Introduction
Gastroenteropancreatic neuroendocrine neoplasms
(GEP-NENs) are a group of rare tumors originating from
neuroendocrine cells of the pancreas or of the gastrointestinal
tract and comprise well-differentiated neuroendocrine tumors (NETs)
and poorly-differentiated neuroendocrine carcinomas (NECs)
(1). Moreover, depending on the
hormone and amine secretion activity, GEP-NENs can be classified
into functional and non-functional neoplasms (2). They represent the second most common
cancer of the digestive system as their incidence has increased
over the last decades (3). GEP-NENs
are characterized by an indolent behavior in terms of tumor growth
and the treatment of choice in locally defined tumors remains
surgical resection (4). However, at
presentation, ~80% of patients have already developed liver or
lymph node metastases (5). The
number of targeted and theranostic options for patients with NETs
have expanded significantly over the last decade; however, the
cytoreductive capacity of these agents remains quite modest
(6). Thus far, chemotherapy has
demonstrated to be clinically useful in patients with metastatic or
unresectable pancreatic NETs and grade 3 NET disease, however,
novel combination regimens and targeted therapy are necessary to
increase the number of cytotoxic options that may be relevant for
patients with NETs (6,7).
Numerous anticancer studies have focused lately on
natural compounds, present in fruits, vegetables and spices, that
exhibit pro-apoptotic and anti-inflammatory activity by targeting
diverse molecules including transcription factors, cytokines,
chemokines, growth factor receptors and inflammatory enzymes
(8). In addition, for their easy
availability, safety and relative low-cost, natural compounds are
particularly attractive for chemoprevention (8). Among the natural compounds, curcumin,
a hydrophobic polyphenol has been revealed to possess anticancer
properties along with an excellent safety profile although it
presents low solubility and bioavailability (9,10). To
circumvent this latter problem, a number of strategies have been
developed, involving the modification of its structure or
application of drug delivery agents, such as nanoparticles,
liposomes and micelles (11).
Another approach is based on the interaction of curcumin ligands
with inorganic or organometallic ruthenium moieties to provide more
soluble and assimilable compounds (12–14)
that are more attractive for clinical practice. Those compounds are
stable and water-soluble and their frameworks provide considerable
scope for optimizing the design in terms of their biological
activity and for minimizing side-effects. For these reasons,
organometallic ruthenium compounds are promising anticancer
molecules to be tested in clinical practice especially in
combination with other drugs (15–17).
It was recently demonstrated by the authors the anticancer effects
of ruthenium(II)-curcumin compounds in vitro in different
cancer cell lines including colon, breast and glioblastoma
(18,19), as well as their lack of toxicity in
normal cells (16). However, to the
best of the authors' knowledge, these compounds have never been
used in neuroendocrine tumors.
Different from normal cells, cancer cells often
activate an adaptive response to resist to the anticancer
treatments (20). In this regard,
the authors' recent studies demonstrated the role of the nuclear
factor erythroid 2-related factor 2 (NRF2)-induced pathway as key
determinant in cancer cells' resistance to therapies (18,19,21).
NRF2 transcription factor is the master regulator of the oxidative
stress and its detoxifying activity is often hijacked by cancer
cells as a protective mechanism, particularly in the course of
anti-cancer treatments, leading to cancer cell resistance to
therapies (22). Among the NRF2
targets, involved in cancer progression and chemoresistance, is
heme-oxygenase 1 (HO-1), catalase, NAD(P)H quinone oxidoreductase 1
(NQO1) and p62/SQSTM (herein p62) (22,23).
Intriguingly, NRF2 induces p62 expression and p62 stabilizes NRF2
by triggering the degradation of NRF2 inhibitor Kelch-like
ECH-associated protein 1 (Keap1), creating a positive feedback loop
between NRF2 and p62 that induces cancer progression and resistance
to therapies (24,25). NRF2 is often overexpressed in
pancreatic cancer which may reflect a greater intrinsic capacity of
the tumor cells to respond to stress signals and resist to the
chemotherapeutic agents (26).
However, to the best of the authors' knowledge, NRF2 activity has
never been evaluated in the pancreatic NET cell line BON-1, one of
the two commonly used human cell line GEP-NET-derived, which also
carries an endogenous dysfunctional p53 that inhibits apoptosis
(27–29). Among the oncogenic pathways that
establish a cross-talk with NRF2, other than p62, is mutant (mut)
p53 (30). Tumor suppressor p53
plays a central role in tumor prevention and in response to
anticancer therapies and, for these reasons, is the most
inactivated oncosuppressor in human tumors, by gene mutation or by
protein deregulation (31,32). TP53 gene mutations have been found
in pancreatic cancer affecting both cancer progression and response
to therapies (33), and also in
poorly differentiated NEC (34). In
BON-1 cell line, a homozygous stop-gain g.7574003A>G mutation
has been found in exon 10 of TP53 with possible inhibition of the
p53 apoptotic activity (28).
On the basis of the aforementioned background, in
the present study it was aimed to evaluate the anticancer effects
of the cationic Ruthenium (Ru)(II)-Bisdemethoxycurcumin compound
(Ru-bdcurc) in the cell line BON-1. The present study highlighted
the key role of NRF2 in BON-1 resistance to the cytotoxic activity
of the curcumin compound and the interplay of NRF2 with a
dysfunctional endogenous p53, supporting the monitoring of this
potential biomarker in neuroendocrine tumors for future assessment
of response to therapies.
Materials and methods
Cell culture and reagents
In the present study, the BON-1 cell line (RRUD:
CVCL_3985) (NCL2110P096; DBA Italia s.r.l.) (https:www.cellosaurus.org/CVCL_3985), established from
a lymph node metastasis of a human pancreatic carcinoid tumor
(27), was used. Cells arrived at
passage 1 and were used for additional 6 passages. Cells were
maintained in Dulbecco's modified Eagle's medium (DMEM)
(Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10%
heat-inactivated fetal bovine serum (FBS) (Corning, Inc.), plus
glutamine and antibiotics [Penicillin-Streptomycin-L-Glutamine,
100× (+) 29.2 mg/ml L-glutamine; cat. no. 30-009-CI; Merck KGaA] in
a humidified atmosphere with 5% CO2 at 37°C. Cells
underwent routine testing to ensure that they were mycoplasm
negative. The cationic Ruthenium (Ru)(II) compound containing
bisdemethoxycurcumin and the hydrosoluble PTA phosphine
([(cym)Ru(bdcurc)(PTA)]SO3CF3) (where cym=cymene,
bdcurc=bisdemethoxycurcumin and
PTA=1,3,5-triaza-7-phosphaadamantane) (herein Ru-bdcurc), with the
chemical formula: C36H42F3N3O7PRuS and the molecular weight: 849,8
g/mol, was synthesized as previously reported (16,17).
The Ru-bdcurc compound was dissolved in DMSO and stored at −20°C
before using it at 50 and 100 µM for the indicated times, as
previously reported (19). The
inhibitor of the antioxidant response Brusatol (cat. no. SML1868;
Sigma-Aldrich; Merck KGaA) (35,36)
was used at 100 µM for 4 h pre-treatment, as previously reported
(37).
Cell viability and colony assays
Cell viability was measured by Trypan blue (cat. no.
72571; Sigma-Aldrich; Merck KGaA) assay. Subconfluent cells were
plated in six-well plates and, the day after, treated with
different concentration of Ru-bdcurc for 24 and 48 h or in
combinations with a 4 h pre-treatment of NRF2 inhibitor Brusatol
(100 nM). After treatments, both floating and adherent cells were
collected and stained with Trypan blue. Cell viability of
triplicates was assessed by counting blue (dead)/total cells with a
Neubauer hemocytometer using light microscopy. For long-term cell
survival, cells were plated in 60 mm Petri dishes until
subconfluence. Then, cells were mock-treated or treated with
Ru-bdcurc (50 or 100 µM) for 16 h. After treatments, cells were
washed, trypsinized, counted and equal cell number re-plated in
duplicate with fresh medium in 60-mm Petri dishes for determining
cell survival. Death-resistant colonies (with >50 cells) were
stained with crystal violet (cat. no. 46364; Sigma-Aldrich; Merck
KGaA) (diluted 1:2 with the cell culture) 14 days later. Plates
underwent scanning and the intensity of the cell staining was
quantified by ImageJ software.
