
was ~1.7×1011 particles/ml obtained from
~2×107 cells following 48 h culture. On electron
microscopy, the vesicles had the typical round morphology of EVs
(Fig. 2C). To examine whether these
EVs contained ADAR1-HiBiT, 1×1010 vesicles were lysed
and incubated with recombinant LgBiT protein, yielding
significantly higher luciferase values (Fig. 2D), suggesting that ADAR1-HiBiT was
included in the EVs released from Huh7 cells.
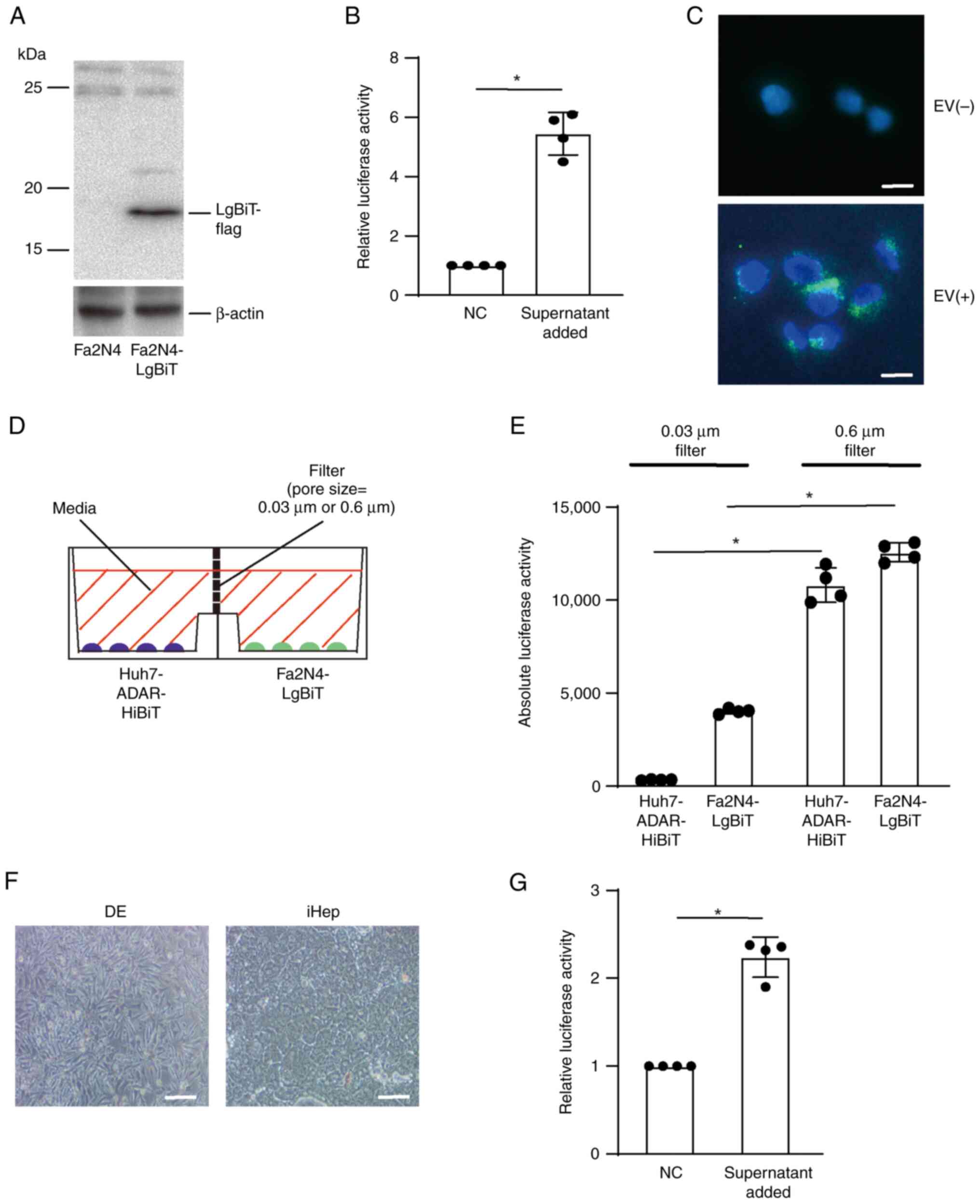
EVs containing ADAR1 are taken up by
neighboring cells
To examine whether the released EVs were taken by
neighboring cells, polyclonal immortalized human normal
hepatocytes, Fa2N4 cells, constitutively expressing LgBiT protein
were established (Fig. 3A). When
culture medium collected from Huh7-ADAR1-HiBiT cells was added to
Fa2N4-LgBiT cells, luciferase activities were significantly higher
than when using culture medium from control Huh7 cells (Fig. 3B), suggesting that the EVs
containing ADAR1-HiBiT were taken up by Fa2N4 cells. EVs captured
by Fa2N4 cells were mostly localized in the cytoplasmic and
perinuclear spaces (Fig. 3C). To
determine the transport of ADAR1 by EVs, horizontal co-culture
system with filters of two different pore sizes was used. As the
sizes of EVs from Huh7 cells were primarily 100–200 nm (Fig. 2B), pore sizes of 0.03 and 0.6 µm
were used to block and allow exchange, respectively, between
