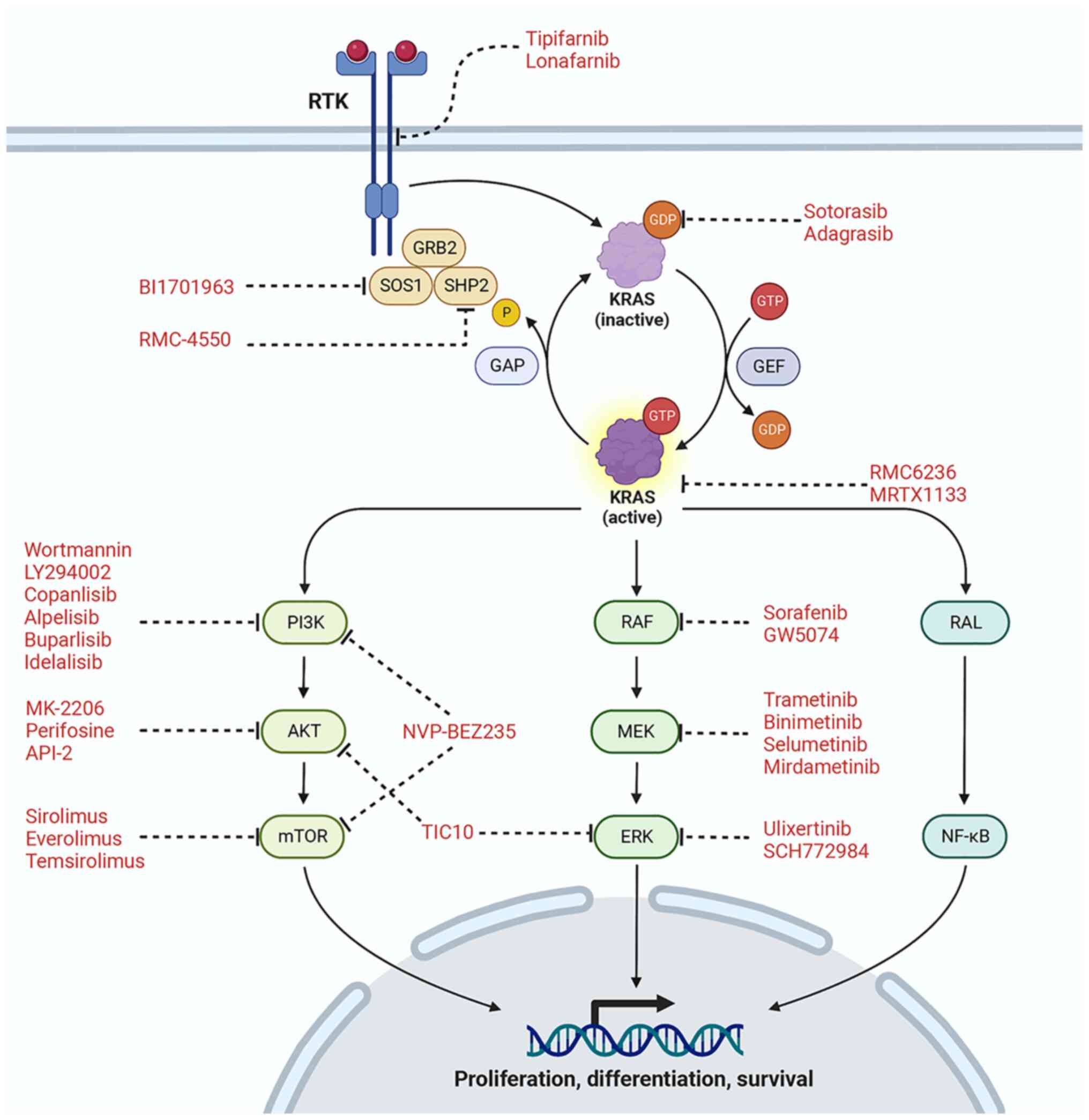
KRAS is a small GTPase signal transduction protein
that cycles between active guanosine triphosphate (GTP)-bound and
inactive guanosine diphosphate (GDP)-bound states. In normal
quiescent cells, RAS is largely GDP-bound and inactive, but the
GTP-bound state is formed through extracellular stimuli activation
of receptor tyrosine kinases (RTKs) as well as other cell-surface
receptors. Activated KRAS maintains the engagement of effector
proteins that then regulate several intracellular signaling
networks that control mitogenic processes. KRAS has been shown to
play a central role in controlling tumor metabolism (2). Oncogenic KRAS genes are
characterized by missense mutations that encode single amino acid
substitutions at three primary locations: Glycine-12 (G12),
glycine-13 (G13), or glutamine-61 (Q61). Among these, G12 mutations
comprise 83% of all KRAS mutations, followed by G13 (14%)
and Q61 (2%) mutations (3). The
mutation subtypes at KRAS are mainly classified as
KRAS G12D, G12V, G12C, G13D, G12R and G12A. KRAS
mutations are most common in pancreatic cancer, NSCLC and
colorectal cancer, and the profiles of KRAS mutation
subtypes differ in different types of cancer. For example, G12C
mutation is the most common subtype in NSCLC (41%), whereas G12D
and G12V are the major subtypes in pancreatic cancer and colorectal
cancer (4). All KRAS
mutations render KRAS constitutively bound to GTP and active,
overstimulating effector signaling pathways to drive uncontrolled
growth of cells leading to cancer formation. This suggests that
blocking KRAS has high therapeutic potential for several cancers.
In the present review, the critical role of oncogenic KRAS
signaling, challenges in its targeting, and preclinical and
clinical studies targeting KRAS signaling, including its downstream
signaling effectors or direct inhibitors, in pancreatic ductal
adenocarcinoma (PDAC) were investigated. In the present review, a
comprehensive literature search was performed using three
databases, PubMed, Medline and Web of Science.
Comprising >90% of cases, the most common form of
pancreatic cancer is PDAC, which has a 5-year overall survival (OS)
rate of 11% (5). PDAC is the 3rd
leading cause of cancer-related deaths in the US and has the
highest frequency of KRAS mutations (>90%). PDAC displays
two major KRAS mutations at G12D (41%) and G12V (34%), while
other less frequent mutations are G12R (16%), Q61H (4%) and G12C
(~1%) (6).
PDAC has four types of pre-neoplastic precursors:
Intraductal papillary mucinous neoplasm, pancreatic mucinous cystic
neoplasm, intraductal tubular papillary neoplasm, and pancreatic
intraepithelial neoplasia (PanIN). PanIN is the most common
precursor lesion and is classified as low-grade (PanIN-1A and
PanIN-1B), intermediate-grade (PanIN-2), or high-grade (PanIN-3)
before progressing to PDAC. KRAS mutation is an early event
in the genetic onset of PDAC. The progression from normal
pancreatic tissue to PDAC is typically initiated by advancing
stages on noninvasive microscopic ductal lesions, PanINs.
KRAS mutations are considered to be the driving force in the
development of human PanINs and >90% frequency of KRAS
mutations are identified in PanIN-1 lesions. Furthermore, Aguirre
et al reported that the KRASG12D mutation
alone formed PanIN and the protracted onset of PDAC (7).
PanIN-to-PDAC progression is initiated by subsequent
inactivation of tumor suppressor gene cyclin-dependent kinase
inhibitor 2A, identified in PanIN-2, and then followed by
inactivation of two other tumor suppressor genes, TP53 and
SMAD4, proteins unique to PanIN-3. KRAS-mediated signaling
then further leads to the development of PDAC through several
metabolic processes, providing the energy and biosynthetic building
blocks necessary to drive uncontrolled tumor growth. Furthermore,
previous studies revealed that KRAS-mutant PDAC tumor cells
are capable of regulating autophagy to meet metabolic demand
(8,9). Although KRAS mutations are the
initiating genetic step, mutant KRAS is still involved in
every step to maintain the growth of metastatic PDAC (2). The gradual progression of
KRAS-mediated PDAC progression ultimately results in rapid growth
and metastasis. Thus, there is high therapeutic potential in
targeting the KRAS pathway for improving clinical PDAC therapy.
In theory, targeting KRAS signaling could
potentially provide an effective approach to block the growth of
KRAS-dependent tumors, but this was deemed unrealistic due to
several barriers. First, KRAS is a member of a large family of
related proteins sharing similar GTP/GDP binding domains, causing
the development of specific targeted drugs very difficult. Second,
KRAS has a very high picomolar affinity for GTP and GDP, both of
which have very high intracellular concentrations. Third, the KRAS
protein lacks accessible binding sites for the high-affinity
binding of small-molecule inhibitors (18). Thus, not only do KRAS protein
inhibitors have limited effectiveness due to chemical affinity, but
they are also hindered by the structure of the target proteins. Due
to these challenges, KRAS is widely considered undruggable,
shifting focus to blocking downstream effector signaling.
Targeting downstream effector signaling has emerged
as a promising alternative approach to block oncogenic KRAS
signaling pathways. Oncogenic-activating KRAS mutations
regulate an intricate complex of cytoplasmic signaling networks.
There are at least 11 different effector families, and
tumorigenesis is likely the result of multiple integrated
effector-signaling pathways (19).
Thus, while it shows great promise, targeting downstream KRAS
effector signaling is more complex than it seemed at first glance.
Therapeutic approaches must decide which effector pathways are the
best to target and if co-inhibition of multiple effectors is
required. Based on the prominent role of KRAS in PDAC progression
and difficulties in the direct targeting of KRAS, much of the
efforts have centered on indirect strategies in hopes of finding a
more effective treatment for PDAC.
