Introduction
Prostate cancer (PCa) is the second leading cause of
death from cancer in men in the United States (1). The majority of men who are diagnosed
with localized PCa and who elect to undergo therapy are treated
with surgery or radiation, by which they expect to be cured
(2). A number of patients
experience a prolonged period without evidence of biochemical
recurrence or obvious disease, but some relapse with single or
multiple metastases frequently detected in bone (3,4).
Notably, ~90% of patients who succumb to advanced-stage PCa have
bone metastases, whereas only ~10% of these individuals had bone
metastases at the time of diagnosis (5–7). These
observations suggest that disseminated tumor cells (DTCs) may have
left the primary tumor site in the prostate and took up residence
in distant sites. For PCa, it has been shown that DTCs may be
present in the bone marrow at the time of initial diagnosis
(8–11). In the bone marrow, DTCs maintain the
ability to become reactivated and generate metastases by poorly
understood processes (8–11). Further studies are needed in the
context of PCa DTC dormancy to identify and develop improved
therapeutic strategies to treat metastatic diseases.
One well-delineated pathway in the understanding of
cellular dormancy is p38/ERK signaling. High levels of p38MAPK
activity function as an inhibitory regulator of ERK, thus
preventing cell proliferation by inducing
G0/G1 phase arrest, or by initiating
senescence and apoptosis (12–14).
Transforming growth factor-β2 (TGF-β2), which is secreted from bone
marrow-derived cells, is known to induce expression of a
p38high/ERKlow phenotype in cancer (15). The subsequent activation of Smad1/5
and increased expression of p27 results in the downregulation of
cyclin-dependent kinase (CDK)4, which collectively facilitates the
transition into cellular quiescence (15,16).
Additionally, p38high/ERKlow facilitates
G0/G1 phase arrest via the regulation of
various factors, including NR2F1, and CDK inhibitors p27 and p21.
Therefore, the combined regulation of transcription factors by a
p38high/ERKlow phenotype is responsible for
quiescence (17). A second
well-described pathway that regulates PCa dormancy in the bone
marrow is activated by growth arrest-6 (GAS6) signaling. In bone
marrow, GAS6 secreted by osteoblasts activates the TAM family of
receptors (Tyro3, Axl and MerTK) on PCa cells, which serve as
molecular switches to turn on (Tyro3, Axl) and off (MerTK) the
dormancy programs (18). Similarly,
bone morphogenetic protein (BMP)7, which is produced by bone
stromal cells, induces dormancy in PCa cells by activating p38
signaling (19). These studies have
suggested that cells in the bone marrow microenvironment, including
osteoblasts, serve key roles in regulating PCa dormancy.
The present study explored the signaling pathways
activated by the dormancy-inducing phytohormone ABA in PCa cells.
ABA has long been known as a regulator of plant growth and survival
under stress conditions, but has more recently been detected in rat
brain tissues (20). Since its
discovery in mammals, ABA has been identified in blood plasma
(21), and has been demonstrated to
be produced by pancreatic β cells (22), adipocytes (21), keratinocytes (23), hematopoietic immune cells, such as
granulocytes (24), monocytes
(25) and macrophages (26), and human mesenchymal stem cells
(27). ABA has also been shown to
be linked to several human diseases, including type-2 diabetes and
colitis (28,29). ABA signaling is mediated by two
known receptors, lanthionine synthetase C-like protein 2 (LANCL2)
and peroxisome proliferator activated receptor γ (PPARγ) (28–30).
Numerous studies have demonstrated that LANCL2 serves a role in the
regulation of stress responses, inflammation, and in metabolic and
immune-related diseases (31,32).
It has also been demonstrated that LANCL2 is expressed by DTCs in
the bone marrow of patients with breast cancer (30). PPARγ is a nuclear hormone receptor,
the signaling of which results in anti-proliferative activities by
decreasing the levels of cyclin D1 and E, and increasing the
expression of p21 and p27 in PCa (33–35).
The present study further explored the role of ABA in dormancy and
the molecular pathways activated by its signaling in PCa.
Materials and methods
Reagents
A number of the reagents used, their source and
catalogue numbers are presented in Table I; short hairpin RNA (shRNA)
targeting sequences are presented in Table II; primer sequences are presented
in Table III.
 | Table I.Reagents used in the present
study. |
Table I.
Reagents used in the present
study.
| A, Animals |
|---|
|
|---|
| Reagent or
resource | Supplier | Identifier |
|---|
| Male 4–6 week old
C57BL/6J mice | Jackson
Laboratory | Strain no.
000664 |
|
| B,
Antibodies |
|
| Reagent or
resource |
Supplier |
Identifier |
|
| β-actin | Cell Signaling
Technology, Inc. | 4970 |
| Tubulin | ABclonal Biotech
Co., Ltd. | AC021 |
| p21 | Cell Signaling
Technology, Inc. | 2947 |
| p27 | Cell Signaling
Technology, Inc. | 3686 |
| LANCL2 | Abcam | ab237520 |
| PPARγ | Cell Signaling
Technology, Inc. | 2443 |
| p-p38 | Cell Signaling
Technology, Inc. | 4511 |
| t-p38 | Cell Signaling
Technology, Inc. | 9212 |
| NR2F1 | Abcam | ab181137 |
|
| C, Chemicals,
peptides and recombinant proteins |
| Reagent or
resource |
Supplier |
Identifier |
|
| DMEM | Cytiva | SH30022.01 |
| RPMI | Cytiva | SH30027.01 |
| α-MEM | Gibco; Thermo
Fisher Scientific, Inc. | 12571-063 |
| Fetal bovine
serum | Gemini Bio
Products | 50-753-2984 |
| 0.25%
Trypsin-EDTA | Gibco; Thermo
Fisher Scientific, Inc. | 25200-056 |
|
Penicillin-streptomycin | Gibco; Thermo
Fisher Scientific, Inc. | 15140-122 |
| Effectene
transfection reagent | Qiagen, Inc. | 301425 |
| ABA | Abcam | ab120860 |
|
| D, Experimental
cell lines |
|
| Reagent or
resource |
Supplier |
Identifier |
|
| 293T | ATCC | CRL-3216 |
| PC3 | ATCC | CRL-1435 |
| C4-2B | ATCC | CRL-3315 |
| DU145 | ATCC | HTB-81 |
| Myc-CaP | ATCC | CRL-3255 |
| Table II.Short hairpin RNA sequences. |
Table II.
Short hairpin RNA sequences.
| Gene/shRNA | Clone | ID sequence | Sequence,
5′-3′ | Supplier |
|---|
| Nonspecific
sequence | - | #RHS6848 | - | Horizon
Discovery |
| LANCL2 | 1 | TRCN0000045403 |
TAAGGCATACAGATAACCTGC | Horizon
Discovery |
| LANCL2 | 2 | TRCN0000045404 |
AAATTATGAATGATCTTCCCG | Horizon
Discovery |
| PPARγ | 1 | TRCN0000001670 |
GCCAACATTTCCCTTCTTCCA | MilliporeSigma |
| PPARγ | 2 | TRCN0000001671 |
CTGGCCTCCTTGATGAATAAA | MilliporeSigma |
| Table III.Primer sequences. |
Table III.
Primer sequences.
| Gene | Sequence,
5′-3′ |
|---|
| ACTB | F:
TCAGGACGGGAAGATCATTCA |
|
| R:
CAGAGCAGTCATGGGGATCAG |
| LANCL2 | F:
TCAGGACGGGAAGATCATTCA |
|
| R:
CAGAGCAGTCATGGGGATCAG |
| PPARγ | F:
GGGATCAGCTCCGTGGATCT |
|
| R:
TGCACTTTGGTACTCTTGAAGTT |
| p21 | F:
TGTCCGTCAGAACCCATGC |
|
| R:
AAAGTCGAAGTTCCATCGCTC |
| p27 | F:
AACGTGCGAGTGTCTAACGG |
|
| R:
CCCTCTAGGGGTTTGTGATTCT |
| p16 | F:
GATCCAGGTGGGTAGAAGGTC |
|
| R:
CCCCTGCAAACTTCGTCCT |
| E-cadherin | F:
CGAGAGCTACACGTTCACGG |
|
| R:
GGGTGTCGAGGGAAAAATAGG |
| Vimentin | F:
GACGCCATCAACACCGAGTT |
|
| R:
CTTTGTCGTTGGTTAGCTGGT |
| ZEB2 |
F:CAAGAGGCGCAAACAAGCC |
|
| R:
GGTTGGCAATACCGTCATCC |
Cell culture
As the present study aimed to study bone marrow
DTCs, the following metastatic cell lines were used. The human
androgen-independent bone metastatic PCa cell lines PC3 and LnCaP
subline C4-2B were predominantly used in the present study, as they
metastasize to bone (36–38). When injected into bone, PC3 cells
result in predominantly lytic lesions, whereas C4-2B cells may
produce mixed osteoblastic and osteolytic lesions (36–38).
In some experiments, the human dura metastasis cell line DU145 and
murine Myc-CaP cells were used. We recently demonstrated that
Myc-CaP cells can also metastasize to bone (39). The human PCa cells were routinely
grown in RPMI 1640, whereas the murine Myc-CaP cells were grown in
DMEM. All cultures were supplemented with 10% fetal bovine serum
(FBS) and 1% penicillin-streptomycin, and were maintained in an
incubator set to 5% CO2 atmosphere at 37°C (Marshall
Scientific).