Huh7-ADAR1-HiBiT and Fa2N4-LgBiT cells (Fig. 3D). Compared with pore size of 30 nm,
luciferase activities from both Huh7 and Fa2N4 cells were
significantly higher when using 600 nm filter, suggesting that both
ADAR1-HiBiT and LgBiT proteins were exchanged after release from
the cells into the culture medium (Fig.
3E). To confirm the uptake of ADAR1-containing EVs from HCC
cells by normal human hepatocytes, normal human hepatocyte-like
cells induced from iPS cells were used (Figs. 3F and S1). After adding culture medium of
Huh7-ADAR1-HiBiT cells to normal hepatocyte-like cells, luciferase
activity was significantly increased when cell lysate was mixed
with recombinant LgBiT protein (Fig.
3G), suggesting that normal hepatocyte-like cells also took up
ADAR1-containing EVs from HCC cells. Similarly, cell lysate from
LgBiT-expressing Huh7 cells after adding the culture medium of
Huh7-ADAR1-HiBiT showed significantly increased luciferase activity
(Fig. S2), suggesting that ADAR1
released from HCC cells spread to neighboring HCC cells as well as
non-cancerous hepatocytes.
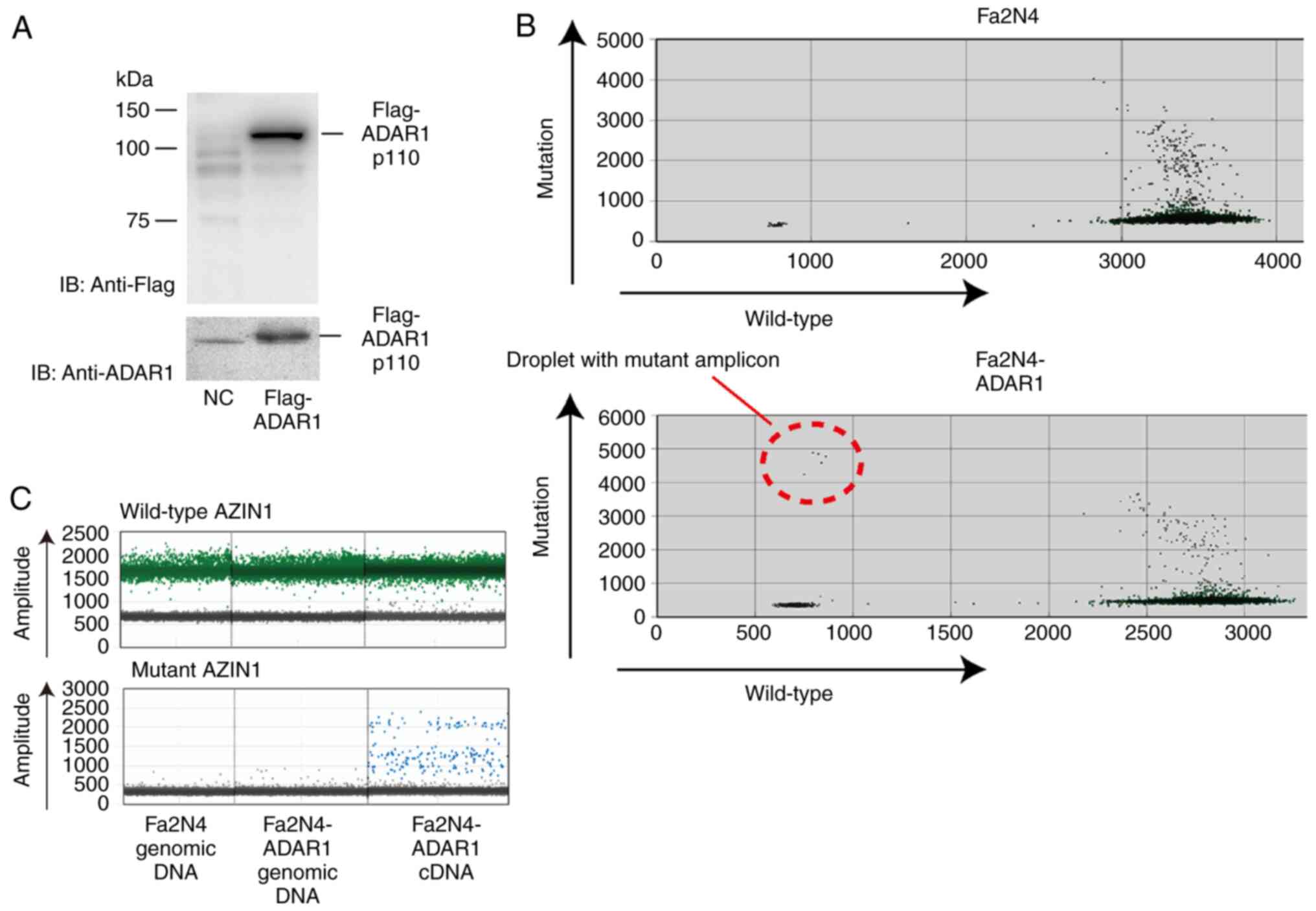
ADAR1 induces AZIN1 mutation
ADAR1 is an RNA editing enzyme that converts
adenosine residues to inosine in double-stranded RNA (7). Especially in the liver, editing at
residue 367 of AZIN1 RNA by ADAR1 is frequently linked to tumor
initiation and development (10,12–15).
