Introduction
C-X-C motif chemokine (CXCL)12 and its receptors,
C-X-C chemokine receptor type (CXCR)4 and CXCR7/atypical chemokine
receptor 3, are central to the control of cell migration and cell
positioning during development (1),
as well as under various pathophysiological conditions, such as
cancer (2). In a large number of
human malignancies, including prostate, breast, lung, pancreas and
colon/rectum cancer, the CXCL12 pathway not only controls tumor
growth and angiogenesis, but also acts as a key regulator of tumor
metastasis (3,4). In tumors, several sources of CXCL12
have been identified, including tumor cells themselves,
cancer-associated fibroblasts (CAFs) and endothelial cells, among
others (3,5). In addition, most tumor cells also
express detectable levels of both CXCR4 and CXCR7 (6). However, the tumorigenic effects of
CXCL12 are mediated by either CXCR4 and/or CXCR7 depending on the
cell type, the reasons of which are currently unknown (6). Thus, the signaling mechanisms
underlying the tumorigenic effects of CXCL12 remain unclear.
The two CXCL12 receptors show similarities, as well
as distinct differences in their signaling behavior. CXCR4 is a
classical G protein-coupled chemokine receptor that preferentially
activates cell signaling through Gi, but also through Gq and
G12/G13 (7).
However, in certain cell types, such as HeLa cells, 293 cells and
podocytes, CXCR4-dependent cell signaling and subsequent effects on
cell migration and adhesion appear to be mediated by β-arrestin1
and/or β-arrestin2 (8–10).
Depending on the context and cell type, CXCR7 has
different functions. For example, CXCR7 can act as a scavenging
receptor, allowing CXCR4-dependent cell migration by shaping the
extracellular CXCL12 gradient (11). CXCR7 also acts as an active
signaling receptor, affecting the same cell functions as CXCR4,
including cell proliferation, migration and invasion (12). To date, several studies have
provided evidence that CXCR7 activates cell signaling through
β-arrestin2 (13–15), while known exceptions are primary
astrocytes and certain glioma cells. In these cells,
ligand-activated CXCR7 binds and activates G proteins and thus
appears to function as a classical chemokine receptor (16,17).
Another known mechanism by which CXCL12 receptors
activate cell signaling is through transactivation of EGFR family
members via Src. This mechanism is currently best documented for
CXCR4 (18–24) in cancer cells, but has also been
suggested for CXCR7 in embryonic cells (25).
The fact that CXCR4 and CXCR7 can form homomers or
heteromers adds further complexity to CXCL12 signaling (12). CXCR4/CXCR7 heteromers allow for
enhanced recruitment of β-arrestin to the receptor complex compared
with receptor monomers/homodimers (26), ultimately altering the kinetics of
activated signaling pathways (27).
In addition, CXCR4 homomers have been shown to be more efficient
than CXCR4 monomers in promoting the migration of a variety of cell
lines, such as 293 and HeLa cells (28). Finally, CXCR4 dimers appear to form
nanoclusters involving specific transmembrane residues.
Nanoclusters are considered essential for full activation of CXCR4,
and thus for maximal cellular responses (29).
The outlined concept of CXCR4 and CXCR7 signaling is
additionally complicated by several novel roles of β-arrestins in G
protein-coupled receptor (GPCR) signaling. Initially, β-arrestins
were believed to facilitate endocytosis of GPCRs. More recently,
β-arrestin1 was found to additionally mediate the interaction
between the plasma membrane and internal pools of CXCR4 (30), thereby defining the final cellular
response by coordinating receptor signaling at the cell surface and
at internal sites (30). In
addition, evidence has emerged that GPCRs also activate cell
signaling through Gαi/β-arrestin complexes (31).
To better understand CXCL12 signaling in
tumorigenesis, the present study investigated whether CXCL12/CXCR4-
and CXCL12/CXCR7-dependent control of cancer cell migration, and
thus metastatic behavior, depends on G proteins, arrestins or both.
It was also examined whether CXCL12-induced cell migration involves
transactivation of EGFR. To this end, several human tumor cell
lines were used, which have recently been characterized in terms of
the receptors activated by CXCL12 to control cell migration in our
previous study (6). Tumor cells
were analyzed for CXCL12-dependent cell migration following
pharmacological inhibition of either Gα proteins, EGFR or Src
kinase, as well as following depletion of arrestins by RNA
interference.
Materials and methods
Cell culture
The human tumor cell lines A549 (lung
adenocarcinoma), C33A (cervical carcinoma), DLD-1 (colorectal
adenocarcinoma), MDA-MB-231 (breast adenocarcinoma) and PC-3
(prostate carcinoma) were purchased from American Type Culture
Collection. These cell lines were selected because the respective
tumors are known to be affected by the CXCL12 pathway (3). The cell line authenticity was verified
by the supplier using short tandem repeat profiling. Cell lines
were regularly tested for mycoplasma contamination using
MycoSpy® Master Mix (Biontex Laboratories GmbH). Cells
were plated on 12-well culture plates (TPP Techno Plastic Products
AG), and grown in either DMEM (4.5 g/l glucose) for C33A,
MDA-MB-231 and PC-3 or RPMI-1640 for A549 and DLD-1, supplemented
with 0.05% gentamycin and 10% fetal bovine serum (FBS) (all from
Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a water-saturated
atmosphere of 95% air and 5% CO2. For experiments, cell
cultures at 60% confluence were starved for 24 h in their
corresponding serum-free culture medium at 37°C, supplemented with
or without pertussis toxin (PTX; 100 ng/ml pre-dissolved; cat. no
P2980; Sigma-Aldrich; Merck KGaA), followed by addition of the
following compounds for 1 h: CCX771 (CXCR7 antagonist; 100 nM
dissolved in DMSO; Amgen, Inc.), AMD3100 (CXCR4 antagonist; 10 µM
dissolved in double-distilled water; Sigma-Aldrich; Merck KGaA),
Tyrphostin AG1478 (EGFR inhibitor; 2 µM dissolved in DMSO; cat. no.
T4182; Sigma-Aldrich; Merck KGaA), Src inhibitor-1 (Src-I1; Src
kinase inhibitor; 10 µM dissolved in DMSO; Sigma-Aldrich; Merck
KGaA). For control purposes, untreated cells were supplemented with
appropriate concentrations of DMSO.