Cell viability assays
To evaluate viability, 3,000 PCa cells/well (PC3,
C4-2B, Du145 and Myc-CaP) in 100 µl medium were added to 96-well
plates in quintuplicate, and allowed to adhere overnight. The next
day, the cells were washed in PBS (Thermo Fisher Scientific, Inc.)
and fresh medium was added. ABA was diluted in dimethyl sulfoxide
(DMSO), and was added at the following doses: 0 µM (DMSO only), 25,
50, 100 and 200 µM for 72 h at 37°C. To determine viability, cells
were either recovered from culture with trypsin (MilliporeSigma)
and counted using a hemocytometer under a Nikon Eclipse Ts2
inverted phase light microscope (Nikon Corporation) following
trypan blue staining (at 23°C for 3 min), or were assessed using a
colorimetric assay. For the colorimetric assays, a total of 70 h
after the addition of ABA or control, 20 µl CellTiter 96® Aqueous
Non-Radioactive Cell Proliferation Reagent (cat. no. G5421; Promega
Corporation) was added to the cultures for 2 h at 37°C. The optical
density (O.D.) of the cell culture plates was then measured using a
plate reader (DU530 UV/Vis scanning spectrophotometer; BD
Biosciences) at a wavelength of 490 nm. To determine if ABA
permanently affected viability, cells were treated with ABA for 72
h at 37°C, washed, trypsinized and counted. An equal number of
cells (1×105 cells/well in 12-well plates, 1 ml of
culture media) were then added back to culture for an additional 72
h at 37°C and cell viability was evaluated by cell counting using a
hemocytometer. Cell viability (%) was calculated as follows:
(treatment group-background)/(control group-background)x100. The
data are presented as the mean ± SEM.
Cytotoxicity assays
The cytotoxicity of ABA was assessed using a
CytoTox96 Non-Radioactive Cytotoxicity Assay kit (Promega
Corporation) according to the manufacturer's instructions. Briefly,
PCa cell lines (PC3, C4-2B, DU145 and Myc-CaP) were seeded on
96-well plates (1×104 cells/well) in quintuplicate per
group in 100 µl complete culture medium. After overnight
incubation, the cells were treated with 25, 50, 100 and 200 µM ABA
for 4 h at 37°C; the control group was treated with DMSO. To
measure lactate dehydrogenase (LDH) release, 50 µl supernatant was
mixed with 50 µl reconstituted substrate mix solution in a new
96-well plate and was incubated for 30 min in the dark at room
temperature. The reaction was stopped with 50 µl stop buffer and
the O.D. was then measured at a wavelength of 490 nm. Each
cytotoxicity assay was independently repeated two times.
shRNA and lentivirus preparation
pLKO.1 lentiviral vector-based shRNAs targeting
specific candidate genes and the non-specific (NS) control shRNA
were obtained from Horizon Discovery (NS control and shRNA LANCL2)
and MilliporeSigma (shRNA PPARγ) (Table II). Lentivirus particles were
prepared by transfecting 293T cells in 12-well plates with 0.5 µg
either gene-specific shRNA plasmids (LANCL2, clone ID no.
TRCN0000045403; and PPARγ, clone ID no. TRCN0000001670) or NS shRNA
plasmids along with lentiviral packaging plasmids [2nd
generation psPAX2 packaging (plasmid #12260; Addgene, Inc.) and
pMD2.G envelope (plasmid #12259; Addgene, Inc.) in a 1:1 ratio for
48 h at 37°C. All lentiviral transfections were performed using
Effectene Transfection Reagent. Stable cell lines were generated by
infecting PCa cells (PC3 and C4-2B) with a multiplicity of
infection of ~250 viral particles per cell in 200 µl (total
collection volume, 1 ml) in 12-well plates for 24 h, followed by
selection in puromycin (1 µg/ml) at 37°C for 1 week. Thereafter
reverse transcription-quantitative PCR (RT-qPCR) was used to assess
the mRNA expression changes and western blotting was used to
validate protein expression changes. After the initial assessment
of the efficiency of shRNA knockdown (KD) of the target genes,
clone 1 was selected for both LANCL2 and PPARγ for further studies.
Thereafter, frozen stocks were established and cells were used for
experimentation after validation of the targeted KD within 2 weeks
of transfection or thaw.
RNA extraction, cDNA preparation and
RT-qPCR
Total RNA was extracted from target cells using
TRIzol® Reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and
was purified using the RNeasy Mini Kit (Qiagen, Inc.). cDNA was
synthesized using the ProtoScript First Strand cDNA Synthesis Kit
(New England Biolabs, Inc.) according to the manufacturer's
instructions. qPCR was performed with gene-specific primers using
the iTaq Universal SYBR® Green Supermix (Bio-Rad Laboratories,
Inc.) on an Applied Biosystems 7500 thermocycler system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling
conditions were as follows: Initial denaturation at 95°C for 15
min; 40 cycles at 95°C for 15 sec, annealing at 60°C for 30 sec and
72°C for 30 sec; and a final extension step at 72°C for 7 min.
Results were normalized against β-actin levels using the formula
ΔCq=Cq of target gene-Cq of β-actin. The mRNA expression levels of
the control group were used to establish the baseline; therefore,
ΔΔCq was calculated using the formula ΔΔCq=ΔCq of target gene-ΔCq
of the baseline. The fold change of mRNA expression was calculated
as fold=2−ΔΔCq (40).
The primer sequences for all the genes analyzed in the present
study are provided in Table
II.
Migration and invasion assays
Migration assays were performed using a Transwell
with 8.0 µm pore polycarbonate membrane inserts (cat. no. 3422;
Corning, Inc.), and invasion assays were performed using BioCoat
Growth Factor Reduced Matrigel Invasion Chambers (cat. no. 354483;
Corning, Inc.) in Transwell inserts. For these studies, PC3 and
C4-2B cells infected with NS, LANCL2 and PPARγ shRNAs were
serum-starved for 6 h, and were then recovered from culture and
seeded in triplicate in the upper chamber of Transwell inserts
(5×104 cells/well). The lower chambers were filled with
medium containing 10% FBS as a chemoattractant. The cells in the
upper chambers were treated with 100 µM ABA or DMSO as a control at
37°C. After 24 h for PC3 cells and 48 h for C4-2B cells, the cells
remaining in the top chambers (those cells that had not migrated or
invaded) were removed with cotton-tipped applicators. The number of
cells that had migrated or invaded into the lower chambers were
quantified following DAPI staining at 23°C for 3 min; 5–8
fields/membrane were evaluated under an All-in-one Fluorescence
Microscope (BZ-X800/BZ-X810; Keyence Corporation; ×10
magnification) and nuclei quantification was performed using ImageJ
1.53K software (National Institutes of Health). The percentage of
migration or invasion was calculated as follows: (number of cells
that migrated or invaded in the treated group)/(number of cells
that migrated or invaded in the control group) ×100.
Soft agar assays
PC3 and C4-2B cells (5×103/well) infected
with LANCL2, PPARγ or NS shRNAs were seeded into 6-well plates on
0.4% low gelling point agarose (cat. no. A9045; MilliporeSigma) and
layered on top of 0.8% agarose. The cultures were maintained for
15–18 days, with fresh medium replenished every third day
containing DMSO (control) or 100 µM ABA. At the end of the
experiment, the images of the cell colonies (>30 cells) in soft
agar were captured using an inverted light microscope (Olympus
Corporation; ×10 magnification). Colony size was measured using
ImageJ 1.53K software (National Institutes of Health) plotted as
relative colony size (%) when compared with control cells.
Western blot analysis
Whole cell protein extracts were prepared using RIPA
lysis buffer (Pierce; Thermo Fisher Scientific, Inc.) containing a
Protease Inhibitor Cocktail (Roche Diagnostics) and Phosphatase
Inhibitor (Thermo Fisher Scientific, Inc.). Lysed samples were
centrifuged at 18,000 RCF for 15 min at 4°C, and the clarified
supernatants were collected and stored at −80°C. Protein
concentrations were determined using the Bradford Protein Assay
Reagent (Bio-Rad Laboratories Inc.). Equal amounts of protein
samples (25 µg) were separated by 10 or 12% SDS-PAGE and
transferred onto polyvinylidene difluoride membranes (Thermo Fisher
Scientific, Inc.) using wet-transfer apparatus (Bio-Rad
Laboratories, Inc.). The membranes were blocked with 5% skim milk
(AmericanBio) and probed with primary antibodies (1:1,000 dilution)
overnight at 4°C. After washing with TBST buffer (25 mM Tris-HCl,
125 mM NaCl, 0.1% Tween 20), the membranes were incubated with
IRDye®800CW- or IRDye®680-conjugated secondary antibodies (cat.
nos. 926-32211 and 926-68071; 1:5,000 dilution; LI-COR Biosciences)
for 1 h at room temperature. The results were visualized using an
Odyssey Infrared Imager (LI-COR Biosciences). For loading controls,
the membranes were stripped and reprobed for β-actin and/or
tubulin. All primary antibodies used for western blotting are
listed in Table I.