To determine the outcome of the spread of ADAR1 to neighboring
non-cancerous hepatocytes from ADAR1-expressing HCC cells, the
present study established Fa2N4 cells constitutively expressing
ADAR1 p110 to mimic the surrounding non-cancerous hepatocytes after
taking up ADAR1 from neighboring cancerous cells (Fig. 4A). Mutation at residue 367 in AZIN1
RNA was detected in ADAR1-expressing cells, while this mutation was
not observed in the control Fa2N4 cells (Fig. 4B). Although this mutation was
detected in cDNA, it was not detected in the genomic DNA (Fig. 4C), suggesting that elevated ADAR1
levels in hepatocytes resulted in induction of the mutation in
AZIN1 RNA.
Heterogeneous expression of ADAR1 in
HCC tissue
According to the Protein Atlas Database (21), high expression levels of ADAR1
protein in HCC tissues result in poorer prognosis (Fig. 5A). To examine the expression status,
intracellular localization of ADAR1 was assessed in HCC and
surrounding non-cancerous tissues. Although nuclear staining was
prominent in HCC tissue, no nuclear staining was observed in the
surrounding non-cancerous tissue (Fig.
5B; Table SI). Cytoplasmic
staining was more intense in the surrounding non-cancerous liver
tissue close to the HCC lesions (Fig.
5C). In addition, nuclear and cytoplasmic staining were
heterogeneously observed within the same HCC nodule (Fig. 5D).