Western blot analysis
Cells were harvested and centrifuged and the
resulting pellets were lysed in lysis buffer (50 mM Tris-HCl, pH
7.5, 150 mM NaCl, 5 mM EDTA, 150 mM KCl, 1 mM dithiothreitol and 1%
Nonidet P-40) (all from Sigma-Aldrich; Merck KGaA) containing
protease inhibitors (CompleteTM, Mini Protease Inhibitor Cocktail;
Merck Life Science S.r.l.). Protein concentration was determined by
the Bio-Rad protein assay kit (cat. no. 5000001; Bio-Rad
Laboratories, Inc.), a simple colorimetric assay for measuring
total protein concentration based on the Bradford dye-binding
method. Proteins were separated by loading 10–30 µg of total cell
lysates on denaturing 8–15% SDS-PAGE (polyacrylamide gel
electrophoresis) gels (Bio-Rad Laboratories, Inc.), following
semidry blotting to polyvinylidene difluoride (PVDF) membranes
(Immobilon-P; Merck KGaA). Unspecific signals were blocked by
incubating the membranes in Tris-buffered saline containing 0.1%
Tween 20 (TBS) and 3% BSA (Sigma-Aldrich; Merck KGaA) for 1 h at
room temperature. Membranes were then probed with the primary
antibodies and subsequently with the following secondary
antibodies: Goat Anti-Mouse IgG (1:10,000; cat. no. 1706516) and
Goat Anti-Rabbit IgG (1:10,000; cat. no. 1706515; both from Bio-Rad
Laboratories, Inc.). The enzymatic signal was visualized by
chemiluminescence (ECL Detection system; Amersham; Cytiva). The
following antibodies were used: Mouse monoclonal anti-p62/SQSTM1
(D-3; 1:1,000; cat. no. sc-28359), mouse monoclonal anti-catalase
(H-9; 1:1,000; cat. no. sc-271803), mouse monoclonal anti-p53
(DO-1; 1:1,000; cat. no. sc-126), mouse monoclonal anti-Bcl-2
(1:1,000; cat. no. sc-509) and mouse monoclonal anti-Mcl1 (G-7;
1:1,000; cat. no. sc-74437) (all from Santa Cruz Biotechnology,
Inc.), rabbit polyclonal anti-NRF2 (1:1,000; cat. no. ab62352;
Abcam), mouse monoclonal anti-phospho-Histone H2AX (Ser139 clone
JBW301; 1:1,000; cat. no. 05-636; Sigma-Aldrich; Merck KGaA),
rabbit polyclonal anti-phospho-4E-BP1 (Thr37/46; 1:200; ca.t no.
2855), rabbit polyclonal anti-4E-BP1 (1:200; cat. no. 9452; both
from Cell Signaling Technology, Inc.), mouse monoclonal
anti-poly(ADP-ribose) polymerase (PARP, cleavage site-214-215;
1:1,000; cat. no. AB3565; Sigma-Aldrich; Merck KGaA). Mouse
monoclonal β-actin (Ab-1; 1:10,000; (cat. no. CP01; Calbiochem;
Merck KGaA), was used as protein loading control.
Densitometric analysis
Densitometry was performed on ECL results with
ImageJ software (1.47 version; National Institutes of Health) which
was downloaded from the NIH website (http://imagej,nih.gov/ij) and the relative band
intensity was normalized to β-actin signals and plotted as protein
expression/β-actin ratio.
Small interference (si)RNA
transfection
Cells were plated at subconfluency in 35-mm Petri
dishes and, the day after plating, were transfected with the Nrf2
small interference (si)RNA (cat. no. sc-37030) or control siRNA
(cat. no. sc-37007) (both from Santa Cruz Biotechnology, Inc.; the
siRNA sequences are not available) using LipofectaminePLus reagent
(cat. no. 11514015; Thermo Fisher Scientific, Inc.) as previously
reported (18). The concentration
used was 10 nmol. For p53 knockdown, cells were transfected with
sip53 plasmid (sip53) or an empty vector (si-ctr) (38,39)
using LipofectaminePLus reagent according to the manufacturer's
instructions. A total of 24 h after transfection, cells were
trypsinized and replated for the indicated experiments.
RNA extraction and semiquantitative
reverse transcription (RT)-polymerase chain reaction (PCR)
analysis
Total RNA extraction was performed by using TRIzol
Reagent (Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. cDNA was synthesized by using MuLV
reverse transcriptase kit according to the manufacturer's
instructions (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Semiquantitative RT-PCR was carried out with 2 µl cDNA reaction and
genes specific oligonucleotides under conditions of linear
amplification. by using Hot-Master Taq polymerase (Thermo Fisher
Scientific, Inc.). Primer sequences specific to the target genes
were as follows: HO-1 forward, 5′AAGATTGCCCAGAAAGCCCTGGAC-3′ and
reverse, 5′-AACTGTCGCCACCAGAAAGCTGAG-3′ (40) (annealing temperature: 58°C for 30
cycles); p62 forward, 5′-CTGCCCAGACTACGACTTGTGT-3′ and reverse,
5′-TCAACTTCAATGCCCAGAGG-3′ (19,40)
(annealing temperature: 58°C for 28 cycles); NRF2 forward,
5′-TCCATTCCTGAGTTACAGTGTCT-3′ and reverse,
5′-TGGCTTCTGGACTTGGAACC-3′ (18,40,41)
(annealing temperature: 58°C for 30 cycles); MDR1 forward,
5′-AACGGAAGCCAGAACATTCC-3′ and reverse, 5′-AGGCTTCCTGTGGCAAAGAG-3′
(42,43) (annealing temperature: 60°C for 29
cycles); 28S forward, 5′-GTTCACCCACTAATAGGGAACGTGA-3′ and reverse,
5′-GGATTCTGACTTAGAGGCGTTCAGT-3′ (39,40,44,45)
(annealing temperature: 58°C for 15 cycles); Bcl-2 forward,
5′-AGGATTGTGGCCTTCTTTGAG-3′ and reverse,
5′-GAGACAGCCAGGAGAAATCAAA-3′ (46)
(annealing temperature: 58°C for 30 cycles); and NOXA forward,
5′-AGGACTGTTCGTGTTCAGCTC-3′ and reverse 5′-GTCCACCTCCTGAGAAAACTC-3′
(47) (annealing temperature: 55°C
for 28 cycles). The denaturation and extension temperatures were,
respectively 98 and 72°C for all the mRNA amplifications. PCR
products were run on a 2% agarose gel and visualized with GelRed
Nucleic Acid gel stain (Biotium, Inc.). The housekeeping 28S gene,
used as internal standard, was amplified from the same cDNA
reaction mixture. Densitometric analysis was applied to quantify
mRNA levels compared with 28S control gene expression.
Statistical analysis
The results are expressed as the mean ± standard
deviation (SD) of at least three independent experiments and
statistical analyses were performed using GraphPad Prism® software
(Version 9.0.0; Dotmatics). The unpaired two-tailed Student t-test
(for data containing two groups) and the nonparametric 1-way
analysis of variance (ANOVA) followed by Tukey's HSD test (for
multiple comparisons tests) were used to demonstrate statistical
significance. A difference was considered statistically significant
when P≤0.05.