Western blot analysis
For western blot analysis, cultured A549, C33A,
DLD-1, MDA-MB-231 and PC3 cells were harvested and immediately
placed in liquid nitrogen. Total protein was isolated in RIPA
protein extraction buffer (cat. no. 89901; Thermo Fisher
Scientific, Inc.) supplemented with protease and phosphatase
inhibitors (cat. no. 78442; Thermo Fisher Scientific, Inc.) to
prevent protein dephosphorylation. Protein content was determined
using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Proteins (10–20
µg/lane) were separated using 10% SDS-PAGE and transferred to
nitrocellulose membranes by electroblotting. After blocking
non-specific binding sites with Pierce Fast Blocking Buffer (cat.
no. 37575; Thermo Fisher Scientific, Inc.) for 60 min at room
temperature, the membranes were incubated overnight at 4°C with one
of the following primary antibodies: Rabbit anti-EGFR (1:1,000;
cat. no. 4267; Cell Signaling Technology Europe, B.V.), rabbit
anti-β-arrestin2 (1:1,000; cat. no. 3857; Cell Signaling Technology
Europe, B.V.) and mouse monoclonal anti-β-arrestin1 (1:1,000; cat.
no. MA1-183; Thermo Fisher Scientific, Inc.). Subsequently, the
membranes were incubated at room temperature for 1 h with
HRP-conjugated anti-mouse (1:10,000; cat. no. PI-2000-1; Vector
Laboratories, Inc.) or anti-rabbit (1:10,000; cat. no. 711-035-152;
Jackson ImmunoResearch Laboratories Europe, Ltd.) secondary
antibodies. Antibody-labeling was visualized using Pierce™ ECL
Western Blotting Substrate (Thermo Fisher Scientific, Inc.). To
control for protein loading, membranes were re-probed with rabbit
anti-β-actin (1:5,000; cat. no. 4970; Cell Signaling Technology
Europe, B.V.) or mouse anti-GAPDH (1:5,000; cat. no. 10R-G109A;
Fitzgerald Industries International) antibodies. Chemiluminescence
was captured on a Biostep Celvin S imager (Biostep; Bionis) and
immunoreactive protein bands were quantified using the Snap and Go
1.8.3 software (Biostep; Bionis).
Chemotaxis assay
Chemotactic responses of A549, C33A, DLD-1,
MDA-MB-231 and PC3 cells to CXCL12 were determined in a modified
12-well or 48-well Boyden chamber (Neuro Probe Inc.), in which the
upper and lower wells were separated by polyornithine-coated
Nucleopore® PVP-free polycarbonate filter (Whatman plc;
Cytiva; 8-µm pore size). For seeding, pretreated cells were counted
using an improved Neubauer chamber and ~10,000 cells were placed in
serum-free DMEM or RPMI medium into the upper well of the Boyden
chamber. A total of 150 µl of the respective serum-free medium
supplemented with CXCL12 (100 ng/ml; PeproTech, Inc.) were added to
the lower chamber. The chamber was incubated at 37°C in a
water-saturated atmosphere of 95% air and 5% CO2 for 4
h. After incubation, non-migrated cells attached to the upper part
of the membrane were wiped off, and migrated cells attached to the
lower part of the membrane were fixed with ice-cold methanol (100%)
at room temperature for 5 min, stained with DAPI (1:5,000; AAT
Bioquest, Inc.; 5 min, room temperature), and counted on an Olympus
BX40 microscope using the Olympus cellSens Dimension software
(Olympus Corporation) at a final magnification of 50×. The number
of cells migrating in the absence of CXCL12 was set to 1 and the
migration index was calculated as the ratio of cells migrating in
the presence and absence of CXCL12.
RNA interference
Predesigned human β-arrestin1 small interfering
(si)RNA (cat. no. 4392420; Assay ID, s1624) and human β-arrestin2
(cat. no. 4392420; Assay ID, s1625), as well as control siRNA (cat.
no. 4390844) were purchased from Thermo Fisher Scientific, Inc. The
sequences of the siRNAs are presented in Table I. Transfection of A549, C33A, DLD-1,
MDA-MB-231 and PC3 cells with siRNA (75 pmol/well) was performed in
6-well plates (TPP Techno Plastic Products AG) at 37°C in the
presence of serum-free DMEM or RPMI-medium using the siPORT Amine
Transfection Agent (cat. no. AM4503; Thermo Fisher Scientific,
Inc.) for 16 h. Transfected cells were further maintained with
serum-free DMEM or RPMI-medium and subjected to experiments after
24 h. The success of RNA interference was validated by western
blotting and reverse transcription-quantitative (RT-q)PCR. Cells
treated with PTX (100 ng/ml, 24 h) were used as a positive control
for inhibited cell migration.
 | Table I.Sequences of small interfering RNAs
used. |
Table I.
Sequences of small interfering RNAs
used.
| Gene | Sense (5′-3′) | Antisense
(5′-3′) |
|---|
| Human
β-arrestin1 |
CCAAUCUCAUAGAACUUGATT |
UCAAGUUCUAUGAGAUUGGTA |
| Human
β-arrestin2 |
AAGUCUCUGUGAGACAGUATT |
UACUGUCUCACAGAGACUUTG |
| Control | Sequence
confidential, not provided by the supplier |
|
RT-qPCR
Briefly, total RNA was extracted from A549, C33A,
DLD-1, MDA-MB-231 and PC3 cells using InviTrap Spin Universal RNA
Mini Kit (Invitek Diagnostics), followed by reverse transcription
of 1 µg RNA using Protoscript First Strand cDNA Synthesis Kit (New
England BioLabs, Inc.) as per the manufacturer's instructions. For
quantification of gene expression, qPCR analysis was performed with
PowerUp SYBR Green Master Mix (cat. no. A25742; Thermo Fisher
Scientific, Inc.) on a CFX96 thermal cycler system (Bio-Rad
Laboratories, Inc.). PCR conditions consisted of an initial
denaturation step at 95°C for 10 min, followed by 40 cycles of 95°C
for 15 sec, 59.5°C for 30 sec and 72°C for 30 sec. Gene expression
was calculated using the 2−ΔΔCq method (32) and normalized to β-actin. Primer
sequences are listed in Table
II.
| Table II.Sequences of primers used. |
Table II.