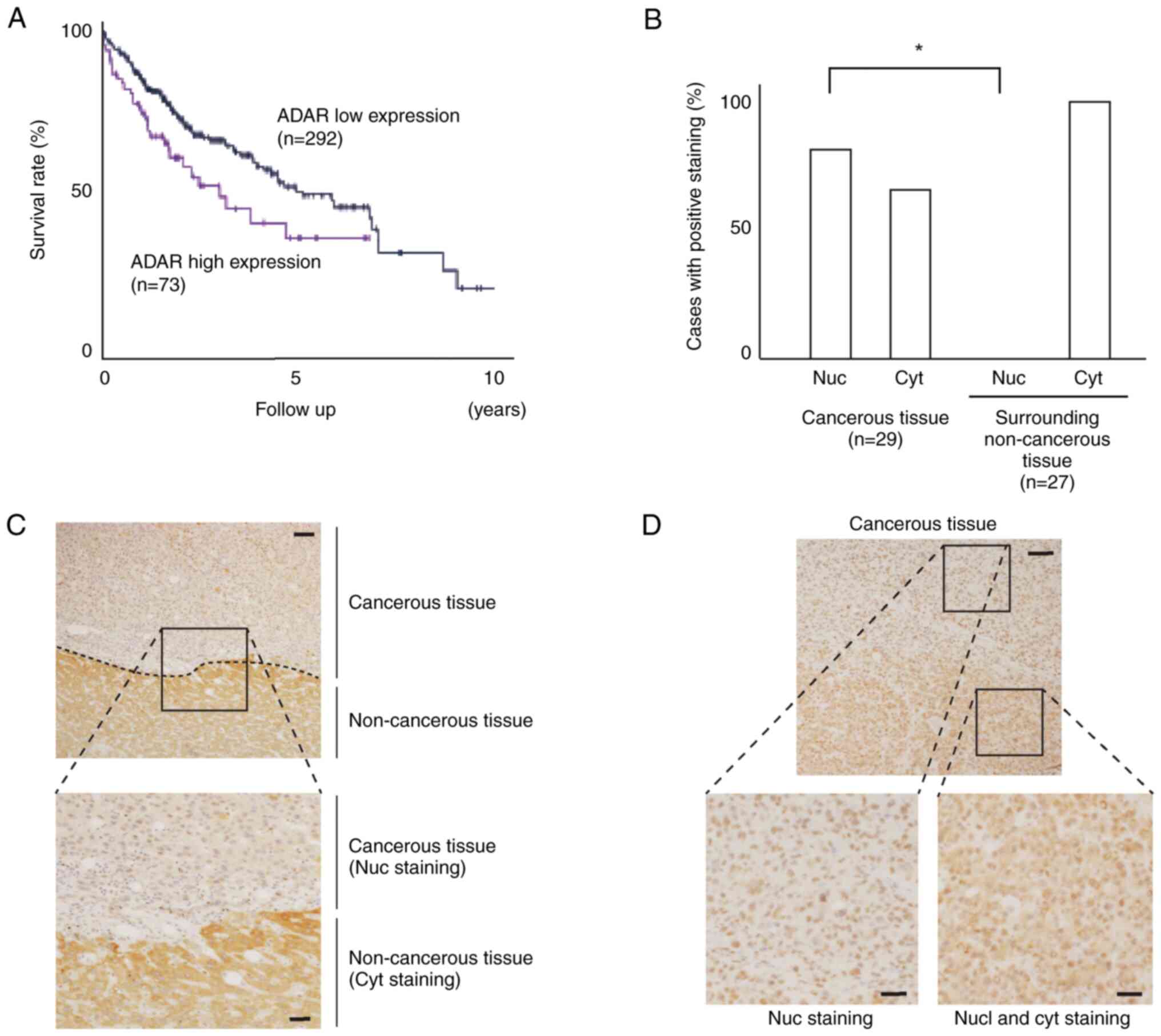 | Figure 5.Heterogeneous ADAR1 expression in
clinical HCC samples. (A) Prognostic survival curve of HCC cases
with high and low ADAR1 expression from the Protein Atlas database.
