Thyroid hormone receptor interactor 13 (TRIP13) is a
member of the AAA+ ATPase family able to generate mechanical
stresses through ATP hydrolase activities (1). TRIP13 was initially identified as a
protein that interacts with the E1 protein of the human
papillomavirus (2). In 1999, TRIP13
was found to be required for the meiotic checkpoint in yeast
(3). Studies have showed
involvement of TRIP13 in meiotic recombination and DNA repair in
several organisms, including rice (4), yeast (5), Drosophila melanogaster
(6), Caenorhabditis elegans
(7) and mice (8–10).
Additionally, it has been identified as a constituent of the
spindle assembly checkpoint (SAC) pathway (11–13),
which is involved in the accurate segregation of chromosomes
(14). TRIP13 [and its homolog
pachytene checkpoint 2 (PCH2) is an AAA+ ATPase that produces
homohexamers and uses ATP as a substrate (3,8,9,
15–19). PCH2 binds to Hop1 and alters its
structure, which displaces Hop1 from DNA (20). Hydrolysis of ATP by TRIP13/PCH2
provides energy necessary to undergo conformational changes that
exert mechanical force on Hop1 (21). In addition to its role as a
kinetochore protein, TRIP13/PCH2 interacts with the
p31comet protein, which is responsible for gene
silencing (11). TRIP13/PCH2 is
linked to various malignancies due to its involvement in ensuring
proper biorientation of chromosomes during mitosis; TRIP13 has been
shown to be overexpressed in a number of malignancies, including
colorectal (22–24), head and neck (25), breast (26,27),
lung (28–30), liver (31,32)
and prostate cancer (33,34), multiple myeloma (35), bladder cancer (36,37)
and human chronic lymphoblastic leukemia (38). Importantly, overexpression of TRIP13
promotes advancement of head and neck squamous cell carcinoma
(HNSCC) (25) and lung
adenocarcinoma (39). Depletion of
TRIP13 or suppression of its activity has been demonstrated to
diminish tumor development in head and neck and colon cancer and
hepatocellular carcinoma (HCC) (32,40).
TRIP13 also serves an essential role in the survival and spread of
tumor stem cells in cutaneous melanoma (41), prostate cancer (33) and lung adenocarcinoma (39). Based on these findings, TRIP13
serves a key role in tumor development.
Effective cancer therapy is hampered significantly
by the development of resistance to anticancer medications.
Overexpression of TRIP13 has been linked to decreased sensitivity
to anticancer medicines (such as bortezomib and cisplatin)
(25,42). TRIP13 facilitates development of
nedaplatin resistance in esophageal squamous cell carcinoma
(43). In addition, synergistic
anti-HCC efficacy is achieved by mixing TRIP13 inhibitor DCZ0415
with the PARP1 inhibitor olaparib (44). Therefore, TRIP13 is involved in drug
resistance in cancer cells. The present study evaluated the
function of TRIP13 expression in anticancer drug resistance and
potential methods to overcome this resistance. Furthermore, the
present study explored the underlying mechanism of targeting TRIP13
to overcome anticancer drug resistance and summarized the roles
TRIP13 plays in cancer treatment. The present study also reviewed
the effects of combination treatment, which include a TRIP13
inhibitor in addition to other inhibitors.
TRIP13/PCH2 has been shown to have a role in
mitosis, namely in the transition from metaphase to anaphase, as
well as the SAC (19). It also
releases anaphase-promoting complex (APC) from checkpoint
inhibition (19). Before anaphase,
the cell must make sure its chromosomes are appropriately organized
and bioriented for separation of sister chromatids. Numerous
proteins such as spindle checkpoint, securin and cyclin B, are
needed for this process to maintain accurate timing and reliable
separation. The activation of the APC is required for mitosis.
Protein CDC20, which is typically suppressed by the mitotic
checkpoint complex (MCC), activates the APC. The TRIP13-related
gene Mad2 exists in two isoforms, the open (O-) form and the closed
(C-) form (19). O-Mad2 changes
into C-Mad2 when kinetochores detach and C-Mad2 may then hook onto
CDC20 and sequester it, blocking mitotic progression (46). MCC must be disassembled, and this
process is mediated by p31comet (47). This is hypothesized to take place in
part via structural mimicry as p31comet has structural
similarities to C-Mad2 (48).
Nevertheless, ATP is needed for this step. TRIP13 uses
p31comet as an adaptor protein to convert C-Mad2 into
O-Mad2, then induces activation of the SAC and the formation of the
MCC. To conclude, TRIP13/PCH2 is key for SAC activation and MCC
formation.
HNSCC exhibits TRIP13 overexpression, which promotes
proliferation and invasion (25).
Overexpression of both TRIP13 and Mad2 are associated with various
types of cancer, including multiple myeloma (42), head and neck cancer (25), colorectal cancer (22–24),
chronic lymphocytic leukemia (50),
lung adenocarcinoma (39) and
prostate cancer (33).
Overexpression of TRIP13 attenuates the mitotic delay caused by
Mad2 overexpression, but downregulation of TRIP13 compounds the
effects of Mad2 overexpression. In addition, downregulating TRIP13
and overexpressing Mad2 suppresses proliferation in cells and tumor
xenografts, suggesting therapeutic potential for inhibition of
TRIP13 (47). Mad2 promotes drug
resistance in ovarian cancer (51).
The expression of TRIP13 is markedly increased in epithelial
ovarian cancer (EOC) cell lines (SKOV-3, HEY and OVCAR-3) compared
with normal ovarian cell lines (52). Additionally, knockdown of TRIP13 in
EOC cells inhibits cell proliferation, decreases cell invasion and
migration and stimulates apoptosis (52). This demonstrates that TRIP13
promotes the development of tumor cells and may be a potential
target for tumor therapy.
Anticancer regimens have the potential to kill most
cancer cells at first, but some malignant cells survive because
they have either already developed mechanisms of drug resistance
(intrinsic resistance) or have acquired them through random
mutation and genetic alteration (acquired resistance). Resistance
mechanisms include: Decreased expression or dysfunction of influx
drug transporters, increased activity of multidrug-resistant (MDR)
efflux pumps of the ATP-binding cassette (ABC) superfamily, such as
P-glycoprotein, multidrug resistance associated protein 1 (MRP1)
and breast cancer resistance protein (ABCG2) (53), qualitative and quantitative changes
in the drug target and drug sequestration within intracellular
compartments. MDR efflux pumps remove numerous anticancer
medications from cancer cells or store them in organelles such as
lysosomes, where they are inaccessible to the cell target sites
(54). The emergence of resistance
to anticancer drugs is associated with genomic instability, which
may include mutations, amplifications, deletion and/or
translocations (55). MDR is caused
by a combination of variables, including genetics (gene mutations,
amplification and epigenetic modifications), growth hormones and
higher DNA repair ability (56).