Ethics approval and consent to
participate
Animal studies
The University of Alabama at Birmingham (UAB)
Institutional Animal Care and Use Committee (IACUC) (Birmingham,
AL, USA), approved the work (approval no. IACUC-21928). Male
C57B/6J mice (age, 4–6 weeks; weight, 22 g) were housed for 2–3
weeks to permit acclimatization and were monitored daily. The
animals were housed at 25°C, under a 12-h light/dark cycle, with
ad libitum access to food and water at 50% relative
humidity. The ~22 g animals (n3) were exposed to 5% isoflurane for
induction of anesthesia. Thereafter, the animals were maintained in
3% isoflurane until cardiac puncture and removal of the required
blood volume (1.0 ml) was performed. To ensure euthanasia, the
heart was removed. The long bone marrow of three animals was
isolated by sterile dissection, and the cells from three animals
were pooled.
Human studies
The studies were evaluated by the UAB Human Subjects
Committee under IRB-300004457 ‘The Biology of Prostate Cancer
Skeletal Metastases’. Given that no subject interactions were
planned, and cell lines were to be purchased from commercial
vendors, the studies were not deemed Human Subjects investigations
and were therefore considered exempt.
Co-cultures of PC3-GFP cells with bone
marrow cells and fluorescence-activated cell sorting (FACS)
analyses
Murine primary bone marrow stromal cells were
isolated by crushing the long bones from ~2-month-old male C57BL/6J
mice. The cell clumps and debris were removed by filtering the
cells using a 70-µm cell strainer (Thermo Fisher Scientific, Inc.).
PC3 cells infected with GFP-labeled LANCL2 and PPARγ shRNA
lentiviral vectors were placed into co-culture with confluent mouse
primary bone marrow cells. Co-culture of PC3-GFP and bone marrow
cells, with and without 100 µM ABA treatments were carried out for
3 days in an atmosphere containing 5% CO2 at 37°C. The
proliferating PC3-GFP cells in the co-cultured (PC3-GFP/bone
marrow) system were recovered using trypsin in PBS, and sorted
using a BD FACS Melody™ Cell Sorter (BD Biosciences) to distinguish
PC3 cells from murine bone marrow cells based upon GFP expression.
To distinguish live versus dead cells or cellular debris, parallel
cultures were used to establish gating parameters. A minimum of
20,000 events were recorded for each condition. The data were
analyzed using BD FACSChorus™ 3.0 software (BD Biosciences). Cell
proliferation percentages were established by comparing to control
cells, and histograms were plotted using GraphPad Prism 5
(Dotmatics).
Statistical analyses
GraphPad Prism 5 was used for statistical analyses.
All experiments were repeated at least three times and the results
are presented as the mean ± SEM. For each data point, a two-tailed,
unpaired, Student's t-test was performed to determine the
significance of the differences between two groups, and one-way
analysis of variance (ANOVA) followed by Bonferroni's post hoc test
was performed to compare three or more groups. When analyzing
multiple variables a two-way ANOVA analysis followed by
Bonferroni's post hoc test was performed to determine significance.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ABA treatment inhibits PCa cell
viability
To determine the effects of ABA on PCa DTC
viability, human and mouse PCa cells were exposed to increasing
doses of ABA (0, 25, 50, 100 and 200 µM) for 72 h in culture
medium. ABA significantly reduced the viability of the PCa cell
lines in a dose-dependent manner (Fig.
1A-D). Specifically, ABA at concentrations of 100 and 200 µM
decreased the viability of the PC3, C4-2B, DU145 and Myc-CaP cells
by ~23, 33, 26 and 25%, and by ~24, 52, 36 and 48% compared with
the control groups, respectively (Fig.
1A-D).
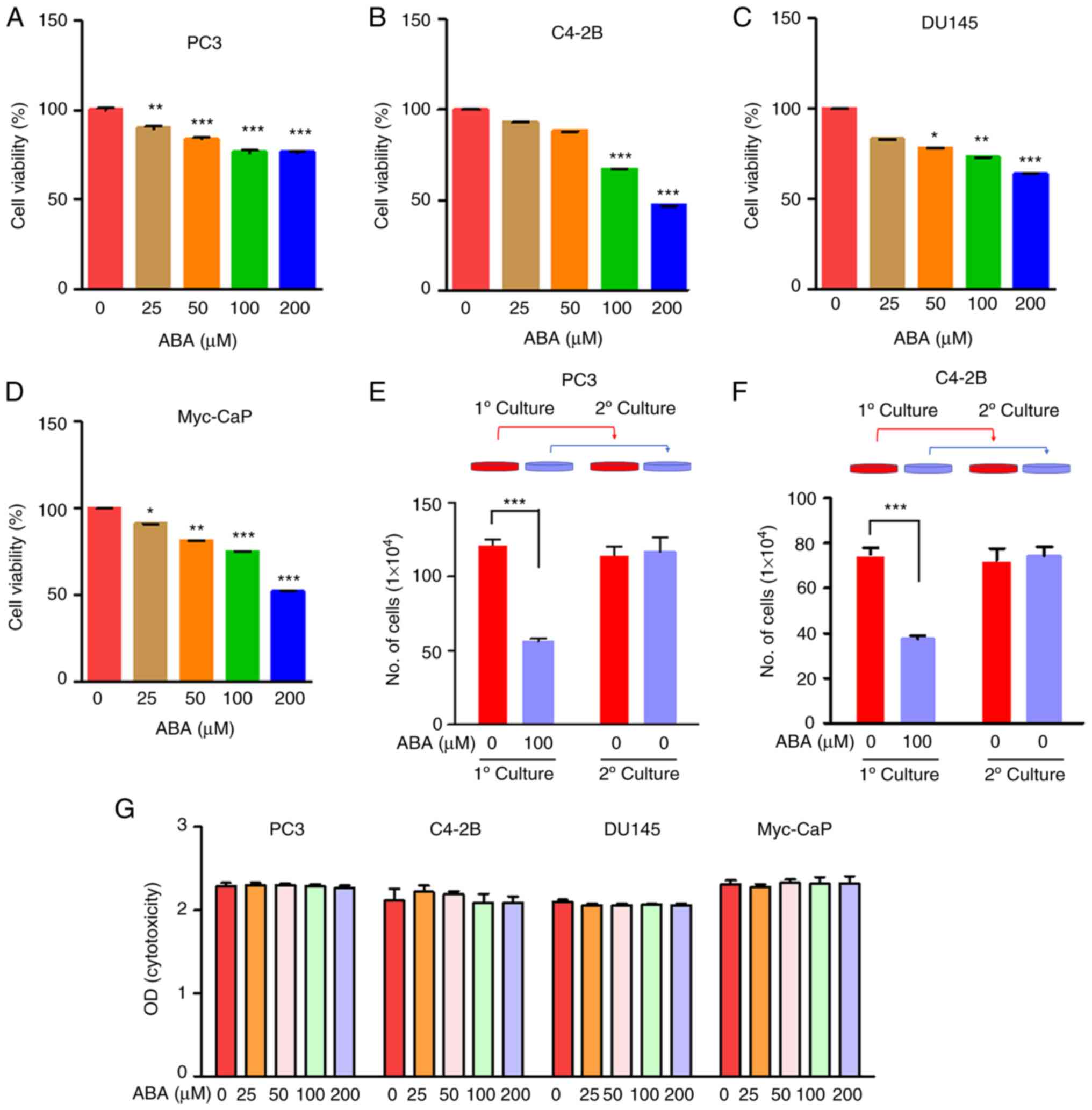 | Figure 1.ABA inhibits PCa cell viability. (A)
PC3, (B) C4-2B, (C) DU145 human and (D) Myc-CaP mouse PCa cells
were plated in 96-well plates, and were treated with 0, 25, 50, 100
and 200 µM ABA for 72 h in quintuplicate. Changes in cell density
were determined colorimetrically using the CellTiter 96 AQueous
Non-Radioactive Cell Proliferation Assay. Data are presented as the
mean ± SEM. Significance was calculated by one-way ANOVA followed
by Bonferroni's post hoc test (*P<0.05, **P<0.01,
***P<0.001 vs. control). (E) PC3 and (F) C4-2B cells were seeded
in 12-well plates and treated with 100 µM ABA for 72 h. Viable cell
numbers were assessed using trypan blue exclusion staining and a
hemocytometer. To determine if ABA-induced inhibition of viability
was reversible, ABA-treated and control cultures were established.
After 72 h, the cells were recovered from culture and equal numbers
of cells were plated in secondary cultures for an additional 72 h
without ABA supplementation and were quantified. Data are presented
as the mean ± SEM. Significance was calculated by one-way ANOVA
followed by Bonferroni's post hoc test (***P<0.001). (G)
Cytotoxicity assays on PCa cells were conducted by treating them
with ABA at concentrations of 0, 25, 50, 100 and 200 µM for 4 h,
and subsequently assessing toxicity using the CytoTox96
Non-Radioactive Cytotoxicity Assay to detect lactate dehydrogenase
release into the medium. ABA, abscisic acid; O.D., optical density;
PCa, prostate cancer. |
To determine if the inhibition of viability by ABA
was reversible, a pulse-chase type of investigation was performed.
Here, the metastatic PCa cells were treated with vehicle or ABA
(100 µM) for 72 h. After the initial culture, the cells were
washed, trypsinized and secondary cultures were established without
additional ABA treatment. To account for the differences in
viability that ABA induces, the secondary cultures were established
with equal numbers of viable cells. As expected, the ABA-treated
PC3 and C4-2B primary cultures had fewer cells relative to the
vehicle-treated cultures (Fig. 1E and
F). In the secondary cultures, the cells grew equally as well,
regardless of their prior exposure in primary culture to ABA, thus
demonstrating that the ABA-induced inhibition of viability was
reversable (Fig. 1E and F).