(B) Immunohistochemical staining of ADAR1 protein in clinically
resected human HCC and surrounding non-cancerous tissue. Among 30
cases examined, HCC tissue from one case and non-cancerous tissue
from three cases were excluded due to poor sample quality. While
both nuclear and cytoplasmic staining were observed in cancerous
tissue, no nuclear staining was observed in non-cancerous tissue.
*P<0.05. (C) Representative border of cancerous and
non-cancerous tissue. Although nuclear staining was seen in
cancerous tissues, cytoplasmic staining was predominant in
non-cancerous tissues and the expression levels decreased with
distance from the border. (D) Representative cancerous tissue.
While nuclear staining of ADAR1 was dominant, cytoplasmic staining
was detected in some parts within the cancerous lesions, resulting
in heterogeneous staining. Scale bar, 10 µm. ADAR, adenosine
deaminase acting on RNA-1; HCC, hepatocellular carcinoma; nuc,
nucleus; cyt, cytoplasm. |
Discussion
The present study showed that ADAR1 in HCC cells was
released in the EVs, which may be taken up by neighboring cells. As
ADAR1 induces RNA editing of genes, including oncogenic genes such
as AZIN1, in hepatocytes (10,23),
these phenomena may contribute to spread of HCC as well as to the
induction of heterogeneity in cancerous tissues, leading to poor
prognosis of HCC.
Studies have shown that EVs are associated with
numerous features of cancer (6,16). EVs
secreted by cancer cells transmit various types of cargo, such as
proteins, nucleic acids and small molecules, to neighboring cells,
resulting in the promotion of tumor growth and metastasis (6). Using a sensitive reporter system, the
present study found that ADAR1 protein was encapsulated in EVs
secreted from HCC cells and taken up by the surrounding normal
hepatocytes as well as neighboring cancer cells. The reporter
system established by gene editing insertion of HiBiT peptide into
the endogenous locus was a sensitive and useful method to monitor
the behavior of proteins in EVs.
The incorporation of cargo into EVs is a non-random
event (24). One mechanism by which
cargo is sorted into EVs involves tetraspanin-enriched
microdomains, which sequester RNA-binding proteins in the membrane
subdomains (24). As ADAR1 is an
RNA-binding protein (23,25), it is possible that ADAR1 protein is
sorted into EVs and released from HCC cells. Following
incorporation into EVs, ADAR1 released from HCC cells in EVs may be
taken up by neighboring HCC cells or surrounding non-cancerous
hepatocytes, as suggested by the present in vitro results as
well as distribution of positive immunohistochemical staining in
resected liver tissue. While ADAR1 has two isoforms, p110 and p150,
it remains undetermined which isoform plays key biological roles in
these contexts. Nonetheless, similar to the EV communication
observed in the present, it was recently reported that glioma cells
communicate with non-glioma cells, including glial cells, via EVs
(26). EVs released from cancer
cells may serve active biological roles in cancer survival and
spread (26).
According to the Protein Atlas data, higher
expression levels of ADAR1 are associated with poorer prognosis of
HCC, consistent with previous reports (9,10).
Although most RNA mutations are likely to be passengers, certain
editing events may serve as driver mutations, leading to cancer
progression. One such mutation occurs in AZIN1. A-to-I RNA editing
results in a serine to glycine conversion at residue 367 in AZIN1
protein (10). Compared with
wild-type AZIN1, mutant AZIN1 has greater antizyme binding
activity, inhibiting antizyme-mediated degradation of ornithine
decarboxylase, facilitating entry into the cell cycle and
increasing malignancy of cancer cells (10). Although this may represent one of
the mechanisms underlying poor prognosis of HCC, there are other
possible target genes of ADAR1 (27,28)
that may be involved in cancer progression, heterogeneity and
malignant behavior. There may also be indirect mechanisms, such as
suppression of anti-tumor immunity due to the decreased endogenous
non-coding dsRNA by RNA editing mediated by ADAR1 (29). Further studies are required to
elucidate the precise downstream mechanisms involved following the
increase in intracellular ADAR1 levels by transport via EVs.