Results
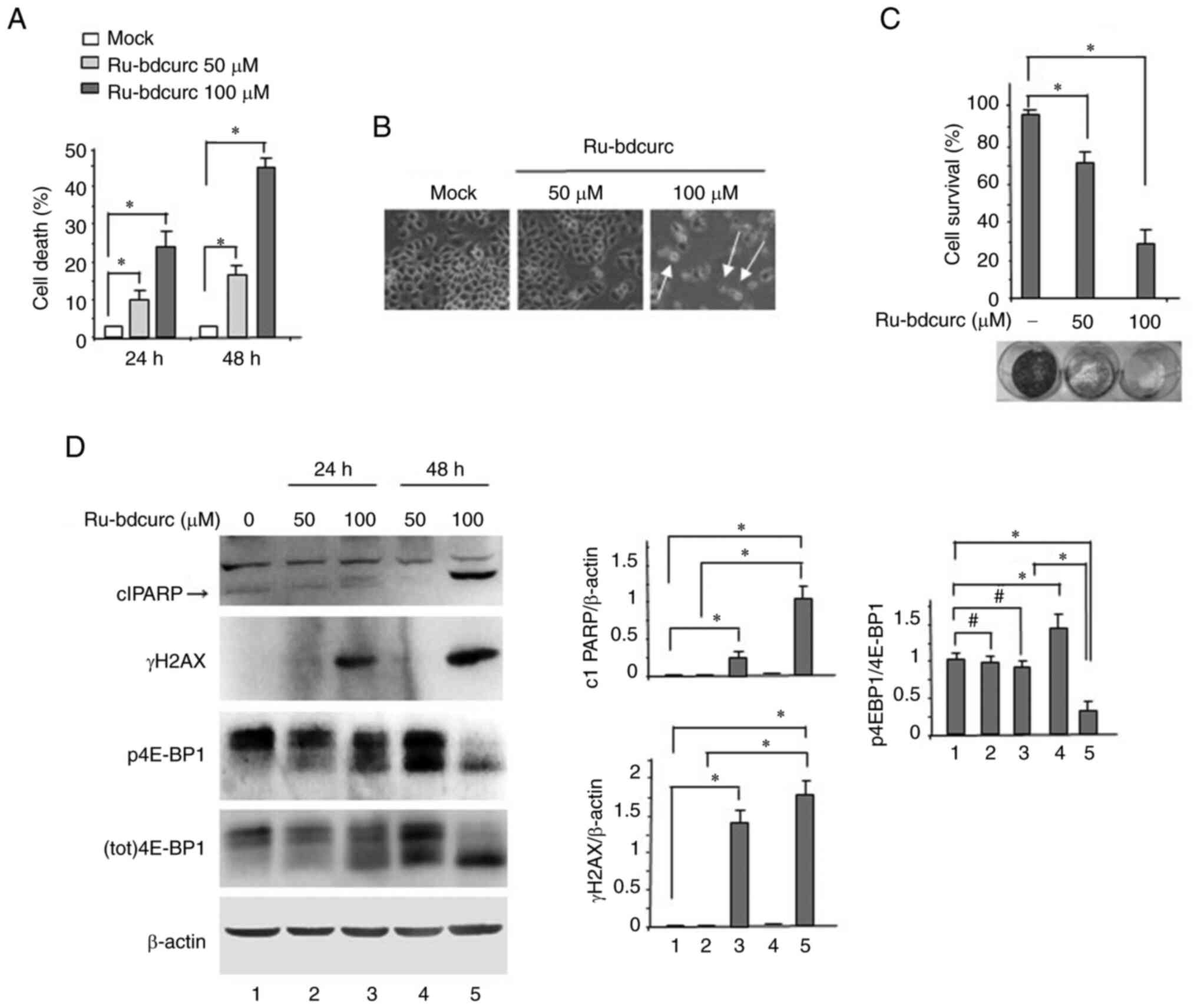
Ru-bdcurc compound induces dose- and
time-dependent BON-1 cells' death
BON-1 cells were treated with two concentrations of
Ru-bdcurc compound that were recently demonstrated by the authors
to have different cytotoxic activity against colon cancer cells
(19). The results revealed a dose-
and time-dependent cell death, as indicated by the trypan blue
assay (Fig. 1A). Cell death was
also evidenced microscopically where distinct signs of cell
shrinkage were observed, in particular at 100 µM dose of Ru-bdcurc
(Fig. 1B). The EC50 for the
Ru-bdcurc compound was 100 µM (AAT Bioquest, Inc; http://www.aatbio.com/tools/ic50-calculator). Then the
long-term survival was analyzed by colony formation assay. The
results of the densitometric analysis demonstrated that 100 µM dose
of Ru-bdcurc significantly reduced BON-1 cell survival, compared
with 50 µM dose (Fig. 1C). At the
biochemical level, the occurrence of an apoptotic cell death, as
indicated by the cleaved fragment of PARP (clPARP) and by the
increased clPARP/PARP ratio, was evident only with the 100 µM dose
treatment, compared with 50 µM dose (Fig. 1D). The apoptotic cell death
associated with the appearance of the phosphorylated form of H2AX
(γH2AX) (Fig. 1D), a marker of DNA
damage and apoptosis (48). Then
the phosphorylation of 4EBP1 (p4E-BP1), a mTOR target whose
activation can sustain cancer cell survival and predict poor
prognosis (49,50), was investigated. In agreement, mTOR
pathway has been found dysregulated in GEP-NET and involved in
tumor development (51). As shown
in Fig. 1D, p4E-BP1 was induced by
50 µM dose, on the other hand, p4E-BP1 was impaired by 100 µM dose
of Ru-bdcurc. This latter result associated with the greater
induction of cell death that was observed by using 100 µM dose of
Ru-bdcurc, compared with 50 µM dose, strengthening the
antiapoptotic role of p4E-BP1. Collectively, these results
indicated that 100 µM dose of Ru-bdcurc induced BON-1 cell death
while 50 µM dose activated survival pathways that potentially
reduced the compound cytotoxic effects.
NRF2 pathway activation in response to
50 µM dose of Ru-bdcurc
Several lines of evidence suggest the important role
of NRF2 in the chemoresistance of different types of cancer cells
(22), as also assessed by the
authors' recent studies using curcumin compounds (18,19).
However, to the best of the authors' knowledge, NRF2 has never been
evaluated in BON-1 cells. Therefore, the NRF2 pathway in BON-1
cells in response to the Ru-bdcurc treatment was next assessed. To
this aim, the two doses of the compound, that were revealed to have
different outcome in terms of cell death as aforementioned, were
used. The experimental data demonstrated that 50 µM dose of
Ru-bdcurc significantly increased the levels of NRF2 protein,
compared with the 100 µM dose, and induced the expression of its
targets such as catalase and p62 (Fig.
2A). To evaluate if the NRF2 induction was at the
transcriptional or post-transcriptional level, mRNA analysis was
performed. It was identified that 50 µM dose of Ru-bdcurc did not
induce NRF2 gene expression (Fig.
2B), suggesting rather stabilization of NRF2 at the protein
level. In addition, only the 50 µM dose increased the mRNA
expression of the NRF2 targets HO-1 and p62 (Fig. 2B), indicative of NRF2
transcriptional activation, in this setting. Thus, the increased
p62 gene expression was in accordance with the finding that p62 is
a NRF2 transcriptional target (23–25).
Interestingly, it has been reported that p62 activates pro-survival
pathways including mTOR (52), in
agreement with the aforementioned results in Fig. 1 and with the cross-talk among
oncogenic pathways such as p62/mTOR/NRF2 to increase tumor
development and resistance to therapies (30). In agreement with the different
extent of cell death in response to different doses of Ru-bdcurc,
the results showed that only 50 µM dose of Ru-bdcurc increased the
antiapoptotic Bcl-2 protein levels while the 100 µM dose
significantly reduced the levels of Mcl1 protein (Fig. 2A), a pro-survival member of the
Bcl-2 family (53). Moreover, 50 µM
dose of Ru-bdcurc treatment, compared with 100 µM dose, increased
the endogenous p53 protein level (Fig.
2A), in agreement with the paradigm of NRF2/mutp53 interplay to
sustain their oncogenic activities (41,54).
The endogenous p53 is reported to be dysfunctional in BON-1 cells
and inhibit apoptosis (28). In
agreement, the present results revealed that 50 µM dose of
Ru-bdcurc, compared with 100 µM dose, induced the expression of
multidrug resistant gene 1 (MDR1), a target of some mutant p53
proteins (55), as well as of the
antiapoptotic gene Bcl-2 (Fig. 2B),
which associated with a potential oncogenic activity of the
endogenous dysfunctional p53 in BON-1 cells. On the other hand, 100
µM dose of Ru-bdcurc induced the expression of Noxa (Fig. 2B), a pro-apoptotic gene induced by
p53 family members (56), that has
been shown to inhibit the antiapoptotic Mcl1 protein (57). This latter result is in consistency
with the reduction of Mcl1 protein levels in Fig. 2A and with induction of the apoptotic
cell death (Fig. 1A). Taken
together, these findings indicated that the Ru-bdcurc treatment was
able to induce pro-apoptotic (PARP cleavage, Fig. 1) or cell death-resistant pathways
(NRF2-induced targets, mTOR target 4E-BP1, Bcl-2 and dysfunctional
p53; Fig. 2) according to the low
(50 µM) or high (100 µM) dose used.
