Introduction
Glioma is a malignant tumor that develops from glial
cells and accounts for ~10% of all primary brain tumors.
Glioblastoma (GBM) is the most aggressive type of glioma, with an
extremely poor prognosis (1).
Several treatments, including surgery, radiation therapy, and
chemotherapy (temozolomide), are currently available for GBM
(2). The combination of radiation
therapy and chemotherapy (temozolomide) is the standard treatment
that prolongs the survival and improves the quality of life of
patients with GBM (2). However, the
advantages of standard treatment are limited as recurrence is
observed in most patients with GBM within one year of treatment,
and their five-year survival rate is only 7.2% (3). GBM is a highly proliferative and
invasive disease (4,5), and therapeutic resistance to radiation
therapy and temozolomide critically contributes to the poor
prognosis of affected patients (6,7).
Temozolomide resistance is a major cause of GBM treatment failure
(8). GBM stem-like cells (GSCs)
play key roles in the therapeutic resistance of patients with GBM
(9–11). As cancer stem cells are
undifferentiated and self-replicating malignant cells that act as a
source of cancer cells (9,10), conventional treatment with
temozolomide is insufficient to inhibit GSC functions (11). Therefore, development of novel
alternative treatments that suppress the malignant characteristics
of GBM, including proliferation, metastasis, and GSC function, is
necessary for the prognostic improvement of affected patients.
However, the detailed molecular pathogenesis and key molecules
regulating the malignant transformation of cells in GBM remain
unknown.
Cylindromatosis (CYLD), a tumor suppressor gene, was
originally discovered as a causative gene in familial
cylindromatosis (12). CYLD serves
as a deubiquitinating enzyme that cleaves the Lys63-bound ubiquitin
chain to regulate various cell signaling pathways (13). Previous studies have revealed that
CYLD negatively controlled the activation of various signaling
pathways, such as nuclear factor kappa-light-chain-enhancer of
activated B cells (NF-κB) (14),
transforming growth factor (TGF)-β (15,16),
c-Jun N-terminal kinase (JNK) (17), and tumor necrosis factor (TNF)
receptor-associated factor 2-p38 mitogen-activated protein kinase
(TRAF2-p38MAPK) signaling (18),
and that CYLD played important roles in various physiological
processes, including immune response, inflammation, and the cell
cycle, through regulating those signaling pathways (13,15,19–25).
It has been reported that there is a strong association between
excessive activation of signaling pathways, and the loss of CYLD
function by its downregulation, in various tumors (26,27).
CYLD downregulation was revealed to promote chemoresistance and
invasion via NF-κB and TGF-β signaling in oral squamous cell
carcinoma (16,28). Moreover, CYLD downregulation was
demonstrated to be significantly associated with the malignant
characteristics and poor prognosis of breast cancer (29). CYLD downregulation was also
correlated with worsening grade and poor prognosis in CYLD-negative
patients with GBM (30).
Pathological analysis of GBM tissues has revealed that CYLD
expression is reduced under hypoxic conditions, which is closely
related to various malignant transformations, and that CYLD
downregulation is involved in resistance to angiogenesis inhibitors
(30). Despite the numerous studies
strongly suggesting CYLD downregulation as a crucial factor
responsible for poor prognosis, effective therapeutic targets and
agents are still not available for CYLD-negative patients with
GBM.
In the present study, CYLD knockdown in GBM was
investigated to determine the molecular pathological mechanism of
malignant transformation in CYLD-negative GBM cells and identify
the crucial cell signaling pathways responsible for CYLD
downregulation-dependent malignant transformation using
comprehensive proteomic analysis.
Materials and methods
Antibodies and reagents
Wnt/β-catenin signaling inhibitor (ICG-001) was
purchased from Selleck Chemicals. All other reagents were of the
commercially available grade.
Cell lines and culture
Human GBM cell line (U251MG) was obtained from the
Japanese Collection of Research Bio Resources Cell Bank. Cells were
cultured in the Dulbecco's modified Eagle's medium (DMEM) with 10%
heat-inactivated fetal bovine serum (FBS; both from Thermo Fisher
Scientific, Inc.) at 5% CO2 and 37°C.
Transfection with small interfering
RNA (siRNA) and plasmid DNA
For siRNA transfection, U215MG cells were incubated
in a six-well plate (2×105 cells/well) or 24-well plate
(1×104 cells/well) for 24 h and transiently transfected
with siRNA (20 nM) using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 48 h,
according to the manufacturer's protocol. Following transfection of
the cells and incubation for 48 h, the experiments were performed.
Silencer Negative Control siRNA (cat. no. AM4636; Ambion/Applied
Biosystems; Thermo Fisher Scientific, Inc.) was used as a control
(siN, https://genesdev.cshlp.org/content/suppl/2019/06/04/gad.324814.119.DC1/Supplemental_methods.pdf)
(31). Sequences of the siRNAs
targeting CYLD (siCYLD) were sense, 5′-GAUUGUUACUUCUAUCAAAtt-3′ and
antisense, 5′-UUUGAUAGAAGUAACAAUCtt-3′.
For plasmid DNA transfection, U251MG cells were
incubated in a 12-well plate (1.6×105 cells/well;
Corning, Inc.) at 37°C for 24 h and transiently transfected at 37°C
for 48 h with the control vector (pcDNA3) or wild-type CYLD
expression plasmid (pcDNA3) (500 ng/well) (15,32)
using Lipofectamine® 2000, according to the
manufacturers' protocol. The efficacy of transfections was
confirmed in the present study (Fig.
S1).
Cell viability assay
After 72–120 h of transfection, 50 µl/well of the
Cell Counting Kit-8 solution (Dojindo Laboratories, Inc.) was added
to the cells and incubated at 37°C for 2 h, and the absorbance at
450 nm was measured using EMax SOFTmaxPRO (Molecular Devices, LLC).
To evaluate the effect of ICG-001 on cell viability, cells were
treated with ICG-001 (0–50 µM) and control reagent [dimethyl
sulfoxide (DMSO)] in serum-free DMEM after transfection at 37°C,
and the absorbance 72 h after treatment was measured.
Transwell migration assay
Following transfection in a six-well plate, U251MG
cells were recovered by trypsin addition and suspended in
serum-free DMEM, and reseeded (1.0×104 cells/well) in
the upper part of an 8-µm Transwell insert (Corning, Inc.) with a
total volume of 200 µl/well. Cell migration was induced using DMEM
containing 10% heat-inactivated FBS at a total volume of 600
µl/well. To evaluate the effect of ICG-001, ICG-001 (50 µM) or
control (DMSO) with serum-free DMEM was added to the upper
Transwell plate. After incubation at 37°C for 24 h, the migrating
cells were stained with crystal violet at room temperature for 20
min, and the ratio of the stained area observed by light microscope
was quantified using the ImageJ software (version 1.51j8; National
Institutes of Health).
Sphere-formation assay
Following transfection in a six-well plate, U251MG
cells were recovered by trypsin addition and suspended in
serum-free neural stem cell (NSC) medium, and reseeded
(1.0×104 cells/well) in an ultra-low adhesion 96-well
plate (Corning, Inc.). NSC medium including serum-free DMEM/F12,
human leukemia thinner (Sigma Aldrich; Merck KGaA), human basic
fibroblast growth factor (human FGF-basic; PeproTech, Inc.), human
epithelial cell growth factor (human EGF; PeproTech.Inc.), heparin
(Sigma Aldrich; Merck KGaA), insulin (Sigma Aldrich; Merck KGaA),
N2 (Gibco), B27 (Gibco), GlutaMax (Gibco), and
penicillin/streptomycin was used, as previously described (33,34).
Cells with masses ≥50 µm observed by light microscope were counted
as spheres.