Notably, it was determined that ABA did not induce cell death of
any of the PCa cells using a non-radioactive cytotoxicity assay to
measure LDH release (Fig. 1G).
These data collectively suggested that ABA induces reversible
arrest of metastatic PCa cells.
ABA inhibits PCa cell viability by
signaling through LANCL2 and PPARγ
To assess the molecular mechanisms by which ABA
signaling influences PCa viability, stable KD of the ABA receptors
LANCL2 and PPARγ were performed in metastatic PCa cell lines using
shRNAs. To evaluate the efficiency of the KD, LANCL2 or PPARγ mRNA
expression levels were confirmed by RT-qPCR (Fig. 2A-D) and protein expression levels
were detected using western blotting (Fig. 2E-H). The targeting sequence #1 for
LANCL2 and PPARγ were selected for all subsequent studies. After
validation of the KD, the sensitivity of PC3 and C4-2B cells to ABA
in vitro was assessed using cell viability assays. shRNA KD
of LANCL2 or PPARγ conferred partial resistance to ABA in both PC3
and C4-2B cells (Fig. 2I and
J).
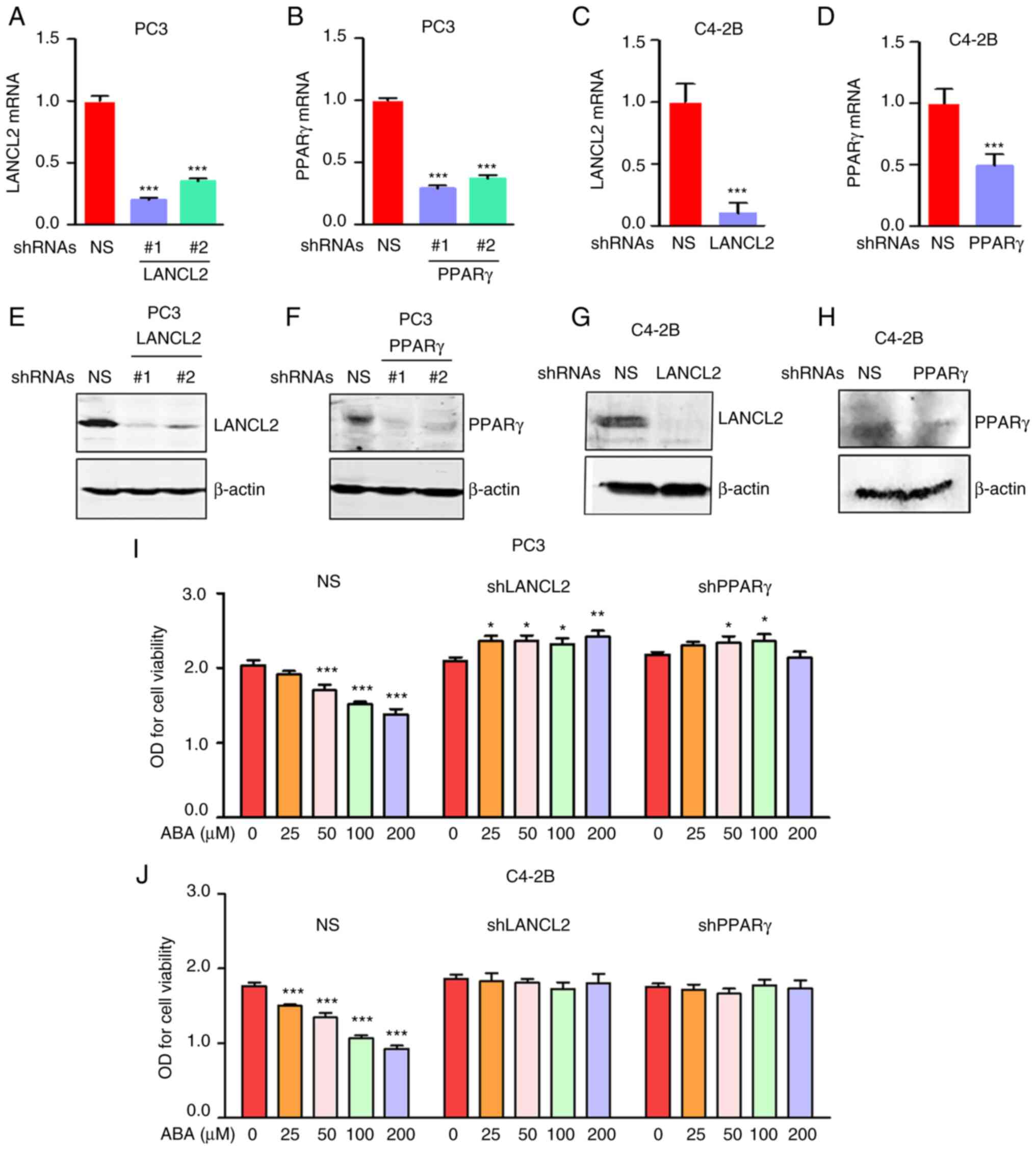 | Figure 2.ABA inhibits PCa cell viability
through LANCL2 and PPARγ. For all studies, PCa cells were infected
with NS shRNA, or shRNAs targeting (A) LANCL2 or (B) PPARγ for PC3
cells, and (C) LANCL2 or (D) PPARγ for and C4-2B cells. The cells
were then analyzed for LANCL2 and PPARγ mRNA expression by reverse
transcription-quantitative PCR. Expression levels of LANCL2 and
PPARγ mRNA were plotted relative to those in the NS group. β-actin
was used for normalization. Data are presented as the mean ± SEM.
(A and B) Significance was calculated by one-way ANOVA followed by
Bonferroni's post hoc test (***P<0.001 vs. shRNA NS). (C and D)
Significance was calculated using a two-tailed, unpaired Student's
t-test, (***P<0.001 vs. shRNA NS). (E) LANCL2 and (F) PPARγ
expression in PC3 cells, and (G) LANCL2 and (H) PPARγ expression in
C4-2B cells were analyzed via western blotting after SDS-PAGE and
blot transfer to membranes with specific antibodies to the
receptors. β-actin was used as a loading control. (I) Viability of
PC3 and (J) C4-2B cells infected with NS shRNA, or shRNAs targeting
LANCL2 or PPARγ, and treated with ABA (0, 25, 50, 100 and 200 µM)
for 72 h. Cell density was enumerated colorimetrically using
CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay Kit.
Data are presented as the mean ± SEM. Significance was calculated
by one-way ANOVA followed by Bonferroni's post hoc test
(*P<0.05, **P<0.01, ***P<0.001 vs. control). ABA, abscisic
acid; LANCL2, lanthionine synthetase C-like protein 2; NS,
non-specific; O.D., optical density; PCa, prostate cancer; PPARγ,
peroxisome proliferator activated receptor γ; shRNA, short hairpin
RNA. |
As a surrogate assay for estimating in vivo
tumorigenesis, anchorage-independent growth in soft-agar assays was
performed to evaluate the effects of ABA and its signaling through
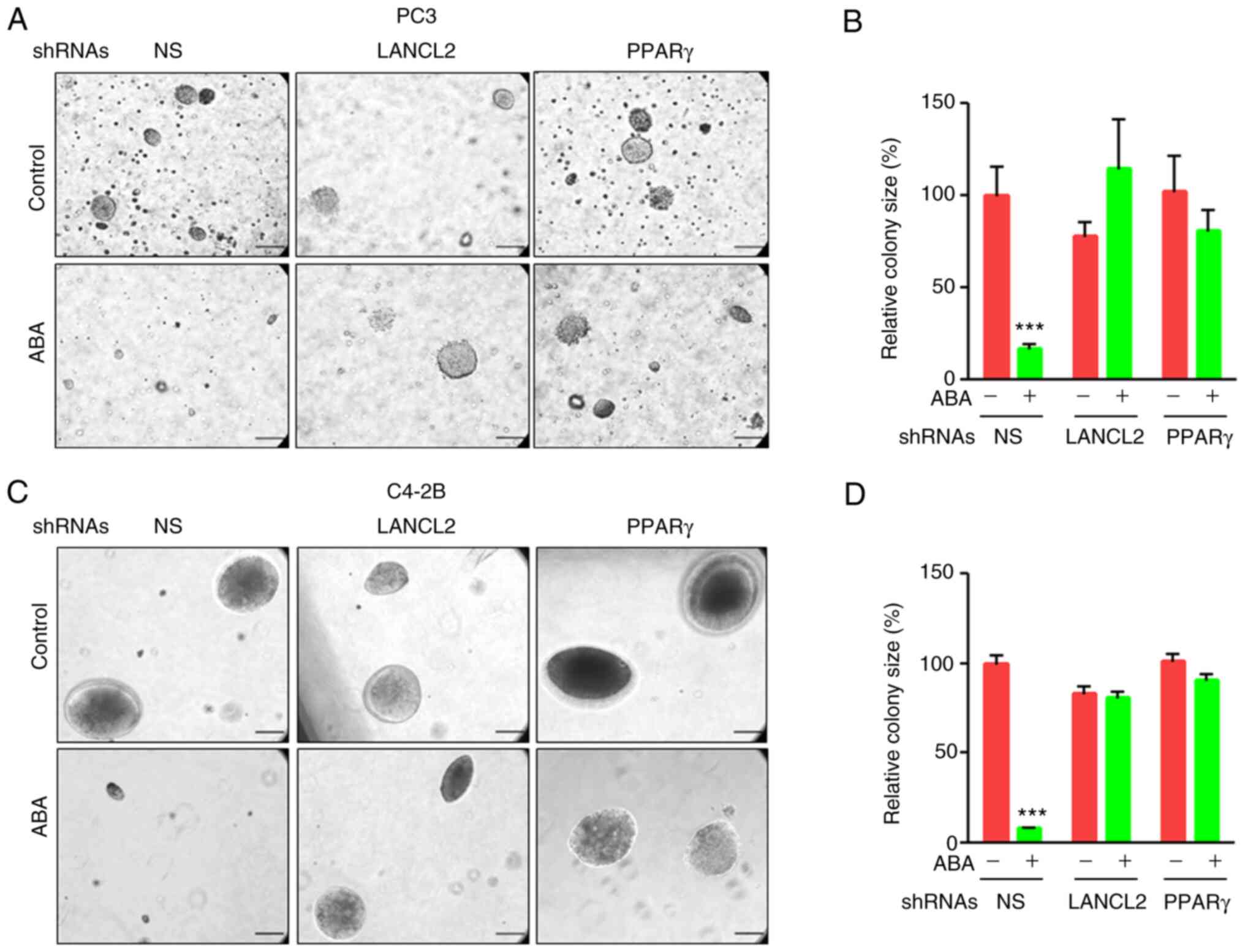
its receptors (41,42). ABA significantly decreased the
ability of PCa PC3 and C4-2B cells to form colonies in the NS
groups; however, when LANCL2 or PPARγ expression was downregulated
by shRNA, ABA treatment did not affect colony formation in soft
agar (Fig. 3A-D). These
observations suggested that LANCL2 or PPARγ are required for ABA
signaling to induce anchorage-independent growth. Given the
functional link between colony formation and cancer-stem cell (CSC)
activities (43), these data also
indicated that ABA, and its signaling though its receptors, may
downregulate CSC proliferation; however, the impact on CSCs was not
formally tested.