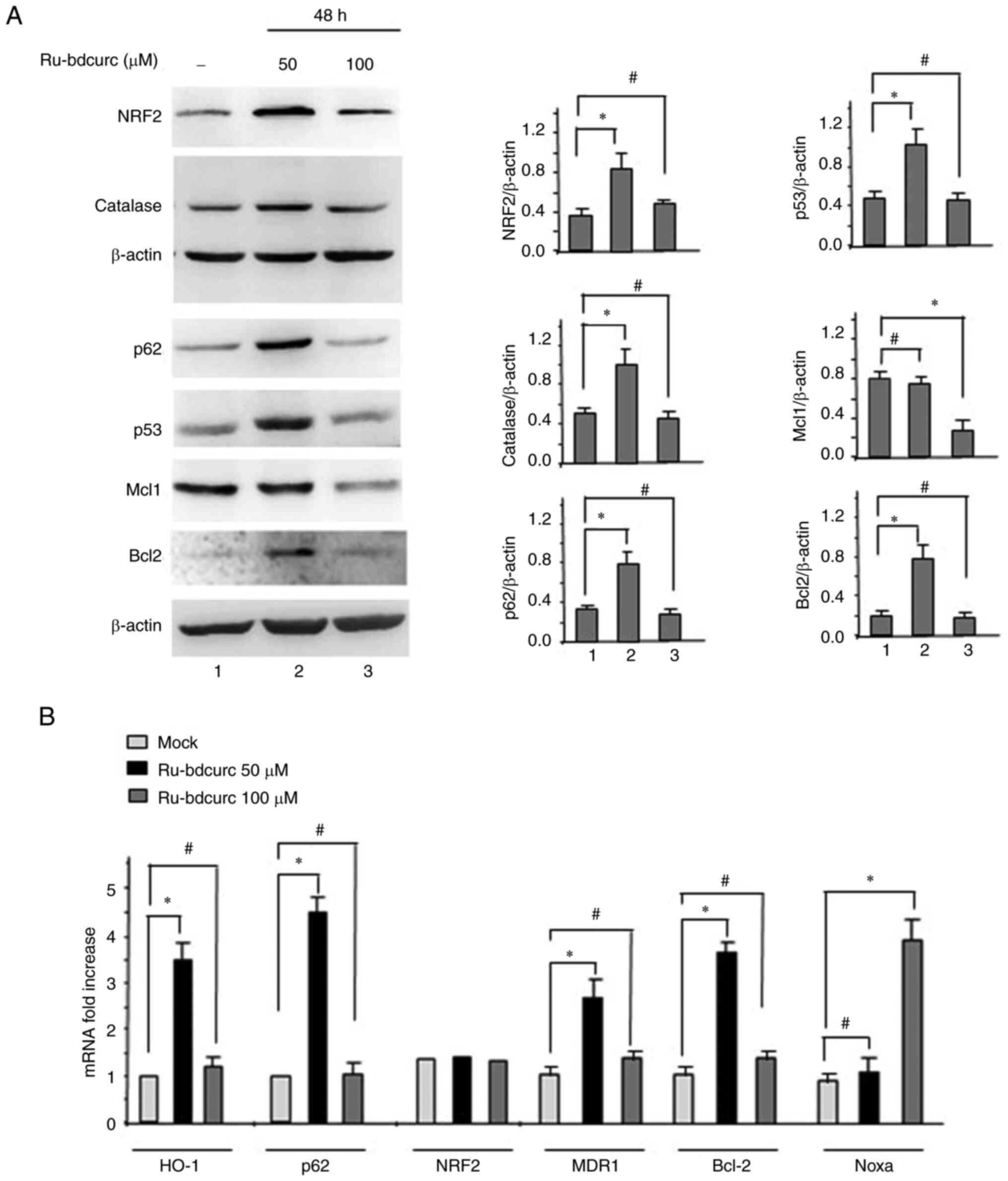 | Figure 2.NRF2 activation in response to
Ru-bdcurc. (A) BON-1 cells were treated with Ru-bdcurc (50 and 100
µM) for 48 h and the protein levels of NRF2, catalase, p62, p53,
Mcl1 and Bcl2 proteins were evaluated by western blot analysis;
β-actin was used as protein loading control and one representative
experiment is shown. The ratio of the protein levels vs. β-actin,
following densitometric analysis, is reported as histograms plus
SD. Statistics was measured between treated (50 or 100 µM) and the
untreated cells (−). (B) Total mRNA was extracted from cells
treated as in (A) to evaluate HO-1, NRF2, p62, MDR1, Bcl-2 and Noxa
gene expression by semiquantitative reverse transcription PCR of
reverse transcribed cDNA. Histograms represent the mean ± SD of
three independent experiments. Statistics was measured between
treated (50 or 100 µM) and the untreated cells (Mock).
*P≤0.05 and #, not statistically significant.
NRF2, nuclear factor erythroid 2-related factor 2; Ru-bdcurc,
Ruthenium (Ru)(II)-Bisdemethoxycurcumin. |
Targeting NRF2 improves the cytotoxic
activity of 50 µM dose of Ru-bdcurc
Then, to evaluate the biological role of NRF2 in
this setting, it was attempted to inhibit it by pharmacologic or
genetic means. NRF2 pharmacologic inhibitor Brusatol (35,36)
was used before exposing BON-1 cells to 50 µM of Ru-bdcurc. The
results demonstrated that the Ru-bdcurc/Brusatol combination
increased BON-1 cell death (Fig.
3A, upper panel), compared with the 50 µM dose Ru-bdcurc alone.
At the biochemical level, the Ru-bdcurc/Brusatol combination
counteracted the Ru-bdcurc-induced upregulation of NRF2 as well as
of its target p62 (Fig. 3A, lower
panels); in addition, the Ru-bdcurc/Brusatol combination
significantly increased the expression of γH2AX compared with the
50 µM dose of Ru-bdcurc alone (Fig.
3A, lower panels). Similarly, knocking down NRF2 by specific
siRNA (Fig. 3B) increased the cell
death induced by 50 µM dose of Ru-bdcurc compound (Fig. 3C, left panels). At the biochemical
level, the NRF2 silencing reduced catalase and p62 expression, as
well as the p53 levels that were induced in response to 50 µM dose
of Ru-bdcurc (Fig. 3C, right
panels), suggesting that an interplay between NRF2 and
dysfunctional p53 exists in BON-1 cells. In addition, the
Ru-bdcurc/Brusatol combination significantly increased the
expression of γH2AX compared with the 50 µM dose of Ru-bdcurc alone
(Fig. 3C, lower panels).
Collectively, these results suggested that inhibiting the NRF2
pathway, induced by the lower dose of Ru-bdcurc, increased the
cytotoxic effect of Ru-bdcurc compound.