Sequences of primers used.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| Human
β-arrestin1 |
GACCATGGGCGACAAAGG |
GGTAGACGGTGAGCTTTCCATT |
| Human
β-arrestin2 |
GGAAGCTGGGCCAGCAT |
TGTGACGGAGCATGGAAGATT |
| Human CXCL12 |
CACAGAAGGTCCTGGTGGTA |
CATTGAAAAGCTGCAATCAC |
| Human β-actin |
GGCCTCGCTGTCCACCTT |
TGTCACCTTCACCGTTCCAGTTTT |
Statistical analysis
Data are presented as the mean ± SD Experiments were
replicated at least three times. When results showed large
variations, a total of up to 15 replicates were used. GraphPad
Prism 9 (Dotmatics) was used for statistical analysis. One-way
ANOVA followed by Tukey's post hoc test was employed for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
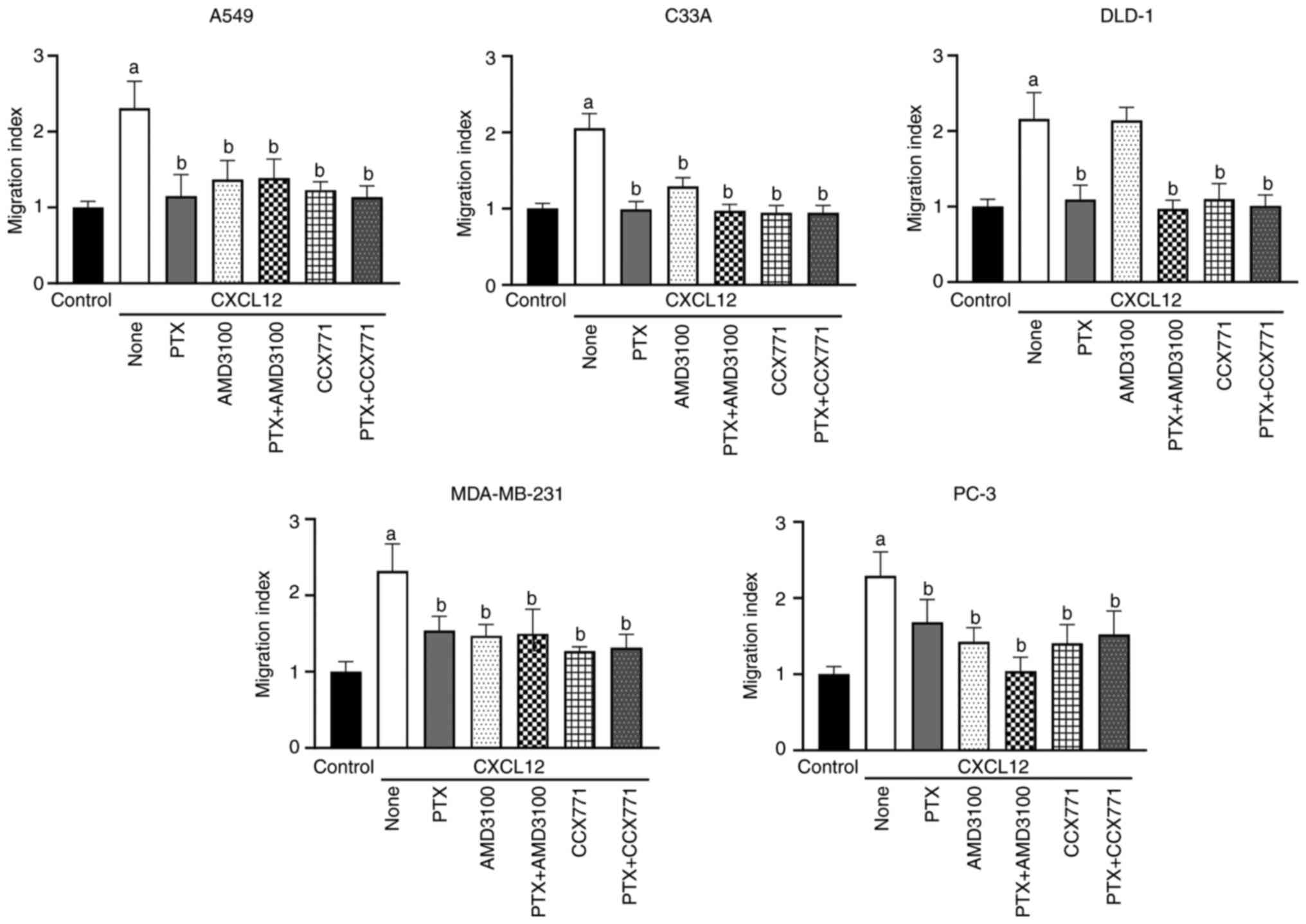
CXCL12 induces cancer cell migration
via the differential activation of CXCR4 and/or CXCR7
Consistent with our previous study (6), CXCL12 at 100 ng/ml induced chemotaxis
of all tumor cell lines as evidenced by a 2- to 3-fold increase in
their migration index (Fig. 1). In
addition, the CXCR4 antagonist AMD3100 and the CXCR7 antagonist
CCX771 attenuated the CXCL12-induced migration of C33A, MDA-MB-231
and PC-3 cells (Fig. 1), suggesting
that the chemotactic responses of these cells to CXCL12 are
mediated by both CXCR4 and CXCR7. Furthermore, the CXCL12-induced
migration of DLD-1 cells was sensitive to CCX771, but not to
AMD3100, confirming the previously suggested primary role of CXCR7
in the CXCL12-dependent migration of DLD-1 cells (6). In further accordance with our previous
study, CXCL12-induced migration of A549 cells was sensitive to
CCX771. However, in contrast to the previous observation showing a
tendency of AMD3100 to attenuate CXCL12-induced migration of A549
cells (6), a statistically
significant decrease in the numbers of migrating A549 cells treated
with AMD3100 and CXCL12 compared with CXCL12 alone was observed in
the present study (Fig. 1). This
discrepancy likely reflects the use of a 12-well Boyden chamber for
chemotactic analysis in the present experiments compared with a
48-well Boyden chamber plated with 5,000 cells per well in our
previous study (6). Comparison of
the chemotactic responses of A549 cells to CXCL12 in the presence
and absence of receptor antagonists under the different
experimental set-ups indicated more pronounced inhibitory responses
in the 12-well Boyden chamber compared with the 48-well Boyden
chamber assay, the reason of which is currently unknown (Fig. S1). The mechanism responsible for
this discrepancy is currently unknown, but may well reflect
differences in cell adhesion in the different sized chambers. These
data imply that contrary to the conclusion of our previous study,
CXCL12-dependent chemotaxis of A549 cells is mediated by both CXCR7
and CXCR4 and not solely CXCR7.
Tumor cells have been reported to express CXCL12
(5). The present RT-qPCR analysis
revealed low levels of expression of CXCL12 mRNA in C33A cells
which became detectable only after 30 PCR cycles. By contrast, in
A549, DLD-1, MDA-MB-231 and PC3 cells, CXCL12 mRNA was virtually
undetectable after 30 PCR cycles (data not shown). These findings
suggest that CXCL12 in respective tumors is predominantly derived
from non-tumor cells, such as CAFs or endothelial cells (5). These findings further argue against
the possibility that the current analyses are biased by endogenous
CXCL12.
Evaluation of the role of G proteins
and arrestins in CXCR4 and CXCR7 signaling
It is has been demonstrated that CXCR4 and CXCR7
initiate cell signaling through either G proteins (preferentially
Gα protein) or β-arrestin1/β-arrestin2, depending on the cell type
and context (7–10,12).