According to Vasan et al (57), the primary factors that determine
treatment resistance include: Tumor burden and growth dynamics;
tumor heterogeneity; physical barriers; the immune system and the
tumor microenvironment. All of these factors contribute to the
diminished effectiveness of the medications used to treat the
tumor.
TRIP13 expression is shown to be higher in samples
from patients with multiple myeloma compared with control samples
(42,58). Overexpression of TRIP13 is linked to
decreased sensitivity to bortezomib and cisplatin (25,42,59).
TRIP13 is shown to be responsible for the development of nedaplatin
resistance in esophageal squamous cell carcinoma (43). Furthermore, considering the role of
cisplatin (60–62) and PARP (63–65)
inhibitors in the treatment of ovarian cancer, further study is
needed to elucidate the role of TRIP13 in treatment-induced drug
resistance caused by cisplatin and PARP therapy. These
aforementioned results suggest a connection between elevated TRIP13
levels and drug resistance.
Targeting TRIP13 is a potential strategy for
overcoming drug resistance. Inhibiting TRIP13 makes HNSCC cells
more sensitive to effects of radiation and chemotherapy (25). Recent research found that
TRIP13−/− HCC cells are more vulnerable to the effects
of chemotherapy than normal HCC cells (44). DCZ0415 may stimulate antimyeloma
activity in primary cells generated from individuals with myeloma
who are resistant to several drugs (35). Inhibition or depletion of TRIP13 may
thus constitute a viable approach to circumventing resistance that
anticancer drugs cause.
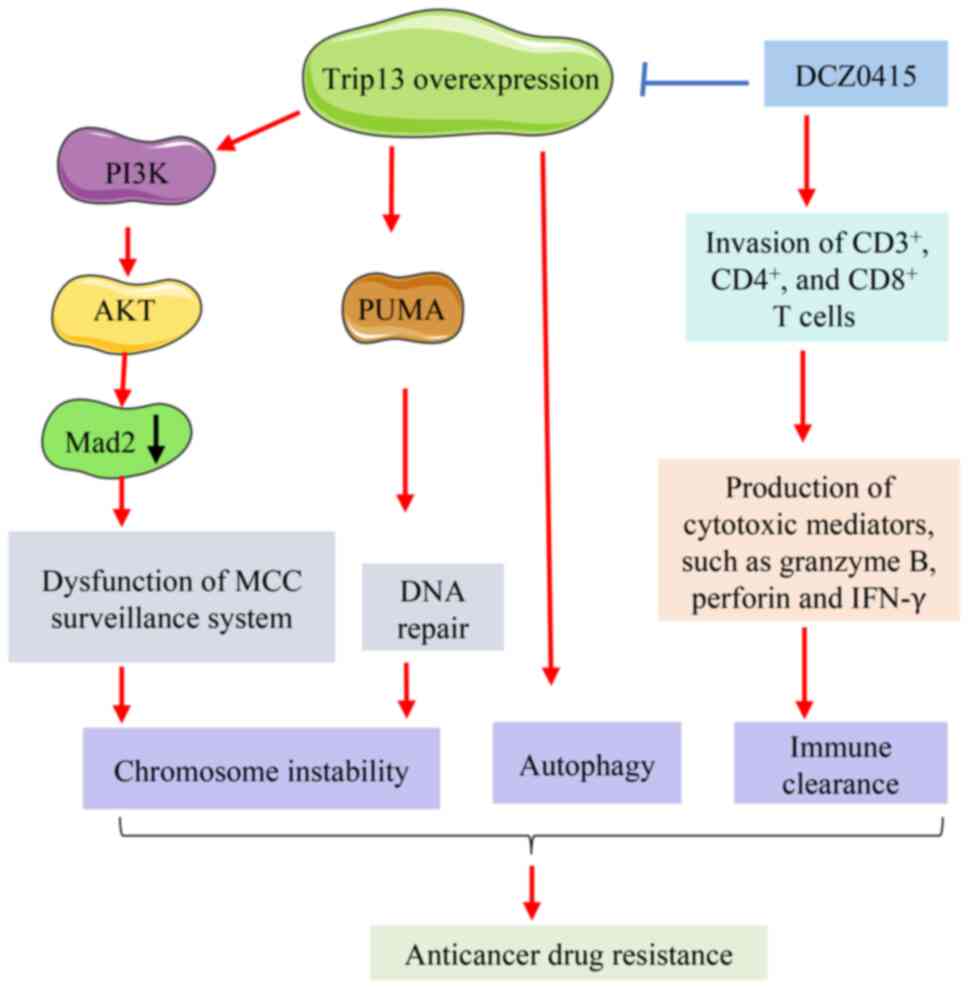
There are primarily three molecular mechanisms
involved in TRIP13-mediated drug resistance. These mechanisms
include promoting chromosomal instability, enhancing autophagy and
modulating the tumor microenvironment (Fig. 2).
Previous studies have suggested that SAC is a
universal safeguard that guarantees the integrity of chromosomal
separation in cell division (66–68).
Multiple malignancies exhibit overexpression of SAC proteins, which
is linked to chromosome instability (CIN) in tumors (69–71).
Additionally, telomere dysfunction (72–74)
and faulty DNA repair mechanism response (75) contribute significantly to CIN in
cancer. Not only does CIN serve a role in the origin, maintenance
and growth of tumors, but it also stimulates the development of
treatment resistance in cancer cells. TRIP13 is associated with CIN
in human malignancies such as multiple myeloma (12,76,77)
and treatment resistance. Previous studies have documented that
TRIP13 plays a role in the induction of CIN and drug resistance via
SAC signaling and modulating DNA damage repair (25,42,43,58,78).
To prevent genomic instability in the host, DNA
damage response and repair mechanisms have been conserved
throughout evolution (85) of both
prokaryotes and eukaryotes. In mammalian cells, dysregulation of
proteins involved in these processes may increase genomic changes,
which lead to genomic instability, a well-established hallmark of
cancer (86,87). There has been emergence of new and
promising techniques for targeting the DNA damage response and
repair pathways to increase cancer cell sensitivity to existing
therapeutic drugs (88). Targeting
DNA damage is signaling and repair may interrupt the compensatory
activation of DNA repair pathways that may function as a drug
resistance mechanism. For example, targeting DNA repair has become
a legitimate therapeutic approach. This method uses PARP inhibitors
to treat breast, ovarian, pancreatic and prostate cancers that have
DNA repair deficiencies (89–95).