KRAS-GTP is known to activate several downstream
oncogenic signaling pathways including PI3K/AKT/mTOR, MAPK,
RAL-PLD1 and T1AM1-Rac, and crosstalk exists among these pathways
(Fig. 1). Thus, the concept of
targeting KRAS effector signaling is not without complications.
Previous studies have shown that PI3K subunits play
a critical role as effectors of mutant KRAS-driven oncogenesis
(20,21). PI3K phosphorylates PIP2 and
stimulates the formation of PIP3, which then phosphorylates and
activates a multitude of proteins, including AKT 1/2/3, localizing
it in the plasma membrane. AKT phosphorylates numerous other
proteins that promote cell growth, in particular the mammalian
target of rapamycin (mTOR) (22,23).
The PI3K/AKT/mTOR pathway plays a pivotal role in a number of
cellular processes such as proliferation, survival, and growth.
Abnormalities in this pathway such as PI3K mutation/amplification,
loss of PTEN, AKT mutation or RTK activation, have been
implicated in cancer cell proliferation, invasion, survival,
metastasis, epithelial-mesenchymal transition (EMT) and drug
resistance (24,25). There have been conflicting studies
regarding PI3K activation in driving PDAC development. A previous
study revealed that 93% of rare PIK3CA mutations co-occur
with a KRAS mutation, suggesting that activated KRAS
is not sufficient to effectively activate PI3K (26). However, another study revealed that
KRAS suppression did not alter AKT activation levels in a majority
of KRAS-mutated PDAC cell lines (27).
Several drugs have been developed to target this
pathway, including PI3K, PI3K/mTOR, mTOR, and AKT inhibitors
(Table I). Wortmannin was the first
PI3K inhibitor developed which was identified to be extremely
effective in inhibiting PI3K, but it was revealed to be
nonspecific, unstable and toxic in animals (28). In orthotopic PDAC xenografts,
wortmannin promoted the antitumor activity of gemcitabine (tumor
weight reduction relative to control was 1.4-fold by gemcitabine
and 5-fold by gemcitabine plus wortmannin; P<0.001) supporting
its potential as an adjuvant to conventional chemotherapy
treatments of PDAC (29). LY294002,
however, is a very specific synthetic inhibitor of PI3K and is
chemically more stable but has been identified to be less potent
than wortmannin in preclinical studies. In PDAC models, LY294002
inhibited in vitro cell proliferation, induced apoptosis and
reduced in vivo tumor growth. Furthermore, LY294002 enhanced
the effects of cisplatin both in vitro and in vivo
(tumor volume decreased to 77, 70 or 44% of the volume in the
controls by cisplatin, LY294002 or combination; P<0.05)
(30). Subsequently, Wang et
al reported the opposite effect of LY294002 by demonstrating
that LY294002 (but not wortmannin) enhanced AKT phosphorylation in
the gemcitabine-resistant PDAC cell lines (31). This study suggested that the PI3K
inhibitors can be counterproductive with gemcitabine-resistant PDAC
cells.
There are three major mTOR inhibitors currently
approved by the FDA: Sirolimus, everolimus and temsirolimus.
Sirolimus (rapamycin) induced autophagy and apoptosis in PDAC cell
lines (35) but it also led to
resistance mediated by AKT phosphorylation (36). The second-generation mTOR
inhibitors, such as KU63794 and PP242, also led to treatment
resistance due to an increase in ERK activation (36). Rapamycin exhibited a dose-dependent
radiosensitization effect (37) and
synergistic antiproliferative and antiangiogenic effects in
combination with EGFR inhibitor gefitinib on PDAC cells (38). In vivo studies in mice
demonstrated that rapamycin was specifically effective on
KRAS-mutant PDAC tumors that have loss of PTEN
(median survival in controls, gemcitabine, rapamycin and
gemcitabine plus rapamycin was 10, 14, 56 and 32 days,
respectively), while KRAS-mutant tumors with mutant
p53 (KPC) did not respond (39). Metformin causes diverse effects on
PDAC tumorigenesis in both mTOR-dependent and -independent manner.
In the syngeneic mouse model using C57BL/6 mice and Pan02 cells,
metformin and rapamycin both exhibited significant tumor growth
reduction (tumor burden compared with control 0.90 g was 0.62 g
with metformin and 0.25 g with rapamycin) (40). Everolimus, an analog of rapamycin,
sensitized PDAC cells to the effects of gemcitabine in an in
vitro study (41). In
vitro studies demonstrated that gemcitabine-resistant PDAC
cells were more sensitive to everolimus, in contrast to various
EGFR, AKT and PI3K inhibitors (42). In another in vitro study in
PDAC cell lines, everolimus had an antiproliferative effect, while
its combination with sorafenib exhibited an antagonistic effect
(43). Wei et al
demonstrated that neither everolimus nor AZD8055, a 2nd-generation
mTOR inhibitor, exerted any cell viability inhibitory effect on
PDAC cell lines (44). In PDAC PDX
models, everolimus displayed higher antitumor efficacy when
combined with either sorafenib (the tumor/control ratio was 0.6,
0.5 and 0.2 for treatment with everolimus, sorafenib and the
combination) (45) or trametinib
(46). Temsirolimus (CCI-779), a
water-soluble, more stable and specific mTOR inhibitor, showed a
significant antiproliferative effect on PDAC cells (47) and exhibited synergistic antitumor
response in combination with gemcitabine in PDAC xenograft models
(compared with control, decrease in tumor volume was 68%, P=0.0009
with a high dose of CCI-779; and 41%, P=0.0002 with CCI-779 plus
gemcitabine (48).
Dual inhibition of PI3K-mTOR holds arguably the most
potential. NVP-BEZ235, the primary proponent of this class of
drugs, demonstrated significant delay in tumor growth (56, 36 and
46%) in three different orthotopic PDX models (P<0.05) (49). In the peritoneal dissemination
animal survival model, NVP-BEZ235 enhanced gemcitabine response
(median survival in days was 16, 21, 28 and 30 in control,
NVP-BEZ235, gemcitabine, and combination; P<0.05) (50). In PDAC subcutaneous xenografts,
NVP-BEZ235 exhibited synergistic tumor growth inhibition in
combination with pan-histone deacetylase inhibitor panobinostat
(51) or inRas37 antibody (52). In vitro study using PANC-1
and MIA PaCa-2 PDAC cells revealed that NVP-BEZ235 markedly induced
the ERK/MEK pathway. The MEK inhibitors U126 and PD0325901
prevented ERK overactivation induced by NVP-BEZ235 and the
combination of MEK inhibitors with NVP-BEZ235 produced a further
inhibition of PDAC cell proliferation (53).
Another hallmark downstream pathway through which
KRAS signaling occurs is the Ras-Raf-MEK-ERK (MAPK) pathway which
is the key effector in the initiation, progression, and maintenance
of KRAS-dependent tumors. Thus, several drugs targeting different
components of this pathway have been explored extensively for
therapeutic intervention in KRAS-mutant PDAC (Table I).
Ras proteins undergo farnesylation by the enzyme
farnesyl-protein transferase for their biological or transforming
functions. Therefore, farnesyltransferase inhibitors (FTIs),
tipifarnib (R115777) and lonafarnib (SCH66336) have been assessed
in PDAC preclinical models. Tipifarnib suppressed the growth of
human PDAC cell lines through modulation of the STAT3 and ERK
pathways (54). The FTI,
lonafarnib, synergized with taxanes to inhibit cell proliferation
in KRAS-mutant and KRAS-wild-type PDAC cells
(55).
Raf kinases (ARaf, BRaf, and CRaf/Raf1) comprise the
most significant effectors of KRAS-driven PDAC. In PDAC cell lines,
Raf inhibitor sorafenib (BAY 43-9006) that also targets VEGFR2 and
PDGFR-b, demonstrated strong antiproliferative and proapoptotic
effects, either alone (56) or in
combination with vitamin K (57).
Sorafenib also synergized with melatonin to suppress the growth of
PDAC both in vitro and in vivo (58). Sorafenib alone or in combination
with gemcitabine and EMAP inhibited PDAC cell proliferation. This
study also showed enhancement in animal survival by combination
treatment of sorafenib, gemcitabine and EMAP (median survival in
controls, gemcitabine, sorafenib, EMAP and gemcitabine + sorafenib
+ EMAP was 22, 29, 23, 25 and 36 days; P=0.004) (59). In PDAC orthotopic xenografts, a
selective c-RAF inhibitor, GW5074, exhibited a significant decrease
in tumor weight, either alone or in combination with polyamine
biosynthesis inhibitor difluoromethylornithine, but it had no
effect on improving animal survival (60).
Downstream to RAF, inhibitors for MEK and ERK have
been assessed extensively. In KRAS-mutant and wild-type
orthotopically implanted patient-derived tumors, the MEK-inhibitor,
trametinib, showed a significant reduction in tumor growth and
liver metastasis that was increased by the EGFR/HER-2 inhibitor
lapatinib (61). In
KRAS-mutant PDAC cell-derived xenograft (CDX) models,
trametinib increased the effects of nab-paclitaxel-based
chemotherapy by inhibiting tumor growth and enhancing animal
survival (compared with controls, increase in animal survival was
95 and 145% by nab-paclitaxel + gemcitabine and nab-paclitaxel +
gemcitabine + trametinib) (62).