To assess the role of Gα proteins in CXCL12-dependent tumor cell
migration, the migratory responses of tumor cells previously
treated with PTX (100 ng/ml) for 24 h were analyzed. PTX partially
or completely prevented the CXCL12-induced chemotaxis of A549,
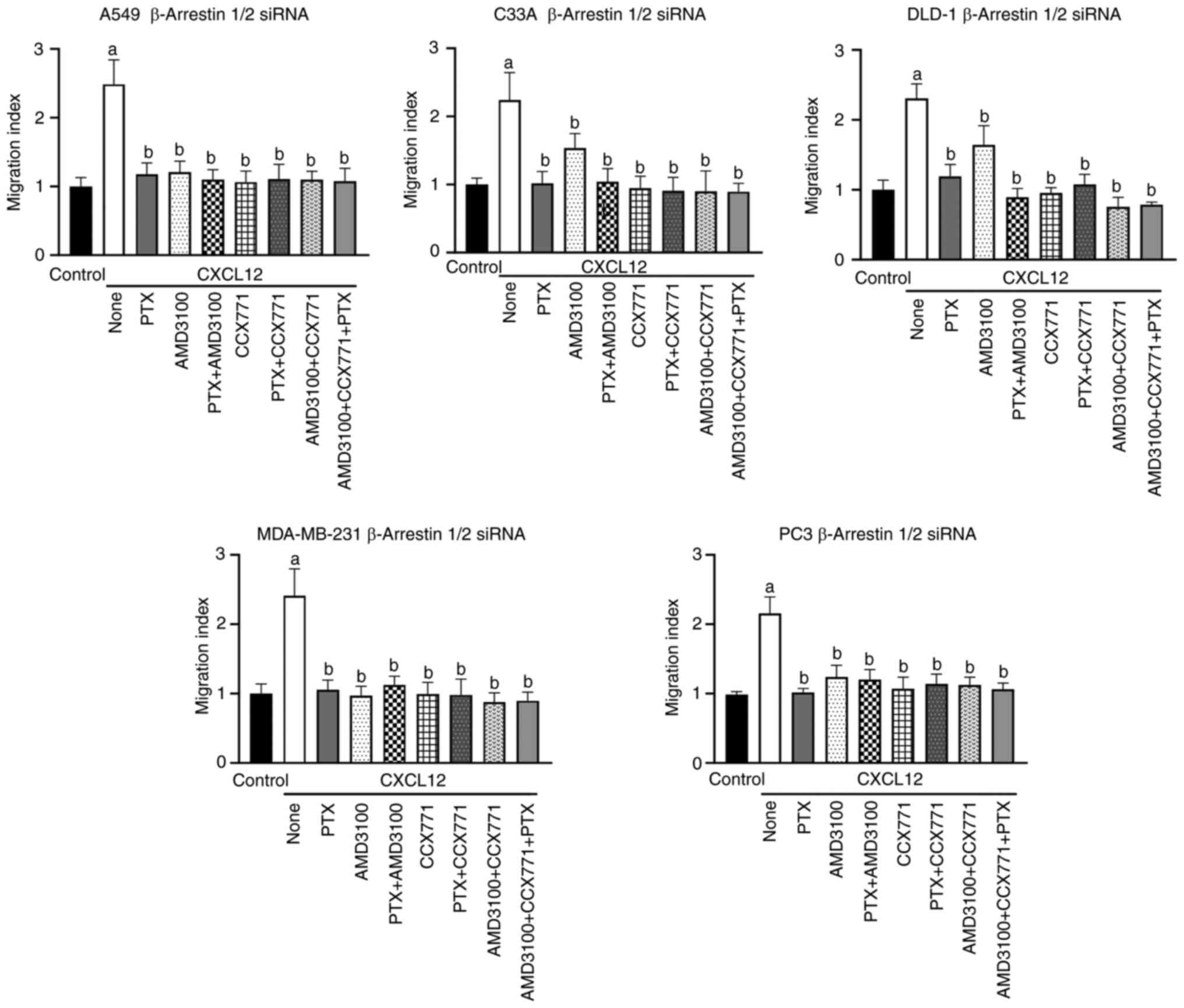
C33A, DLD-1, MDA-MB-231 and PC-3 cells (Figs. 1 and S2). To determine whether arrestins are
additionally involved in CXCL12-induced tumor cell migration, cells
were transfected with siRNA recognizing β-arrestin1 mRNA,
β-arrestin2 mRNA or both. Western blot and RT-qPCR analyses
confirmed a substantial 60–98% decrease in both β-arrestin1 and
β-arrestin2 protein as well as mRNA levels on day 2 post
transfection (Figs. S3 and
S4). The depletion of β-arrestins
had no apparent effect on the chemotactic responses of tumor cells
to CXCL12, which were similar to those observed with wild-type
cells (Figs. 2 and S5). Furthermore, in A549, C33A,
MDA-MB-231 and PC-3 cells neither PTX treatment nor β-arrestin
depletion affected the sensitivity of CXCL12-induced chemotaxis to
AMD3100 and/or CCX771 (Figs. 1 and
2). Similarly, the sensitivity of
CXCL12-induced chemotaxis to these inhibitors was unaffected after
transfection of cells with control siRNA (Figs. S6 and S7). Collectively, these findings
suggested that inhibition of CXCR4 or CXCR7 is not associated with
a switch from the inactive to the active state of the receptor
downstream CXCL12 signaling. However, it was also observed that
transfection of DLD-1 cells with control siRNA or β-arrestin siRNA
rendered CXCL12-dependent chemotaxis partially sensitive to
AMD3100, the reason of which is unclear (Figs. 2 and S6). Collectively, these findings
indicated that CXCL12-induced tumor cell migration mediated by
CXCR4 and/or CXCR7 is typically dependent on Gαi/o
signaling.
CXCR4 and CXCR7 control cancer cell
migration via the transactivation of EGFR
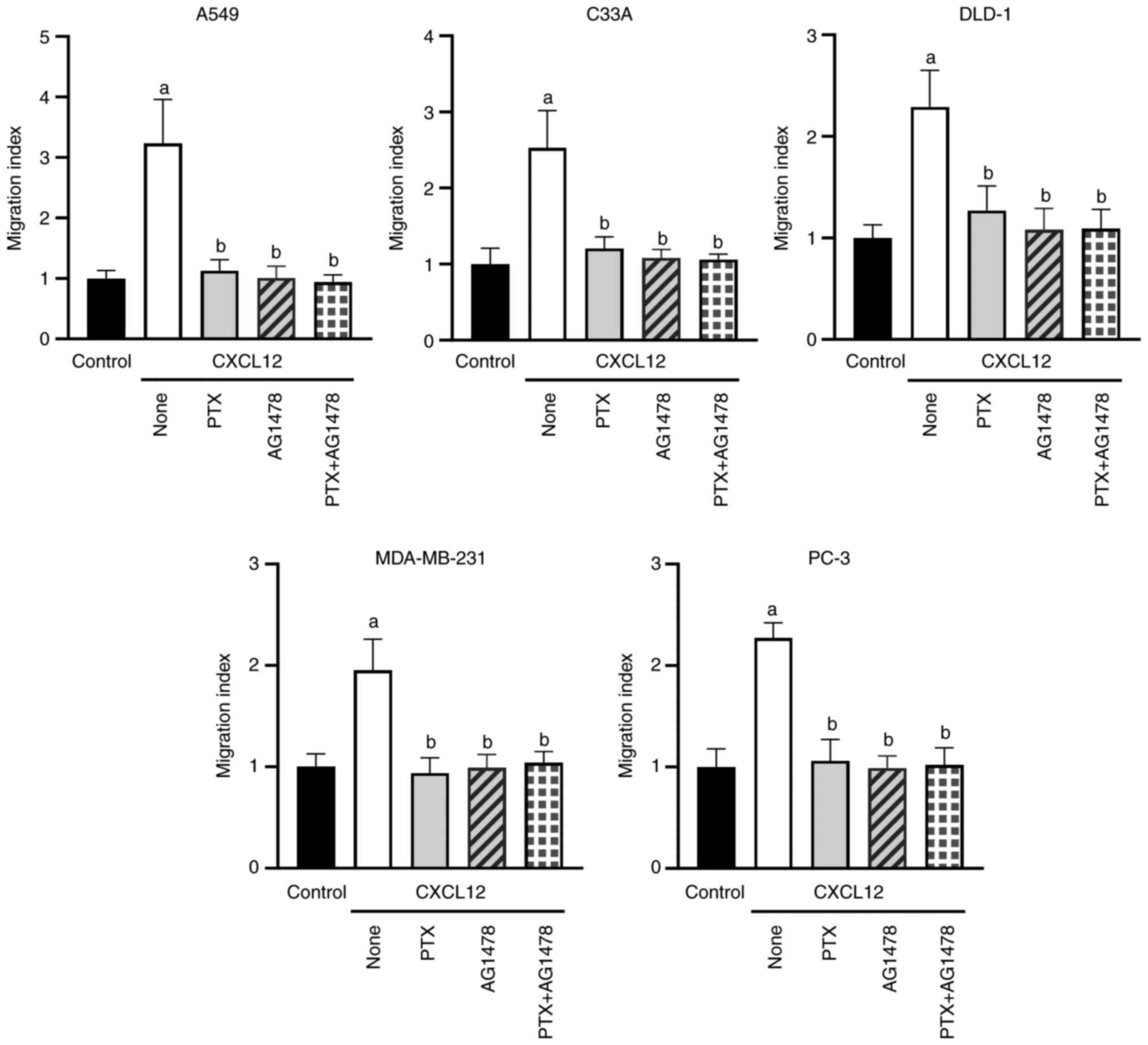
Among others, G proteins activate cell signaling
through Src-dependent transactivation of EGFR family members,
including EGFR/ErbB1 (18). Western
blotting demonstrated that the tumor cell lines used in the present
study expressed EGFR at varying levels, with the lowest levels
observed in C33A cells (Fig. S8).