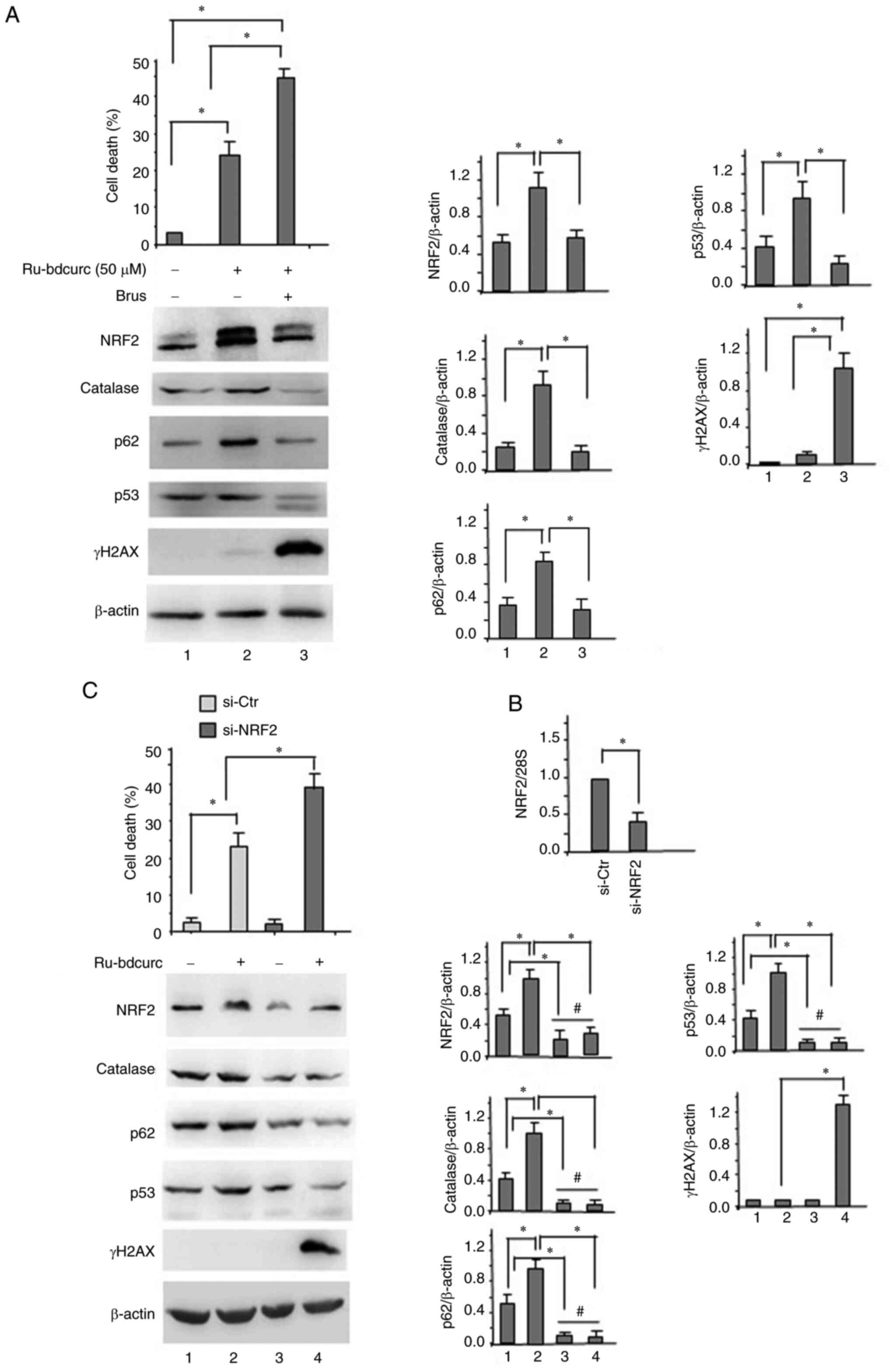 | Figure 3.Pharmacologic or genetic inhibition
of NRF2 improves the 50 µM Ru-bdcurc cytotoxic activity. (A) BON-1
cells were left untreated or pre-treated with 100 nM Brus for 4 h
and then treated with 50 µM Ru-bdcurc for 48 h, before cell
viability was measured by Trypan blue exclusion assay (upper
panel). Protein levels were evaluated by western blot analysis
(lower panel). β-actin was used as protein loading control and one
representative experiment is shown. The ratio of the protein levels
vs. β-actin, following densitometric analysis, is reported as
histograms plus SD (right panels). Statistics was measured between
treated (50 µM) and untreated cells (−) with or without Brus
co-treatment. (B) BON-1 cells were transfected with siRNA control
(si-ctr) or siNRF2 and, 24 h after transfection, NRF2 mRNA was
evaluated by semiquantitative reverse transcription PCR. (C) BON-1
cells, transfected as in (B) for siRNA interference, were treated,
24 h after siRNA transfection, with Ru-bdcurc (50 µM) for 48 h
before cell viability was measured by Trypan blue exclusion assay
(upper panel). Protein levels were evaluated by western blot
analysis (lower panel). β-actin was used as protein loading control
and one representative experiment is shown. The ratio of the
protein levels vs. β-actin, following densitometric analysis is
reported as histograms plus SD (right panels). Statistics was
measured between treated (50 µM) and untreated cells (−) and
compared according to NRF2 interference. *P≤0.05 and
#, not statistically significant. NRF2, nuclear factor
erythroid 2-related factor 2; Ru-bdcurc, Ruthenium
(Ru)(II)-Bisdemethoxycurcumin; Brus, brusatol; siRNA, small
interfering RNA. |
Targeting p53 reduces NRF2 activation
and increases the cytotoxic effect of 50 µM dose of Ru-bdcurc
Then, to evaluate the role of p53 in BON-1 cell
response to the Ru-bdcurc compound and its interplay with NRF2, it
was attempted to inhibit it by specific siRNA (38,39).
The results demonstrated that knocking down p53 (Fig. 4A) reduced the Ru-bdcurc-induced
upregulation of NRF2 and of its targets p62 and catalase (Fig. 4B). At the biological level, knocking
down p53 increased the cell death induced by 50 µM dose of
Ru-bdcurc (Fig. 4C). Taken
together, these results indicated that a dysfunctional endogenous
p53 in BON-1 contributed to cellular resistance to Ru-bdcurc
cytotoxicity, likely in an interplay with NRF2.
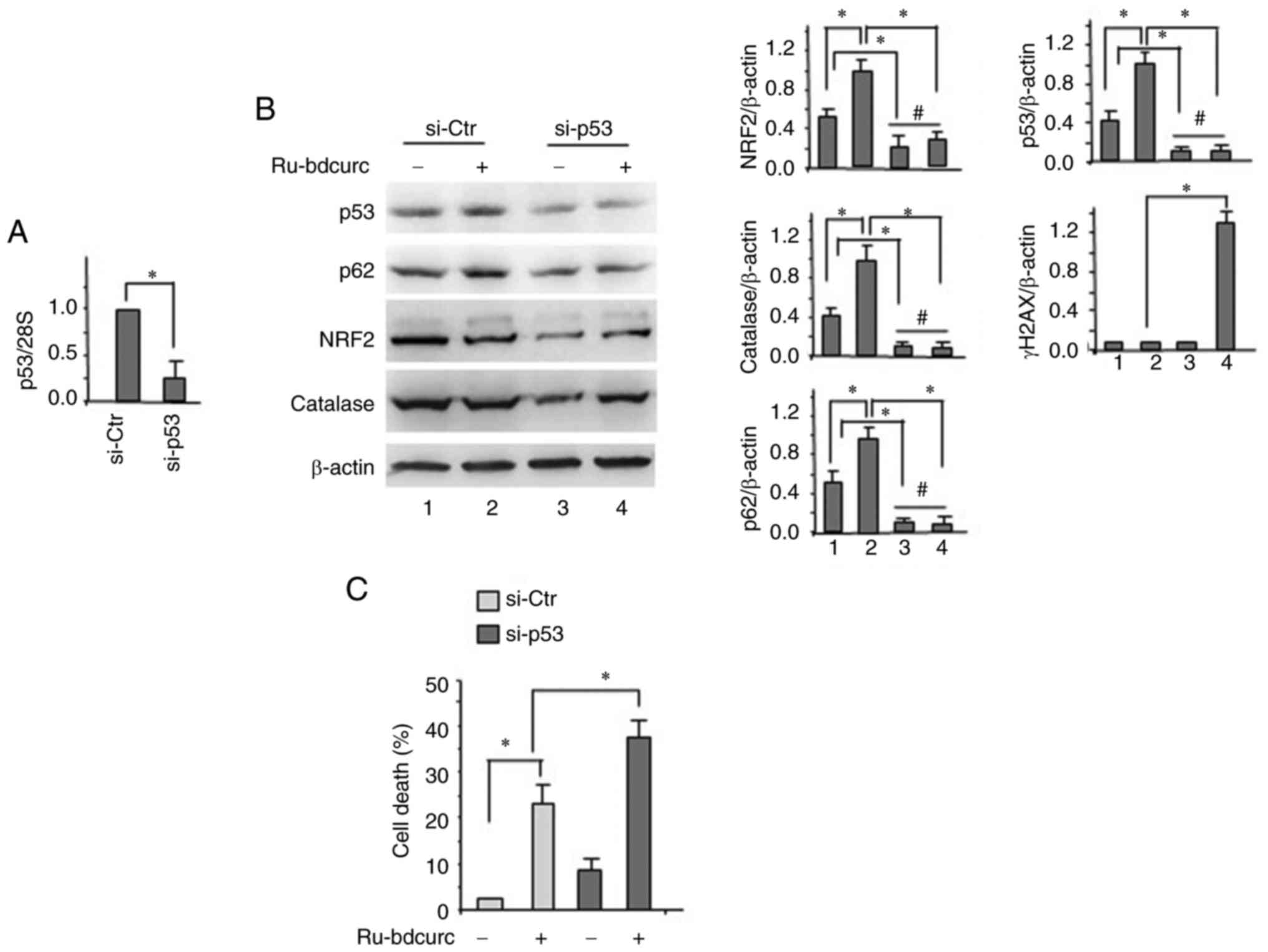 | Figure 4.Dysfunctional-p53 cross-talks with
NRF2 to reduce BON-1 chemosensitivity. (A) BON-1 cells were
transfected with siRNA control (si-ctr) and sip53 and, 24 h after
transfection, p53 mRNA was evaluated by semiquantitative reverse
transcription PCR. (B) BON-1 cells, transfected as in (A) for siRNA
interference were treated, 24 h after siRNA transfection, with
Ru-bdcurc (50 µM) for 48 h. Protein levels were evaluate by western
blot analysis. β-actin was used as protein loading control and one
representative experiment is shown. The ratio of the proteins level
vs. b-actin, following densitometric analysis, is reported as
histograms plus SD (right panels). (C) Trypan blue exclusion assay
was performed to asses viability of cells treated as in. (B)
Statistics was measured between treated (50 µM) and untreated cells
(−) and compared according to p53 interference. *P≤0.05 and
#, not statistically significant. NRF2, nuclear factor
erythroid 2-related factor 2; siRNA, small interfering RNA;
Ru-bdcurc, Ruthenium (Ru)(II)-Bisdemethoxycurcumin. |
Discussion
In the present study, it was revealed that the novel
Ru-bdcurc compound (16) was able
to induce cell death in the pancreatic NET cell line BON-1, in a
dose-dependent way (Fig. 5A).