Limiting dilution assay
Following transfection in a six-well plate, U251MG
cells were recovered by trypsin addition, suspended in the NSC
medium, and reseeded (1,500-2,000 cells/well) in the ultra-low
adhesion 96-well plate (Corning, Inc.) using the serial dilution
method. To evaluate the effect of ICG-001, after 24 h of incubation
at 37°C in NSC medium, ICG-001 (10 µM) or control (DMSO) was added
to the cells, cell masses ≥50 µm were counted as spheres, and the
proportion of wells in which the spheres were formed was calculated
to be the percentage/4 wells after 72 h.
GBM database analysis
Patient clinical data (RNA-sequencing data of 270
samples) were obtained from a previous study (35), and RNA sequencing (RNA-seq) data
were obtained from the Ivy Glioblastoma Atlas Project (GAP)
database made publicly available by the Allen Institute (2015 Allen
Institute for Brain Science, Ivy Glioblastoma Atlas Project 2;
http://glioblastoma.alleninstitute.org/). The Ivy
Glioblastoma Atlas Project is a collaborative partnership between
the Ben and Catherine Ivy Foundation, Allen Institute for Brain
Science, and Ben and Catherine Ivy Center for Advanced Brain Tumor
Treatment. The aforementioned study provides a foundation for GBM
research (35). The Ivy
Glioblastoma Atlas Project provided RNA-seq data to study the gene
expression patterns of anatomical structures (identified by
hematoxylin and eosin staining) and cancer stem cell clusters
(identified by ISH investigation) in GBM with 270 samples in both
analyses combined. The expression levels of CYLD were compared with
each marker or signaling-activating factor (Ki-67, fibronectin,
nestin, CK2β, and CKIε) in these samples. Correlation coefficient
(r) was calculated, and | r | ≥0.2 indicated a correlation.
Proteomic analysis using liquid
chromatograph-tandem mass spectrometry (LC-MS/MS)
Whole cell lysates of U251MG cells were prepared
using the phase transfer surfactant (PTS) method, as previously
described (36,37). Sodium deoxycholate (SDC), sodium
N-lauroylsarcosinate (SLS), ammonium bicarbonate, dithiothreitol,
iodoacetamide, mass spectrometry grade lysyl endoprotease, ethyl
acetate, acetonitrile, acetic acid, methanol, trifluoroacetic acid
(FUJIFILM Wako Pure Chemical Corporation), modified trypsin
(Promega Corporation), and 4-(2-aminoethyl) benzenesulfonyl
fluoride hydrochloride (Nacalai Tesque, Inc.) were used in this
study. Proteins were extracted using the PTS solution [12 mM SDC,
12 mM SLS, and 100 mM Tris-HCl (Ph 9.0)] and ultrasonically crushed
for 20 min. After incubating for 5 min at 95°C, proteins in the
supernatant solution were quantified using the BCA method with the
BCA Protein Assay Kit (Thermo Fisher Scientific, Inc.). The
proteins were reduced with 10 mM dithiothreitol for 30 min and
alkylated with 50 mM iodoacetamide in the dark for 30 min at room
temperature. The protein mixture was 2-fold diluted with 50 mM
ammonium bicarbonate, digested with Lys-C (1/50 sample weight) at
room temperature for 3 h prior to the addition of trypsin (1/50
sample weight), and incubated at 37°C for 20 h. An equal volume of
ethyl acetate was added to the sample solution, and the mixture was
acidified with 0.5% trifluoroacetic acid (final concentration). The
mixture was shaken for 2 min and centrifuged at 15,600 × g for 2
min at room temperature. The upper layer was removed using a
pipette and dried using a vacuum evaporator. The sample was then
suspended in 100 µl buffer A (5% acetonitrile, 0.1% TFA) and
desalted with GL-Tip SDB (GL Sciences, Inc.) and ODS-A-HG AAG12S50
(YMC CO., LTD.), as previously described (38,39).
Peptides were eluted with buffer B (80% acetonitrile and 0.1% TFA).
To concentrate the phosphorylated proteins, the Titasphere Phos-TiO
Kit (GL Sciences, Inc.), was used according to the manufacturer's
protocol. Subsequently, a 5% pyrrolidine solution was used for
elution, as previously described (40). The eluted fraction was acidified
with TFA, desalted using GL-Tip SDB, and concentrated in a vacuum
evaporator. TripleTOF 5600 (SCIEX) equipped with Dionex Ultimate
3000 RSLS (Thermo Fisher Scientific, Inc.) was used for nano
LC-MS/MS measurements. The injection volume was 5 µl, and the flow
rate was 300 ml/min. A nanotrap column (100 µm ID, 2-cm length,
packed with 5 µm Acclaim PepMap100C18; Thermo Fisher Scientific,
Inc.) and an analytical nanocolumn (75 µm ID, 25-cm length, packed
with 2 µm Acclaim PepMap C18; Thermo Fisher Scientific, Inc.) were
used. MS data were acquired using Analyst Software TF 1.7 (SCIEX).
For peptide identification, data were acquired in a data-dependent
acquisition mode and analyzed using ProteinPilot 4.5 (SCIEX)
connected to the UniProt human reference proteome database (Release
2017_11). Phosphorylation was set as the ‘Special Factor’ in the
sample description. The protein identification confidence for the
dataset was evaluated based on the false-discovery rate. For
protein and peptide quantification, data were acquired in a
data-independent acquisition mode (sequential window acquisition of
all theoretical fragment ion spectra; SWATH-MS) with a variable
window for the precursor ions. The proteomic datasets have been
submitted to jPOSTrepo
[https://repository.jpostdb.org/preview/187456970364aca64292d78;
Access key: 4555; Accession no. JPST002236 (PXD043537)].
Proteomic data analysis
The results were analyzed using Kinase Enrichment
Analysis 2 (https://www.maayanlab.net/KEA2/). By setting the
library as ‘Literature Based Kinase-Substrate Library with
Phosphosites’ and analyzing it using Kinase Enrichment Analysis 2,
the combination of protein and phosphorylation sites was clarified.
In addition, the kinases that interacted predominantly with these
proteins were inferred and mapped. The results were visualized
based on the Wnt/β-catenin signaling pathway-Homo sapiens
(humans) in the Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway.
Statistical analysis
Unpaired Student's t-test was used to evaluate the
differences between the two groups. Statistical analysis was
performed using Statcel (ver. 4; OMS Publishing Co., Ltd.). Data
are presented as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
Involvement of CYLD knockdown in the
proliferation and migration of GBM cells
To develop clinically effective treatments for
CYLD-negative patients with GBM, first the involvement of CYLD
downregulation in the malignant characteristics of GBM cells was
determined. As revealed in Fig. 1A,
GBM cell proliferation was significantly promoted in CYLD-silenced
GBM cells. Consistently, RNA-seq data obtained from the Ivy GAP
database further revealed that, in the tissues of patients with
GBM, CYLD expression was negatively correlated with the expression
of Ki-67, a proliferation marker in GBM tissues (Fig. 1B). Next, a Transwell migration assay
was performed to evaluate GBM cell migration, an
epithelial-mesenchymal transition (EMT)-like change. CYLD knockdown
significantly enhanced GBM cell migration (Fig. 1C), whereas CYLD overexpression
markedly suppressed this effect (Fig.
1D). The Ivy GAP database further revealed a significant
negative correlation between the expression levels of CYLD and
fibronectin, a mesenchymal marker, in the infiltrating area of GBM
tissues (Fig. 1E), indicating that
CYLD downregulation enhanced the EMT-like changes and infiltration
in GBM. These results indicated the promotion of cell proliferation
and migration in CYLD-silenced GBM cells.