To assess the putative role of EGFR transactivation in the
chemotactic responses of A549, C33A, DLD-1, MDA-MB-231 and PC-3
cells to CXCL12, chemotaxis was re-analyzed after pretreatment of
the cells with the EGFR inhibitor AG1478 (2 µM) for 1 h. AG1478
completely abolished the CXCL12-dependent chemotactic responses of
the different cell lines (Figs. 3
and S9). These inhibitory effects
were not affected by the additional treatment of the cells with PTX
(Fig. 3). Transactivation of EGFR
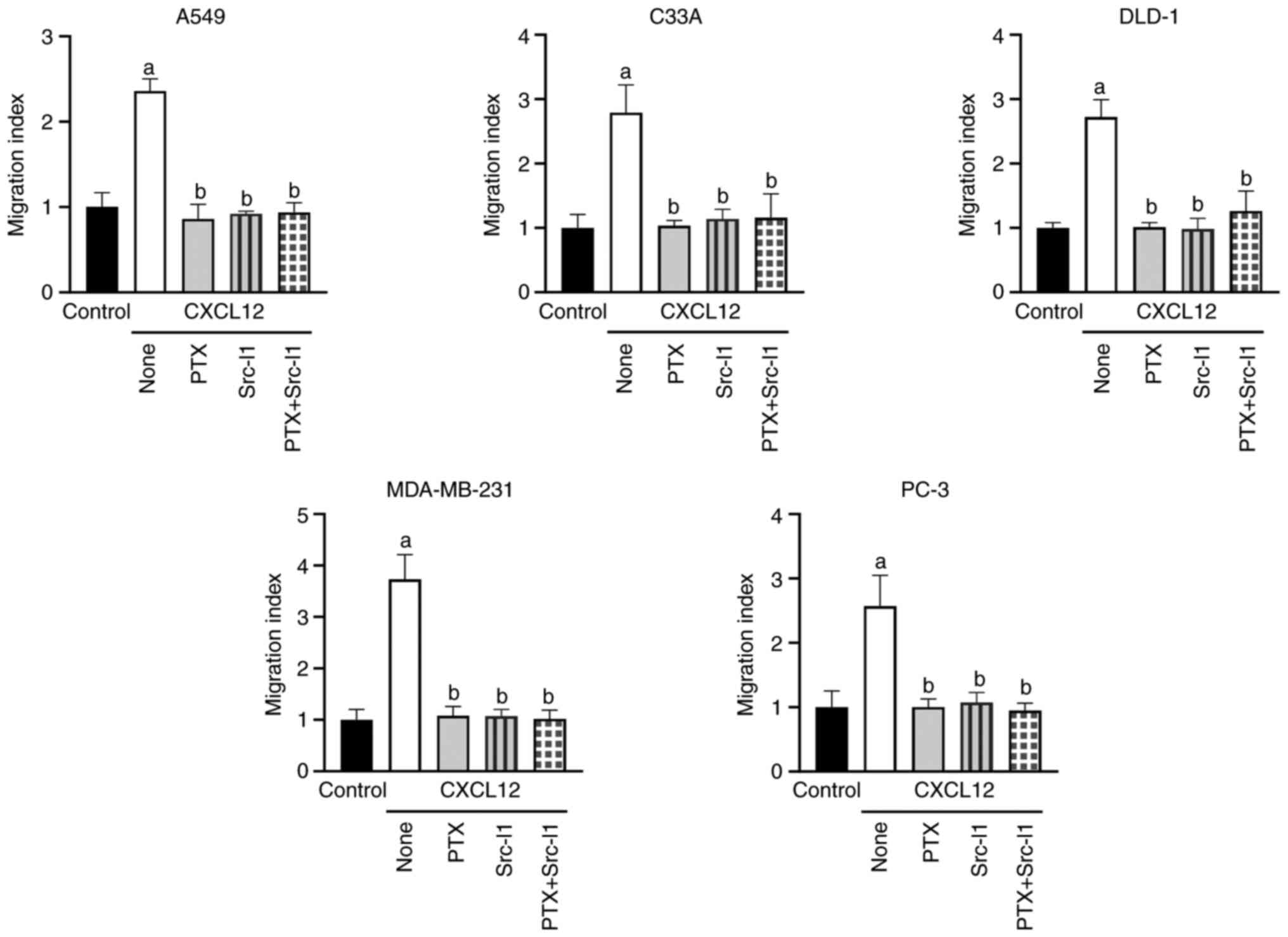
typically occurs via Src according to previous reports (18–23).
The chemotactic responses of the different cell lines to CXCL12
were also prevented after pretreatment with the Src kinase
inhibitor Src-I1 (10 µM) for 1 h (Figs.
4 and S10). Again, these
inhibitory effects were not further modulated by PTX treatment
(Fig. 4). To evaluate the putative
effects of the different inhibitors on EGFR expression, tumor cells
were treated with either PTX (100 ng/ml) for 24 h or AG1478 (2 µM)
and Src-I1 (10 µM) for 1 h, and EGFR expression levels were then
analyzed by western blotting. None of the treatments resulted in
obvious changes in EGFR protein, implying that the observed
inhibitory effects are not due to the loss or reduction of EGFR
expression (Fig. S8).
Taken together, these findings identified
Src-dependent transactivation of EGFR as a common mechanism
underlying CXCL12-induced migration of tumor cells from a number of
organs. In addition, the present results showed that both CXCR4 and
CXCR7 signal through EGFR transactivation in tumor cells.
Discussion
In recent years, CXCL12 and its receptors, CXCR4 and
CXCR7, have emerged as critical regulators of tumor metastasis, and
have therefore attracted considerable attention as potential
therapeutic targets in numerous human malignancies, including
prostate, breast, lung and pancreatic cancer, gliomas and multiple
myeloma (3,4,33). An
ongoing controversy is whether CXCR4 and CXCR7 control cell
migration and thus metastasis by activating cell signaling via G
proteins and/or arrestins (12).