Clinical research and development of small compounds that target
key components of the DNA damage response and repair pathways,
including DNA-dependent protein kinase (DNA-PK), ataxia
telangiectasia mutated (ATM) and Rad3-related kinase (ATR), ATM and
checkpoint kinase 1 (CHK1), have been accelerated (96). In addition, accurate targeting of
these key molecules offers the possibility of using biomarkers of
DNA repair deficiency to choose the most effective therapy for each
individual patient to achieve the highest possible therapeutic
index (96).
Recent research has provided support for the
hypothesis that TRIP13-induced anti-apoptosis may contribute to
drug resistance in cancer cells (97). This is because one of the primary
strategies anticancer medications utilize to trigger cell death is
induction of apoptosis. When TRIP13 is overexpressed, cancer cells
are less sensitive to bortezomib and cisplatin (25,42).
When multiple myeloma cells transfected with TRIP13 are treated
with bortezomib and etoposide, cell viability experiment indicates
that the number of viable tumor cells is greater in the
TRIP13-transfected cells compared with control cells (42). Comparatively, HNSCC cells that
overexpress TRIP13 display a decreased sensitivity to cisplatin
when compared with control cells (25). Therefore, TRIP13 plays a role in the
development of drug resistance in cancer cells. When treated with
increasing doses of bortezomib, TRIP13-overexpressing ARP1 and
OCIMy5 multiple myeloma cell lines are less likely to undergo
apoptosis and better able to withstand the cytotoxic effects
compared with cells transfected with empty vectors. In
TRIP13-overexpressing ARP1 multiple myeloma cell line, the G2/M
cell cycle arrest induced by bortezomib is consistently suppressed
compared with control cells (42).
In addition, TRIP13 small interfering (si)RNA knockdown in multiple
myeloma cells eliminates doxycycline treatment resistance and
triggers apoptosis both in vitro and in a mouse model of
xenograft myeloma (35,42). In ARP1 and OCIMy5 multiple myeloma
cells, downregulation of TRIP13 leads to an increase in levels of
cleaved PARP and activation of caspase 3, both of which indicate a
potential role for knockdown of TRIP13 in inhibiting the apoptotic
pathway (42). Similarly, in human
chronic lymphocytic leukemia, microarray data evaluated using the
‘canonical pathway’ module of Ingenuity route analysis reveal that
TRIP13 participates in numerous apoptosis-associated pathways,
including ‘induction of apoptosis by HIV1’, ‘p53 signaling’ and
‘PPAR signaling’ (50). In
addition, knocking down TRIP13 causes a notable increase in the
activity of caspase 3/7 in Granta-519 and JVM-2 B cell lymphocytic
leukemia cell lines (50). The
modulation of the c-Myc/TRIP13/p53 upregulated modulator of
apoptosis (PUMA) axis is the mechanism by which TRIP13 serves a
role in the development of chronic lymphocytic leukemia (50). In HNSCC, suppression of TRIP13 also
induces cell cycle arrest (25). In
cells transfected with TRIP13 siRNA, there is a greater
accumulation of phosphorylated histone H2A histone family member X
(the marker of DSBs) (25).
According to western blot analysis, DSBs caused by TRIP13 siRNA
occur prior to apoptosis (25).
These results provided compelling evidence that TRIP13 increase DNA
repair, which in turn leads to treatment resistance.
Autophagy is a natural method of cell survival that
is effectively employed by tumor cells to prevent cell death and
generate drug resistance (98–100).
Both of these may be accomplished by tumor cells via autophagy, a
macromolecular process in which cells break down and recycle
intracellular substrates and damaged organelles to reduce cell
stress caused by factors such as nutritional deficiency, hypoxia,
irradiation and cytotoxic chemicals. When cancer is in the early
stages, autophagy has been shown to protect against malignant
disease; nevertheless, transformed cells exhibit increased
autophagy to promote survival, proliferation and metastasis
(101–103). Although the precise function of
autophagy in cell death and survival remains unclear, autophagy is
enhanced in cancer cells exposed to stressful situations, such as
anticancer treatment, which may result in anticancer drug
resistance (104–106). Previous research examined TRIP13
role in autophagy by treating cells with DMSO (control), gefitinib
(an autophagy agonist) and 3-methyladenine (3-MA; an autophagy
inhibitor) (107); TRIP13 induces
autophagy in non-small cell lung cancer (NSCLC) cells, shown by an
increase in the number of LC3B (autophagy marker)-positive puncta
seen by immunofluorescence examination (107). Overexpression of TRIP13 in NSCLC
cells leads to an increase in LC3B and decrease in autophagy marker
P62 expression (107). The
opposite effects are seen in tumor cells when TRIP13 expression is
suppressed (107). All of the
aforementioned effects are mitigated by the gefitinib therapy, but
3-MA has the opposite effect (107). Therefore, TRIP13 may be
responsible for inducing gefitinib resistance in NSCLC cells via
increasing autophagy (107).
Therefore, TRIP13 can be used as a biomarker and therapeutic target
autophagy for overcoming drug resistance.
The tumor microenvironment, which comprises immune
cells, stroma and vasculature, may promote drug resistance via
numerous methods, including inhibiting immune clearance of tumor
cells, preventing drug absorption and increasing paracrine growth
factors, to promote cancer cell development (108). DCZ0415 prevents proliferation
inhibition of multiple myeloma cells even in the presence of bone
marrow stromal cells and the cytokines IL6 and IGF1 in a cellular
experiment that mimics multiple myeloma in its microenvironment
(35). In addition to cytotoxicity
against multiple myeloma cells, DCZ0415 also targets the bone
marrow microenvironment and overcomes the proliferative effects on
bone marrow stromal cells (35).
Additionally, suppression of TRIP13 stimulates an
anticancer immune response by increasing production of cytotoxic
mediators. Recent studies show that TRIP13 inhibition promotes the
invasion of CD3+, CD4+ and CD8+ T
cells (35). DCZ0415 considerably
increases the production of cytotoxic mediators such as granzyme B,
perforin and IFN-γ, which may contribute to the cytotoxic effect
against murine MC38 cells. In addition, blocking immunological
checkpoints, such as PD-1 and cytotoxic T-lymphocyte-associated
protein 4 (CTLA4), in tumors treated with DCZ0415 has the potential
to increase cytotoxicity and regression of the tumor (109). Hence, decreased expression of
TRIP13 stimulates immune responses in the microenvironment of the
tumor, which may help overcome drug resistance to anticancer
medications.
DNA DSBs and DNA damage responses induced by certain
anticancer drugs, such as example, β-emitter iodine-131,
O-6-methylguanine-DNA methyltransferases and cisplatin, make cancer
cells more vulnerable to further treatment (110). DNA damage response followed by
effective repair of DSBs is key to maintain genomic integrity. On
the other hand, in cancer, the repair of anticancer agent-induced
DSBs via the non-homologous end joining (NHEJ) or homologous
recombination (HR) repair pathways enhances treatment resistance
and recurrence in patients (111).