Although it is possible that different ADAR1
isoforms were expressed and showed differential intracellular
localization, cytoplasmic ADAR1 protein in HCC lesions and
surrounding non-cancerous tissue may have been transported from
neighboring ADAR1-expressing HCC cells and retained in cytoplasm.
As RNA mutations induced by ADAR1 are associated with more
aggressive behavior of HCC (10),
these phenomena may contribute to worsening malignant biological
behavior of HCC and spread of cancerous lesions, resulting in
poorer prognosis.
In general, EVs released from cancer cells serve
crucial roles in cancer progression, such as priming metastatic
niches, regulating the tumor microenvironment and chemoresistance
(30). Although the present study
identified ADAR1 as one of the functional proteins in EVs released
from HCC cells, >4,400 proteins, RNA species and lipids have
been detected in exosomes (31).
Therefore, to understand the functional roles of EVs from cancer
cells, it is necessary to identify the cargo in EVs and determine
their functions in recipient cells. The HiBiT system used in the
present study may be useful in such studies because of its
convenience and high sensitivity.
Our previous study demonstrated that senescent cells
produce interferon (IFN)-associated genes without increased IFN
levels or exogenous IFN stimulation (32). As ADAR1 is an IFN-inducible gene,
ADAR1 expression may be increased in senescent cells, such as
stellate cells in the liver, with aging (33) or non-alcoholic steatohepatitis
(34). This may cause oncogenesis
in neighboring parenchymal hepatocytes due to the transfer of ADAR1
via EVs. Further studies are required to determine whether this
hypothesis can also explain other aging-associated disease.
Although the present study demonstrated that the
EV-mediated ADAR1 in HCC tissue may be the underlying cause of
tumor heterogeneity, there are limitations. First, the downstream
events after cells receive ADAR1 in the EVs were not fully
clarified. Second, the carcinogenesis processes after EV transfer
were not fully elucidated. Third, biological phenomenon in which
ADAR1 isoforms (p110 and p150) serve crucial roles were not
determined and require further investigation.
In summary, the present study suggested that ADAR1
in EVs released from HCC cells may serve a significant role in
cancer spread and heterogeneity. RNA mutations induced by
ADAR1-mediated RNA editing may be responsible for transition from
normal to malignant cells, as well as causing tumor heterogeneity
within HCC tissues, leading to poor prognosis. Further elucidation
of the mechanisms underlying transfer of ADAR1 into EVs and
downstream events may facilitate the development of novel methods
for prevention of HCC progression and improve its prognosis.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Ms Sayaka Ito (The
University of Tokyo, Tokyo, Japan) for technical assistance.
Funding
The present study was supported by Grants-in-Aid from the
Ministry of Education, Culture, Sports, Science and Technology,
Japan (grant nos. #20J20625, #22H02828, #22K15958 and #21H02893),
Japan Society of Technology Core Research for Evolutionary Science
and Technology (grant no. #JPMJCR19H5) and Research Program on
Hepatitis from Japan Agency for Medical Research and Development
(grant nos. JP23fk0210092 and JP23fk0310506).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CS, MO and MF designed the methodology and wrote the
manuscript. CS, TSe and TK performed experiments. TSh and TA
collected clinical samples and performed immunostaining. CS and MO
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
Surgically resected HCC samples were obtained from
the Department of Surgery, Dokkyo Medical University (Tochigi,
Japan). Written informed consent was obtained from all patients and
the study protocols were approved by the Ethics Committee of Dokkyo
Medical University (approval no. 28110).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EV
|
extracellular vesicle
|
|
HCC
|
hepatocellular carcinoma
|
|
AZIN1
|
adenosine to inosine RNA editing of
antizyme inhibitor 1
|
|
ADAR1
|
adenosine deaminase acting on
RNA-1
|
|
ddPCR
|
droplet digital PCR
|
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wolf E, Rich NE, Marrero JA, Parikh ND and
Singal AG: Use of hepatocellular carcinoma surveillance in patients
with cirrhosis: A systematic review and meta-analysis. Hepatology.