Chao et al observed that MEK inhibitors (PD98059 or
trametinib) increased the antiproliferative and proapoptotic
effects of HDAC inhibitors (MPT0E028 or SAHA) in KRAS-mutant
and wild-type PDAC cell in vitro studies. Furthermore, in
AsPC-1 subcutaneous xenografts, MPT0E028 and PD98059 combination
revealed enhanced antitumor activity (63). Recently, MEK inhibitor mirdametinib
(PD0325901) exerted antitumor efficacy in MIA PaCa-2 PDAC
xenografts at day 4 and became refractory within a week after
treatment due to the involvement of clusterin expression (64). SCH772984 was the first ERK inhibitor
studied in PDAC, and it displayed the ability to suppress PDAC
xenograft growth (27). SCH772984
in combination with cucurbitacin, a natural tetracyclic triterpene,
synergized to induce growth inhibition and apoptosis of PDAC cells
in vitro and reducing tumor growth in PDAC subcutaneous
xenografts (percent inhibition in tumor volume by cucurbitacin,
SCH772984 and combination treatment was 63.8, 54.7 and 85,
respectively) (65). Furthermore,
SCH772984, in combination with autophagy inhibition,
synergistically reduced PDAC growth in vitro and in
vivo models (66,67). Recently, SCH772984 has been
demonstrated to combine synergistically with CDK4/6 inhibitor
palbociclib in KRAS-mutant PDAC cell lines and organoid
models (68). Another ERK
inhibitor, ulixertinib, has been shown to effectively inhibit the
growth of multiple PDAC cell lines and potentiate the cytotoxic
effect of gemcitabine in vitro and in vivo. This
study also showed that concurrent inhibition of HER (with afatinib)
or PI3K proteins (with GDC-0941) synergizes with ulixertinib in
suppressing PDAC cell growth in subcutaneous xenograft models
(69). Thus, MEK and ERK inhibitors
showed promise in preclinical studies in mitigating the progression
of PDAC.
Due to the extensive crosstalk and compensatory
mechanism between PI3K/AKT and MAPK pathways, simultaneous
targeting of these two pathways has been explored in PDAC. In PDAC
subcutaneous xenografts, a combination treatment with MEK inhibitor
mirdametinib and AKT inhibitor (API-2) induced activation of
apoptotic pathways, radiosensitized pancreatic cancer cells and
maximized tumor growth inhibition (70). Tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) has been shown to induce
apoptosis of cancer cells by engaging its cell surface death
receptors (71). A TRAIL-inducing
compound TIC10 (ONC201) that causes dual inhibition of AKT and
ERK-inducing TRAIL pathway-mediated cell death, inhibited PDAC cell
survival and proliferation in vitro, and demonstrated potent
antitumor activity in PDAC xenografts as well as synergized
gemcitabine activity (72).
Subsequently, Awasthi et al demonstrated that the combined
inhibition of PI3K and MAPK signaling with MK-2206 and trametinib,
respectively, displayed an enhanced nab-paclitaxel plus gemcitabine
(NPT+Gem) chemotherapy antitumor response in PDAC in vitro
and in vivo models. In this study, compared with controls,
enhancement in animal survival by NPT + Gem and NPT + Gem + MK-2206
+ trametinib treatment was 67 and 129%, respectively (73). These findings indicate the potential
of dual inhibition of downstream targets of
KRAS-mutation-driven signaling in combination with current
treatments for clinical PDAC therapy.
Dense desmoplastic stroma which comprises >90% of
the tumor mass is one of the hallmarks of PDAC. This stroma plays a
major role in PDAC progression, metastasis, and therapy resistance
(74). The TME in PDAC preclinical
models and patients with PDAC have several differences including
composition, oncogenic mutations and expression of oncogenic growth
factors and cytokines (75,76). Therefore, inhibiting the major
signaling pathways downstream of KRAS sometimes has different
effects on PDAC tumors in preclinical settings compared with their
effect on the progression and survival of patients with PDAC
(76).
Copanlisib (BAY 80-6946), a pan-class I PI3K
inhibitor, in combination with gemcitabine or cisplatin plus
gemcitabine in solid tumors including PDAC demonstrated favorable
clinical efficacy (out of 4 evaluated patients with PDAC, 1 partial
response, 2 stable disease) with an acceptable toxicity profile in
a phase I study (77). In a phase I
trial of advanced patients with PDAC who have not received any
cytotoxic chemotherapy except as adjuvant therapy, alpelisib
(BYL719), an α-specific PI3K inhibitor, in combination with
nab-paclitaxel plus gemcitabine, was safely administered and the
median progression-free survival (PFS) and OS were 5.36 and 8.74
months (78). Another pan-class I
PI3K inhibitor buparlisib (BKM120), in a phase I study which
included patients with metastatic PDAC with RAS- or
BRAF-mutation, in combination with trametinib showed minimum
clinical activity (best overall response was stable disease)
(79). In a phase I study of
patients with refractory solid tumors including PDAC, buparlisib
combination with mFOLFOX6 caused an increase in toxicity and one
patient with stage IV PDAC exhibited a 47% decrease in measurable
disease from baseline (80). A
phase Ib study of PI3K inhibitor idelalisib alone or in combination
with nab-paclitaxel or mFOLFOX6 in patients with PDAC was
terminated prematurely due to severe toxicity issues observed in a
phase III clinical study of idelalisib for hematological
malignancies (81).
Perifosine (KRX-0401) and MK-2206 are the most
studied AKT inhibitors for PDAC clinical therapy. Although
perifosine exhibited significant activity in PDAC preclinical
studies, two phase II clinical trials using this drug in patients
with advanced PDAC who were previously untreated (82) or had one prior systemic therapy
(83) were halted prematurely due
to a lack of efficacy and high toxicity. A phase II study compared
MK-2206 plus MEK inhibitor selumetinib with mFOLFOX in
gemcitabine-refractory patients with metastatic PDAC (84). The median OS was shorter in the
MK-2206 plus selumetinib versus mFOLFOX (3.9 vs. 6.7 months;
P=0.15) and the median PFS was also inferior (1.9 vs. 2.0 months;
P=0.02) (84). Subsequently, a
phase I study of MK-2206 plus CDK inhibitor dinaciclib in patients
with previously treated metastatic PDAC demonstrated disappointing
results (median survival 2.2 months; survival rates at 6 and 12
months 11 and 5%, respectively) (85).
The mTOR inhibitor everolimus in a phase II study in
gemcitabine refractory metastatic PDAC patients demonstrated
minimal clinical activity and median OS and PFS were 4.5 and 1.8
months, respectively (86). A phase
II study of temsirolimus in gemcitabine refractory PDAC patients
was closed to accrual due to significant adverse effects and median
OS and PFS were 44 and 19 days, respectively (87). Considering the disappointing results
of mTOR inhibitors as monotherapy, several clinical studies
evaluated mTOR inhibitors in combination with other cytotoxic or
targeted agents.
A phase II study of everolimus plus erlotinib in
patients with advanced PDAC who received at least one prior
gemcitabine-based regimen showed minimal clinical activity with an
OS of 87 days and a PFS of 49 days (87). The oral regimen with the combination
of everolimus and capecitabine in a phase II trial of patients with
advanced PDAC who were untreated (first-line) or had prior
chemotherapy (second-line) demonstrated a moderate activity (median
OS of 8.9 months and a PFS of 3.6 months) with an acceptable
toxicity profile (88).
Temsirolimus plus gemcitabine in previously untreated patients with
advanced PDAC failed to show any meaningful clinical response in a
phase II study (OS, 4.95 months; PFS, 2.69 months) (89). Furthermore, the combination of
temsirolimus and docetaxel in patients with refractory solid tumors
including PDAC did not meet its primary objective due to
dose-limiting toxicities in a phase I study (90).
The combination of PI3K/AKT/mTOR inhibitors with
other targeted therapies has also been evaluated in several
clinical studies. A phase I study evaluated everolimus and the
CDK4/6 inhibitor ribociclib as a third-line therapy in patients
with PDAC after disease progression on both 5-fluorouracil and
gemcitabine. This study demonstrated favorable tolerability with a
decrease in CDK4/6-regulated genes but median OS and PFS were only
3.7 and 1.8 months, respectively (91). A phase I clinical study is currently
ongoing to evaluate the combination of the PI3K/mTOR inhibitor,
gedatolisib, and the CDK4/6 inhibitor, palbociclib, in patients
with solid tumors (including PDAC) that is metastatic or
unresectable and resistant to standard therapy (NCT03065062). Based
on suggestive crosstalk between PI3K/AKT/mTOR and hedgehog (Hh)
signaling, the mTOR inhibitor sirolimus was assessed in combination
with the Hh inhibitor vismodegib in patients with advanced PDAC.
However, in contrast with preclinical results, the combination of
vismodegib and sirolimus only showed stable disease in a subgroup
of patients (92). EGFR
overexpression has been reported in up to 95% of patients with
PDAC. The small molecule EGFR inhibitor erlotinib in combination
with gemcitabine is an approved therapy for PDAC (93). Because PI3K/mTOR signaling is a
well-established resistance mechanism to erlotinib, the combination
of mTOR inhibitors and erlotinib was assessed in PDAC clinical
trials. However, the combination of erlotinib with everolimus
(87) or temsirolimus (94) did not demonstrate any meaningful
clinical benefit. In addition, a phase I/II study of the
combinations of temsirolimus with the EGFR monoclonal antibody
cetuximab and capecitabine in patients with advanced PDAC
demonstrated no clinical benefit with a median OS of 5 months
(95).
Among FTIs, tipifarnib (R115777) has been assessed
in PDAC clinical trials. In a phase II study, treatment with
tipifarnib in previously untreated patients with metastatic PDAC
resulted in partial inhibition of farnesyltransferase activity but
it did not exhibit antitumor activity (median OS, 19.7 weeks; PFS,
4.9 weeks; and the estimated 6-month survival rate was 25%)
(96). Another phase II study of
tipifarnib in previously untreated patients with metastatic PDAC
did not show any clinical efficacy and the median OS, PFS and
6-month survival rate were 2.6 months, 1.4 months and 19%,
respectively (97). In a phase III
trial, the tipifarnib combination with gemcitabine did not exhibit
any clinical benefit compared with gemcitabine alone (median OS,
193 vs. 182 days; median PFS, 112 vs. 109 days; 6-month and 1-year
survival rates were 53 and 27 vs. 49 and 24%, respectively)
(98). In a phase II trial, weekly
paclitaxel, gemcitabine and radiation (CXRT) was compared with CXRT
plus maintenance tipifarnib. This study demonstrated that the
addition of tipifarnib was associated with a broad range of
toxicities and there was no clinical benefit (median OS, 11.5 vs.