ABA inhibits PCa cell migration and
invasion through LANCL2 and PPARγ receptors
Given the effects of ABA on viability and colony
formation, the impact of ABA on PCa invasion was next evaluated.
For these studies, the migration of cells treated with or without
ABA was detected in response to FBS as a chemoattractant in
Transwell plates. ABA decreased the invasion of PCa cells, whereas
targeted KD of LANCL2 or PPARγ expression reduced the effects of
ABA treatment on migration (Fig.
4A-D). To evaluate the effects of ABA and the respective
receptor KD on invasion, Transwell plates covered in Matrigel were
utilized. As with the migration assays, ABA decreased cell invasion
in response to FBS, whereas KD of LANCL2 or PPARγ expression
abrogated this effect (Fig.
4E-H).
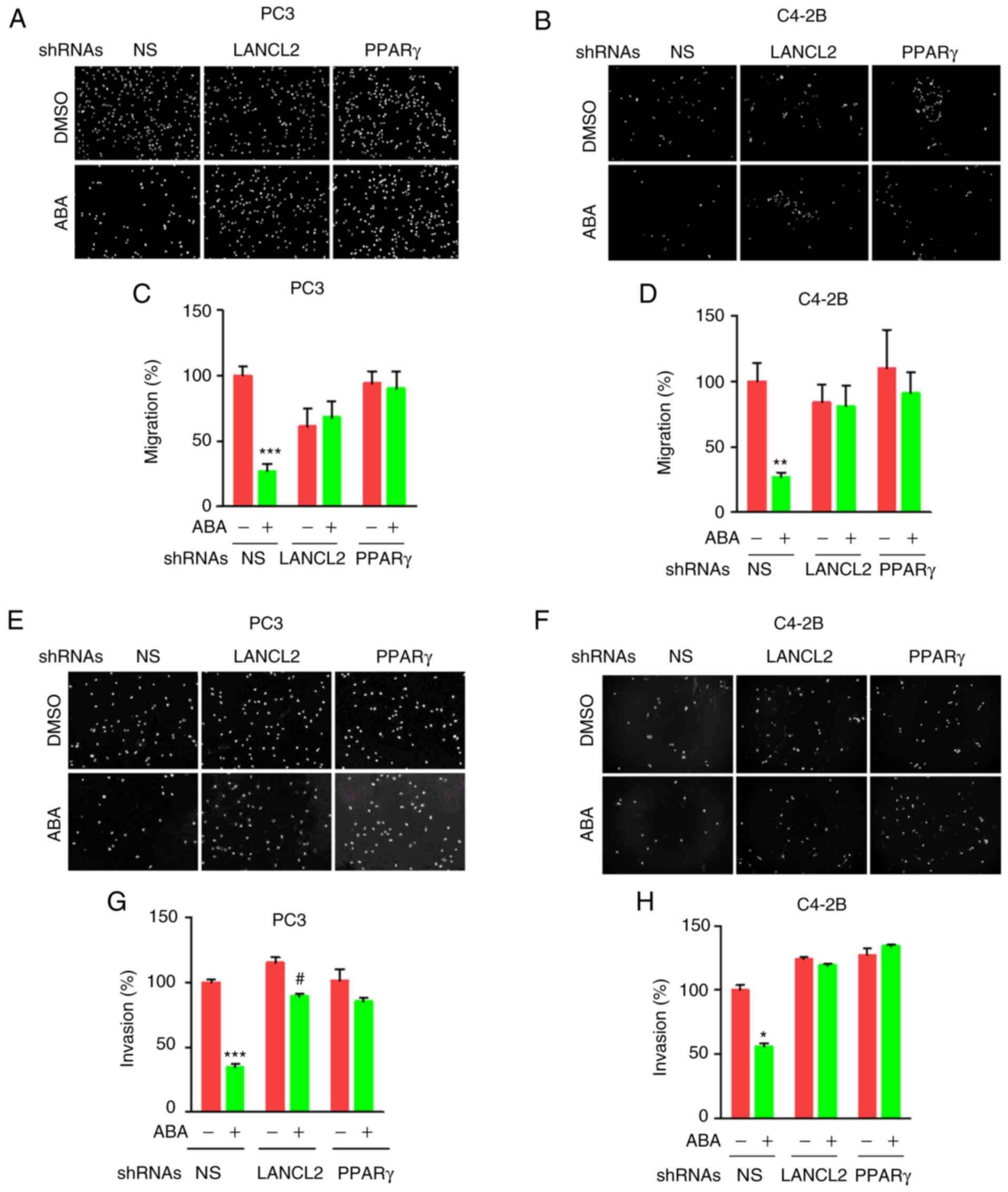 | Figure 4.ABA inhibits prostate cancer cell
migration and invasion through LANCL2 or PPARγ. Migration of (A)
PC3 or (B) C4-2B cells infected with NS shRNA, or shRNAs targeting
LANCL2 or PPARγ, across a Transwell membrane in response to 10%
FBS. Cells were treated with ABA (100 µM) for 24 h (PC3) and 48 h
(C4-2B), and were analyzed at the end of the culture period.
Representative images (×10 magnification) at the indicated times
are shown. Relative cell migration (%) of (C) PC3 and (D) C4-2B
cells. Invasion assay of vehicle- and ABA-treated (E) PC3 and (F)
C4-2B cells across a Transwell membrane covered with Matrigel (×10
magnification). Invasion through the matrix was evaluated at (G) 24
h (PC3) and (H) 48 h (C4-2B). Invasion (%) is shown. Data are
presented as the mean ± SEM. Significance was calculated by two-way
ANOVA followed by Bonferroni's post hoc test (*P<0.05,
**P<0.01, ***P<0.001 vs. shRNA NS control;
#P<0.05 vs. shRNA LANCL2 control). ABA, abscisic
acid; LANCL2, lanthionine synthetase C-like protein 2; NS,
non-specific; PPARγ, peroxisome proliferator activated receptor γ;
shRNA, short hairpin RNA. |
To evaluate the mechanisms by which ABA affects
invasion, the present study evaluated the mRNA expression levels of
E-cadherin, Vimentin and ZEB2 by RT-qPCR. The results showed that
ABA increased E-cadherin, and decreased Vimentin and ZEB2
expression levels in PCa cells in the NS group (Fig. S1). However, shRNA KD of LANCL2 or
PPARγ receptors mitigated the effects of ABA on the majority of the
cells. The exception being that the mRNA expression levels of
Vimentin in ABA-treated shRNA PPARγ KDC4-2B cells were reduced
compared with in those cells not treated with ABA, which could be
due to a compensatory effect of the LANCL2 receptor (Fig. S1). Collectively, these results
indicated that LANCL2 and PPARγ are necessary for the effects of
ABA on PCa cell migration and invasion, and suggested that these
receptors play a critical role in the metastatic attributes of PCa
cells.
Inhibition of PCa cell viability in
co-culture is enhanced by ABA
To explore the role that ABA and its receptors play
in promoting dormancy in bone marrow, in vitro studies were
performed. For these studies, PC3 cells were cultured in the
presence of confluent monolayers of bone marrow stromal cells
isolated from C57BL/6J mice. The co-cultures were grown for 3 days
in the presence or absence of ABA. To distinguish the PC3 cells
from the stromal layers, PC3-GFP cells were used. The viability of
PC3-GFP cells was evaluated upon recovery of the cells from culture
with trypsin by FACS probing for GFP. In co-culture, PC3-GFP
viability was significantly decreased when ABA was included in the
co-cultures (Fig. 5A, B and G).