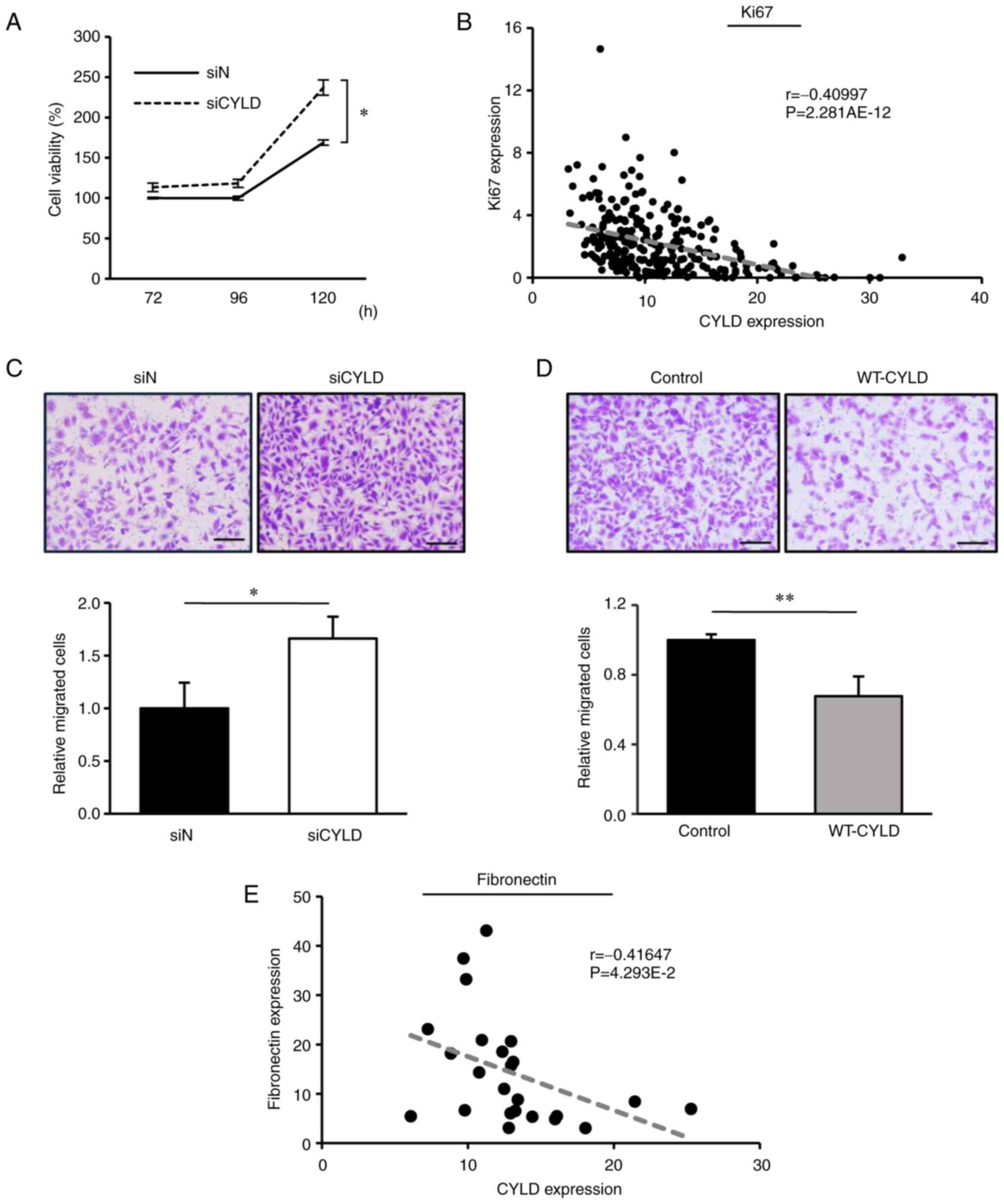 | Figure 1.Involvement of CYLD knockdown in cell
proliferation and migration of GBM cells. (A) Human GBM (U215MG)
cells were transfected with the control siRNA (siN) or
CYLD-specific siRNA (siCYLD), and cell viability was assessed
72–120 h after transfection. Values are expressed as the mean ± SD
of triplicate samples. *P<0.05 vs. the siN group, via Student's
t-test. (B) RNA-seq data of 270 samples using the Ivy GAP database
revealed a correlation between CYLD and Ki-67 expression. The
correlation coefficient is r, and the P-value indicates the
significance of the correlation coefficient. (C and D) Transwell
migration assays were performed using (C) CYLD-silenced and (D)
CYLD-overexpressed cells. Cells were transfected, reseeded into the
Transwell insert for 24 h, and migrating cells were stained with
crystal violet. Scale bars, 500 µm. Values are expressed as the
means ± SD. of triplicate samples. *P<0.05 and **P<0.01 vs.
siN or the control group via Student's t-test. (E) RNA-seq data of
24 samples from the Ivy GAP database revealed a correlation between
CYLD and fibronectin expression in the infiltrating area of GBM
tissues. CYLD, cylindromatosis; GBM, glioblastoma; siRNA or si,
small interfering RNA; SD, standard deviation; RNA-seq, RNA
sequencing; GAP, Glioblastoma Atlas Project; WT, wild-type. |
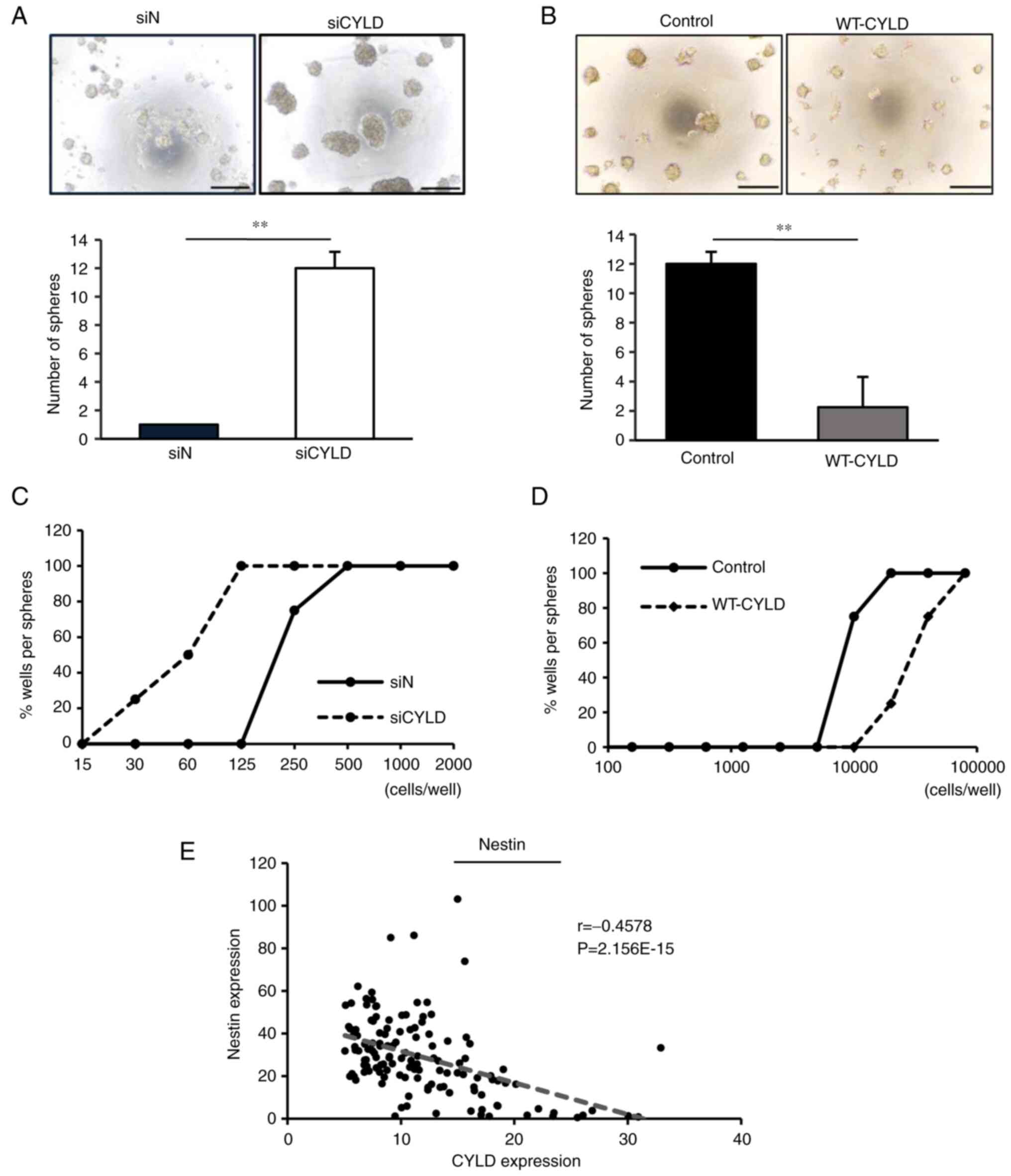
CYLD knockdown induces stem cell-like
characteristics in glioma cells
As cancer stem cell-like cells critically contribute
to malignancy in patients with GBM (41), the effects of CYLD knockdown on stem
cell-like characteristics were determined in GBM cells. The
sphere-forming ability, a characteristic of cancer stem cell-like
cells, was evaluated. Notably, CYLD knockdown significantly
increased the sphere-forming ability of GBM cells (Fig. 2A), whereas CYLD overexpression
markedly suppressed this effect (Fig.