8.9 months for the CXRT and CXRT + tipifarnib, respectively)
(99).
Raf, a signaling protein downstream of Ras, is an
important target for cancer therapy. Sorafenib (BAY 43-9006) was
extensively assessed in combination with chemotherapy in several
clinical studies of metastatic PDAC. A phase I study of sorafenib
plus gemcitabine in advanced unresectable or metastatic PDAC
revealed favorable tolerability with 56.6% of patients (n=13)
achieving disease stabilization (100). Subsequently, a phase II trial of
sorafenib alone versus sorafenib plus gemcitabine in patients with
metastatic PDAC who received no prior chemotherapy or completed
prior adjuvant chemotherapy >6 months before study entry,
demonstrated no clinical benefit (median OS, 4.3 vs. 6.5 months;
median PFS, 2.3 vs. 2.9 months) (101). A phase III trial of gemcitabine
plus sorafenib and gemcitabine plus placebo in previously untreated
patients with advanced PDAC exhibited a median OS of 9.2 and 8
months, and a median PFS of 5.7 and 3.8 months (102). Another phase II study of cisplatin
plus gemcitabine with and without sorafenib in patients with
metastatic PDAC also showed no clinical activity of sorafenib
addition (median OS, 7.5 vs. 8.3 months; median PFS, 4.3 vs. 4.5
months) (103). A phase I study
demonstrated modest clinical activity of sorafenib with concurrent
radiation therapy and gemcitabine in advanced PDAC patients. The
median OS and PFS for 25 evaluable patients were 12.6 and 10.6
months, respectively (104).
A phase II trial of gemcitabine plus trametinib or
placebo in previously untreated patients with PDAC revealed no
significant clinical benefits with an observed median OS of 8.4 vs.
6.7 months (P=0.453), a median PFS of 16 vs. 15 weeks (P=0.349),
and an overall response rate 22 vs. 18%. These outcomes were
independent of KRAS mutations determined by circulating free
DNA (105). A phase I study of
patients with KRAS-mutant solid tumors including PDAC showed
manageable toxicity of trametinib plus lapatinib but there was no
preliminary sign of antitumor activity in patients with PDAC
(106). Based on preclinical
evidence of synergistic antitumor response of MEK and EGFR
inhibition in PDAC, a phase II study evaluated trametinib plus
erlotinib in chemotherapy-refractory PDAC patients. This trial
demonstrated a modest clinical efficacy of this combination in PDAC
(median OS, 7.3 months; median PFS, 1.9 months) (107). Recently, a phase II study in
locally recurrent, KRAS-mutant and PD-1 positive PDAC
patients after surgery followed by chemotherapy (mFOLFIRINOX or
5-FU) demonstrated promising clinical efficacy after receiving the
combination of radiotherapy plus pembrolizumab and trametinib
(median OS and PFS of 14.9 and 8.2 months, respectively) compared
with radiotherapy plus gemcitabine (median OS and PFS of 12.8 and
5.4 months, respectively) (108).
Recently, a phase II trial of patients with advanced PDAC whose
disease progressed after first-line chemotherapy with the
combination treatment of trametinib and an oral FAK inhibitor,
GSK2256098, demonstrated dismal clinical activity with a median PFS
of 1.6 months and an OS of 3.6 months (109). A phase I trial is currently
ongoing to evaluate trametinib and hydroxychloroquine in patients
with PDAC (NCT03825289).
The MEK inhibitor, selumetinib, was compared with
capecitabine in a phase II study in patients with PDAC who failed
first-line gemcitabine therapy. In this trial, selumetinib was well
tolerated with a manageable safety profile but showed no
significant difference in OS compared with capecitabine (5.4 vs.
5.0 months) (110). A recent phase
II study assessing selumetinib in KRASG12R-mutant
PDAC patients who received two or more lines of systemic
chemotherapy (~87.5% patients) demonstrated dismal clinical
activity, revealing a 3-month median PFS and a 9-month median OS
(111). A phase I study evaluating
the MEK inhibitor, binimetinib plus the anti-PD-L1 antibody
avelumab in patients with metastatic PDAC after 1–2 prior lines of
therapy was terminated due to toxicity issues (112). A phase I study of binimetinib plus
hydroxychloroquine in patients with metastatic PDAC who had at
least one line of systemic therapy and harbor a KRAS
mutation is currently ongoing (113).
The ERK inhibitor ulixertinib in combination with
gemcitabine plus nab-paclitaxel in untreated patients with
metastatic PDAC showed potentially similar efficacy (the median PFS
and OS were 5.46 and 12.23 months, respectively) as chemotherapy
and the study was terminated due to treatment-related adverse
events (114). A phase I study of
ulixertinib and palbociclib is currently ongoing in patients with
advanced solid tumors including patients with PDAC who have
received at least one line of therapy in the metastatic setting
(115).
Attributed to the synergistic antitumor response in
preclinical studies, simultaneous targeting of MAPK and PI3K
pathways was assessed in several clinical studies in patients with
KRAS-mutant PDAC. A phase I clinical trial of patients with
solid tumors including PDAC with the PI3K/mTOR inhibitor GSK2126458
and trametinib demonstrated poor tolerability and limited antitumor
efficacy (only 2 out of 7 patients with PDAC had stable disease)
(116). As aforementioned in the
previous section, a phase II study on gemcitabine-refractory PDAC
patients demonstrated no clinical benefit of MK-2206 plus
selumetinib therapy compared with mFOLFOX (84). Furthermore, a phase I study
evaluating the combination of BKM120 plus binimetinib did not
report any clinical efficacy in patients with advanced PDAC
(117).
Until recently, direct KRAS targeting by small
molecule inhibitors was not possible and indirect therapeutic
strategies including targeting RAS signaling and metabolic pathways
demonstrated disappointing results in clinical trials in treating
patients with KRAS-mutant cancers. Therefore, recently,
numerous drugs with the potential of direct KRAS targeting have
been evaluated in KRAS-mutant cancers including PDAC
(Table III).
Advancements in drug discovery and significant
efforts by several groups led to a breakthrough by successfully
developing compounds to target the
KRASG12C-mutant allele (118), paving the way for the development
of two inhibitors of this class, sotorasib (AMG510) (119) and adagrasib (MRTX849) (120). These two inhibitors are more
relevant in NSCLC where the mutation frequency of
KRASG12C is 13.8% but less relevant in colorectal
and pancreatic cancer where KRASG12C mutation is
3.2% and <1%, respectively.
Targeting KRAS signaling has great potential for
therapeutic intervention in several solid tumors including PDAC.
While downstream effector signal inhibitors have shown promise in
preclinical studies, these drugs displayed limited clinical
benefits, largely due to differences in the TME in these settings
leading to enhanced toxicity and induction of adaptive resistance
mechanisms in patients. Recent advances in preclinical models such
as PDX and tumor organoids which better recapitulate patient tumor
biology have helped design promising combination therapies
targeting downstream KRAS effectors to overcome toxicity and
resistance issues and improve PDAC clinical therapy. A combination
of BRAF inhibitor vemurafenib plus sorafenib (phase II),
binimetinib plus hydroxychloroquine, and binimetinib plus
palbociclib are currently under clinical investigation
(NCT05068752, NCT04132505, NCT04870034) in KRAS-mutant PDAC
patients. Due to limited success with downstream KRAS inhibition
strategy, attention has recently shifted to direct mutant-specific
KRAS inhibitors and pan-KRAS inhibitors. Novel drugs such as
sotorasib and adagrasib have been effective at inhibiting
KRAS at G12C, but these are largely ineffective in PDAC as
the major mutant isoforms are found at other locations. Recent
advancements in KRASG12D-mutant-specific
inhibitors (such as MRTX1133) are very encouraging and their
success could be a game changer in PDAC clinical therapy. A phase I
study is evaluating mesenchymal stromal cells-derived exosomes with
KrasG12DsiRNA (iExosomes) in metastatic PDAC
patients with KRASG12D mutation (NCT03608631). In
addition, the upstream pan-KRAS inhibitors RMC-6236, RMC-4550 and
BI 1701963 are currently under clinical investigation in
KRAS-mutant PDAC (NCT05379985, NCT04916236, NCT04975256).
These pan-KRAS inhibitors that have the ability to target a broad
range of oncogenic KRAS variants, including all major G12
and G13 subtypes, could make a marked clinical impact in the
majority of patients with PDAC irrespective of their KRAS
mutation subtypes. Additionally, KRAS targeting therapeutic
vaccines including peptides, dendritic cells and mRNA vaccines have
recently emerged as a promising therapy and these are currently
under clinical investigation (NCT05013216, NCT03592888,
NCT03948763) for potential improvement of PDAC clinical
therapy.
Not applicable.
The present study was financially supported by Indiana
University School of Medicine funds.
Not applicable.