Intriguingly, the higher dose of Ru-bdcurc (i.e., 100 µM) induced
apoptotic cell death while the lower dose (i.e, 50 µM) induced
chemoresistant pathways that reduced the cytotoxic activity of the
compound. The 100 µM dose of Ru-bdcurc treatment increased the
clPARP/PARP ratio, compared with 50 µM dose, and correlated with
the appearance of the phosphorylated form of H2AX (γH2AX), a marker
of DNA damage and apoptosis (48).
Moreover, 100 µM dose of Ru-bdcurc induced the expression of Noxa,
a pro-apoptotic gene induced by p53 family members (56), that has been reported to inhibit the
antiapoptotic Mcl1 protein (57),
in agreement with the reduction of Mcl1 protein levels in Fig. 2A. On the other hand, 50 µM dose of
Ru-bdcurc induced the phosphorylation of 4EBP1 (p4E-BP1), a mTOR
target whose activation can sustain cancer cell survival and
predict poor prognosis (49,50).
In agreement, mTOR pathway has been found to be dysregulated in
GEP-NET and involved in tumor development (51). The 50 µM dose of Ru-bdcurc activated
the NRF2 pathway with upregulation of its targets p62 and catalase,
increased the antiapoptotic Bcl-2 protein levels and the endogenous
dysfunctional p53 protein levels. The increased p62 gene expression
was in accordance with the finding that p62 is a NRF2
transcriptional target (23–25).
The reason why only the lower dose of Ru-bdcurc induced NRF2
activity remains not completely resolved at the molecular level.
From the literature it is known that curcumin can induce NRF2
through activation of p62 by phosphorylation (58), therefore it can be hypothesized that
only the lower dose of Ru-bdcurc might induce the kinases involved
in p62 posttranslational modifications in order to be activated.
Interestingly, it has been revealed that p62 activates pro-survival
pathways including mTOR (52), in
agreement with the aforementioned results in Fig. 1 and with the cross-talk among
oncogenic pathways such as p62/mTOR/NRF2 to increase tumor
development and resistance to therapies (30). Moreover, only the 50 µM dose of
Ru-bdcurc treatment, compared with 100 µM dose, increased the
endogenous p53 protein level, in agreement with the paradigm that
the interplay between NRF2 and mutp53 stabilizes each other
proteins to sustain their oncogenic activities (41,54).
The endogenous p53 has been reported to be dysfunctional in BON-1
cells and to inhibit apoptosis (28). Thus, in this study the wild-type p53
target genes were not detected after cell treatment with 50 µM dose
of Ru-bdcurc (data not shown), suggesting lack of p53 wild-type
activity. On the other hand, 50 µM dose of Ru-bdcurc, compared with
100 µM, induced the expression of MDR1, a target of some mutant p53
proteins (55), as well as of the
antiapoptotic gene Bcl-2, which associated with a potential
oncogenic activity of the endogenous dysfunctional p53 in BON-1
cells. Interestingly, 100 µM dose of Ru-bdcurc induced cell death
that correlated with the expression of Noxa a pro-apoptotic gene
induced by p53 but also by p53 family members such as p73 (56). The fact that p53 protein expression
did not increase after 100 µM dose of Ru-bdcurc but rather
decreased (Fig. 4B) can suggest
that mutant/dysfunctional p53 in BON-1 cells was downregulated in
agreement with NRF2 downregulation but that this downregulation did
not reactivate a potential endogenous wild-type p53 gene,
suggesting that p53 family members, rather than wild-type p53, were
inducing Noxa mRNA expression. However, further experiments are
necessary to validate the endogenous p53 status in BON-1 cells and
its or not reactivation after curcumin treatment. Mutations in p53
gene are reported in ~90–95% of GEP-NEC and in 3% of GEP-NET
(59); however, the endogenous
mutant p53 activity in BON-1 needs to be explored in future
studies. Inhibiting the NRF2 pathway by genetic or pharmacologic
means increased the cytotoxic effect of the lower dose of the
Ru-bdcurc compound. Similarly, silencing of the endogenous
dysfunctional p53 counteracted the NRF2 activation in response to
the lower dose of Ru-bdcurc and increased the cytotoxic effect of
the compound, strongly supporting the oncogenic interplay between
NRF2 and p53 in this setting (Fig.
5B). GEP-NEN are extremely heterogeneous tumors and identifying
potential biomarkers for therapeutic purpose is difficult. In the
present study, one potential pathway of chemoresistance to be
validated in tumor samples, was suggested. However, the limitation
of the present study is the use of only one cell line, despite the
cell lines available for this type of tumors are only two (27–29).
It was also attempted to perform the experiments in the QGP-1 cell
line but it was not possible not obtain the same results as in
BON-1. QGP-1 cell line carries a wtp53 but does not present NRF2
induction after treatment, as observed in BON-1 cells, at least in
the present experiments. Given that these tumors are genetically
different it becomes very difficult to generalize the results
between the two cell lines. Therefore, additional studies are
necessary to clearly establish a role for NRF2 and the
interconnected oncogenic molecular pathways in tumors in
vivo.
The results of the present study can be discussed in
view of the potential effects of curcumin treatment in patients; In
particular, the low versus the high dose of curcumin that can be
achieved in vivo in patients. Thus, in clinical trials high
dose of curcumin ends often in low plasma level of curcumin
(60,61) likely due to curcumin low solubility
and bioavailability and low curcumin doses have been shown to act
as an antioxidant agent and do not induce cell death, thus
contributing to acquired chemoresistance (62). For this reason, the use of more
soluble and assimilable compounds such as the organometallic
ruthenium ones (12–14), used in the present study, could be
taken in consideration in clinical practice. On the other side,
combination therapies with curcumin compounds and more classical
chemotherapeutic agents should include also molecules that target
the NRF2 pathway to inhibit not only NRF2 but also the
interconnected oncogenic pathways that often interact with it to
increase tumor resistance to therapies. In agreement, previous
studies have reported that targeting NRF2 is a promising strategy
for the treatment of several aggressive cancers where the
activation of the NRF2 pathway protects cancer cells from the
cytotoxic effects of the chemotherapeutic drugs (63–66).
In conclusion, the results of the present study demonstrated for
the first time, to the best of the authors' knowledge, that NRF2
may play a role in chemoresistance of the pancreatic neuroendocrine
BON-1 cancer cells and suggested to evaluate the NRF2-dependent
pathway as a potential biomarker in GEP-NENs tissues. If this
hypothesis is confirmed in GEP-NENs, NRF2 pathway could become a
novel biomarker to be taken in consideration to tailor more
effective cytotoxic therapies against this type of rare tumor that
remains an orphan-drug cancer.