2B).
In the limiting dilution assay, CYLD-knockdown GBM
cells were able to form spheres with lower cell numbers than the
control GBM cells (Fig. 2C),
whereas CYLD-overexpressed GBM cells required more cells (Fig. 2D). Moreover, RNA-seq data obtained
from the Ivy GAP database further revealed that CYLD expression was
negatively correlated with the expression of nestin, a cancer stem
cell marker, in GBM tissues (Fig.
2E), suggesting that GBM cells acquire stem-like
characteristics via CYLD knockdown.
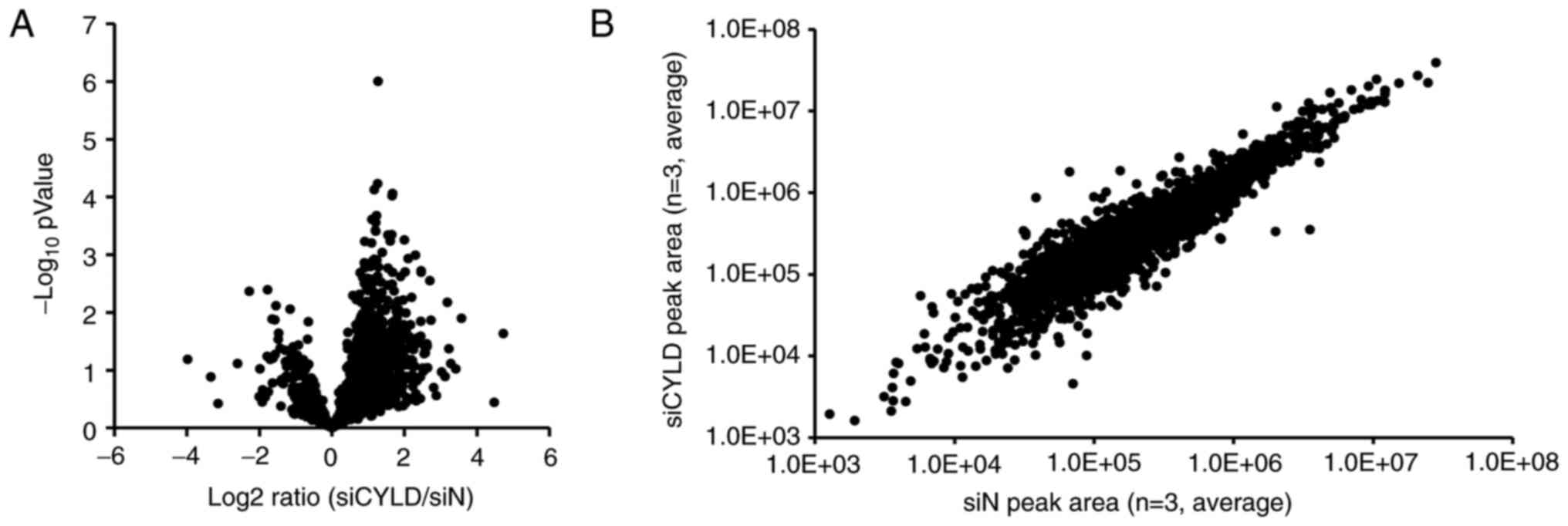
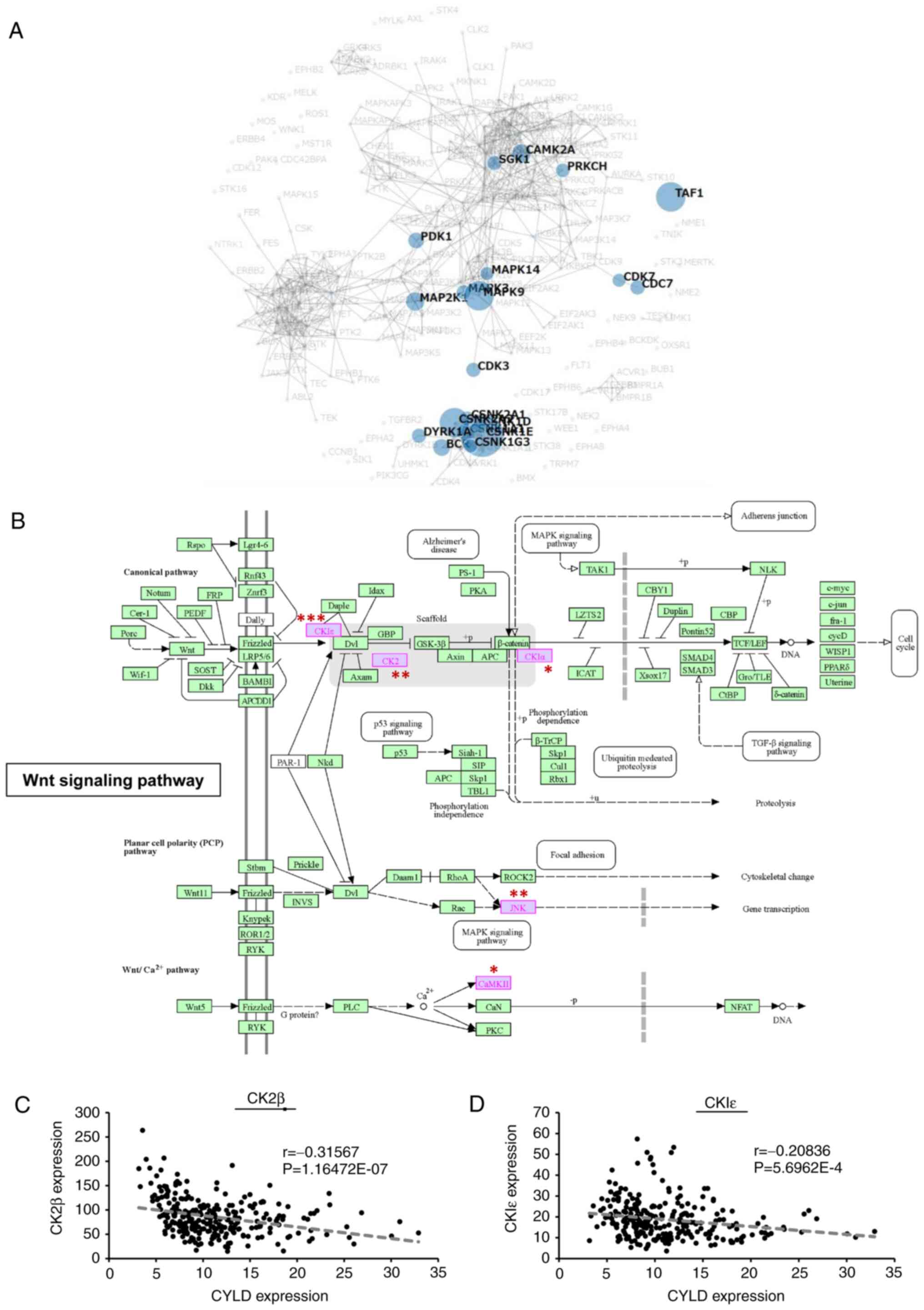
Activation of Wnt/β-catenin signaling
is identified in CYLD-knockdown GBM cells
To identify novel therapeutic target signals in
CYLD-knockdown cells, a proteomic analysis was performed to
comprehensively identify the intracellular signaling pathways
involved in the malignant characteristics caused by CYLD
downregulation. In total 2,914 phosphorylated proteins were
identified, among which, 337 proteins were phosphorylated more than
double due to CYLD downregulation (Fig.