According to recent research, TRIP13 improves NHEJ repair and
generates treatment resistance in head and neck cancer via binding
to NHEJ proteins Ku protein with molecular weight of 70 KDa (KU70,
encoded by the X-ray repair cross-complementing protein 6 gene
located on chromosome 22), Ku protein with molecular weight of 80
KDa, (KU80,encoded by the X-ray repair cross-complementing protein
5 gene on chromosome 2) and DNA-dependent protein kinase catalytic
subunit (DNA-PKcs) (25).
Additionally, GFP-based reporter experiment demonstrates that
DCZ0415 inhibits DNA repair by the NHEJ repair pathway (35). Hence, combining TRIP13 inhibitor
with other inhibitors may be an option to overcome anticancer drug
resistance (Table I).
In addition, the upregulation of the TRIP13-mediated
NHEJ repair pathway is the cause of the resistance to anticancer
drugs (such as bortezomib and cisplatin). Therefore, cancer cells
that have high levels of TRIP13 expression will be more susceptible
to DNA-PKcs inhibitor than cancer cells with low levels of TRIP13
expression (25). Banerjee et
al (25) showed that tumor
cells that overexpress TRIP13 are more sensitive to the DNA-PKcs
inhibitor Nu7026 (25). Hence,
combining the TRIP13 inhibitor with DNA-PKcs inhibitor may be an
effective treatment approach for TRIP13-mediated drug
resistance.
TRIP13/PCH2 is associated with a number of
malignancies as it ensures proper alignment of chromosomes during
mitosis. As a result, TRIP13 may be a target for the development of
an anticancer medication. In addition, overexpression of TRIP13 is
associated with decreased susceptibility to bortezomib and
cisplatin (25,42). In esophageal squamous cell
carcinoma, TRIP13 causes nedaplatin resistance (43). In addition, combination of DCZ0415
with olaparib results in a synergistic effect on the activity of
HCC (44). Thus, TRIP13 serves a
role in cancer cell drug resistance.
To determine how TRIP13 contributes to the
development of anticancer drug resistance, it is vital to research
the mechanism that controls the regulation of TRIP13 expression. In
perihilar cholangiocarcinoma cells, c-Myc stimulates the
transcription of TRIP13 (117). In
addition, transcription factor specificity protein 1 (SP1) serves a
role in controlling the amount of TRIP13 that is expressed
(44). TRIP13 expression is
drastically reduced by SP1 inhibition or knockdown, whereas
overexpression of SP1 notably increases TRIP13 expression (44). However, it is unclear whether c-Myc
or SP1 are involved in development of TRIP13-mediated drug
resistance. Because of this, identifying the transcription factors
that govern expression of TRIP13 is required, as well as genes
controlled by TRIP13, which may to act as targets for the
development of cancer treatment.
To create anticancer treatments that are effective
for people with cancer with high levels of TRIP13, it is necessary
to identify new TRIP13-binding proteins and the TRIP13 binding
domain. There is a possibility that peptides with a structure
identical to the binding domain of TRIP13 may facilitate overcoming
resistance to anti-cancer medications. For example, TRIP13 binds to
ubiquitin-specific protase-7 (USP7), then TRIP13-induced resistance
to proteasome inhibition can be overcome by a USP7 inhibitor
(25,42,59).
Additionally, the microenvironment of a tumor
comprises innate and adaptive immune cells (B and T, dendritic and
myeloid-derived suppressor cells and M1/2 macrophages), as well as
cancer cells and endothelial cells and cancer-associated
fibroblasts. The tumor microenvironment is essential for the
development and progression of cancer. There is evidence that
interactions between cancer cells and immune cells may enhance the
growth of cancer cells (108).
Depletion of TRIP13 facilitates activation of immune responses in
the tumor microenvironment (35),
which improves anticancer treatment resistance. In immune cells, it
is important to discover the genes controlled by the TRIP13 protein
to develop potential immunotherapeutic targets.
TRIP13 inhibitor resistance may develop in future.
Because of this, it will be required to identify genes that confer
resistance to TRIP13 inhibitors as well as genes controlled by
TRIP13 inhibitors. It is possible that immune evasion, drug efflux,
the overexpression of oncogenes and the downregulation of tumor
suppressor genes mediate resistance to TRIP13 inhibitors.
The authors would like to thank Dr Peng Zhao
(Universiti Pendidikan Sultan Idris, Tanjong Malim, Malaysia) for
critical review of manuscript.
The present study was supported by National Natural Science
Foundation of China (grant no. 82100557), Science Foundation of
Nantong (grant no. JC2021018), and Large Instruments Open
Foundation of Nantong University (grant no. KFJN2375).
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
LZ, NJ and SY wrote the manuscript. TH and YJG
performed the literature review and edited the manuscript. All
authors have read and approved the final manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Jeong H, Wie M, Baek IJ, Sohn G, Um SH,
Lee SG, Seo Y, Ra J, Lee EA, Kim S, et al: TRIP13 participates in
immediate-early sensing of DNA strand breaks and ATM signaling
amplification through MRE11. Cells. 11:40952022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee JW, Choi HS, Gyuris J, Brent R and
Moore DD: Two classes of proteins dependent on either the presence
or absence of thyroid hormone for interaction with the thyroid
hormone receptor. Mol Endocrinol. 9:243–254. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
San-Segundo PA and Roeder GS: Pch2 links
chromatin silencing to meiotic checkpoint control. Cell.