Notably, when the PCa cells with KD of LANCL2 or PPARγ were
cultured with the stromal cells, they exhibited increased viability
relative to the NS control group (Fig.
5C-G). As expected, the addition of exogenous ABA in the
absence of LANCL2 or PPARγ expression led to little to no impact on
cellular viability in the co-cultures (Fig. 5C-F). These results indicated that
ABA inhibited PCa cell viability in co-culture, whereas when LANCL2
or PPARγ expression was reduced, either the PCa cells were no
longer sensitive to ABA expressed by the bone marrow cells
(44), or they become insensitive
to another negative cell growth regulator produced by stomal cells
which signals through LANCL2 or PPARγ.
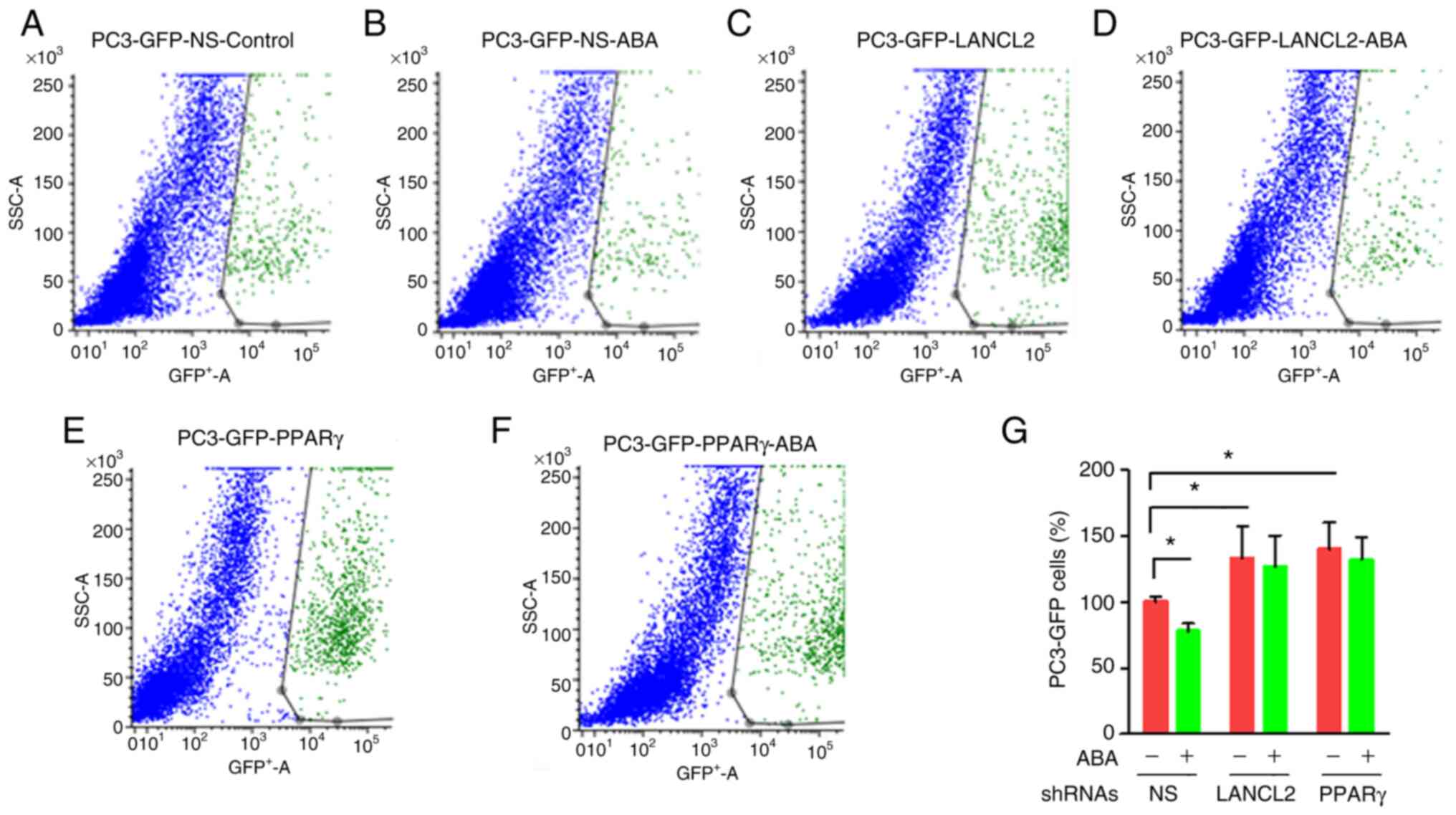 | Figure 5.Effect of bone marrow cells on the
viability of prostate cancer cells with and without ABA. Murine
bone marrow stromal cells were cocultured with GFP-labeled PC3
cells with reduced expression of LANCL2 or PPARγ in the presence or
absence of exogenous ABA. At 72 h, the cultures were trypsinized
and the GFP-labeled PC3 cells were distinguished and enumerated by
FACS gating on GFP. Representative FACS plots of the recovered
cells, with PC3 cells shown in green and stromal cells in blue. In
each case the shRNA contained a GFP expression cassette. (A)
Control NS PC3 cells treated with vehicle, (B) control PC3 cells
treated with ABA, (C) KD LANCL2 PC3 cells treated with vehicle, (D)
KD LANCL2 PC3 cells treated with ABA, (E) KD PPARγ PC3 cells
treated with vehicle and (F) KD PPARγ PC3 cells treated with ABA
(G) Percentage of PC3-GFP cells. Data are presented as the mean ±
SEM. Significance was calculated by two-way ANOVA followed by
Bonferroni's post hoc test (*P<0.05). ABA, abscisic acid; FACs,
fluorescence-activated cell sorting; LANCL2, lanthionine synthetase
C-like protein 2; NS, non-specific; PPARγ, peroxisome proliferator
activated receptor γ; shRNA, short hairpin RNA. |
ABA regulates transcription of p21,
p27 and p16 through LANCL2 and PPARγ in PCa cells
The role of p21, p27 and p16 in cell cycle
progression is well established. Activation of p21, p27 and p16
inhibits phosphorylation of CDK1 and CDK2 resulting in cell cycle
arrest. To explore whether ABA signaling through LANCL2 and PPARγ
impacts the expression of p21, p27 or p16, the PCa control or
LANCL2 and PPARγ KD cells were treated with ABA and were examined
by RT-qPCR analysis. Unexpectedly, when PPARγ expression was
knocked down by shRNA ABA increased the expression levels of LANCL2
(Fig. 6A and F), and had variable
impact on PPARγ expression when LANCL2 expression was knocked down
by shRNA (Fig. 6B and G), thus
suggesting that the expression of these ABA receptors establish a
feedback loop in PC3 (Fig. 6A and
B) and C4-2B cells (Fig. 6F and
G). Notably, ABA induced the mRNA expression levels of p21, p27
and p16 in both PC3 (Fig. 6C-E) and
C4-2B (Fig. 6H-J) cells. In the
predominance of the studies, KD of LANCL2 and PPARγ by shRNA
markedly abrogated the effect of ABA on the mRNA expression levels
of p21, p27 and p16 (Fig. 6C-E and
H-J). Collectively, these results suggested that ABA induces
transcriptional regulation of several cell cycle inhibitor proteins
in PCa cells through LANCL2 and PPARγ signaling.