3A and B). Kinase Enrichment Analysis 2 was used to analyze the
337 proteins. It revealed 44 combinations of proteins and
phosphorylation sites, and the identified proteins were likely to
interact with 20 kinases (Table I
and Fig. 4A). Among the 20 kinases,
9 kinases exhibited significant differences, and the majority of
them (CKIε, CK2, MAPK9, CAMK2A, and CKIα) were involved in
Wnt/β-catenin signaling (Fig. 4B).
RNA-seq data from the Ivy GAP database further confirmed that CYLD
expression was negatively correlated with the expression of CK2β
and CKIε, Wnt/β-catenin signaling activators, in GBM tissues
(Fig. 4C and D). These results
indicated that Wnt/β-catenin signaling plays key roles in the
malignancy of cells via CYLD knockdown.
 | Table I.Twenty kinases identified by Kinase
Enrichment Analysis 2. |
Table I.
Twenty kinases identified by Kinase
Enrichment Analysis 2.
| Node name | Original
P-value | Total genes in gene
set | Total genes
intersected | Intersecting
genes |
|---|
| CSNK1E | 8.27814E-05 | 186 | 7 | YWHAQ_S232 | EIF4B_S597 | PKP3_S238 | DDX21_S171 | GTF2A1_S316 | SMN1_S28 | SRRM2_S1326 |
|
| TAF1 | 0.001325212 | 8 | 2 | GTF2A1_S316 | GTF2A1_S321 |
|
|
|
|
|
|
| CSNK2A2 | 0.001947237 | 412 | 8 | GTF2A1_S316 | GTF2A1_S321 | HNRNPC_S260 | EIF4B_S597 | RPLP1_S104 | RPLP1_S101 | MCRS1_S282 | DDX46_S804 |
| MAPK9 | 0.002454397 | 166 | 5 | CTTN_T401 | CTTN_S405 | CTTN_S418 | SLC9A1_S726 | EIF3G_S42 |
|
|
|
| CAMK2A | 0.019347493 | 100 | 3 | EGFR_S1071 | EGFR_S1081 | EGFR_S1166 |
|
|
|
|
|
| MAP2K1 | 0.019702853 | 37 | 2 | CTTN_S405 | CTTN_S418 |
|
|
|
|
|
|
| CSNK1A1 | 0.027572079 | 115 | 3 | YWHAQ_S232 | HNRNPC_S260 | HNRNPC_S253 |
|
|
|
|
|
| BCR | 0.032953797 | 5 | 1 | YWHAQ_S232 |
|
|
|
|
|
|
|
| PDK1 | 0.043737813 | 58 | 2 | PDPK1_S241 | PRKACA_T198 |
|
|
|
|
|
|
| CDC7 | 0.059602559 | 10 | 1 | MCM2_S108 |
|
|
|
|
|
|
|
| CDK3 | 0.070059921 | 12 | 1 | YLPM1_S634 |
|
|
|
|
|
|
|
| SGK1 | 0.074449031 | 79 | 2 | DNAJC5_S10 | EBAG9_S36 |
|
|
|
|
|
|
| DYRK1A | 0.080403609 | 14 | 1 | CCNL2_S330 |
|
|
|
|
|
|
|
| MAPK3 | 0.089542329 | 188 | 3 | EGFR_T693 | CTTN_S405 | CTTN_S418 |
|
|
|
|
|
| CSNK2A1 | 0.095808202 | 435 | 5 | GTF2A1_S316 | GTF2A1_S321 | HNRNPC_S260 | IGF2R_S2409 | MCM2_S108 |
|
|
|
| PRKCH | 0.105773401 | 19 | 1 | PRKD2_S710 |
|
|
|
|
|
|
|
| CSNK1D | 0.113731726 | 102 | 2 | YWHAQ_S232 | GRLF1_S1179 |
|
|
|
|
|
|
| MAPK14 | 0.138835423 | 630 | 6 | SLC9A1_S726 | YLPM1_S634 | TCEA1_S100 | LMO7_S988 | SMARCA5_S66 | EGFR_T693 |
|
|
| CDK7 | 0.149726421 | 28 | 1 | MCM2_S108 |
|
|
|
|
|
|
|
| CSNK1G3 | 0.149726421 | 28 | 1 | YWHAQ_S232 |
|
|
|
|
|
|
|
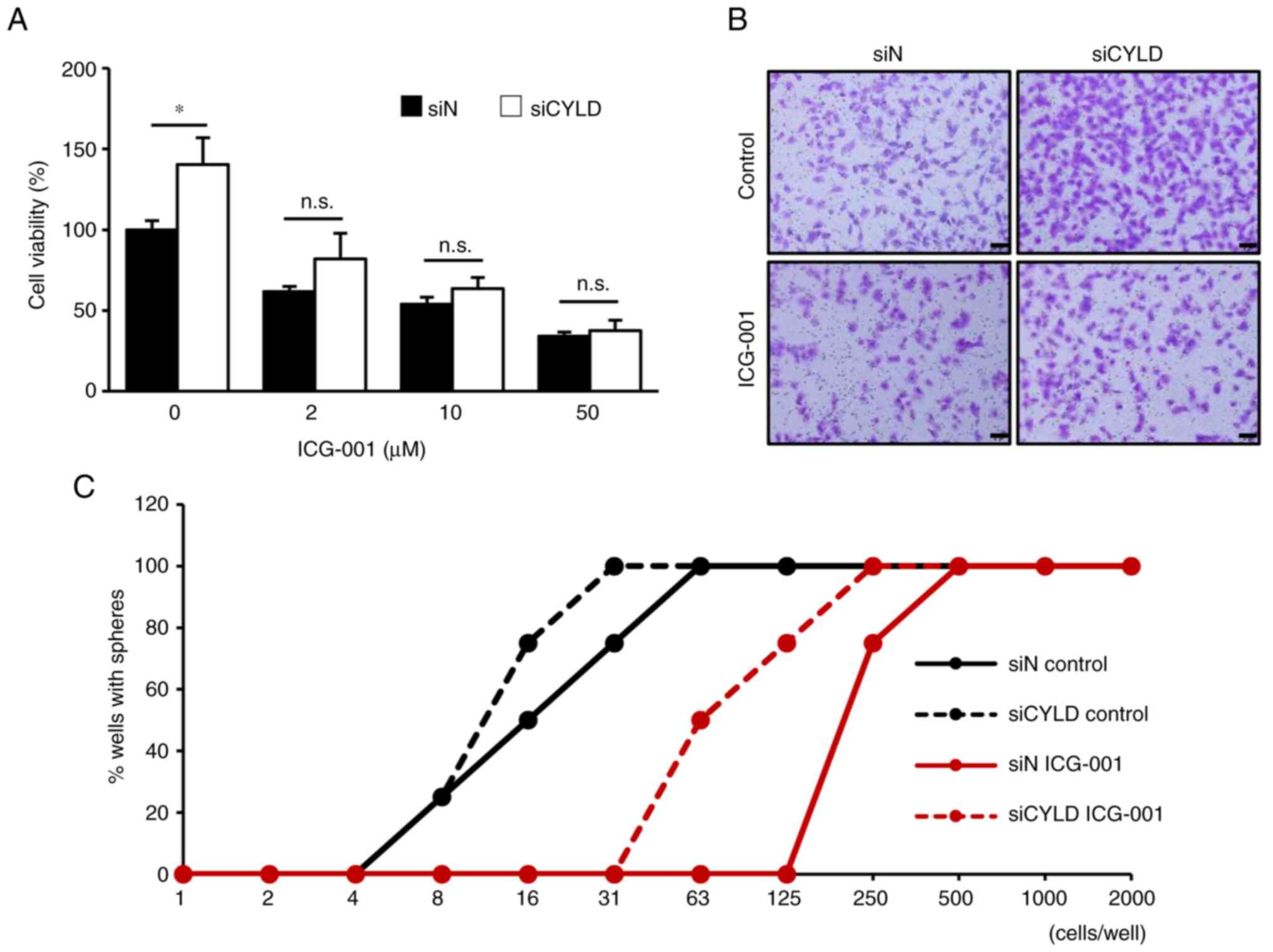
Therapeutic effect targeting
Wnt/β-catenin signaling on CYLD-knockdown GBM cells
Finally, the therapeutic effects of inhibiting
Wnt/β-catenin signaling on the malignant characteristics of
CYLD-knockdown cells were determined. As shown in Fig. 5A and B, the promotion of cell
proliferation and migration by CYLD silencing was significantly
suppressed by treatment with ICG-001, a Wnt/β-catenin signaling
inhibitor. Furthermore, in the limiting dilution assay, ICG-001
treatment significantly inhibited the sphere-forming ability of
CYLD-knockdown GBM cells (Fig. 5C),
suggesting that the inhibition of Wnt/β-catenin signaling may
suppress the stem cell-like characteristics caused by CYLD
knockdown. Taken together, the results of the present study suggest
targeting Wnt/β-catenin signaling as a potential therapeutic
strategy for CYLD-negative patients with GBM.