This issue is further complicated by the fact that CXCL12 mediates
its chemotactic effects through either CXCR4 and/or CXCR7,
depending on the cell type (6).
Notably, the selective use of CXCR4 and/or CXCR7 receptors by tumor
cells is not reflected by their cellular expression levels, and the
mechanism of selective receptor activation remains to be further
elucidated (6). The present
analyses of five different human tumor cell lines in which
CXCL12-induced chemotaxis is mediated either by CXCR4 and CXCR7
(A549, C33A, MDA-MB-231 and PC-3) or only by CXCR7 (DLD-1)
unraveled the dependence of CXCL12-induced cancer cell migration on
G proteins. In addition, the present study provided evidence that
ligand-dependent activation of CXCR4 and/or CXCR7 induced cell
signaling via Src-mediated transactivation of EGFR, as judged from
the absence of CXCL12-dependent chemotaxis following
pharmacological inhibition of EGFR or Src kinase and the
established role of Src in EGFR (trans)activation (18–23).
These additional insights into the molecular mechanisms of
CXCL12-mediated cancer cell migration may have implications for
current efforts to establish CXCR4 or CXCR7 as therapeutic targets
in cancer (34).
Ligand-bound CXCR4 has been reported to activate
both G protein- and arrestin-dependent cell signaling, whereas
ligand-bound CXCR7 is believed to activate arrestin-dependent
signaling (12). Currently known
exceptions are primary cortical rat astrocytes and certain glioma
cells, in which CXCR7 activates G proteins (16,17).
In the present study, CXCL12-induced migration of the various tumor
cells required activation of Gαi/o, but not arrrestins,
as evidenced by the attenuated cell migration in the presence of
PTX and sustained cell migration following siRNA-mediated cellular
depletion of β-arrestin1 and β-arrestin2. In addition, the
Gαi/o-dependent mechanism was observed in tumor cells,
in which CXCL12 cell migration was induced through both CXCR4 and
CXCR7 (A549, C33A, MDA-MB-231 and PC-3) as well as CXCR7 alone
(DLD-1). These findings expand the range of cells in which CXCR7
activates or at least assists in the activation of G proteins. It
should be emphasized that the observed lack of effects of arrestin
depletion on cell migration cannot be attributed to residual low
levels of β-arrestin in β-arrestin siRNA-transfected cells. Our
previous study has demonstrated that reducing β-arrestin2 levels in
primary astrocytes to 20% by siRNA is sufficient to abolish
CXCL11-signaling (17). The
observed persistence of the migratory responses of
β-arrestin-depleted tumor cells to CXCL12 further excludes the
involvement of previously proposed alternative mechanisms by which
β-arrestin may control cell function, such as i)
β-arrestin-mediated communication between the cell
surface-associated pool and the intracellular pool of CXCL12
receptors (30); and ii) the
induction of cell signaling by Gαi/β-arrestin complexes. (31). It could be hypothesized that CXCR4
and CXCR7 cooperate in A549, C33A, MDA-MB-231 and PC-3 cells
through the formation of receptor heteromers (35). However, the observed dependence of
CXCL12-induced chemotaxis on G proteins argues against this
mechanism. Previous studies have shown that the formation of
CXCR4/CXCR7 heteromers attenuates G protein-dependent cell
signaling and promotes arrestin signaling (26,36).
As these results were obtained in cells with ectopic overexpression
of CXCR4 and CXCR7, the possibility that intrinsic CXCR4 and CXCR7
function differentially cannot be excluded (12).
Activated G proteins induce cell signaling through
Src-mediated transactivation of EGFR family members, among other
mechanisms. This mechanism is currently best studied for
ligand-activated CXCR4, which transactivates EGFR via
Gαi and subsequently promotes tumor cell migration and
invasion (18,19,21,22,24)
and proliferation (20,23). To date, only one study has shown
that EGFR transactivation also occurs through ligand-activated
CXCR7 (25). The present findings
identified Src-dependent transactivation of EGFR as the common
molecular mechanism via which CXCL12 controls tumor cell migration
and potentially metastasis. Importantly, EGFR transactivation
occurred in tumor cells in which either CXCR7 alone or together
with CXCR4 mediated CXCL12-dependent chemotaxis. In addition, both
the EGFR inhibitor AG1478 and the Src kinase inhibitor Src-I1
attenuated the CXCL12-induced chemotactic responses in all tumor
cells. Notably, our previous study has demonstrated that CXCL12
controls the migration of various tumor cells through ERK- and/or
PI3K/Akt-mediated signaling (37),
the major signaling pathways activated by EGFR (38). Although EGFR transactivation emerged
as a common molecular downstream event in CXCL12-induced migration
of the tumor cells examined in the present study, not all malignant
and non-malignant cells may utilize this mechanism. For example,
CXCR4-dependent migration and homing of endothelial colony-forming
cells requires G protein-induced calcium activation (39). By contrast, CXCR7-dependent
migration of cholangiocarcinoma cells and melanocytes requires
arrestin signaling (13,15). However, these studies did not
exclude the involvement of EGFR, which is also transactivated by
arrestins (40–42). The interplay between CXCL12
receptors and ErbB family members is not limited to receptor
transactivation, but is more complex. For example, EGFR and other
ErbB family members (such as ErbB3) control the expression of
CXCL12 receptors and vice versa (43–52).
In addition, CXCR7 can directly interfere with the ligand-activated
signaling of EGFR, probably by its direct coupling to EGFR
(53–55). The extent to which this interplay
also occurs in tumors and further modulates CXCL12-induced tumor
cell migration remains to be elucidated. CXCL12 and its receptors
appear to affect different cell functions through different
receptors/molecular mechanisms (12). For example, CXCL12 induces
chemotaxis of MDA-MB-341 cells via CXCR4 and CXCR7, but promotes
their proliferation only through CXCR4 (6). Hence, future studies are needed to
define whether the effects of CXCL12 on other tumor cell functions,
such as cell survival and proliferation, are also dependent on EGFR
transactivation.
Taken together, the present study revealed that the
CXCL12-CXCR4-CXCR7 chemokine system controls tumor cell migration
and potentially tumor metastasis through a common signaling pathway
involving Gα/Scr-dependent transactivation of EGFR. This points to
EGFR inactivation as a favorable therapeutic approach to prevent
the CXCL12-induced expansion of various tumor types. The success of
this approach is independent of whether CXCL12 affects cancer cell
migration through CXCR4 or CXCR7. Several inhibitors of wild-type
EGFR are currently used in the clinic (56). However, their application to the
CXCL12-dependent tumor cell expansion requires further verification
in vivo.
Supplementary Material
Supporting Data
Acknowledgements
We would like to thank Dr James Campbell (Amgen,
Inc.) for donating the CCX771 compound and Mr. Florian Kirmse
(Institute of Anatomy, University of Leipzig) for technical
assistance.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KZN and JE contributed to the study conception and
design and interpretation of the data. FK acquired and analyzed the
data. JE drafted the manuscript. KZN and FK revised the manuscript.