97:313–324. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miao C, Tang D, Zhang H, Wang M, Li Y,
Tang S, Yu H, Gu M and Cheng Z: Central region component1, a novel
synaptonemal complex component, is essential for meiotic
recombination initiation in rice. Plant Cell. 25:2998–3009. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Farmer S, Hong EJ, Leung WK, Argunhan B,
Terentyev Y, Humphryes N, Toyoizumi H and Tsubouchi H: Budding
yeast Pch2, a widely conserved meiotic protein, is involved in the
initiation of meiotic recombination. PLoS One. 7:e397242012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Joyce EF and McKim KS: Drosophila PCH2 is
required for a pachytene checkpoint that monitors
double-strand-break-independent events leading to meiotic crossover
formation. Genetics. 181:39–51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ye Q, Rosenberg SC, Moeller A, Speir JA,
Su TY and Corbett KD: TRIP13 is a protein-remodeling AAA+ ATPase
that catalyzes MAD2 conformation switching. Elife. 4:e073672015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li XC and Schimenti JC: Mouse pachytene
checkpoint 2 (trip13) is required for completing meiotic
recombination but not synapsis. PLoS Genet. 3:e1302007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roig I, Dowdle JA, Toth A, de Rooij DG,
Jasin M and Keeney S: Mouse TRIP13/PCH2 is required for
recombination and normal higher-order chromosome structure during
meiosis. PLoS Genet. 6:e10010622010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wojtasz L, Daniel K, Roig I, Bolcun-Filas
E, Xu H, Boonsanay V, Eckmann CR, Cooke HJ, Jasin M, Keeney S, et
al: Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal
proteins, are depleted from synapsed chromosome axes with the help
of TRIP13 AAA-ATPase. PLoS Genet. 5:e10007022009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tipton AR, Wang K, Oladimeji P, Sufi S, Gu
Z and Liu ST: Identification of novel mitosis regulators through
data mining with human centromere/kinetochore proteins as group
queries. BMC Cell Biol. 13:152012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang K, Sturt-Gillespie B, Hittle JC,
Macdonald D, Chan GK, Yen TJ and Liu ST: Thyroid hormone receptor
interacting protein 13 (TRIP13) AAA-ATPase is a novel mitotic
checkpoint-silencing protein. J Biol Chem. 289:23928–23937. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eytan E, Wang K, Miniowitz-Shemtov S,
Sitry-Shevah D, Kaisari S, Yen TJ, Liu ST and Hershko A:
Disassembly of mitotic checkpoint complexes by the joint action of
the AAA-ATPase TRIP13 and p31(comet). Proc Natl Acad Sci USA.
111:12019–12024. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Silva RD, Mirkovic M, Guilgur LG, Rathore
OS, Martinho RG and Oliveira RA: Absence of the spindle assembly
checkpoint restores mitotic fidelity upon loss of sister chromatid
cohesion. Curr Biol. 28:2837–2844.e2833. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bhalla N and Dernburg AF: A conserved
checkpoint monitors meiotic chromosome synapsis in Caenorhabditis
elegans. Science. 310:1683–1686. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Börner GV, Barot A and Kleckner N: Yeast
Pch2 promotes domainal axis organization, timely recombination
progression, and arrest of defective recombinosomes during meiosis.
Proc Natl Acad Sci USA. 105:3327–3332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Joshi N, Barot A, Jamison C and Börner GV:
Pch2 links chromosome axis remodeling at future crossover sites and
crossover distribution during yeast meiosis. PLoS Genet.
5:e10005572009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vader G, Blitzblau HG, Tame MA, Falk JE,
Curtin L and Hochwagen A: Protection of repetitive DNA borders from
self-induced meiotic instability. Nature. 477:115–119. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vader G: Pch2(TRIP13): Controlling cell
division through regulation of HORMA domains. Chromosoma.
124:333–339. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen C, Jomaa A, Ortega J and Alani EE:
Pch2 is a hexameric ring ATPase that remodels the chromosome axis
protein Hop1. Proc Natl Acad Sci USA. 111:E44–E53. 2014.PubMed/NCBI
|
|
21
|
Yedidi RS, Wendler P and Enenkel C:
AAA-ATPases in protein degradation. Front Mol Biosci. 4:422017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sheng N, Yan L, Wu K, You W, Gong J, Hu L,
Tan G, Chen H and Wang Z: TRIP13 promotes tumor growth and is
associated with poor prognosis in colorectal cancer. Cell Death
Dis. 9:4022018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kurita K, Maeda M, Mansour MA, Kokuryo T,
Uehara K, Yokoyama Y, Nagino M, Hamaguchi M and Senga T: TRIP13 is
expressed in colorectal cancer and promotes cancer cell invasion.
Oncol Lett. 12:5240–5246. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Agarwal S, Behring M, Kim HG,
Chandrashekar DS, Chakravarthi BVSK, Gupta N, Bajpai P, Elkholy A,
Al Diffalha S, Datta PK, et al: TRIP13 promotes metastasis of
colorectal cancer regardless of p53 and microsatellite instability
status. Mol Oncol. 14:3007–3029. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Banerjee R, Russo N, Liu M, Basrur V,
Bellile E, Palanisamy N, Scanlon CS, van Tubergen E, Inglehart RC,
Metwally T, et al: TRIP13 promotes error-prone nonhomologous end
joining and induces chemoresistance in head and neck cancer. Nat
Commun. 5:45272014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lan J, Huang J, Tao X, Gao Y, Zhang L,
Huang W, Luo J, Liu C, Deng Y, Liu L and Liu X: Evaluation of the
TRIP13 level in breast cancer and insights into potential molecular
pathways. J Cell Mol Med. 26:2673–2685. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu L, Zhang Z, Xia X and Lei J: KIF18B
promotes breast cancer cell proliferation, migration and invasion
by targeting TRIP13 and activating the Wnt/β-catenin signaling
pathway. Oncol Lett. 23:1122022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li ZH, Lei L, Fei LR, Huang WJ, Zheng YW,
Yang MQ, Wang Z, Liu CC and Xu HT: TRIP13 promotes the
proliferation and invasion of lung cancer cells via the Wnt
signaling pathway and epithelial-mesenchymal transition. J Mol
Histol. 52:11–20. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cai W, Ni W, Jin Y and Li Y: TRIP13
promotes lung cancer cell growth and metastasis through
AKT/mTORC1/c-Myc signaling. Cancer Biomark. 30:237–248. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Q, Dong Y, Hao S, Tong Y, Luo Q and
Aerxiding P: The oncogenic role of TRIP13 in regulating
proliferation, invasion, and cell cycle checkpoint in NSCLC cells.
Int J Clin Exp Pathol. 12:3357–3366. 2019.PubMed/NCBI
|
|
31
|
Yao J, Zhang X, Li J, Zhao D, Gao B, Zhou
H, Gao S and Zhang L: Silencing TRIP13 inhibits cell growth and
metastasis of hepatocellular carcinoma by activating of
TGF-β1/smad3. Cancer Cell Int. 18:2082018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garcia MR, Meissburger B, Chan J, de Guia
RM, Mattijssen F, Roessler S, Birkenfeld AL, Raschzok N, Riols F,
Tokarz J, et al: Trip13 depletion in liver cancer induces a
lipogenic response contributing to plin2-dependent mitotic cell
death. Adv Sci (Weinh). 9:e21042912022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dong L, Ding H, Li Y, Xue D, Li Z, Liu Y,
Zhang T, Zhou J and Wang P: TRIP13 is a predictor for poor
prognosis and regulates cell proliferation, migration and invasion
in prostate cancer. Int J Biol Macromol. 121:200–206. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zeng L, Liu YM, Yang N, Zhang T and Xie H:
Hsa_circRNA_100146 promotes prostate cancer progression by
upregulating TRIP13 via sponging miR-615-5p. Front Mol Biosci.