Discussion
To date, the correlation between CYLD downregulation
and poor prognosis has been revealed in a variety of malignant
tumors (16,29,42,43).
Although effective treatments for CYLD-downregulated patients with
poor prognosis have not yet been established, epidermal growth
factor receptor-targeted molecular therapies are effective against
CYLD-downregulated oral squamous cell carcinoma cells (44). In the present study, CYLD-silenced
GBM cells were investigated and it was determined that
Wnt/β-catenin signaling was significantly activated by CYLD
knockdown. The results of the present study suggest that inhibiting
Wnt/β-catenin signaling may be an effective therapeutic strategy
for CYLD-downregulated patients with GBM with poor prognosis.
Wnt/β-catenin signaling plays important roles in
cell development, regeneration, and homeostasis, and modulates the
proliferation, migration, and stem cell-like characteristics of
cells (45). In GBM, Wnt/β-catenin
signaling is involved in the molecular pathogenesis and progression
of GBM (46–48). In the present study it was revealed
that Wnt/β-catenin signaling played key roles in GBM malignant
transformation caused by CYLD downregulation. Since CYLD was
originally identified as a negative regulator of NF-κB signaling,
numerous studies have focused on the effects of CYLD downregulation
on NF-κB signaling in GBM (49–51).
Song et al reported that CYLD suppression promotes the NF-κB
signaling pathway and induces an aggressive phenotype in glioma
cells (51). In addition, it was
previously reported by the authors, that CYLD suppression under
hypoxia promotes inflammation in GBM via NF-κB signaling (30). In the present study, both proteomic
and RNA sequence analyses revealed that Wnt/β-catenin signaling, a
novel critical signaling pathway, was activated and associated with
CYLD downregulation in GBM (Fig.
4). Notably, all the malignant characteristics of GBM caused by
CYLD knockdown were significantly suppressed by treatment with
ICG-001, a Wnt/β-catenin signaling inhibitor (Fig. 5). In clinical settings, temozolomide
is the only antitumor drug currently approved for GBM treatment
(2). Notably, temozolomide
treatment did not exert any therapeutic effects on CYLD
knockdown-induced cell proliferation, migration, and GSC formation
in the present study (Fig. S2),
suggesting that targeting Wnt/β-catenin signaling may be a
potential therapeutic strategy for CYLD-downregulated patients with
GBM. Loss of CYLD expression has been revealed to enhance
Wnt/β-catenin signaling via K63-linked ubiquitination of the
Dishevelled (Dvl) protein (52). In
the present study, KEGG pathway analysis also revealed the
involvement of Dvl (Fig. 4B) in
GBM, and suggested that CYLD may regulate Wnt/β-catenin signaling
via the ubiquitination of Dvl. Although further investigation is
necessary to clarify the precise mechanisms, including the
relationship between Wnt/β-catenin and NF-κB signaling, in
CYLD-downregulated GBM, Wnt/β-catenin inhibition may improve the
prognosis of CYLD-downregulated patients with GBM.
Although GSCs, which exhibit chemoradiotherapy
resistance and recurrence characteristics, are key prognostic
factors in GBM patients (9–11), the molecular mechanisms of GSC
development remain unknown. Another interesting finding in the
present study is that CYLD knockdown played key roles in
development of GSCs through Wnt/β-catenin signaling. The results of
the present study clearly demonstrated the impact of CYLD
expression in the sphere-forming ability of GBM cells (Fig. 2). Clinical data from GBM tissues
further revealed that CYLD expression was significantly associated
with the cancer stem cell marker expression (Fig. 2). Moreover, Wnt/β-catenin signaling
activator (CKIε) expression was correlated with CYLD expression
(Fig. 4D). Consistently, a higher
correlation coefficient was observed in regions with more cancer
stem cells than in the entire tumor region (r=−0.235402318; data
not shown). Furthermore, the expression levels of other
Wnt/β-catenin signaling activators (CK2α and CK1α) were also
correlated with CYLD downregulation in regions with more cancer
stem cells (CK2α, r=−0.207300535; CK1α, r=−0.223871685; data not
shown) in RNA-seq data. These results suggest that Wnt/β-catenin
signaling, activated by CYLD knockdown, may be involved in the
formation and maintenance of GSCs. As GSCs are clinically
associated with chemo-radiotherapy resistance, the stem-like
characteristics induced by CYLD knockdown may induce resistance to
temozolomide treatment in patients with GBM (Fig. S2). In addition to the possible
involvement of NF-κB signaling in GSC formation (50), the authors have previously revealed
that ribosomal protein S6 (RPS6) promotes the stem-like
characteristics of glioma cells (41,53,54).
As RPS6 is predominantly expressed in GSC niches, concurrent with
data from the Ivy GAP database, this suggests an association
between CYLD and RPS6 expression. However, the detailed molecular
mechanisms underlying the development and maintenance of GSCs
induced by CYLD downregulation require further investigation.
The present study has some limitations. First, as
the number of patients with GBM was relatively small, clinical
evidence showing an association between the activation of
Wnt/β-catenin signaling and CYLD expression at the protein level
was limited. Second, the therapeutic effects of Wnt/β-catenin
signaling inhibitor in an in vivo CYLD-silenced GBM model
was not verified, which is necessary for the practical application
of the findings of the present study. To address these limitations,
the collection of more GBM specimens and the performance of in
vivo experiments will be undertaken in future studies.
In summary, it was revealed in the present study
that Wnt/β-catenin signaling was critically responsible for CYLD
silenced-induced malignant characteristics, such as proliferation,
migration, and GSC formation, in GBM cells. Therefore, targeting
Wnt/β-catenin signaling may be a novel effective therapeutic
strategy for CYLD-downregulated patients with GBM with poor
prognosis.