Acknowledgements
The authors would like to thank Dr Elisa Melucci
(Regina Elena National Cancer Institute; Rome, Italy) for technical
support.
Funding
The present study was supported by the Italian Association for
Cancer Research (AIRC) (grant nos. Id16742 and Id23040) and by the
University of Camerino (Fondo di Ateneo per la Ricerca-University
Research; grant no: 2018).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GDO, AG, MC, SS, MA, RP and FM conceptualized the
study. RP and FM developed methodology. AG, LM, GP, IV and GDO
curated data. GDO wrote the original draft. GDO, AG. MC, SS and FM
wrote, reviewed and edited the manuscript. GDO, SS and FM
supervised the study. GDO, MC and FM acquired funding. AG and LM
confirm the authenticity of all the raw data. All authors have read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rindi G, Mete O, Uccella S, Basturk O, La
Rosa S, Brosens LAA, Ezzat S, de Herder WW, Klimstra DS, Papotti M
and Asa SL: Overview of the 2022 WHO Classification of
Neuroendocrine Neoplasms. Endocr Pathol. 33:115–154. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cives M and Strosberg JR:
Gastroenteropancreatic neuroendocrine tumors. CA Cancer J Clin.
68:471–487. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huguet I, Grossman AB and O'Toole D:
Changes in the epidemiology of neuroendocrine tumours.
Neuroendocrinology. 104:105–111. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Norton JA: Surgery for primary pancreatic
neuroendocrine tumors. J Gastrointest Surg. 10:327–331. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Akirov A, Larouche V, Alshehri S, Asa SA
and Ezzat S: Treatment options for pancreatic neuroendocrine
tumors. Cancers. 11:8282019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Das S, Al-Toubah T and Strosberg J:
Chemotherapy in neuroendocrine tumors. Cancers. 13:48722021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dai M, Mullins CS, Lu L, Alsfasser G and
Linnebacher M: Recent advances in diagnosis and treatment of
gastroenteropancreatic neuroendocrine neoplasms. World J
Gastrointest Surg. 14:383–396. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haque A, Brazeau D and Amin AR:
Perspectives on natural compounds in chemoprevention and treatment
of cancer: An update with new promising compounds. Eur J Cancer.
149:165–183. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patel SS, Acharya A, Ray RS, Agrawal R,
Raghuwanshi R and Jain P: Cellular and molecular mechanisms of
curcumin in prevention and treatment of disease. Crit Rev Food Sci
Nutr. 60:887–939. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zoi V, Galani V, Lianos GD, Voulgaris S,
Kyritsis AP and Alexiou GA: The role of curcumin in cancer
treatment. Biomedicines. 9:10862021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stohs SS, Chen O, Ray SD, Ji J, Bucci LR
and Preuss HG: Highly bioavailable forms of curcumin and promising
avenues for Curcumin-Based research and application. Molecules.
25:13972020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wanninger S, Lorenz V, Subhan A and
Edelmann FT: Metal complexes of curcumin-synthetic strategies,
structures and medicinal applications. Chem Soc Rev. 44:4986–5002.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pagliaricci N, Pettinari R, Marchetti F,
Pettinari C, Cappellacci L, Tombesi A, Cuccioloni M, Hadiji M and
Dyson PJ: Potent and selective anticancer activity of half-sandwich
ruthenium and osmium complexes with modified curcuminoid ligands.
Dalton Trans. 51:13311–13321. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gobbo A, Pereira SAP, Biancalana L,
Zacchini S, Saraiva MLMFS, Dyson PJ and Marchetti F: Anticancer
ruthenium(II) tris(pyrazolyl)methane complexes with bioactive
co-ligands. Dalton Trans. 51:17050–17063. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bonfili L, Pettinari R, Cuccioloni M,
Cecarini V, Mozzicafreddo M, Angeletti M, Lupidi G, Marchetti F,
Pettinari C and Eleuteri AM: Arene-RuII complexes of curcumin exert
antitumor activity via proteasome inhibition and apoptosis
induction. Chem Med Chem. 7:2010–2020. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pettinari R, Marchetti F, Condello F,
Pettinari C, Lupidi G, Scopelliti R, Mukhopadhyay S, Riedel T and
Dyson PJ: Ruthenium(II)-Arene RAPTA type complexes containing
curcumin and bisdemethoxycurcumin display potent and selective
anticancer activity. Organometallics. 33:3709–3715. 2014.
View Article : Google Scholar
|
|
17
|
Caruso F, Pettinari R, Rossi M, Monti E,
Gariboldi MB, Marchetti F, Pettinari C, Caruso A, Ramani MV and
Subbaraju GVJ: The in vitro antitumor activity of
arene-ruthenium(II) curcuminoid complexes improves when decreasing
curcumin polarity. J Inorg. Biochem. 162:44–51. 2016.
|
|
18
|
Garufi A, Baldari S, Pettinari R,
Gilardini Montani MS, D'Orazi V, Pistritto G, Crispini A, Giorno E,
Toietta G, Marchetti F, et al: A ruthenium(II)-curcumin compound
modulates NRF2 expression balancing the cancer cell death/survival
outcome according to p53 status. J Exp Clin Cancer Res. 39:1222020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Garufi A, Pettinari R, Marchetti F, Cirone
M and D'Orazi G: NRF2 and Bip interconnection mediates resistance
to the organometallic ruthenium-cymene bisdemethoxycurcumin complex
cytotoxicity in colon cancer cells. Biomedicines. 11:5932023.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
D'Orazi G and Cirone M: Interconnected
adaptive responses: A way out for cancer cells to avoid cellular
demise. Cancers. 14:27802022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Garufi A, Pistritto G, D'Orazi V, Cirone M
and D'Orazi G: The impact of NRF2 inhibition on drug-induced colon
cancer cell death and p53 activity: A pilot study. Biomolecules.
2:4612022. View Article : Google Scholar
|
|
22
|
Rojo de la Vega M, Chapman E and Zhang DD:
NRF2 and the hallmarks of cancer. Cancer Cell. 34:21–43. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jain A, Lamark T, Sjottem E, Bowitz Larsen
K, Awuh JA, Overvatn A, McMahon M, Hayes JD and Johansen T:
p62/SQSTM1 is a target gene for transcription factor NRF2 and
creates a positive feedback loop by inducing antioxidant response
element-driven gene transcription. J Biol Chem. 285:22576–22591.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Komatsu M, Kurokawa H, Waguri S, Taguchi
K, Kobayashi A, Ichmura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et
al: The selective autophagy substrate p62 activates the stress
response transcription factor Nrf2 through inactivation of Keap1.
Nat Cell Biol. 12:213–23. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang T, Harder B, Rojo de la Vega M, Wong
PK, Chapman E and Zhang DD: p62 links autophagy and Nrf2 signaling.
Free Rad Biol Med. 88:199–204. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lister A, Nedjadi T, Kitteringham NR,
Campbell F, Costello E, Lloyd B, Copple IM, Williams S, Owen A,
Neoptolemos JP, et al: Nrf2 is overexpressed in pancreatic cancer:
Implications for cell proliferation and therapy. Mol Cancer.
10:372011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Evers BM, Ishizuka J, Townsend CM Jr and
Thompson JC: The human carcinoid cell line, BON. A model system for
the study of carcinoid tumors. Ann N Y Acad Sci. 733:393–406. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vandamme T, Peeters M, Dogan F, Pauwels P,
Van Assche E, Beyens M, Mortier G, Vandeweyer G, de Herder W, Van
Camp G, et al: Whole-exome characterization of pancreatic
neuroendocrine tumor cell lines BON-1 and QGP-1. J Mol Endocrinol.
54:137–47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luley KB, Biedermann SB, Kunstner A, Busch
H, Franzenburg S, Schrader J, Grabowsi P, Wellner U, Keck T,
Brabant G, et al: A comprehensive molecular characterization of the
pancreatic neuroendocrine tumor cell lines BON-1 and QGP-1. Cancers
(Basel). 12:6912020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cirone M and D'Orazi G: NRF2 in cancer:
Cross-talk with oncogenic pathways and involvement in
gammaherpesviruses-driven carcinogenesis. Int J Mol Sci.