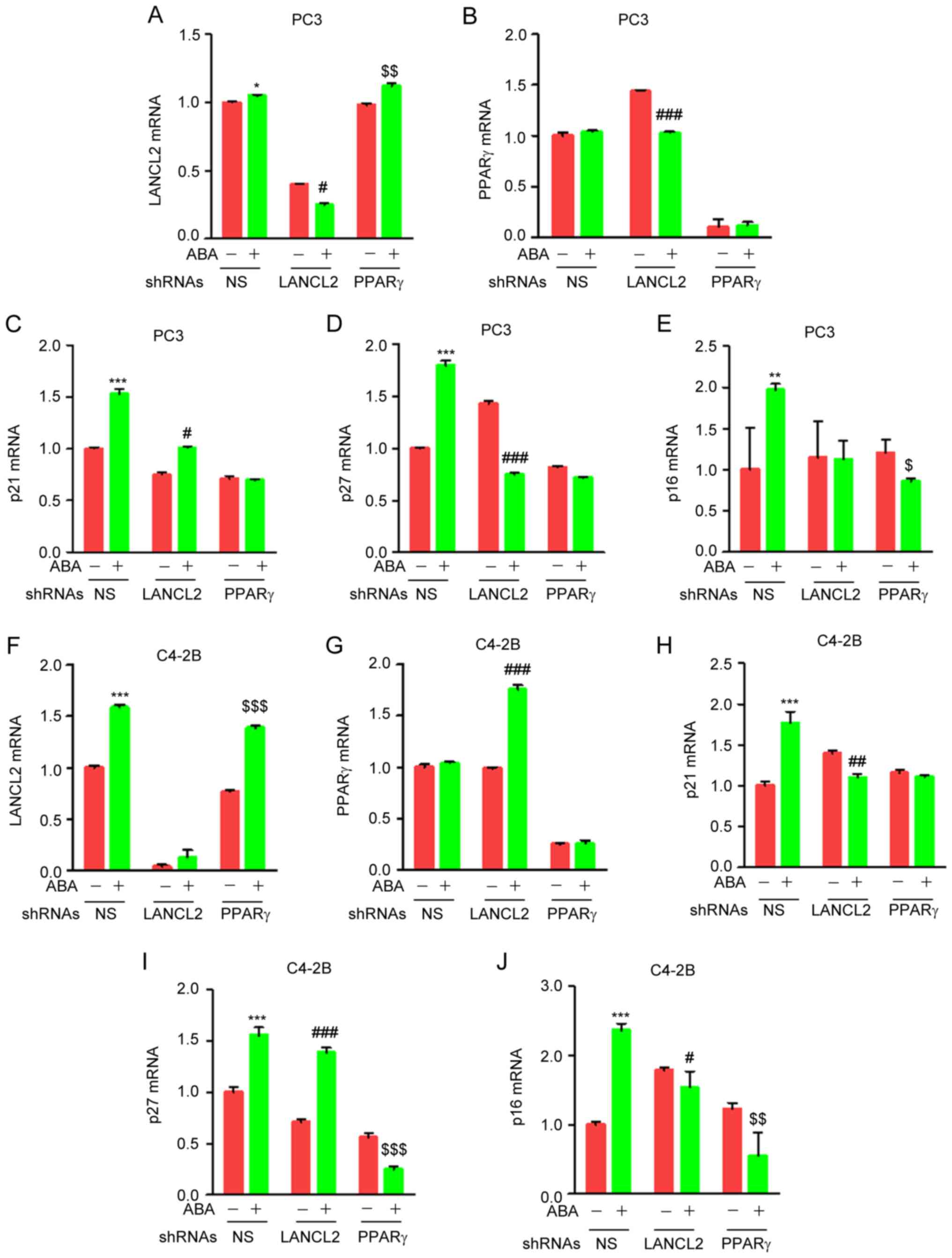 | Figure 6.ABA increases the mRNA expression
levels of cell cycle-regulating genes through LANCL2 and PPARγ. PC3
prostate cancer cells infected with NS shRNA, or shRNAs targeting
LANCL2 or PPARγ, were treated with 100 µM ABA and (A) LANCL2, (B)
PPARγ, (C) p21, (D) p27 and (E) p16 expression was evaluated. The
mRNA expression levels of the indicated genes were analyzed by
quantitative PCR. For C4-2B prostate cancer cells, they too were
infected with NS shRNA, or shRNAs targeting LANCL2 or PPARγ, and
were treated with 100 µM ABA, and (F) LANCL2, (G) PPARγ, (H) p21,
(I) p27 and (J) p16 expression was evaluated. Fold changes were
calculated based on normalization to β-actin levels and using the
untreated control. Data are presented as the mean ± SEM.
Significance was calculated by two-way ANOVA followed by
Bonferroni's post hoc test (*P<0.05, **P<0.01, ***P<0.001
vs. shRNA NS control; #P<0.05,
##P<0.01, ###P<0.001 vs. shRNA LANCL2
control; $P<0.05, $$P<0.01,
$$$P<0.001 vs. shRNA PPARγ control). ABA, abscisic
acid; LANCL2, lanthionine synthetase C-like protein 2; NS,
non-specific; PPARγ, peroxisome proliferator activated receptor γ;
shRNA, short hairpin RNA. |
ABA induces PCa dormancy through
LANCL2 and PPARγ and p38MAPK pathway activation
Phosphorylation of p38MAPK is one of the major
signaling pathways involved in the induction of dormancy by
molecules that inhibit cell proliferation, including TGFβ, BMPs and
GAS6 (15,17). Notably, activation of p38MAPK
signaling by phosphorylation, and enhanced expression of the cell
cycle inhibitors p21 and p27, are considered as dormancy markers in
cancer cells (15,19,45).
Therefore, the present study evaluated the effects of ABA signaling
on p38MAPK activation. Treatment of the PCa cells with ABA resulted
in phosphorylation 38MAPK (Fig. 7A, B,
D and E), as well as stimulation of NR2F1 expression, a
dormancy marker (Fig. 7A, C, D and
F). ABA also enhanced the expression levels of the downstream
targets of p38MAPK, including p21 and p27 (Fig. 7G and H). As expected, KD of the ABA
receptors LANCL2 and PPARγ negated the ABA-induced effects on p21,
p27 and NR2F1 expression (Fig.
7A-H). Collectively, these findings indicated that ABA affects
the viability arrest of PCa cells through LANCL2 and PPARγ
receptors, in part by signaling through p38MAPK.
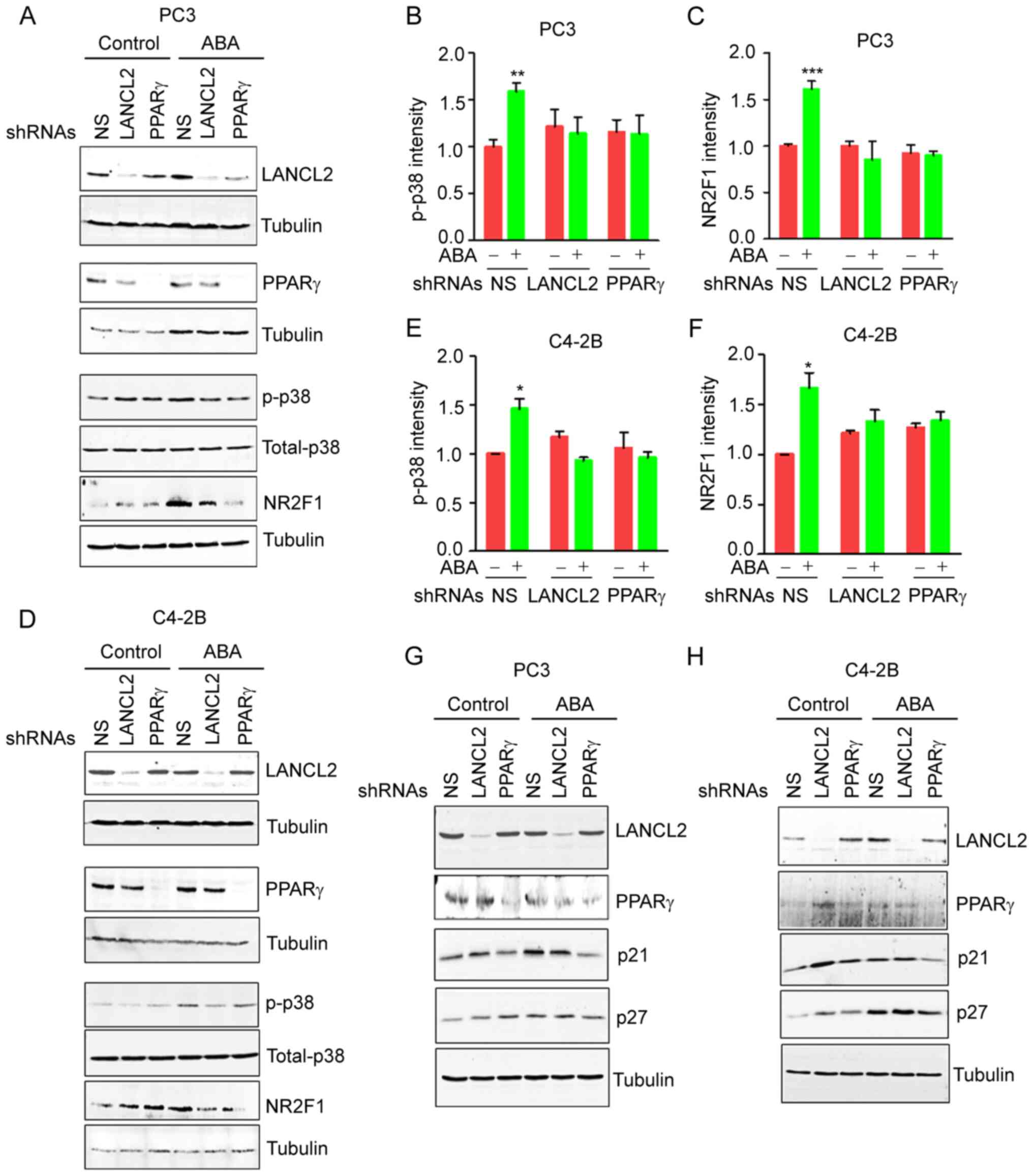 | Figure 7.ABA induces changes in PCa cell
dormancy via signaling through LANCL2 or PPARγ to activate the
p38MAPK pathway in PCa. PC3 and C4-2B cells infected with either NS
shRNA, or shRNAs targeting LANCL2 or PPARγ, were treated with ABA
and analyzed by western blotting for the indicated proteins. (A)
Expression of LANCL2, total p38 and p-p38 in PC3 cells. (B) p-p38
expression was semi-quantified, and (C) NR2F1 band intensity was
measured and normalized to the loading control tubulin using ImageJ
software in PC3 cells. (D) Expression of LANCL2, total p38 and
p-p38 expression in C4-2B cells. (E) p-p38 was semi-quantified, and
(F) NR2F1 band intensity was evaluated using ImageJ software in
C4-2B cells. In (G) PC3 and (H) C4-2B cells, the expression levels
of LANCL2, PPARγ, p21 and p27 were detected in response to ABA.