Supplementary Material
Supporting Data
Acknowledgements
We would like to thank Mr Shota Uchino, Ms Hitomi
Arakaki, Mr Taiki Katsume, Ms Kaho Matsuyama, and Mr Yoshiki Mori
(Department of Clinical Pharmaceutical Sciences, Graduate School of
Pharmaceutical Sciences, Kumamoto University, Kumamoto, Japan) for
their technical assistance in the present study.
Funding
The present study was supported by Grants-in-Aid for Scientific
Research (B) (grant no.18H02591) to H.J. and Young Scientists (A)
(grant no. 26713006) to H.J. from MEXT KAKENHI, Ministry of
Education, Culture, Sports, Science, and Technology, Japan.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The proteomics datasets have been submitted to jPOSTrepo
[https://repository.jpostdb.org/preview/187456970364aca64292d78;
Access key: 4555; Accession number: JPST002236 (PXD043537)].
Authors' contributions
TH and HJ made substantial contributions to the
conception and design of the study. AK, YI and MY performed most of
the experiments, acquired and analyzed the data, and confirm the
authenticity of all the raw data. YS, KY, SM, MA, and MO designed
the experimental procedure. TM performed the proteomic analysis
using LC-MS/MS. AM, JDL and HS supervised and conceptualized the
study. All the authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CYLD
|
cylindromatosis
|
|
GBM
|
glioblastoma
|
|
GSCs
|
GBM stem-like cells
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Hide T, Komohara Y, Miyasato Y, Nakamura
H, Makino K, Takeya M, Kuratsu JI, Mukasa A and Yano S:
Oligodendrocyte progenitor cells and macrophages/microglia produce
glioma stem cell niches at the tumor border. EBioMedicine.
30:94–104. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Troike KM, Acanda de la Rocha AM, Alban
TJ, Grabowski MM, Otvos B, Cioffi G, Waite KA, Barnholtz Sloan JS,
Lathia JD, Guilarte TR and Azzam DJ: The Translocator Protein
(TSPO) genetic polymorphism A147T is associated with worse survival
in male glioblastoma patients. Cancers (Basel). 13:45252021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lara-Velazquez M, Al-Kharboosh R,
Jeanneret S, Vazquez-Ramos C, Mahato D, Tavanaiepour D, Rahmathulla
G and Quinones-Hinojosa A: Advances in brain tumor surgery for
glioblastoma in adults. Brain Sci. 7:1662017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Barros A, Attal J, Roques M, Nicolau J,
Sol JC, Cohen-Jonathan-Moyal E and Roux FE: Impact on survival of
early tumor growth between surgery and radiotherapy in patients
with de novo glioblastoma. J Neurooncol. 142:489–497. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Karachi A, Dastmalchi F, Mitchell DA and
Rahman M: Temozolomide for immunomodulation in the treatment of
glioblastoma. Neuro Oncol. 20:1566–1572. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gujar AD, Le S, Mao DD, Dadey DY, Turski
A, Sasaki Y, Aum D, Luo J, Dahiya S, Yuan L, et al: An NAD +
-dependent transcriptional program governs self-renewal and
radiation resistance in glioblastoma. Proc Natl Acad Sci USA.
113:E8247–E8256. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perazzoli G, Prados J, Ortiz R, Caba O,
Cabeza L, Berdasco M, Gónzalez B and Melguizo C: Temozolomide
resistance in glioblastoma cell lines: Implication of MGMT, MMR,
P-glycoprotein and CD133 expression. PLoS One. 10:e01401312015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Colwell N, Larion M, Giles AJ, Seldomridge
AN, Sizdahkhani S, Gilbert MR and Park DM: Hypoxia in the
glioblastoma microenvironment: Shaping the phenotype of cancer
stem-like cells. Neuro Oncol. 19:887–896. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun H, Zhang M, Cheng K, Li P, Han S, Li
R, Su M, Zeng W, Liu J, Guo J, et al: Resistance of glioma cells to
nutrient-deprived microenvironment can be enhanced by
CD133-mediated autophagy. Oncotarget. 7:76238–76249. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lathia JD, Mack SC, Mulkearns-Hubert EE,
Valentim CLL and Rich JN: Cancer stem cells in glioblastoma. Genes
Dev. 29:1203–1217. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Blake PW and Toro JR: Update of
cylindromatosis gene (CYLD) mutations in Brooke-Spiegler syndrome:
Novel insights into the role of deubiquitination in cell signaling.
Hum Mutat. 30:1025–1036. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun SC: CYLD: A tumor suppressor
deubiquitinase regulating NF-kappaB activation and diverse
biological processes. Cell Death Differ. 17:25–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Urbanik T, Köhler BC, Boger RJ, Wörns MA,
Heeger S, Otto G, Hövelmeyer N, Galle PR, Schuchmann M, Waisman A
and Schulze-Bergkamen H: Down-regulation of CYLD as a trigger for
NF-κB activation and a mechanism of apoptotic resistance in
hepatocellular carcinoma cells. Int J Oncol. 38:121–131.
2011.PubMed/NCBI
|
|
15
|
Lim JH, Jono H, Komatsu K, Woo CH, Lee J,
Miyata M, Matsuno T, Xu X, Huang Y, Zhang W, et al: CYLD negatively
regulates transforming growth factor-β-signalling via
deubiquitinating Akt. Nat Commun. 3:7712012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shinriki S, Jono H, Maeshiro M, Nakamura
T, Guo J, Li JD, Ueda M, Yoshida R, Shinohara M, Nakayama H, et al:
Loss of CYLD promotes cell invasion via ALK5 stabilization in oral
squamous cell carcinoma. J Pathol. 244:367–379. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Reiley W, Zhang M and Sun SC: Negative
regulation of JNK signaling by the tumor suppressor CYLD. J Biol
Chem. 279:55161–55167. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tesio M, Tang Y, Müdder K, Saini M, von
Paleske L, Macintyre E, Pasparakis M, Waisman A and Trumpp A:
Hematopoietic stem cell quiescence and function are controlled by
the CYLD-TRAF2-p38MAPK pathway. J Exp Med. 212:525–538. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jono H, Lim JH, Chen LF, Xu H, Trompouki
E, Pan ZK, Mosialos G and Li JD: NF-κB is essential for induction
of CYLD, the negative regulator of NF-κB. J Biol Chem.
279:36171–36174. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yoshida H, Jono H, Kai H and Li JD: The
Tumor suppressor cylindromatosis (CYLD) acts as a negative
regulator for Toll-like Receptor 2 signaling via negative
Cross-talk with TRAF6 and TRAF7. J Biol Chem. 280:41111–41121.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sakai A, Koga T, Lim JH, Jono H, Harada K,
Szymanski E, Xu H, Kai H and Li JD: The bacterium, nontypeable
Haemophilus influenzae, enhances host antiviral response by
inducing Toll-like receptor 7 expression: Evidence for negative
regulation of host anti-viral response by CYLD. FEBS J.
274:3655–3668. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lim JH, Stirling B, Derry J, Koga T, Jono
H, Woo CH, Xu H, Bourne P, Ha UH, Ishinaga H, et al: Tumor
Suppressor CYLD regulates acute lung injury in lethal streptococcus
pneumoniae infections. Immunity. 27:349–360. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lim JH, Jono H, Koga T, Woo CH, Ishinaga
H, Bourne P, Xu H, Ha UH, Xu H and Li JD: Tumor suppressor CYLD
acts as a negative regulator for non-typeable haemophilus
influenza-induced inflammation in the middle ear and lung of mice.
PLoS One. 2:e10322007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koga T, Lim JH, Jono H, Ha UH, Xu H,
Ishinaga H, Morino S, Xu X, Yan C, Kai H and Li JD: Tumor
suppressor cylindromatosis acts as a negative regulator for
streptococcus pneumoniae-induced NFAT signaling. J Biol Chem.