KZN and FK confirm the authenticity of the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Quinn KE, Mackie DI and Caron KM: Emerging
roles of atypical chemokine receptor 3 (ACKR3) in normal
development and physiology. Cytokine. 109:17–23. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Britton C, Poznansky MC and Reeves P:
Polyfunctionality of the CXCR4/CXCL12 axis in health and disease:
Implications for therapeutic interventions in cancer and
immune-mediated diseases. FASEB J. 35:e212602021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi Y, Riese DJ II and Shen J: The Role of
the CXCL12/CXCR4/CXCR7 chemokine axis in cancer. Front Pharmacol.
11:5746672020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luker GD, Yang J, Richmond A, Scala S,
Festuccia C, Schottelius M, Wester HJ and Zimmermann J: At the
Bench: Pre-clinical evidence for multiple functions of CXCR4 in
cancer. J Leukoc Biol. 109:969–989. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu T, Yang W, Sun A, Wei Z and Lin Q: The
Role of CXC chemokines in cancer progression. Cancers (Basel).
15:1672022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Puchert M, Koch C and Engele J: The 5T4
oncofetal glycoprotein does not act as a general organizer of the
CXCL12 system in cancer cells. Exp Cell Res. 364:175–183. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heuninck J, Perpiñá Viciano C, Işbilir A,
Caspar B, Capoferri D, Briddon SJ, Durroux T, Hill SJ, Lohse MJ,
Milligan G, et al: Context-Dependent Signaling of CXC chemokine
receptor 4 and atypical chemokine receptor 3. Mol Pharmacol.
96:778–793. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mo H, Ren Q, Song D, Xu B, Zhou D, Hong X,
Hou FF, Zhou L and Liu Y: CXCR4 induces podocyte injury and
proteinuria by activating β-catenin signaling. Theranostics.
12:767–781. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
D'Agostino G, Artinger M, Locati M, Perez
L, Legler DF, Bianchi ME, Rüegg C, Thelen M, Marchese A, Rocchi
MBL, et al: β-Arrestin1 and β-Arrestin2 Are Required to Support the
Activity of the CXCL12/HMGB1 Heterocomplex on CXCR4. Front Immunol.
11:5508242020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhuo Y, Gurevich VV, Vishnivetskiy SA,
Klug CS and Marchese A: A non-GPCR-binding partner interacts with a
novel surface on β-arrestin1 to mediate GPCR signaling. J Biol
Chem. 295:14111–14124. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Donà E, Barry JD, Valentin G, Quirin C,
Khmelinskii A, Kunze A, Durdu S, Newton LR, Fernandez-Minan A,
Huber W, et al: Directional tissue migration through a
self-generated chemokine gradient. Nature. 503:285–289. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koch C and Engele J: Functions of the
CXCL12 Receptor ACKR3/CXCR7-What has been perceived and what has
been overlooked. Mol Pharmacol. 98:577–585. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gentilini A, Caligiuri A, Raggi C,
Rombouts K, Pinzani M, Lori G, Correnti M, Invernizzi P, Rovida E,
Navari N, et al: CXCR7 contributes to the aggressive phenotype of
cholangiocarcinoma cells. Biochim Biophys Acta Mol Basis Dis.
18652246–2256. 2029.PubMed/NCBI
|
|
14
|
Xu S, Tang J, Wang C, Liu J, Fu Y and Luo
Y: CXCR7 promotes melanoma tumorigenesis via Src kinase signaling.
Cell Death Dis. 10:1912019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee E, Han J, Kim K, Choi H, Cho EG and
Lee TR: CXCR7 mediates SDF1-induced melanocyte migration. Pigment
Cell Melanoma Res. 26:58–66. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fumagalli A, Heuninck J, Pizzoccaro A,
Moutin E, Koenen J, Séveno M, Durroux T, Junier MP, Schlecht-Louf
G, Bachelerie F, et al: The atypical chemokine receptor 3 interacts
with Connexin 43 inhibiting astrocytic gap junctional intercellular
communication. Nat Commun. 11:48552020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Odemis V, Lipfert J, Kraft R, Hajek P,
Abraham G, Hattermann K, Mentlein R and Engele J: The presumed
atypical chemokine receptor CXCR7 signals through G(i/o) proteins
in primary rodent astrocytes and human glioma cells. Glia.
60:372–381. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheng Y, Qu J, Che X, Xu L, Song N, Ma Y,
Gong J, Qu X and Liu Y: CXCL12/SDF-1α induces migration via
SRC-mediated CXCR4-EGFR cross-talk in gastric cancer cells. Oncol
Lett. 14:2103–2110. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Conley-LaComb MK, Semaan L, Singareddy R,
Li Y, Heath EI, Kim S, Cher ML and Chinni SR: Pharmacological
targeting of CXCL12/CXCR4 signaling in prostate cancer bone
metastasis. Mol Cancer. 15:682016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kasina S, Scherle PA, Hall CL and Macoska
JA: ADAM-mediated amphiregulin shedding and EGFR transactivation.
Cell Prolif. 42:799–812. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chinni SR, Yamamoto H, Dong Z, Sabbota A,
Bonfil RD and Cher ML: CXCL12/CXCR4 transactivates HER2 in lipid
rafts of prostate cancer cells and promotes growth of metastatic
deposits in bone. Mol Cancer Res. 6:446–457. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cabioglu N, Summy J, Miller C, Parikh NU,
Sahin AA, Tuzlali S, Pumiglia K, Gallick GE and Price JE:
CXCL-12/stromal cell-derived factor-1alpha transactivates HER2-neu
in breast cancer cells by a novel pathway involving Src kinase
activation. Cancer Res. 65:6493–6497. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Porcile C, Bajetto A, Barbieri F, Barbero
S, Bonavia R, Biglieri M, Pirani P, Florio T and Schettini G:
Stromal cell-derived factor-1alpha (SDF-1alpha/CXCL12) stimulates
ovarian cancer cell growth through the EGF receptor
transactivation. Exp Cell Res. 308:241–253. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan
M, Zhou X, Xia W, Hortobagyi GN, Yu D and Hung MC: Upregulation of
CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell.
6:459–469. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McGinn OJ, Marinov G, Sawan S and Stern
PL: CXCL12 receptor preference, signal transduction, biological
response and the expression of 5T4 oncofoetal glycoprotein. J Cell
Sci. 125:5467–5478. 2012.PubMed/NCBI
|
|
26
|
Décaillot FM, Kazmi MA, Lin Y, Ray-Saha S,
Sakmar TP and Sachdev P: CXCR7/CXCR4 heterodimer constitutively
recruits beta-arrestin to enhance cell migration. J Biol Chem.
286:32188–32197. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Del Molino Del Barrio I, Wilkins GC,
Meeson A, Ali S and Kirby JA: Breast Cancer: An examination of the
potential of ACKR3 to modify the response of CXCR4 to CXCL12. Int J
Mol Sci. 19:35922018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang J, He L, Combs CA, Roderiquez G and
Norcross MA: Dimerization of CXCR4 in living malignant cells:
Control of cell migration by a synthetic peptide that reduces
homologous CXCR4 interactions. Mol Cancer Ther. 5:2474–2483. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Martínez-Muñoz L, Rodríguez-Frade JM,
Barroso R, Sorzano CÓS, Torreño-Pina JA, Santiago CA, Manzo C,
Lucas P, García-Cuesta EM, Gutierrez E, et al: Separating
Actin-dependent chemokine receptor nanoclustering from dimerization
indicates a role for clustering in CXCR4 signaling and function.