JZ and NA contributed to the conception and design
of the article and interpretation of the relevant literature. JZ
and NA prepared the original draft of the manuscript. JZ, LD, MSH
and NA performed formal analysis. JZ, UvH and NA wrote, reviewed,
and edited the manuscript. NA acquired funding. Data authentication
is not applicable. All authors have read and approved the final
version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Cox AD and Der CJ: Ras history: The saga
continues. Small GTPases. 1:2–27. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bryant KL, Mancias JD, Kimmelman AC and
Der CJ: KRAS: Feeding pancreatic cancer proliferation. Trends
Biochem Sci. 39:91–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hobbs GA, Der CJ and Rossman KL: RAS
isoforms and mutations in cancer at a glance. J Cell Sci.
129:1287–1292. 2016.PubMed/NCBI
|
|
4
|
Zhu C, Guan X, Zhang X, Luan X, Song Z,
Cheng X, Zhang W and Qin JJ: Targeting KRAS mutant cancers: From
druggable therapy to drug resistance. Mol Cancer. 21:1592022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Waters AM and Der CJ: KRAS: The critical
driver and therapeutic target for pancreatic cancer. Cold Spring
Harb Perspect Med. 8:a0314352018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aguirre AJ, Bardeesy N, Sinha M, Lopez L,
Tuveson DA, Horner J, Redston MS and DePinho RA: Activated Kras and
Ink4a/Arf deficiency cooperate to produce metastatic pancreatic
ductal adenocarcinoma. Genes Dev. 17:3112–3126. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perera RM, Stoykova S, Nicolay BN, Ross
KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK,
Ferrone CR, et al: Transcriptional control of autophagy-lysosome
function drives pancreatic cancer metabolism. Nature. 524:361–365.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang S, Wang X, Contino G, Liesa M, Sahin
E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al:
Pancreatic cancers require autophagy for tumor growth. Genes Dev.
25:717–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buscail L, Bournet B and Cordelier P: Role
of oncogenic KRAS in the diagnosis, prognosis and treatment of
pancreatic cancer. Nat Rev Gastroenterol Hepatol. 17:153–168. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dey P, Kimmelman AC and DePinho RA:
Metabolic Codependencies in the tumor microenvironment. Cancer
Discov. 11:1067–1081. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen X, Niu N and Xue J: Oncogenic KRAS
triggers metabolic reprogramming in pancreatic ductal
adenocarcinoma. J Transl Int Medicine 0. -. 2022. View Article : Google Scholar
|
|
13
|
Muthalagu N, Monteverde T,
Raffo-Iraolagoitia X, Wiesheu R, Whyte D, Hedley A, Laing S,
Kruspig B, Upstill-Goddard R, Shaw R, et al: Repression of the type
I interferon pathway underlies MYC- and KRAS-dependent evasion of
NK and B cells in pancreatic ductal adenocarcinoma. Cancer Discov.
10:872–887. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dey P, Li J, Zhang J, Chaurasiya A, Strom
A, Wang H, Liao WT, Cavallaro F, Denz P, Bernard V, et al:
Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer
cells utilizes cytokines from the tumor microenvironment. Cancer
Discov. 10:608–625. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Santana-Codina N, Roeth AA, Zhang Y, Yang
A, Mashadova O, Asara JM, Wang X, Bronson RT, Lyssiotis CA, Ying H
and Kimmelman AC: Oncogenic KRAS supports pancreatic cancer through
regulation of nucleotide synthesis. Nat Commun. 9:49452018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Son J, Lyssiotis CA, Ying H, Wang X, Hua
S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et
al: Glutamine supports pancreatic cancer growth through a
KRAS-regulated metabolic pathway. Nature. 496:101–105. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Raho S, Capobianco L, Malivindi R, Vozza
A, Piazzolla C, Leonardis FD, Gorgoglione R, Scarcia P, Pezzuto F,
Agrimi G, et al: KRAS-regulated glutamine metabolism requires
UCP2-mediated aspartate transport to support pancreatic cancer
growth. Nat Metab. 2:1373–1381. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kessler D, Gmachl M, Mantoulidis A, Martin
LJ, Zoephel A, Mayer M, Gollner A, Covini D, Fischer S, Gerstberger
T, et al: Drugging an undruggable pocket on KRAS. Proc Natl Acad
Sci USA. 116:15823–15829. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vigil D, Cherfils J, Rossman KL and Der
CJ: Ras superfamily GEFs and GAPs: Validated and tractable targets
for cancer therapy? Nat Rev Cancer. 10:842–857. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cuesta C, Arévalo-Alameda C and Castellano
E: The importance of being PI3K in the RAS signaling network. Genes
(Basel). 12:10942021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Krygowska AA and Castellano E: PI3K: A
crucial piece in the RAS signaling puzzle. Cold Spring Harb
Perspect Med. 8:a0314502018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hemmings BA and Restuccia DF: PI3K-PKB/Akt
pathway. Cold Spring Harb Perspect Biol. 4:a0111892012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fritsch R and Downward J: SnapShot: Class
I PI3K isoform signaling. Cell. 154:940–940.e941. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peng Y, Wang Y, Zhou C, Mei W and Zeng C:
PI3K/Akt/mTOR pathway and its role in cancer therapeutics: Are we
making headway? Front Oncol. 12:8191282022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rascio F, Spadaccino F, Rocchetti MT,
Castellano G, Stallone G, Netti GS and Ranieri E: The pathogenic
role of PI3K/AKT pathway in cancer onset and drug resistance: An
updated review. Cancers (Basel). 13:39492021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Witkiewicz AK, McMillan EA, Balaji U, Baek
G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et
al: Whole-exome sequencing of pancreatic cancer defines genetic
diversity and therapeutic targets. Nat Commun. 6:67442015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hayes TK, Neel NF, Hu C, Gautam P, Chenard
M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, et al:
Long-term ERK inhibition in KRAS-mutant pancreatic cancer is
associated with MYC degradation and senescence-like growth
suppression. Cancer Cell. 29:75–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Workman P, Clarke PA, Raynaud FI and van
Montfort RL: Drugging the PI3 kinome: From chemical tools to drugs
in the clinic. Cancer Res. 70:2146–2157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ng SS, Tsao MS, Nicklee T and Hedley DW:
Wortmannin inhibits pkb/akt phosphorylation and promotes
gemcitabine antitumor activity in orthotopic human pancreatic
cancer xenografts in immunodeficient mice. Clin Cancer Res.
7:3269–3275. 2001.PubMed/NCBI
|
|
30
|
Fujiwara M, Izuishi K, Sano T, Hossain MA,
Kimura S, Masaki T and Suzuki Y: Modulating effect of the
PI3-kinase inhibitor LY294002 on cisplatin in human pancreatic
cancer cells. J Exp Clin Cancer Res. 27:762008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Y, Kuramitsu Y, Baron B, Kitagawa T,
Tokuda K, Akada J, Maehara SI, Maehara Y and Nakamura K: PI3K
inhibitor LY294002, as opposed to wortmannin, enhances AKT
phosphorylation in gemcitabine-resistant pancreatic cancer cells.
Int J Oncol. 50:606–612. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Z, Luo G and Qiu Z: Akt inhibitor
MK-2206 reduces pancreatic cancer cell viability and increases the
efficacy of gemcitabine. Oncol Lett. 19:1999–2004. 2020.PubMed/NCBI
|
|
33
|
Hu C, Dadon T, Chenna V, Yabuuchi S,
Bannerji R, Booher R, Strack P, Azad N, Nelkin BD and Maitra A:
Combined inhibition of cyclin-dependent kinases (Dinaciclib) and
AKT (MK-2206) blocks pancreatic tumor growth and metastases in
patient-derived xenograft models. Mol Cancer Ther. 14:1532–1539.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Massihnia D, Avan A, Funel N, Maftouh M,
van Krieken A, Granchi C, Raktoe R, Boggi U, Aicher B, Minutolo F,
et al: Phospho-Akt overexpression is prognostic and can be used to
tailor the synergistic interaction of Akt inhibitors with
gemcitabine in pancreatic cancer. J Hematol Oncol. 10:92017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Utomo WK, Narayanan V, Biermann K, van
Eijck CHJ, Bruno MJ, Peppelenbosch MP and Braat H: mTOR is a
promising therapeutical target in a subpopulation of pancreatic
adenocarcinoma. Cancer Lett. 346:309–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith
J and Rozengurt E: Different patterns of Akt and ERK feedback
activation in response to rapamycin, active-site mTOR inhibitors
and metformin in pancreatic cancer cells. PLoS One. 8:e572892013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dai ZJ, Gao J, Kang HF, Ma YG, Ma XB, Lu
WF, Lin S, Ma HB, Wang XJ and Wu WY: Targeted inhibition of
mammalian target of rapamycin (mTOR) enhances radiosensitivity in
pancreatic carcinoma cells. Drug Des Devel Ther. 7:149–159. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Azzariti A, Porcelli L, Gatti G, Nicolin A
and Paradiso A: Synergic antiproliferative and antiangiogenic
effects of EGFR and mTor inhibitors on pancreatic cancer cells.
Biochem Pharmacol. 75:1035–1044. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Morran DC, Wu J, Jamieson NB, Mrowinska A,
Kalna G, Karim SA, Au AYM, Scarlett CJ, Chang DK, Pajak MZ, et al:
Targeting mTOR dependency in pancreatic cancer. Gut. 63:1481–1489.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cifarelli V, Lashinger LM, Devlin KL,
Dunlap SM, Huang J, Kaaks R, Pollak MN and Hursting SD: Metformin
and rapamycin reduce pancreatic cancer growth in obese prediabetic
mice by distinct MicroRNA-regulated mechanisms. Diabetes.
64:1632–1642. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tuncyurek P, Mayer JM, Klug F, Dillmann S,
Henne-Bruns D, Keller F and Stracke S: Everolimus and mycophenolate
mofetil sensitize human pancreatic cancer cells to gemcitabine in
vitro: A novel adjunct to standard chemotherapy? Eur Surg Res.