24:5952022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vousden HK and Prives C: Blinded by the
light: The growing complexity of p53. Cell. 137:413–31. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Voutsadakis IA: Mutations of p53
associated with pancreatic cancer and therapeutic implications. Ann
Hepatobiliary Pancreat Surg. 25:315–327. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vijayvergia N, Boland PM, Handorf E,
Gustafson KS, Gong Y, Cooper HS, Sheriff F, Astsaturov I, Cohen SJ
and Engstrom PF: Molecular profiling of neuroendocrine malignancies
to identify prognostic and therapeutic markers: A Fox Chase Cancer
Center Pilot Study. Br J Cancer. 115:564–570. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ren D, Villeneuve NF, Jiang T, Wu T, Lau A
and Toppin HA: Brusatol enhances the efficacy of chemotherapy by
inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci
USA. 108:1433–1438. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Olayanju A, Copple IM, Bryan HK, Edge GT,
Sison RL, Wong MW, Lai ZQ, Lin ZX, Dunn K, Sanderson CM, et al:
Brusatol provokes a rapid and transient inhibition of Nrf2
signaling and sensitizes mammalian cells to chemical toxicity
implications for therapeutic targeting of Nrf2. Free Rad Biol Med.
78:202–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Garufi A, Traversi G, Gilardini Montani
MS, D'Orazi V, Pistritto G, Cirone M and D'Orazi G: Reduced
chemotherapeutic sensitivity in high glucose condition: implication
of antioxidant response. Oncotarget. 10:4691–4702. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cecchinelli B, Lavra L, Rinaldo C,
Iacovelli S, Gurtner A, Gasbarri A, Ulivieri A, Del Prete F,
Trovato M, Piaggio G, et al: Repression of the antiapoptotic
molecule galectin-3 by homeodomain-interacting protein kinase
2-activated p53 is required for p53-induced apoptosis. Mol Cell
Biol. 26:4746–4757. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brummelkamp TR, Bernards R and Agami R: A
system stable expression of short interfering RNAs in mammalian
cells. Science. 296:550–553. 2020. View Article : Google Scholar
|
|
40
|
Garufi A, Giorno E, Gilardini Montani MS,
Pistritto G, Crispini A, Cirone M and D'Orazi G:
P62/SQSTM1/Keap1/NRF2 axis reduces cancer cells death-sensitivity
in response to Zn(II)-curcumin complex. Biomolecules. 11:3482021.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lisek K, Campaner E, Ciani Y, Walerych D
and Del Sal G: Mutant p53 tunes the NRF2-dependent antioxidant
response to support survival of cancer cells. Oncotarget.
9:20508–20523. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gong C, Yao H, Liu Q, Chen J, Shi J, Su F
and Song E: Markers of tumor-initiating cells predict
chemoresistance in breast cancer. PLoS One. 5:e156302010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Garufi A, Pistritto G, Cirone M and
D'Orazi G: Reactivation of mutant p53 by capsaicin, the major
constituent of peppers. J Exp Clin Cancer Res. 35:1362016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gilles C, Polette M, Mestdagt M,
Nawrocki-Raby B, Ruggeri P, Birembaut P and Foidart JM:
Transactivation of vimentin by beta-catenin in human breast cancer
cells. Cancer Res. 15:2658–2664. 2003.PubMed/NCBI
|
|
45
|
Nodale C, Sheffer M, Jacob-Hirsch J,
Folgiero V, Falcioni R, Aiello A, Garufi A, Rechavi G, Givol D and
D'Orazi G: HIPK2 downregulates vimentin and inhibits breast cancer
cell invasion. Cancer Biol Ther. 13:198–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Garufi A, Trisciuoglio D, Porru M,
Leonetti C, Stoppacciaro A, D'Orazi V, AVantaggiati ML, Crispini A,
Pucci D and D'Orazi G: A fluorescent-based Zn(II)-complex
reactivates mutant (R175H and R272H) p53 in cancer cells. J Exp
Clin Cancer Res. 32:722013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Garufi A, Pucci D, D'Orazi V, Cirone M,
Bossi G, Avantaggiati ML and D'Orazi G: Degradation of mutant
p53H175 protein by Zn(II) through autophagy. Cell Death Dis.
5:e12712014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bonner WM, Redon CE, Dickey JS, Nakamura
AJ, Sedelnikova OA, Solier S and Pommier Y: Gamma H2AX and cancer.
Nat Rev Cancer. 8:957–967. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Miao Y, Chen L, Shi C, Fan R, Chen P, Liu
H, Xia A and Qin H: Increased phosphorylation of 4E-binding protein
1 predicts poor prognosis for patients with colorectal cancer. Mol
Med Rep. 15:3099–3104. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gonnella R, Zarrella R, Santarelli R,
Germano CA, Gilardini Montani MS and Cirone M: Mechanisms of
sensitivity and resistance of primary effusion lymphoma to Dimethyl
Fumarate (DMF). Int J Mol Sci. 23:67732022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zanini S, Renzi S, Giovinazzo F and
Bermano G: mTOR pathway in gastroenteropancreatic neuroendocrine
tumor (GEP-NETs). Front Endocrinol. 11:5625052020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Moscat J, Karin M and Diaz-Meco MT: p62 in
cancer: Signaling adaptor beyond autophagy. Cell. 167:606–609.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Widden H and Placzek WJ: The multiple
mechanisms of Mcl1 in the regulation of cell fate. Commun Biol.
4:10292021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gilardini Montani MS, Cecere N, Granato M,
Romeo MA, Falcinelli L, Ciciarelli U, D'Orazi G, Faggioni A and
Cirone M: Mutant p53, stabilized by its interplay with HSP90,
activates a positive feed-back loop between NRF2 and p62 that
induces chemo-resistance to Apigenin in pancreatic cancer cells.
Cancers. 11:7032019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sampath J, Sun D, Kidd VJ, Grenet J,
Gandhi A, Shapiro LH, Wang Q, Zambetti GP and Schuetz JD: Mutant
p53 cooperates with ETS and selectively up-regulates human MDR1 not
MRP1. J Biol Chem. 276:39359–39367. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Martin AG, Trama J, Crighton D, Ryan KM
and Fearnhead HO: Activation of p53 and induction of Noxa by DNA
damage requires NG-kappa B. Aging (Albany NY). 1:335–349. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chiou JT, Huang NC, Hiang CH, Wang LJ, Lee
YC, Shi YJ and Chang LS: NOXA-mediated degradation of MCL1 and
BCL2L1 causes apoptosis of daunorubicin-treated human acute myeloid
leukemia cells. J Cell Physiol. 236:7356–7375. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Park JY, Sohn HY, Koh YH and Jo C:
Curcumin activates Nrf2 through PKCd-mediated p62 phosphorylation
at Ser351. Sci Rep. 11:84302021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kawasaki K, Fujii M and Sato T:
Gastroenteroepatic neuroendocrine neoplasms: Genes, therapies and
models. Dis Model Mech. 11:dmm0295952018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cao J, Jia L, Zhou HM, Liu Y and Zhong LF:
Mitochondrial and nuclear DNA damage induced by curcumin in human
hepatoma G2 cells. Toxicol. 91:476–483. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kunati SR, Yang SM, William BM and Xu Y:
An LC-MS/MS method for simultaneous determination of curcumin,
curcumin glucuronide and curcumin sulfate in a phase II clinical
trial. J Pharm Biomed Anal. 156:189–198. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Giordano A and Tommonaro G: Curcumin and
Cancer. Nutrients. 11:23762019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Arena A, Romeo MA, Benedetti R, Gilardini
Montani MS, Santarelli R, Gonnella R, D'Orazi G and Cirone M: NRF2
and STAT3: Friends or foes in carcinogenesis? Discov Oncol.
14:372023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sajadimajd S and Khazaei M: Oxidative
stress and cancer: The role of Nrf2. Curr Cancer Drug Targtes.
18:538–557. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
No JH, Kim YB and Song YS: Targeting Nrf2
signaling to combat chemoresistance. J Cancer Prev. 19:111–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Torrente L and DeNicola GM: Targeting NRF2
and its downstream processes: Opportunities and challenges. Annu
Rev Pharmacol Toxicol. 62:279–300. 2022. View Article : Google Scholar : PubMed/NCBI
|