Mol Cell. 70:106–119.e10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
DeNies MS, Smrcka AV, Schnell S and Liu
AP: β-arrestin mediates communication between plasma membrane and
intracellular GPCRs to regulate signaling. Commun Biol. 3:7892020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng K, Smith JS, Eiger DS, Warman A,
Choi I, Honeycutt CC, Boldizsar N, Gundry JN, Pack TF, Inoue A, et
al: Biased agonists of the chemokine receptor CXCR3 differentially
signal through Gαi: β-arrestin complexes. Sci Signal.
15:eabg52032022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Santagata S, Ieranò C, Trotta AM,
Capiluongo A, Auletta F, Guardascione G and Scala S: CXCR4 and
CXCR7 signaling pathways: A focus on the cross-Talk between cancer
cells and tumor microenvironment. Front Oncol. 11:5913862021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Raza S, Rajak S, Tewari A, Gupta P,
Chattopadhyay N, Sinha RA and Chakravarti B: Multifaceted role of
chemokines in solid tumors: From biology to therapy. Semin Cancer
Biol. 86:1105–1121. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luker KE, Gupta M and Luker GD: Imaging
chemokine receptor dimerization with firefly luciferase
complementation. FASEB J. 23:823–834. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Levoye A, Balabanian K, Baleux F,
Bachelerie F and Lagane B: CXCR7 heterodimerizes with CXCR4 and
regulates CXCL12-mediated G protein signaling. Blood.
113:6085–6093. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Koch C, Fischer NC, Puchert M and Engele
J: Interactions of the chemokines CXCL11 and CXCL12 in human tumor
cells. BMC Cancer. 22:13352022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Uribe ML, Marrocco I and Yarden Y: EGFR in
Cancer: Signaling mechanisms, drugs, and acquired resistance.
Cancers (Basel). 13:27482021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zuccolo E, Di Buduo C, Lodola F,
Orecchioni S, Scarpellino G, Kheder DA, Poletto V, Guerra G,
Bertolini F, Balduini A, et al: Stromal cell-derived factor-1α
promotes endothelial colony-forming cell migration through the
Ca2+-Dependent activation of the extracellular signal-regulated
kinase 1/2 and phosphoinositide 3-kinase/AKT pathways. Stem Cells
Dev. 27:23–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jiang Y, Lim J, Wu KC, Xu W, Suen JY and
Fairlie DP: PAR2 induces ovarian cancer cell motility by merging
three signalling pathways to transactivate EGFR. Br J Pharmacol.
178:913–932. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lan L, Wang H, Yang R, Liu F, Bi Q, Wang
S, Wei X, Yan H and Su R: R2-8018 reduces the proliferation and
migration of non-small cell lung cancer cells by disturbing
transactivation between M3R and EGFR. Life Sci. 234:1167422019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hopkins MM, Liu Z and Meier KE: Positive
and negative cross-talk between lysophosphatidic acid receptor 1,
free fatty acid receptor 4, and epidermal growth factor receptor in
human prostate cancer cells. J Pharmacol Exp Ther. 359:124–133.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu J, Liu Y, Ma Y, Wang R, Ji X, Zhang Y
and Du Y: Interaction between CXCR4 and EGFR and downstream
PI3K/AKT pathway in lung adenocarcinoma A549 cells and transplanted
tumor in nude mice. Int J Clin Exp Pathol. 13:132–141.
2020.PubMed/NCBI
|
|
44
|
Liu B, Song S, Setroikromo R, Chen S, Hu
W, Chen D, van der Wekken AJ, Melgert BN, Timens W, van den Berg A,
et al: CX Chemokine Receptor 7 contributes to survival of
KRAS-Mutant Non-Small cell lung cancer upon loss of epidermal
growth factor receptor. Cancers (Basel). 11:4552019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zuo J, Wen M, Li S, Lv X, Wang L, Ai X and
Lei M: Overexpression of CXCR4 promotes invasion and migration of
non-small cell lung cancer via EGFR and MMP-9. Oncol Lett.
14:7513–7521. 2017.PubMed/NCBI
|
|
46
|
Lopez-Haber C, Barrio-Real L,
Casado-Medrano V and Kazanietz MG: Heregulin/ErbB3 signaling
enhances CXCR4-driven Rac1 activation and breast cancer cell
motility via hypoxia-inducible factor 1α. Mol Cell Biol.
36:2011–2026. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tsai MF, Chang TH, Wu SG, Yang HY, Hsu YC,
Yang PC and Shih JY: EGFR-L858R mutant enhances lung adenocarcinoma
cell invasive ability and promotes malignant pleural effusion
formation through activation of the CXCL12-CXCR4 pathway. Sci Rep.
5:135742015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Boudot A, Kerdivel G, Lecomte S, Flouriot
G, Desille M, Godey F, Leveque J, Tas P, Le Dréan Y and Pakdel F:
COUP-TFI modifies CXCL12 and CXCR4 expression by activating EGF
signaling and stimulates breast cancer cell migration. BMC Cancer.
14:4072014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bao W, Fu HJ, Xie QS, Wang L, Zhang R, Guo
ZY, Zhao J, Meng YL, Ren XL, Wang T, et al: HER2 interacts with
CD44 to up-regulate CXCR4 via epigenetic silencing of microRNA-139
in gastric cancer cells. Gastroenterology. 141:2076–2087.e6. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yasumoto K, Yamada T, Kawashima A, Wang W,
Li Q, Donev IS, Tacheuchi S, Mouri H, Yamashita K, Ohtsubo K and
Yano S: The EGFR ligands amphiregulin and heparin-binding egf-like
growth factor promote peritoneal carcinomatosis in CXCR4-expressing
gastric cancer. Clin Cancer Res. 17:3619–3630. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rahimi M, George J and Tang C: EGFR
variant-mediated invasion by enhanced CXCR4 expression through
transcriptional and post-translational mechanisms. Int J Cancer.
126:1850–1860. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Guo Z, Cai S, Fang R, Chen H, Du J, Tan Y,
Ma W, Hu H, Cai S and Liu Y: The synergistic effects of CXCR4 and
EGFR on promoting EGF-mediated metastasis in ovarian cancer cells.
Colloids Surf B Biointerfaces. 60:1–6. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kallifatidis G, Munoz D, Singh RK, Salazar
N, Hoy JJ and Lokeshwar BL: β-Arrestin-2 counters CXCR7-mediated
EGFR transactivation and proliferation. Mol Cancer Res. 14:493–503.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Salazar N, Muñoz D, Kallifatidis G, Singh
RK, Jordà M and Lokeshwar BL: The chemokine receptor CXCR7
interacts with EGFR to promote breast cancer cell proliferation.
Mol Cancer. 13:1982014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Singh RK and Lokeshwar BL: The
IL-8-regulated chemokine receptor CXCR7 stimulates EGFR signaling
to promote prostate cancer growth. Cancer Res. 71:3268–3277. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Abourehab MAS, Alqahtani AM, Youssif BGM
and Gouda AM: Globally approved EGFR inhibitors: Insights into
their syntheses, target kinases, biological activities, receptor
interactions, and metabolism. Molecules. 26:66772021. View Article : Google Scholar : PubMed/NCBI
|