283:12546–12554. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Komatsu K, Lee JY, Miyata M, Hyang Lim J,
Jono H, Koga T, Xu H, Yan C, Kai H and Li JD: Inhibition of PDE4B
suppresses inflammation by increasing expression of the
deubiquitinase CYLD. Nat Commun. 4:16842013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Harsha HC and Pandey A: Phosphoproteomics
in cancer. Mol Oncol. 4:482–495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Massoumi R, Kuphal S, Hellerbrand C, Haas
B, Wild P, Spruss T, Pfeifer A, Fässler R and Bosserhoff AK:
Down-regulation of CYLD expression by Snail promotes tumor
progression in malignant melanoma. J Exp Med. 206:221–232. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Suenaga N, Kuramitsu M, Komure K, Kanemaru
A, Takano K, Ozeki K, Nishimura Y, Yoshida R, Nakayama H, Shinriki
S, et al: Loss of tumor suppressor CYLD expression triggers
cisplatin resistance in oral squamous cell carcinoma. Int J Mol
Sci. 20:51942019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hayashi M, Jono H, Shinriki S, Nakamura T,
Guo J, Sueta A, Tomiguchi M, Fujiwara S, Yamamoto-Ibusuki M,
Murakami K, et al: Clinical significance of CYLD downregulation in
breast cancer. Breast Cancer Res Treat. 143:447–457. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo J, Shinriki S, Su Y, Nakamura T,
Hayashi M, Tsuda Y, Murakami Y, Tasaki M, Hide T, Takezaki T, et
al: Hypoxia suppresses cylindromatosis (CYLD) expression to promote
inflammation in glioblastoma: Possible link to acquired resistance
to anti-VEGF therapy. Oncotarget. 5:6353–6364. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zemke NR, Gou D and Berk AJ:
Dedifferentiation by adenovirus E1A due to inactivation of Hippo
pathway effectors YAP and TAZ. Genes Dev. 33:828–843. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Trompouki E, Hatzivassiliou E, Tsichritzis
T, Farmer H, Ashworth A and Mosialos G: CYLD is a deubiquitinating
enzyme that negatively regulates NF-kappaB activation by TNFR
family members. Nature. 424:793–796. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Balenci L, Clarke ID, Dirks PB, Assard N,
Ducray F, Jouvet A, Belin MF, Honnorat J and Baudier J: IQGAP1
protein specifies amplifying cancer cells in glioblastoma
multiforme. Cancer Res. 66:9074–9082. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Puchalski RB, Shah N, Miller J, Dalley R,
Nomura SR, Yoon JG, Smith KA, Lankerovich M, Bertagnolli D, Bickley
K, et al: An anatomic transcriptional atlas of human glioblastoma.
Science. 360:660–663. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Masuda T, Saito N, Tomita M and Ishihama
Y: Unbiased quantitation of Escherichia coli membrane proteome
using phase transfer surfactants. Mol Cell Proteomics. 8:2770–2777.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Masuda T, Tomita M and Ishihama Y: Phase
transfer surfactant-aided trypsin digestion for membrane proteome
analysis. J Proteome Res. 7:731–740. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rappsilber J, Mann M and Ishihama Y:
Protocol for micro-purification, enrichment, pre-fractionation and
storage of peptides for proteomics using StageTips. Nat Protoc.
2:1896–1906. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rappsilber J, Ishihama Y and Mann M: Stop
and go extraction tips for matrix-assisted laser
desorption/ionization, nanoelectrospray, and LC/MS sample
pretreatment in proteomics. Anal Chem. 75:663–670. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sugiyama N, Masuda T, Shinoda K, Nakamura
A, Tomita M and Ishihama Y: Phosphopeptide enrichment by aliphatic
hydroxy acid-modified metal oxide chromatography for nano-LC-MS/MS
in proteomics applications. Mol Cell Proteomics. 6:1103–1109. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shirakawa Y, Hide T, Yamaoka M, Ito Y, Ito
N, Ohta K, Shinojima N, Mukasa A, Saito H and Jono H: Ribosomal
protein S6 promotes stem-like characters in glioma cells. Cancer
Sci. 111:2041–2051. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Miyake S, Miwa T, Yoneda G, Kanemaru A,
Saito H, Minoda R, Orita Y, Saito H and Jono H: Relationship
between clinicopathological characteristics and CYLD expression in
patients with cholesteatoma. PLoS One. 15:e02402162020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Miyake S, Kanemaru A, Saito H and Jono H:
CYLD: A novel stratification marker for malignant tumors. J Asian
Assoc Sch Pharm. 10:17–22. 2021.
|
|
44
|
Kanemaru A, Shinriki S, Kai M, Tsurekawa
K, Ozeki K, Uchino S, Suenaga N, Yonemaru K, Miyake S, Masuda T, et
al: Potential use of EGFR-targeted molecular therapies for tumor
suppressor CYLD-negative and poor prognosis oral squamous cell
carcinoma with chemoresistance. Cancer Cell Int. 22:3582022.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zuccarini M, Giuliani P, Ziberi S,
Carluccio M, Iorio PD, Caciagli F and Ciccarelli R: The role of wnt
signal in glioblastoma development and progression: A possible new
pharmacological target for the therapy of this tumor. Genes
(Basel). 9:1052018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yun EJ, Kim S, Hsieh JT and Baek ST:
Wnt/β-catenin signaling pathway induces autophagy-mediated
temozolomide-resistance in human glioblastoma. Cell Death Dis.
11:7712020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Boso D, Rampazzo E, Zanon C, Bresolin S,
Maule F, Porcù E, Cani A, Della Puppa A, Trentin L, Basso G and
Persano L: HIF-1α/Wnt signaling-dependent control of gene
transcription regulates neuronal differentiation of glioblastoma
stem cells. Theranostics. 9:4860–4877. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhu H, Chen Z, Shen L, Tang T, Yang M and
Zheng X: Long Noncoding RNA LINC-PINT suppresses cell
proliferation, invasion, and EMT by Blocking Wnt/β-Catenin
signaling in glioblastoma. Front Pharmacol. 11:5866532021.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Song L, Lin C, Gong H, Wang C, Liu L, Wu
J, Tao S, Hu B, Cheng SY, Li M and Li J: miR-486 sustains NF-κB
activity by disrupting multiple NF-κB-negative feedback loops. Cell
Res. 23:274–289. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen Z, Wang S, Li HL, Luo H, Wu X, Lu J,
Wang HW, Chen Y, Chen D, Wu WT, et al: FOSL1 promotes
proneural-to-mesenchymal transition of glioblastoma stem cells via
UBC9/CYLD/NF-κB axis. Mol Ther. 30:2568–2583. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Song L, Liu L, Wu Z, Li Y, Ying Z, Lin C,
Wu J, Hu B, Cheng SY, Li M and Li J: TGF-β induces miR-182 to
sustain NF-κB activation in glioma subsets. J Clin Invest.
122:3563–3578. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tauriello DVF, Haegebarth A, Kuper I,
Edelmann MJ, Henraat M, Canninga-van Dijk MR, Kessler BM, Clevers H
and Maurice MM: Loss of the tumor suppressor CYLD enhances
Wnt/β-catenin signaling through K63-linked ubiquitination of Dvl.
Mol Cell. 37:607–619. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shirakawa Y, Ohta K, Miyake S, Kanemaru A,
Kuwano A, Yonemaru K, Uchino S, Yamaoka M, Ito Y, Ito N, et al:
Glioma cells acquire stem-like characters by extrinsic ribosome
stimuli. Cells. 10:29702021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hide T, Shibahara I, Inukai M, Shigeeda R,
Shirakawa Y, Jono H, Shinojima N, Mukasa A and Kumabe T: Ribosomal
proteins induce stem cell-like characteristics in glioma cells as
an ‘extra-ribosomal function.’. Brain Tumor Pathol. 39:51–56. 2022.
View Article : Google Scholar : PubMed/NCBI
|