8:6934772021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Y, Huang J, Li B, Xue H, Tricot G, Hu
L, Xu Z, Sun X, Chang S, Gao L, et al: A small-molecule inhibitor
targeting TRIP13 suppresses multiple myeloma progression. Cancer
Res. 80:536–548. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao Y, Liu S, Guo Q, Zhang S, Zhao Y, Wang
H, Li T, Gong Y, Wang Y, Zhang T, et al: Increased expression of
TRIP13 drives the tumorigenesis of bladder cancer in association
with the EGFR signaling pathway. Int J Biol Sci. 15:1488–1499.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mohammed aI, Ali ME-H, Mohamed FEA and
Abd-Elrehim DM: Immunohistochemical expression of TRIP13 in
transitional and squamous cell carcinoma of urinary bladder
carcinoma. Minia J Med Res. 2023. View Article : Google Scholar
|
|
38
|
Zhou KS, Zhang Q, Zhang WT, Liu YY, Wu SS,
Zhou J, Wei XD and Song YP: Study on the expression of TRIP13 mRNA
in chronic lymphocytic leukemia B lymphocyte and the molecular
mechanism of TRIP13 mediated JVM-2 cell proliferation and
apoptosis. Zhonghua Xue Ye Xue Za Zhi. 38:618–622. 2017.(In
Chinese). PubMed/NCBI
|
|
39
|
Li W, Zhang G, Li X, Wang X, Li Q, Hong L,
Shen Y, Zhao C, Gong X, Chen Y and Zhou J: Thyroid hormone receptor
interactor 13 (TRIP13) overexpression associated with tumor
progression and poor prognosis in lung adenocarcinoma. Biochem
Biophys Res Commun. 499:416–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ju L, Li X, Shao J, Lu R, Wang Y and Bian
Z: Upregulation of thyroid hormone receptor interactor 13 is
associated with human hepatocellular carcinoma. Oncol Rep.
40:3794–3802. 2018.PubMed/NCBI
|
|
41
|
Lu W, Mengxuan Z, Ming R, Zixu G, Yong Z,
Simin Z, Yang Y, Leqi Q, Kangjie S, Yanlin L, et al: TRIP13/FLNA
complex promotes tumor progression and is associated with
unfavorable outcomes in melanoma. J Oncol. 2022:14191792022.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tao Y, Yang G, Yang H, Song D, Hu L, Xie
B, Wang H, Gao L, Gao M, Xu H, et al: TRIP13 impairs mitotic
checkpoint surveillance and is associated with poor prognosis in
multiple myeloma. Oncotarget. 8:26718–26731. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang LT, Ke LX, Wu XY, Tian HT, Deng HZ,
Xu LY, Li EM and Long L: TRIP13 induces nedaplatin resistance in
esophageal squamous cell carcinoma by enhancing repair of DNA
damage and inhibiting apoptosis. Biomed Res Int.
2022:72954582022.PubMed/NCBI
|
|
44
|
Xu H, Ma Z, Mo X, Chen X, Xu F, Wu F, Chen
H, Zhou G, Xia H and Zhang C: Inducing synergistic DNA damage by
TRIP13 and PARP1 inhibitors provides a potential treatment for
hepatocellular carcinoma. J Cancer. 13:2226–2237. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ye Q, Kim DH, Dereli I, Rosenberg SC,
Hagemann G, Herzog F, Tóth A, Cleveland DW and Corbett KD: The AAA+
ATPase TRIP13 remodels HORMA domains through N-terminal engagement
and unfolding. EMBO J. 36:2419–2434. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mapelli M, Massimiliano L, Santaguida S
and Musacchio A: The Mad2 conformational dimer: Structure and
implications for the spindle assembly checkpoint. Cell.
131:730–743. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Marks DH, Thomas R, Chin Y, Shah R, Khoo C
and Benezra R: Mad2 overexpression uncovers a critical role for
TRIP13 in mitotic exit. Cell Rep. 19:1832–1845. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang M, Li B, Tomchick DR, Machius M, Rizo
J, Yu H and Luo X: p31comet blocks Mad2 activation through
structural mimicry. Cell. 131:744–755. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rosenberg SC and Corbett KD: The
multifaceted roles of the HORMA domain in cellular signaling. J
Cell Biol. 211:745–755. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhou K, Zhang W, Zhang Q, Gui R, Zhao H,
Chai X, Li Y, Wei X and Song Y: Loss of thyroid hormone receptor
interactor 13 inhibits cell proliferation and survival in human
chronic lymphocytic leukemia. Oncotarget. 8:25469–25481. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Furlong F, Fitzpatrick P, O'Toole S,
Phelan S, McGrogan B, Maguire A, O'Grady A, Gallagher M, Prencipe
M, McGoldrick A, et al: Low MAD2 expression levels associate with
reduced progression-free survival in patients with high-grade
serous epithelial ovarian cancer. J Pathol. 226:746–755. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhou XY and Shu XM: TRIP13 promotes
proliferation and invasion of epithelial ovarian cancer cells
through Notch signaling pathway. Eur Rev Med Pharmacol Sci.
23:522–529. 2019.PubMed/NCBI
|
|
53
|
Amawi H, Sim HM, Tiwari AK, Ambudkar SV
and Shukla S: ABC transporter-mediated multidrug-resistant cancer.
Adv Exp Med Biol. 1141:549–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Khatami M: Cancer; an induced disease of
twentieth century! Induction of tolerance, increased entropy and
‘Dark Energy’: Loss of biorhythms (Anabolism v. Catabolism). Clin
Transl Med. 7:202018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang DC, Wang W, Zhu B and Wang X: Lung
cancer heterogeneity and new strategies for drug therapy. Annu Rev
Pharmacol Toxicol. 58:531–546. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bukowski K, Kciuk M and Kontek R:
Mechanisms of multidrug resistance in cancer chemotherapy. Int J
Mol Sci. 21:32332020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Vasan N, Baselga J and Hyman DM: A view on
drug resistance in cancer. Nature. 575:299–309. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lu S, Guo M, Fan Z, Chen Y, Shi X, Gu C
and Yang Y: Elevated TRIP13 drives cell proliferation and drug
resistance in bladder cancer. Am J Transl Res. 11:4397–4410.
2019.PubMed/NCBI
|
|
59
|
Li C, Xia J, Franqui-Machin R, Chen F, He
Y, Ashby TC, Teng F, Xu H, Liu D, Gai D, et al: TRIP13 modulates
protein deubiquitination and accelerates tumor development and
progression of B cell malignancies. J Clin Invest. 131:e1468932021.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ozols RF and Young RC: High-dose cisplatin
therapy in ovarian cancer. Semin Oncol. 12:21–30. 1985.PubMed/NCBI
|
|
61
|
Markman M: Intraperitoneal cisplatin and
carboplatin in the management of ovarian cancer. Semin Oncol.