73:713–725. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chan LK, Tsui YM, Ho DW and Ng IO:
Cellular heterogeneity and plasticity in liver cancer. Semin Cancer
Biol. 82:134–149. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caruso S, O'Brien DR, Cleary SP, Roberts
LR and Zucman-Rossi J: Genetics of hepatocellular carcinoma:
Approaches to explore molecular diversity. Hepatology. 73 (Suppl
1):S14–S26. 2021. View Article : Google Scholar
|
|
5
|
Sun T, Yu Y, Wu X, Acevedo A, Luo JD, Wang
J, Schneider WM, Hurwitz B, Rosenberg BR, Chung H and Rice CM:
Decoupling expression and editing preferences of ADAR1 p150 and
p110 isoforms. Proc Natl Acad Sci USA. 118:e20217571182021.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu R, Rai A, Chen M, Suwakulsiri W,
Greening DW and Simpson RJ: Extracellular vesicles in
cancer-implications for future improvements in cancer care. Nat Rev
Clin Oncol. 15:617–638. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nishikura K: A-to-I editing of coding and
non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 17:83–96. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Paz-Yaacov N, Bazak L, Buchumenski I,
Porath HT, Danan-Gotthold M, Knisbacher BA, Eisenberg E and Levanon
EY: Elevated RNA editing activity is a major contributor to
transcriptomic diversity in tumors. Cell Rep. 13:267–276. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han L, Diao L, Yu S, Xu X, Li J, Zhang R,
Yang Y, Werner HMJ, Eterovic AK, Yuan Y, et al: The genomic
landscape and clinical relevance of A-to-I RNA editing in human
cancers. Cancer Cell. 28:515–528. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen L, Li Y, Lin CH, Chan TH, Chow RK,
Song Y, Liu M, Yuan YF, Fu L, Kong KL, et al: Recoding RNA editing
of AZIN1 predisposes to hepatocellular carcinoma. Nat Med.
19:209–216. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fritzell K, Xu LD, Lagergren J and Öhman
M: ADARs and editing: The role of A-to-I RNA modification in cancer
progression. Semin Cell Dev Biol. 79:123–130. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Okugawa Y, Toiyama Y, Shigeyasu K,
Yamamoto A, Shigemori T, Yin C, Ichikawa T, Yasuda H, Fujikawa H,
Yoshiyama S, et al: Enhanced AZIN1 RNA editing and overexpression
of its regulatory enzyme ADAR1 are important prognostic biomarkers
in gastric cancer. J Transl Med. 16:3662018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shigeyasu K, Okugawa Y, Toden S, Miyoshi
J, Toiyama Y, Nagasaka T, Takahashi N, Kusunoki M, Takayama T,
Yamada Y, et al: AZIN1 RNA editing confers cancer stemness and
enhances oncogenic potential in colorectal cancer. JCI Insight.
3:e999762018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qin YR, Qiao JJ, Chan TH, Zhu YH, Li FF,
Liu H, Fei J, Li Y, Guan XY and Chen L: Adenosine-to-inosine RNA
editing mediated by ADARs in esophageal squamous cell carcinoma.
Cancer Res. 74:840–851. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Peng X, Xu X, Wang Y, Hawke DH, Yu S, Han
L, Zhou Z, Mojumdar K, Jeong KJ, Labrie M, et al: A-to-I RNA
editing contributes to proteomic diversity in cancer. Cancer Cell.
33:817–828.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kalluri R and LeBleu VS: The biology,
function, and biomecial applications of exosomes. Science.
367:eaau69772020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sekiba K, Otsuka M, Ohno M, Yamagami M,
Kishikawa T, Suzuki T, Ishibashi R, Seimiya T, Tanaka E and Koike
K: Inhibition of HBV transcription from cccDNA with nitazoxanide by
targeting the HBx-DDB1 interaction. Cell Mol Gastroenterol Hepatol.
7:297–312. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oishi M, Munesue S, Harashima A, Nakada M,
Yamamoto Y and Hayashi Y: Aquaporin 1 elicits cell motility and
coordinates vascular bed formation by downregulating thrombospondin
type-1 domain-containing 7A in glioblastoma. Cancer Med.