39:380–387. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Peng T and Dou QP: Everolimus inhibits
growth of gemcitabine-resistant pancreatic cancer cells via
induction of caspase-dependent apoptosis and G(2) /M arrest. J Cell
Biochem. 118:2722–2730. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pawaskar DK, Straubinger RM, Fetterly GJ,
Ma WW and Jusko WJ: Interactions of everolimus and sorafenib in
pancreatic cancer cells. AAPS J. 15:78–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wei F, Zhang Y, Geng L, Zhang P, Wang G
and Liu Y: mTOR inhibition induces EGFR feedback activation in
association with its resistance to human pancreatic cancer. Int J
Mol Sci. 16:3267–3282. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pawaskar DK, Straubinger RM, Fetterly GJ,
Hylander BH, Repasky EA, Ma WW and Jusko WJ: Synergistic
interactions between sorafenib and everolimus in pancreatic cancer
xenografts in mice. Cancer Chemother Pharmacol. 71:1231–1240. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Witkiewicz AK, Balaji U, Eslinger C,
McMillan E, Conway W, Posner B, Mills GB, O'Reilly EM and Knudsen
ES: Integrated patient-derived models delineate individualized
therapeutic vulnerabilities of pancreatic cancer. Cell Rep.
16:2017–2031. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL
and Reddy SA: The rapamycin analog CCI-779 is a potent inhibitor of
pancreatic cancer cell proliferation. Biochem Biophys Res Commun.
331:295–302. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ito D, Fujimoto K, Mori T, Kami K, Koizumi
M, Toyoda E, Kawaguchi Y and Doi R: In vivo antitumor effect of the
mTOR inhibitor CCI-779 and gemcitabine in xenograft models of human
pancreatic cancer. Int J Cancer. 118:2337–2343. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cao P, Maira SM, García-Echeverría C and
Hedley DW: Activity of a novel, dual PI3-kinase/mTor inhibitor
NVP-BEZ235 against primary human pancreatic cancers grown as
orthotopic xenografts. Br J Cancer. 100:1267–1276. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Awasthi N, Yen PL, Schwarz MA and Schwarz
RE: The efficacy of a novel, dual PI3K/mTOR inhibitor NVP-BEZ235 to
enhance chemotherapy and antiangiogenic response in pancreatic
cancer. J Cell Biochem. 113:784–791. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Venkannagari S, Fiskus W, Peth K, Atadja
P, Hidalgo M, Maitra A and Bhalla KN: Superior efficacy of
co-treatment with dual PI3K/mTOR inhibitor NVP-BEZ235 and
pan-histone deacetylase inhibitor against human pancreatic cancer.
Oncotarget. 3:1416–1427. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lee JE, Woo MG, Jung KH, Shin SM, Son MK,
Fang Z, Yan HH, Park JH, Yoon YC, Kim YS and Hong SS: Combination
therapy of the active KRAS-targeting antibody inRas37 and a PI3K
inhibitor in pancreatic cancer. Biomol Ther (Seoul). 30:274–283.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Soares HP, Ming M, Mellon M, Young SH, Han
L, Sinnet-Smith J and Rozengurt E: Dual PI3K/mTOR inhibitors induce
rapid overactivation of the MEK/ERK pathway in human pancreatic
cancer cells through suppression of mTORC2. Mol Cancer Ther.
14:1014–1023. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Venkatasubbarao K, Choudary A and Freeman
JW: Farnesyl transferase inhibitor (R115777)-induced inhibition of
STAT3(Tyr705) phosphorylation in human pancreatic cancer cell lines
require extracellular signal-regulated kinases. Cancer Res.
65:2861–2871. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shi B, Yaremko B, Hajian G, Terracina G,
Bishop WR, Liu M and Nielsen LL: The farnesyl protein transferase
inhibitor SCH66336 synergizes with taxanes in vitro and enhances
their antitumor activity in vivo. Cancer Chemother Pharmacol.
46:387–393. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ulivi P, Arienti C, Amadori D, Fabbri F,
Carloni S, Tesei A, Vannini I, Silvestrini R and Zoli W: Role of
RAF/MEK/ERK pathway, p-STAT-3 and Mcl-1 in sorafenib activity in
human pancreatic cancer cell lines. J Cell Physiol. 220:214–221.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wei G, Wang M and Carr BI: Sorafenib
combined vitamin K induces apoptosis in human pancreatic cancer
cell lines through RAF/MEK/ERK and c-Jun NH2-terminal kinase
pathways. J Cell Physiol. 224:112–119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fang Z, Jung KH, Yan HH, Kim SJ, Rumman M,
Park JH, Han B, Lee JE, Kang YW, Lim JH and Hong SS: Melatonin
synergizes with sorafenib to suppress pancreatic cancer via
melatonin receptor and PDGFR-β/STAT3 pathway. Cell Physiol Biochem.
47:1751–1768. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Awasthi N, Zhang C, Hinz S, Schwarz MA and
Schwarz RE: Enhancing sorafenib-mediated sensitization to
gemcitabine in experimental pancreatic cancer through EMAP II. J
Exp Clin Cancer Res. 32:122013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nakkina SP, Gitto SB, Beardsley JM, Pandey
V, Rohr MW, Parikh JG, Phanstiel O IV and Altomare DA: DFMO
improves survival and increases immune cell infiltration in
association with MYC downregulation in the pancreatic tumor
microenvironment. Int J Mol Sci. 22:131752021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Walters DM, Lindberg JM, Adair SJ, Newhook
TE, Cowan CR, Stokes JB, Borgman CA, Stelow EB, Lowrey BT,
Chopivsky ME, et al: Inhibition of the growth of patient-derived
pancreatic cancer xenografts with the MEK inhibitor trametinib is
augmented by combined treatment with the epidermal growth factor
receptor/HER2 inhibitor lapatinib. Neoplasia. 15:143–155. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Awasthi N, Monahan S, Stefaniak A, Schwarz
MA and Schwarz RE: Inhibition of the MEK/ERK pathway augments
nab-paclitaxel-based chemotherapy effects in preclinical models of
pancreatic cancer. Oncotarget. 9:5274–5286. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Chao MW, Chang LH, Tu HJ, Chang CD, Lai
MJ, Chen YY, Liou JP, Teng CM and Pan SL: Combination treatment
strategy for pancreatic cancer involving the novel HDAC inhibitor
MPT0E028 with a MEK inhibitor beyond K-Ras status. Clin
Epigenetics. 11:852019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Amada K, Hijiya N, Ikarimoto S, Yanagihara
K, Hanada T, Hidano S, Kurogi S, Tsukamoto Y, Nakada C, Kinoshita
K, et al: Involvement of clusterin expression in the refractory
response of pancreatic cancer cells to a MEK inhibitor. Cancer Sci.
114:2189–2202. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhou J, Zhao T, Ma L, Liang M, Guo YJ and
Zhao LM: Cucurbitacin B and SCH772984 exhibit synergistic
anti-pancreatic cancer activities by suppressing EGFR,
PI3K/Akt/mTOR, STAT3 and ERK signaling. Oncotarget.
8:103167–103181. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Bryant KL, Stalnecker CA, Zeitouni D,
Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM,
George SD, et al: Combination of ERK and autophagy inhibition as a
treatment approach for pancreatic cancer. Nat Med. 25:628–640.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yan Z, Ohuchida K, Fei S, Zheng B, Guan W,
Feng H, Kibe S, Ando Y, Koikawa K, Abe T, et al: Inhibition of
ERK1/2 in cancer-associated pancreatic stellate cells suppresses
cancer-stromal interaction and metastasis. J Exp Clin Cancer Res.
38:2212019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Goodwin CM, Waters AM, Klomp JE, Javaid S,
Bryant KL, Stalnecker CA, Drizyte-Miller K, Papke B, Yang R, Amparo
AM, et al: Combination therapies with CDK4/6 inhibitors to treat
KRAS-mutant pancreatic cancer. Cancer Res. 83:141–157. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Jiang H, Xu M, Li L, Grierson P,
Dodhiawala P, Highkin M, Zhang D, Li Q, Wang-Gillam A and Lim KH:
Concurrent HER or PI3K inhibition potentiates the antitumor effect
of the erk inhibitor ulixertinib in preclinical pancreatic cancer
models. Mol Cancer Ther. 17:2144–2155. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Williams TM, Flecha AR, Keller P, Ram A,
Karnak D, Galbán S, Galbán CJ, Ross BD, Lawrence TS, Rehemtulla A
and Sebolt-Leopold J: Cotargeting MAPK and PI3K signaling with
concurrent radiotherapy as a strategy for the treatment of
pancreatic cancer. Mol Cancer Ther. 11:1193–1202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Dai X, Zhang J, Arfuso F, Chinnathambi
Zayed ME, Alharbi SA, Kumar AP, Ahn KS and Sethi G: Targeting
TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural
products as a potential therapeutic approach for cancer therapy.
Exp Biol Med (Maywood). 240:760–773. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang Q, Wang H, Ran L, Zhang Z and Jiang
R: The preclinical evaluation of TIC10/ONC201 as an anti-pancreatic
cancer agent. Biochem Biophys Res Commun. 476:260–266. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Awasthi N, Kronenberger D, Stefaniak A,
Hassan MS, von Holzen U, Schwarz MA and Schwarz RE: Dual inhibition
of the PI3K and MAPK pathways enhances nab-paclitaxel/gemcitabine
chemotherapy response in preclinical models of pancreatic cancer.
Cancer Lett. 459:41–49. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Thomas D and Radhakrishnan P:
Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis.