21:17–19; quiz 20. 581994.PubMed/NCBI
|
|
62
|
Zoń A and Bednarek I: Cisplatin in ovarian
cancer treatment-known limitations in therapy force new solutions.
Int J Mol Sci. 24:75852023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Mittica G, Ghisoni E, Giannone G, Genta S,
Aglietta M, Sapino A and Valabrega G: PARP inhibitors in ovarian
cancer. Recent Pat Anticancer Drug Discov. 13:392–410. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Smith M and Pothuri B: Appropriate
selection of PARP inhibitors in ovarian cancer. Curr Treat Options
Oncol. 23:887–903. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jiang X, Li X, Li W, Bai H and Zhang Z:
PARP inhibitors in ovarian cancer: Sensitivity prediction and
resistance mechanisms. J Cell Mol Med. 23:2303–2313. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Musacchio A and Salmon ED: The
spindle-assembly checkpoint in space and time. Nat Rev Mol Cell
Biol. 8:379–393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lara-Gonzalez P, Westhorpe FG and Taylor
SS: The spindle assembly checkpoint. Curr Biol. 22:R966–R980. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chao WC, Kulkarni K, Zhang Z, Kong EH and
Barford D: Structure of the mitotic checkpoint complex. Nature.
484:208–213. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
de Cárcer G and Malumbres M: A centrosomal
route for cancer genome instability. Nat Cell Biol. 16:504–506.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sotillo R, Schvartzman JM, Socci ND and
Benezra R: Mad2-induced chromosome instability leads to lung tumour
relapse after oncogene withdrawal. Nature. 464:436–440. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bargiela-Iparraguirre J, Prado-Marchal L,
Pajuelo-Lozano N, Jiménez B, Perona R and Sánchez-Pérez I: Mad2 and
BubR1 modulates tumourigenesis and paclitaxel response in MKN45
gastric cancer cells. Cell Cycle. 13:3590–3601. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tusell L, Pampalona J, Soler D, Frías C
and Genescà A: Different outcomes of telomere-dependent anaphase
bridges. Biochem Soc Trans. 38:1698–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Stewénius Y, Gorunova L, Jonson T, Larsson
N, Höglund M, Mandahl N, Mertens F, Mitelman F and Gisselsson D:
Structural and numerical chromosome changes in colon cancer develop
through telomere-mediated anaphase bridges, not through mitotic
multipolarity. Proc Natl Acad Sci USA. 102:5541–5546. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bailey SM and Murnane JP: Telomeres,
chromosome instability and cancer. Nucleic Acids Res. 34:2408–2417.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mills KD, Ferguson DO and Alt FW: The role
of DNA breaks in genomic instability and tumorigenesis. Immunol
Rev. 194:77–95. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhou W, Yang Y, Xia J, Wang H, Salama ME,
Xiong W, Xu H, Shetty S, Chen T, Zeng Z, et al: NEK2 induces drug
resistance mainly through activation of efflux drug pumps and is
associated with poor prognosis in myeloma and other cancers. Cancer
Cell. 23:48–62. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Carter SL, Eklund AC, Kohane IS, Harris LN
and Szallasi Z: A signature of chromosomal instability inferred
from gene expression profiles predicts clinical outcome in multiple
human cancers. Nat Genet. 38:1043–1048. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Clairmont CS, Sarangi P, Ponnienselvan K,
Galli LD, Csete I, Moreau L, Adelmant G, Chowdhury D, Marto JA and
D'Andrea AD: TRIP13 regulates DNA repair pathway choice through
REV7 conformational change. Nat Cell Biol. 22:87–96. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Sudakin V, Chan GK and Yen TJ: Checkpoint
inhibition of the APC/C in HeLa cells is mediated by a complex of
BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 154:925–936. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Overlack K, Bange T, Weissmann F, Faesen
AC, Maffini S, Primorac I, Müller F, Peters JM and Musacchio A:
BubR1 promotes Bub3-dependent APC/C inhibition during spindle
assembly checkpoint signaling. Curr Biol. 27:2915–2927.e2917. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Burton JL and Solomon MJ: Mad3p, a
pseudosubstrate inhibitor of APCCdc20 in the spindle assembly
checkpoint. Genes Dev. 21:655–667. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
McGranahan N, Burrell RA, Endesfelder D,
Novelli MR and Swanton C: Cancer chromosomal instability:
Therapeutic and diagnostic challenges. EMBO Rep. 13:528–538. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lischetti T and Nilsson J: Regulation of
mitotic progression by the spindle assembly checkpoint. Mol Cell
Oncol. 2:e9704842015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Sudo T, Nitta M, Saya H and Ueno NT:
Dependence of paclitaxel sensitivity on a functional spindle
assembly checkpoint. Cancer Res. 64:2502–2508. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wang M, Chen S and Ao D: Targeting DNA
repair pathway in cancer: Mechanisms and clinical application.
MedComm. 2020.2:654–691. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Deng S, Vlatkovic T, Li M, Zhan T,
Veldwijk MR and Herskind C: Targeting the DNA damage response and
DNA repair pathways to enhance radiosensitivity in colorectal
cancer. Cancers (Basel). 14:48742022. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Mirza MR, Pignata S and Ledermann JA:
Latest clinical evidence and further development of PARP inhibitors
in ovarian cancer. Ann Oncol. 29:1366–1376. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Schettini F, Giudici F, Bernocchi O,
Sirico M, Corona SP, Giuliano M, Locci M, Paris I, Scambia G, De
Placido S, et al: Poly (ADP-ribose) polymerase inhibitors in solid
tumours: Systematic review and meta-analysis. Eur J Cancer.
149:134–152. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Tutt ANJ, Garber JE, Kaufman B, Viale G,
Fumagalli D, Rastogi P, Gelber RD, de Azambuja E, Fielding A,
Balmaña J, et al: Adjuvant olaparib for patients with BRCA1- or
BRCA2-mutated breast cancer. N Engl J Med. 384:2394–2405. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Carreira S, Porta N, Arce-Gallego S, Seed
G, Llop-Guevara A, Bianchini D, Rescigno P, Paschalis A, Bertan C,
Baker C, et al: Biomarkers associating with PARP inhibitor benefit
in prostate cancer in the TOPARP-B trial. Cancer Discov.