9:3904–3917. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kishikawa T, Otsuka M, Yoshikawa T, Ohno
M, Yamamoto K, Yamamoto N, Kotani A and Koike K: Quantitation of
circulating satellite RNAs in pancreatic cancer patients. JCI
Insight. 1:e866462016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Uhlen M, Zhang C, Lee S, Sjöstedt E,
Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et
al: A pathology atlas of the human cancer transcriptome. Science.
357:eaan2502017. View Article : Google Scholar
|
|
22
|
Shimizu T, Aoki T, Mori S, Iso Y, Kato M,
Ishizuka M and Kubota K: Tumor DNA-dependent protein kinase
catalytic subunit expression is associated with hepatitis B surface
antigen status and tumor progression in patients with
hepatocellular carcinoma. Sci Rep. 8:150192018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chan TH, Lin CH, Qi L, Fei J, Li Y, Yong
KJ, Liu M, Song Y, Chow RK, Ng VH, et al: A disrupted RNA editing
balance mediated by ADARs (Adenosine DeAminases that act on RNA) in
human hepatocellular carcinoma. Gut. 63:832–843. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Perez-Hernandez D, Gutiérrez-Vázquez C,
Jorge I, López-Martín S, Ursa A, Sánchez-Madrid F, Vázquez J and
Yáñez-Mó M: The intracellular interactome of tetraspanin-enriched
microdomains reveals their function as sorting machineries toward
exosomes. J Biol Chem. 288:11649–11661. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi L, Yan P, Liang Y, Sun Y, Shen J, Zhou
S, Lin H, Liang X and Cai X: Circular RNA expression is suppressed
by androgen receptor (AR)-regulated adenosine deaminase that acts
on RNA (ADAR1) in human hepatocellular carcinoma. Cell Death Dis.
8:e31712017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao X, Zhang Z, Mashimo T, Shen B, Nyagilo
J, Wang H, Wang Y, Liu Z, Mulgaonkar A, Hu XL, et al: Gliomas
interact with non-glioma brain cells via extracellular vesicles.
Cell Rep. 30:2489–2500.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang H, Chen S, Wei J, Song G and Zhao Y:
A-to-I RNA editing in cancer: From evaluating the editing level to
exploring the editing effects. Front Oncol. 10:6321872020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tan MH, Li Q, Shanmugam R, Piskol R,
Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et
al: Dynamic landscape and regulation of RNA editing in mammals.
Nature. 550:249–254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Samuel CE: Adenosine deaminase acting on
RNA (ADAR1), a suppressor of double-stranded RNA-triggered innate
immune responses. J Biol Chem. 294:1710–1720. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kosaka N, Yoshioka Y, Fujita Y and Ochiya
T: Versatile roles of extracellular vesicles in cancer. J Clin
Invest. 126:1163–1172. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mathivanan S, Fahner CJ, Reid GE and
Simpson RJ: ExoCarta 2012: Database of exosomal proteins, RNA and
lipids. Nucleic Acids Res. 40:(Database Issue). D1241–D1244. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yamagami M, Otsuka M, Kishikawa T, Sekiba
K, Seimiya T, Tanaka E, Suzuki T, Ishibashi R, Ohno M and Koike K:
ISGF3 with reduced phosphorylation is associated with constitutive
expression of interferon-induced genes in aging cells. NPJ Aging
Mech Dis. 4:112018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maeso-Díaz R, Ortega-Ribera M,
Fernández-Iglesias A, Hide D, Muñoz L, Hessheimer AJ, Vila S,
Francés R, Fondevila C, Albillos A, et al: Effects of aging on
liver microcirculatory function and sinusoidal phenotype. Aging
Cell. 17:e128292018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yoshimoto S, Loo TM, Atarashi K, Kanda H,
Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et
al: Obesity-induced gut microbial metabolite promotes liver cancer
through senescence secretome. Nature. 499:97–101. 2013. View Article : Google Scholar : PubMed/NCBI
|