Mol Cancer. 18:142019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Krempley BD and Yu KH: Preclinical models
of pancreatic ductal adenocarcinoma. Chin Clin Oncol. 6:252017.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yu Y, Yang G, Huang H, Fu Z, Cao Z, Zheng
L, You L and Zhang T: Preclinical models of pancreatic ductal
adenocarcinoma: Challenges and opportunities in the era of
precision medicine. J Exp Clin Cancer Res. 40:82021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kim RD, Alberts SR, Peña C, Genvresse I,
Ajavon-Hartmann A, Xia C, Kelly A and Grilley-Olson JE: Phase I
dose-escalation study of copanlisib in combination with gemcitabine
or cisplatin plus gemcitabine in patients with advanced cancer. Br
J Cancer. 118:462–470. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Soares HP, Al-Toubah TE, Kim RD, Kim J,
Lewis NK and Mahipal A: Final report: A phase I trial of BYL719 in
combination with gemcitabine and nab-paclitaxel in locally advanced
and metastatic pancreatic cancer. J Clin Oncol. 36:398. 2018.
View Article : Google Scholar
|
|
79
|
Bedard PL, Tabernero J, Janku F, Wainberg
ZA, Paz-Ares L, Vansteenkiste J, Cutsem EV, Pérez-García J, Stathis
A, Britten CD, et al: A phase Ib dose-escalation study of the oral
pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral
MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected
advanced solid tumors. Clin Cancer Res. 21:730–738. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
McRee AJ, Sanoff HK, Carlson C, Ivanova A
and O'Neil BH: A phase I trial of mFOLFOX6 combined with the oral
PI3K inhibitor BKM120 in patients with advanced refractory solid
tumors. Invest New Drugs. 33:1225–1231. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Borazanci E, Pishvaian MJ, Nemunaitis J,
Weekes C, Huang J and Rajakumaraswamy N: A Phase Ib study of
single-agent idelalisib followed by idelalisib in combination with
chemotherapy in patients with metastatic pancreatic ductal
adenocarcinoma. Oncologist. 25:e1604–e1613. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Marsh RW, Lima CM, Levy DE, Mitchell EP,
Rowland KM Jr and Benson AB III: A phase II trial of perifosine in
locally advanced, unresectable, or metastatic pancreatic
adenocarcinoma. Am J Clin Oncol. 30:26–31. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hedley D, Moore MJ, Hirte H, Siu L,
Vincent M, Jonker H. Mwang D, Nagai J and Dancey J: A phase II
trial of perifosine as second line therapy for advanced pancreatic
cancer. A study of the princess margaret hospital [PMH] phase II
consortium. J Clin Oncol. 23:4166. 2005. View Article : Google Scholar
|
|
84
|
Chung V, McDonough S, Philip PA, Cardin D,
Wang-Gillam A, Hui L, Tejani MA, Seery TE, Dy IA, Al Baghdadi T, et
al: Effect of Selumetinib and MK-2206 vs Oxaliplatin and
Fluorouracil in patients with metastatic pancreatic cancer after
prior therapy: SWOG S1115 study randomized clinical trial. JAMA
Oncol. 3:516–522. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Murphy AG, Zahurak M, Shah M, Weekes CD,
Hansen A, Siu LL, Spreafico A, LoConte N, Anders NM, Miles T, et
al: A phase I study of dinaciclib in combination with MK-2206 in
patients with advanced pancreatic cancer. Clin Transl Sci.
13:1178–1188. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wolpin BM, Hezel AF, Abrams T, Blaszkowsky
LS, Meyerhardt JA, Chan JA, Enzinger PC, Allen B, Clark JW, Ryan DP
and Fuchs CS: Oral mTOR inhibitor everolimus in patients with
gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol.
27:193–198. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Javle MM, Shroff RT, Xiong H, Varadhachary
GA, Fogelman D, Reddy SA, Davis D, Zhang Y, Wolff RA and Abbruzzese
JL: Inhibition of the mammalian target of rapamycin (mTOR) in
advanced pancreatic cancer: Results of two phase II studies. BMC
Cancer. 10:3682010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kordes S, Klümpen HJ, Weterman MJ,
Schellens JH, Richel DJ and Wilmink JW: Phase II study of
capecitabine and the oral mTOR inhibitor everolimus in patients
with advanced pancreatic cancer. Cancer Chemother Pharmacol.
75:1135–1141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Karavasilis V, Samantas E, Koliou GA,
Kalogera-Fountzila A, Pentheroudakis G, Varthalitis I, Linardou H,
Rallis G, Skondra M, Papadopoulos G, et al: Gemcitabine combined
with the mTOR inhibitor temsirolimus in patients with locally
advanced or metastatic pancreatic cancer. A Hellenic cooperative
oncology group phase I/II study. Target Oncol. 13:715–724. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Amin M, Gao F, Terrero G, Picus J,
Wang-Gillam A, Suresh R, Ma C, Tan B, Baggstrom M, Naughton MJ, et
al: Phase I study of docetaxel and temsirolimus in refractory solid
tumors. Am J Clin Oncol. 44:443–448. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Weinberg BA, Wang H, Witkiewicz AK,
Marshall JL, He AR, Vail P, Knudsen ES and Pishvaian MJ: A phase I
study of ribociclib plus everolimus in patients with metastatic
pancreatic adenocarcinoma refractory to chemotherapy. J Pancreat
Cancer. 6:45–54. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Carr RM, Duma N, McCleary-Wheeler AL,
Almada LL, Marks DL, Graham RP, Smyrk TC, Lowe V, Borad MJ, Kim G,
et al: Targeting of the Hedgehog/GLI and mTOR pathways in advanced
pancreatic cancer, a phase 1 trial of Vismodegib and Sirolimus
combination. Pancreatology. 20:1115–1122. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al:
Erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: A phase III trial of the
national cancer institute of Canada clinical trials group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Park H, Williams K, Trikalinos NA, Larson
S, Tan B, Waqar S, Suresh R, Morgensztern D, Van Tine BA, Govindan
R, et al: A phase I trial of temsirolimus and erlotinib in patients
with refractory solid tumors. Cancer Chemother Pharmacol.
87:337–347. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Kordes S, Richel DJ, Klümpen HJ, Weterman
MJ, Stevens AJ and Wilmink JW: A phase I/II, non-randomized,
feasibility/safety and efficacy study of the combination of
everolimus, cetuximab and capecitabine in patients with advanced
pancreatic cancer. Invest New Drugs. 31:85–91. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Cohen SJ, Ho L, Ranganathan S, Abbruzzese
JL, Alpaugh RK, Beard M, Lewis NL, McLaughlin S, Rogatko A,
Perez-Ruixo JJ, et al: Phase II and pharmacodynamic study of the
farnesyltransferase inhibitor R115777 as initial therapy in
patients with metastatic pancreatic adenocarcinoma. J Clin Oncol.
21:1301–1306. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Macdonald JS, McCoy S, Whitehead RP, Iqbal
S, Wade JL III, Giguere JK and Abbruzzese JL: A phase II study of
farnesyl transferase inhibitor R115777 in pancreatic cancer: A
Southwest oncology group (SWOG 9924) study. Invest New Drugs.
23:485–487. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Van Cutsem E, van de Velde H, Karasek P,
Oettle H, Vervenne WL, Szawlowski A, Schoffski P, Post S, Verslype
C, Neumann H, et al: Phase III trial of gemcitabine plus tipifarnib
compared with gemcitabine plus placebo in advanced pancreatic
cancer. J Clin Oncol. 22:1430–1438. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Rich TA, Winter K, Safran H, Hoffman JP,
Erickson B, Anne PR, Myerson RJ, Cline-Burkhardt VJ, Perez K and
Willett C: Weekly paclitaxel, gemcitabine, and external irradiation
followed by randomized farnesyl transferase inhibitor R115777 for
locally advanced pancreatic cancer. Onco Targets Ther. 5:161–170.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Siu LL, Awada A, Takimoto CH, Piccart M,
Schwartz B, Giannaris T, Lathia C, Petrenciuc O and Moore MJ: Phase
I trial of sorafenib and gemcitabine in advanced solid tumors with
an expanded cohort in advanced pancreatic cancer. Clin Cancer Res.
12:144–151. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
El-Khoueiry AB, Ramanathan RK, Yang DY,
Zhang W, Shibata S, Wright JJ, Gandara D and Lenz HJ: A randomized
phase II of gemcitabine and sorafenib versus sorafenib alone in
patients with metastatic pancreatic cancer. Invest New Drugs.
30:1175–1183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Gonçalves A, Gilabert M, François E, Dahan
L, Perrier H, Lamy R, Re D, Largillier R, Gasmi M, Tchiknavorian X,
et al: BAYPAN study: A double-blind phase III randomized trial
comparing gemcitabine plus sorafenib and gemcitabine plus placebo
in patients with advanced pancreatic cancer. Ann Oncol.
23:2799–2805. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Cascinu S, Berardi R, Sobrero A, Bidoli P,
Labianca R, Siena S, Ferrari D, Barni S, Aitini E, Zagonel V, et
al: Sorafenib does not improve efficacy of chemotherapy in advanced
pancreatic cancer: A GISCAD randomized phase II study. Dig Liver
Dis. 46:182–186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chiorean EG, Schneider BP, Akisik FM,
Perkins SM, Anderson S, Johnson CS, DeWitt J, Helft P, Clark R,
Johnston EL, et al: Phase 1 pharmacogenetic and pharmacodynamic
study of sorafenib with concurrent radiation therapy and
gemcitabine in locally advanced unresectable pancreatic cancer. Int
J Radiat Oncol Biol Phys. 89:284–291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Infante JR, Somer BG, Park JO, Li CP,
Scheulen ME, Kasubhai SM, Oh DY, Liu Y, Redhu S, Steplewski K and
Le N: A randomised, double-blind, placebo-controlled trial of
trametinib, an oral MEK inhibitor, in combination with gemcitabine
for patients with untreated metastatic adenocarcinoma of the
pancreas. Eur J Cancer. 50:2072–2081. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Huijberts S, van Geel R, van Brummelen
EMJ, Opdam FL, Marchetti S, Steeghs N, Pulleman S, Thijssen B,
Rosing H, Monkhorst K, et al: Phase I study of lapatinib plus
trametinib in patients with KRAS-mutant colorectal, non-small cell
lung, and pancreatic cancer. Cancer Chemother Pharmacol.