11:2812–2827. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Mateo J, Porta N, Bianchini D, McGovern U,
Elliott T, Jones R, Syndikus I, Ralph C, Jain S, Varughese M, et
al: Olaparib in patients with metastatic castration-resistant
prostate cancer with DNA repair gene aberrations (TOPARP-B): A
multicentre, open-label, randomised, phase 2 trial. Lancet Oncol.
21:162–174. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Maughan BL and Antonarakis ES: Olaparib
and rucaparib for the treatment of DNA repair-deficient metastatic
castration-resistant prostate cancer. Expert Opin Pharmacother.
22:1625–1632. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Cleary JM, Wolpin BM, Dougan SK, Raghavan
S, Singh H, Huffman B, Sethi NS, Nowak JA, Shapiro GI, Aguirre AJ
and D'Andrea AD: Opportunities for utilization of DNA repair
inhibitors in homologous recombination repair-deficient and
proficient pancreatic adenocarcinoma. Clin Cancer Res.
27:6622–6637. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
van Waardenburg R and Yang ES: Targeting
DNA repair pathways to overcome cancer drug resistance. Cancer Drug
Resist. 4:837–841. 2021.PubMed/NCBI
|
|
97
|
Ghosh S, Mazumdar T, Xu W, Powell RT,
Stephan C, Shen L, Shah PA, Pickering CR, Myers JN, Wang J, et al:
Combined TRIP13 and aurora kinase inhibition induces apoptosis in
human papillomavirus–driven cancers. Clin Cancer Res. 28:4479–4493.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Chang H and Zou Z: Targeting autophagy to
overcome drug resistance: Further developments. J Hematol Oncol.
13:1592020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Ahmadi-Dehlaghi F, Mohammadi P, Valipour
E, Pournaghi P, Kiani S and Mansouri K: Autophagy: A challengeable
paradox in cancer treatment. Cancer Med. 12:11542–11569. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Salimi-Jeda A, Ghabeshi S, Pour ZGM,
Jazaeri EO, Araiinejad M, Sheikholeslami F, Abdoli M, Edalat M and
Abdoli A: Autophagy modulation and cancer combination therapy: A
smart approach in cancer therapy. Cancer Treat Res Commun.
30:1005122022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Levine B and Kroemer G: Biological
functions of autophagy genes: A disease perspective. Cell.
176:11–42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Galluzzi L and Green DR:
Autophagy-independent functions of the autophagy machinery. Cell.
177:1682–1699. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Dikic I and Elazar Z: Mechanism and
medical implications of mammalian autophagy. Nat Rev Mol Cell Biol.
19:349–364. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Wu M and Zhang P: EGFR-mediated autophagy
in tumourigenesis and therapeutic resistance. Cancer Lett.
469:207–216. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye
WC, Zhang DM and Chen ZS: Autophagy and multidrug resistance in
cancer. Chin J Cancer. 36:522017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Amaravadi RK, Kimmelman AC and Debnath J:
Targeting autophagy in cancer: Recent advances and future
directions. Cancer Discov. 9:1167–1181. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Xiao Z, Li M, Zhang X, Rong X and Xu H:
TRIP13 overexpression promotes gefitinib resistance in non-small
cell lung cancer via regulating autophagy and phosphorylation of
the EGFR signaling pathway. Oncol Rep. 49:842023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Sharma P, Hu-Lieskovan S, Wargo JA and
Ribas A: Primary, adaptive, and acquired resistance to cancer
immunotherapy. Cell. 168:707–723. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Agarwal S, Afaq F, Bajpai P, Kim HG,
Elkholy A, Behring M, Chandrashekar DS, Diffalha SA, Khushman M,
Sugandha SP, et al: DCZ0415, a small-molecule inhibitor targeting
TRIP13, inhibits EMT and metastasis via inactivation of the
FGFR4/STAT3 axis and the Wnt/β-catenin pathway in colorectal
cancer. Mol Oncol. 16:1728–1745. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Qie S and Diehl JA: Cyclin D1, cancer
progression, and opportunities in cancer treatment. J Mol Med
(Berl). 94:1313–1326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Lindahl T and Barnes DE: Repair of
endogenous DNA damage. Cold Spring Harb Symp Quant Biol.
65:127–133. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Sarangi P, Clairmont CS, Galli LD, Moreau
LA and D'Andrea AD: p31(comet) promotes homologous recombination by
inactivating REV7 through the TRIP13 ATPase. Proc Natl Acad Sci
USA. 117:26795–26803. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Corbett KD: p31comet and TRIP13 recycle
Rev7 to regulate DNA repair. Proc Natl Acad Sci USA.
117:27761–27763. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Oser MG, Fonseca R, Chakraborty AA, Brough
R, Spektor A, Jennings RB, Flaifel A, Novak JS, Gulati A, Buss E,
et al: Cells lacking the RB1 tumor suppressor gene are
hyperdependent on aurora B kinase for survival. Cancer Discov.
9:230–247. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Gong X, Du J, Parsons SH, Merzoug FF,
Webster Y, Iversen PW, Chio LC, Van Horn RD, Lin X, Blosser W, et
al: Aurora A kinase inhibition is synthetic lethal with loss of the
RB1 tumor suppressor gene. Cancer Discov. 9:248–263. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Schvartzman JM, Duijf PH, Sotillo R, Coker
C and Benezra R: Mad2 is a critical mediator of the chromosome
instability observed upon Rb and p53 pathway inhibition. Cancer
Cell. 19:701–714. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Li Z, Liu J, Chen T, Sun R, Liu Z, Qiu B,
Xu Y and Zhang Z: HMGA1-TRIP13 axis promotes stemness and
epithelial mesenchymal transition of perihilar cholangiocarcinoma
in a positive feedback loop dependent on c-Myc. J Exp Clin Cancer
Res. 40:862021. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zhang X, Zhou J, Xue D, Li Z, Liu Y and
Dong L: MiR-515-5p acts as a tumor suppressor via targeting TRIP13
in prostate cancer. Int J Biol Macromol. 129:227–232. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chen Y, Chen D, Qin Y, Qiu C, Zhou Y, Dai
M, Li L, Sun Q and Jiang Y: TRIP13, identified as a hub gene of
tumor progression, is the target of microRNA-4693-5p and a
potential therapeutic target for colorectal cancer. Cell Death
Discov. 8:352022. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zhu MX, Wei CY, Zhang PF, Gao DM, Chen J,
Zhao Y, Dong SS and Liu BB: Elevated TRIP13 drives the AKT/mTOR
pathway to induce the progression of hepatocellular carcinoma via
interacting with ACTN4. J Exp Clin Cancer Res. 38:4092019.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Arun G, Diermeier SD and Spector DL:
Therapeutic targeting of long non-coding RNAs in cancer. Trends Mol
Med. 24:257–277. 2018. View Article : Google Scholar : PubMed/NCBI
|