85:917–930. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ko AH, Bekaii-Saab T, Van Ziffle J,
Mirzoeva OM, Joseph NM, Talasaz A, Kuhn P, Tempero MA, Collisson
EA, Kelley RK, et al: A multicenter, open-label phase II clinical
trial of combined MEK plus EGFR inhibition for
chemotherapy-refractory advanced pancreatic adenocarcinoma. Clin
Cancer Res. 22:61–68. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhu X, Cao Y, Liu W, Ju X, Zhao X, Jiang
L, Ye Y, Jin G and Zhang H: Stereotactic body radiotherapy plus
pembrolizumab and trametinib versus stereotactic body radiotherapy
plus gemcitabine for locally recurrent pancreatic cancer after
surgical resection: an open-label, randomised, controlled, phase 2
trial. Lancet Oncol. 23:e105–e115. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Aung KL, McWhirter E, Welch S, Wang L,
Lovell S, Stayner LA, Ali S, Malpage A, Makepeace B, Ramachandran
M, et al: A phase II trial of GSK2256098 and trametinib in patients
with advanced pancreatic ductal adenocarcinoma. J Gastrointest
Oncol. 13:3216–3226. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Bodoky G, Timcheva C, Spigel DR, Stella
PJL, Ciuleanu TE, Pover G and Tebbutt NC: A phase II open-label
randomized study to assess the efficacy and safety of selumetinib
(AZD6244 [ARRY-142886]) versus capecitabine in patients with
advanced or metastatic pancreatic cancer who have failed first-line
gemcitabine therapy. Invest New Drugs. 30:1216–1223. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Kenney C, Kunst T, Webb S, Christina D Jr,
Arrowood C, Steinberg SM, Mettu NB, Kim EJ and Rudloff U: Phase II
study of selumetinib, an orally active inhibitor of MEK1 and MEK2
kinases, in KRAS(G12R)-mutant pancreatic ductal adenocarcinoma.
Invest New Drugs. 39:821–828. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Rodon J, Tan DW, Laguna IG, Harb W, Beck
JT, Bahary N, Rottey S, Zhu Z, Deng S, Kowalski K, et al: 344
Avelumab + binimetinib in metastatic pancreatic ductal
adenocarcinoma (mPDAC): Dose-escalation results from the phase 1b/2
JAVELIN PARP MEKi trial. J Immunother Cancer. 9:A371. 2021.
View Article : Google Scholar
|
|
113
|
Surana R, Lee JJ, Smaglo BG, Zhao D, Lee
MS, Wolff RA, Overman MJ, Willis J, Der CJ and Pant S: Phase I
study of hydroxychloroquine plus binimetinib in patients with
metastatic pancreatic cancer (the HOPE trial). J Clin Oncol.
40:TPS634. 2022. View Article : Google Scholar
|
|
114
|
Grierson PM, Tan B, Pedersen KS, Park H,
Suresh R, Amin MA, Trikalinos NA, Knoerzer D, Kreider B, Reddy A,
et al: Phase Ib study of ulixertinib plus gemcitabine and
nab-paclitaxel in patients with metastatic pancreatic
adenocarcinoma. Oncologist. 28:e115–e123. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Raybould AL, Burgess B, Urban C, Naim R,
Lee MS and McRee AJ: A phase Ib trial of ERK inhibition with
ulixertinib combined with palbociclib in patients (Pts) with
advanced solid tumors. J Clin Oncol. 39:3103. 2021. View Article : Google Scholar
|
|
116
|
Grilley-Olson JE, Bedard PL, Fasolo A,
Cornfeld M, Cartee L, Razak ARA, Stayner LA, Wu Y, Greenwood R,
Singh R, et al: A phase Ib dose-escalation study of the MEK
inhibitor trametinib in combination with the PI3K/mTOR inhibitor
GSK2126458 in patients with advanced solid tumors. Invest New
Drugs. 34:740–749. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Bardia A, Gounder M, Rodon J, Janku F,
Lolkema MP, Stephenson JJ, Bedard PL, Schuler M, Sessa C, LoRusso
P, et al: Phase Ib study of combination therapy with MEK inhibitor
Binimetinib and Phosphatidylinositol 3-kinase inhibitor Buparlisib
in patients with advanced solid tumors with RAS/RAF alterations.
Oncologist. 25:e160–e169. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Ostrem JM and Shokat KM: Direct
small-molecule inhibitors of KRAS: From structural insights to
mechanism-based design. Nat Rev Drug Discov. 15:771–785. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Canon J, Rex K, Saiki AY, Mohr C, Cooke K,
Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al: The
clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity.
Nature. 575:217–223. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Hallin J, Engstrom LD, Hargis L, Calinisan
A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA,
et al: The KRAS(G12C) inhibitor MRTX849 provides insight toward
therapeutic susceptibility of KRAS-mutant cancers in mouse models
and patients. Cancer Discov. 10:54–71. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Skoulidis F, Li BT, Dy GK, Price TJ,
Falchook GS, Wolf J, Italiano A, Schuler M, Borghaei H, Barlesi F,
et al: Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl
J Med. 384:2371–2381. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Khan S, Wiegand J, Zhang P, Hu W, Thummuri
D, Budamagunta V, Hua N, Jin L, Allegra CJ, Kopetz SE, et al:
BCL-X(L) PROTAC degrader DT2216 synergizes with sotorasib in
preclinical models of KRAS(G12C)-mutated cancers. J Hematol Oncol.
15:232022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Strickler JH, Satake H, George TJ, Yaeger
R, Hollebecque A, Garrido-Laguna I, Schuler M, Burns TF, Coveler
AL, Falchook GS, et al: Sotorasib in KRAS p.G12C-mutated advanced
pancreatic cancer. N Engl J Med. 388:33–43. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Bekaii-Saab TS, Spira AI, Yaeger R,
Buchschacher GL, McRee AJ, Sabari JK, Johnson ML, Barve NA, Hafez
N, Velastegui K, et al: KRYSTAL-1: Updated activity and safety of
adagrasib (MRTX849) in patients (Pts) with unresectable or
metastatic pancreatic cancer (PDAC) and other gastrointestinal (GI)
tumors harboring a KRASG12C mutation. J Clin Oncol. 40:519. 2022.
View Article : Google Scholar
|
|
125
|
Wang-Gillam A, Hubner RA, Siveke JT, Von
Hoff DD, Belanger B, de Jong FA, Mirakhur B and Chen LT: NAPOLI-1
phase 3 study of liposomal irinotecan in metastatic pancreatic
cancer: Final overall survival analysis and characteristics of
long-term survivors. Eur J Cancer. 108:78–87. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Wang X, Allen S, Blake JF, Bowcut V,
Briere DM, Calinisan A, Dahlke JR, Fell JB, Fischer JP, Gunn RJ, et
al: Identification of MRTX1133, a Noncovalent, potent, and
selective KRAS(G12D) inhibitor. J Med Chem. 65:3123–3133. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Hallin J, Bowcut V, Calinisan A, Briere
DM, Hargis L, Engstrom LD, Laguer J, Medwid J, Vanderpool D, Lifset
E, et al: Anti-tumor efficacy of a potent and selective
non-covalent KRAS(G12D) inhibitor. Nat Med. 28:2171–2182. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Kemp SB, Cheng N, Markosyan N, Sor R, Kim
IK, Hallin J, Shoush J, Quinones L, Brown NV, Bassett JB, et al:
Efficacy of a small molecule inhibitor of KrasG12D in
immunocompetent models of pancreatic cancer. Cancer Discov.
13:298–311. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Bannoura SF, Khan HY and Azmi AS: KRAS
G12D targeted therapies for pancreatic cancer: Has the fortress
been conquered? Front Oncol. 12:10139022022. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Huang L, Guo Z, Wang F and Fu L: KRAS
mutation: From undruggable to druggable in cancer. Signal Transduct
Target Ther. 6:3862021. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Gustafson WC, Wildes D, Rice MA, Lee BJ,
Jiang J, Wang Z, Chang S, Flagella M, Mu Y, Dinglasan N, et al:
Direct targeting of RAS in pancreatic ductal adenocarcinoma with
RMC-6236, a first-in-class, RAS-selective, orally bioavailable,
tri-complex RASMULTI(ON) inhibitor. J Clin Oncol. 40:591. 2022.
View Article : Google Scholar
|
|
132
|
Frank KJ, Mulero-Sánchez A, Berninger A,
Ruiz-Cañas L, Bosma A, Görgülü K, Wu N, Diakopoulos KN, Kaya-Aksoy
E, Ruess DA, et al: Extensive preclinical validation of combined
RMC-4550 and LY3214996 supports clinical investigation for KRAS
mutant pancreatic cancer. Cell Rep Med. 3:1008152022. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Gort E, Johnson ML, Hwang JJ, Pant S,
Dünzinger U, Riemann K, Kitzing T and Janne PA: A phase I,
open-label, dose-escalation trial of BI 1701963 as monotherapy and
in combination with trametinib in patients with KRAS mutated
advanced or metastatic solid tumors. J Clin Oncol. 38:TPS3651.
2020. View Article : Google Scholar
|