Significance was calculated by two-way ANOVA followed by
Bonferroni's post hoc test (*P<0.05, **P<0.01, ***P<0.001
vs. shRNA NS control). ABA, abscisic acid; LANCL2, lanthionine
synthetase C-like protein 2; NS, non-specific; p-, phosphorylated;
PCa, prostate cancer; PPARγ, peroxisome proliferator activated
receptor γ; shRNA, short hairpin RNA. |
Discussion
PCa is a heterogenous disease, which has variable
clinical outcomes ranging from early-stage, curable, advanced and
terminal disease. Although the majority of men are cured soon after
diagnosis at an early stage, a subset of men develop recurrent
disease, or present with de novo metastatic disease
(46). The bone is a common site
for PCa metastasis (47), where
metastatic cells that have escaped the prostate early in disease
are likely to have entered a dormant state for years or even
decades. Notably, ~90% of patients who succumb to metastatic PCa
have some level of bone involvement (48). Several new therapeutic strategies,
including androgen receptor-targeted therapies, chemotherapy, poly
ADP-ribose polymerase inhibitors, sipuleucel-T and radium-223, have
been approved for the treatment of patients with advanced PCa and
bone metastasis. However, this disease still represents the second
most frequent cause of cancer-related death in men in the United
States (49). The challenges in
treating bone metastases at the advanced stage include a high level
of genomic heterogeneity (50), a
pro-immunosuppressive environment (51) and several emerging mechanisms of
androgen independence (52).
Therefore, new therapeutics are urgently needed.
ABA is known to regulate cell responses and
biological processes in plants, mammals and other organisms
(53,54). ABA is a key hormone that promotes
seed dormancy during development in plants and after seed dispersal
(55). ABA controls the induction
of dormancy, and ultimately the release and germination of seeds in
response to environmental signals (55). ABA has also been recognized as an
endogenous hormone in humans (29).
It has a role in the inflammatory response, as well as in
immunoregulatory activities, antioxidant properties and the
maintenance of glycemic control in pre-clinical models of diabetes
and inflammatory diseases (56,57).
Despite the advances in the understanding of the role that ABA
plays in several physiological processes, the role of ABA in tumor
dormancy remains to be elucidated.
The present study demonstrated that ABA suppressed
the viability of four distinct PCa cell lines (PC3, C4-2B, DU-145
and Myc-CaP) in a dose-dependent manner; however, exposure to ABA
was not in and of itself cytotoxic to the cancer cells. Notably,
ABA-induced inhibition of PCa cell viability was reversible, as
cells which were removed from ABA treatment were able to resume
their proliferation. There are at least two receptors that are
responsible for transducing ABA signaling; LANCL2 and PPARγ. To
explore how ABA induced proliferative quiescence, targeted KD of
LANCL2 and PPARγ was established using shRNAs in PC3 and C4-2B cell
lines. In almost every instance where ABA signaling was examined,
including viability, migration, invasion and downstream signaling,
reduced expression of LANCL2 and PPARγ abrogated the impact of ABA
on the cancer cells. Furthermore, it was observed that ABA
inhibited the formation of PCa colonies in soft agar, whereas
LANCL2 and PPARγ KD alone did not. Mechanistically, ABA treatment
induced p21, p27 and p16 mRNA and protein expression, whereas
LANCL2 and PPARγ shRNA KD did not significantly inhibit either p21,
p27 or p16 mRNA or protein expression relative to the controls.
These data suggested that ABA may inhibit viability through
inducing the expression levels of p21, p27 and p16, but when the
expression of ABA receptors is reduced, there is little impact on
p21, p27 and p16 expression.
LANCL2 and PPARγ are known to mediate a range of
physiological responses of ABA in a number of systems, including
granulocytes and rat insulinoma cells (31,58).
ABA signaling in mammalian immune cells, keratinocytes and
pancreatic cells requires LANCL2 expression, as small interfering
RNA targeting of LANCL2 has been shown to abrogate cellular
responses (23,26,58).
Several studies have reported that activation of PPARγ by agonists
markedly reduces cell proliferation, including in hepatocellular
carcinoma (59), PCa (60) and gastric cancer (61). Other studies have demonstrated that
PPARγ agonists promote terminal differentiation, inhibit cell
proliferation and increase the apoptosis of human cancer cell
lines, as well as tumor growth in animal models (33,62).
Notably, in some cases, PPARγ agonists have demonstrated modest
efficacy as chemopreventives in clinical trials (33,62).
It is not surprising that clinical studies have reported that the
overall survival of patients with colorectal cancer is better when
PPARγ expression is detectable in the primary tumors (63). In part, this may be due to the
findings that PPARγ modulates the expression of different cell
cycle regulators, including decreasing the expression of cyclin D1
(64), and increasing the
expression of the CDK inhibitors p21 (65) and p27 (66).
The present study reported on notable findings in
co-culture studies of PCa with bone marrow stromal cells. In the
presence of marrow stroma, ABA is known to inhibit PCa cell
viability (44). However, when PCa
cells with reduced expression of LANCL2 and PPARγ were grown on the
bone marrow stromal cells, the cells proliferated at the same rate
as the control cells. These results suggested that PCa cells with
decreased expression of LANCL2 and PPARγ may no longer be sensitive
to ABA produced by the mesenchymal stromal cells, as shown in a
previous study (67), or are no
longer sensitive to growth inhibitors secreted by the stomal cells,
which also signal through LANCL2 and PPARγ. Very little is known as
to what activates LANCL2. In glioma it has been demonstrated that
LANCL2 expression may be co-regulated by epidermal growth factor
receptor (68). Activation of PPARγ
can inhibit cell proliferation, induce differentiation and promote
metastasis in PCa cells (60). In
other contexts, PPARγ is activated by fatty acids and derivatives
including polyunsaturated fatty acids, derivatives of arachidonic
acid and prostaglandins (69).
However, what other negative regulators of PCa cell viability are
produced by bone marrow stromal cells, which signal through LANCL2
and PPARγ remain unclear.
In the PCa cell lines tested there appears to be
some diversity in signaling in response to ABA through PPARγ and
LANCL2. For example, p27 activity was enhanced by ABA treatment in
both cell lines, but when LANCL2 was knocked down by shRNA, the p27
response was altered in the C4-2B and PC3 cells, although the
response was in the opposite direction. Currently, there is no
specific explanation as to the basis of the differences in these
observations. A reasonable possibility is that the cells have other
receptors for ABA in addition to LANCL2 and PPARγ. In other
studies, LANCL1 and CD38 have been suggested to serve as receptors
or co-stimulatory molecules for LANCL2 (58,70).
Unfortunately, we were not able to achieve functional reductions of
CD38 in the cell lines used in the present study (C4-2B and PC3);
therefore, it is possible that other players participate in the
signaling. Furthermore, there are known ABA inhibitors in plants,
including gibberellins, which can in some cases serve as allergens
and of which derivatives may have anti-neoplastic activities
(71–77). Whether equivalent proteins function
in human systems remains unclear. However, in preliminary studies
we did not observe any significant impact of gibberellin on the
cell cycle of metastatic cancer cell lines (Jung et al,
unpublished data). Further studies are needed to determine the
basis of the variations in cell cycle inhibitors.
To further understand the mechanisms by which ABA
signals through LANCL2 and PPARγ to induce dormancy, western blot
analysis was performed to detect p38 phosphorylation in PCa cells.
It was revealed that KD of LANCL2 and PPARγ inhibited the
activation of p38 by ABA. In addition, NR2F1 has been reported to
act as a master regulator of tumor cell dormancy (78). By binding to DNA and recruiting
coactivator or corepressor complexes, NR2F1 is able to serve as a
cofactor to other nuclear receptors. Notably, NR2F1 expression, one
of the best known dormancy markers, has been reported to be
directly regulated by ABA (78),
and its expression in this previous study was associated with p38
signaling (78). Activation of p38
signaling induces expression of the cell cycle inhibitors p21 and
p27, also considered as dormancy markers in cancer cells (15,19,45).
These findings make sense given the proposed hypothesis that the
ratio between the p38 MAPK stress response signaling pathway and
ERK guides cancer cells into a proliferative or dormant state
(79).
The regulation of cell proliferation in the bone
marrow is a critical function of the marrow microenvironment to
protect and promote proliferation of hematopoietic and mesenchymal
stem cells, and its integration across an organism is critical to
the survival of mammals. The present study assessed the role of ABA
and its signaling pathways in regulating cell
proliferation/quiescence of cells that do not belong in the marrow,
specifically cells of epithelial origin. Given that there are a
number of pathways that regulate the proliferation and dormancy of
stem and progenitor cells in the marrow, it is not surprising that
there are multiple pathways that can regulate stem cell
proliferation (16,80–82).
That tumor cells exploit the niches used by the bone marrow to
regulate stem and progenitor cells is not surprising and makes
sense from an ecological perspective (80). For example, osteoblasts that
participate in forming hematopoietic stem cell (HSC) niches produce
GAS6, which not only regulates HSC function, but limits the
proliferation of PCa cells in vitro (83) and in vivo (84–86).
Other studies have shown that GAS6 and its receptor AXL are
required for TGFβ2-mediated cell proliferation suppression in PCa,
where AXL positively regulates the expression of TGFβ and TGFβ
receptor 2 (18). In the current
study, it was demonstrated that ABA in co-culture of PCa cells with
murine bone marrow significantly inhibited PCa cell viability.
However, LANCL2 and PPARγ KD abrogated the response to ABA.
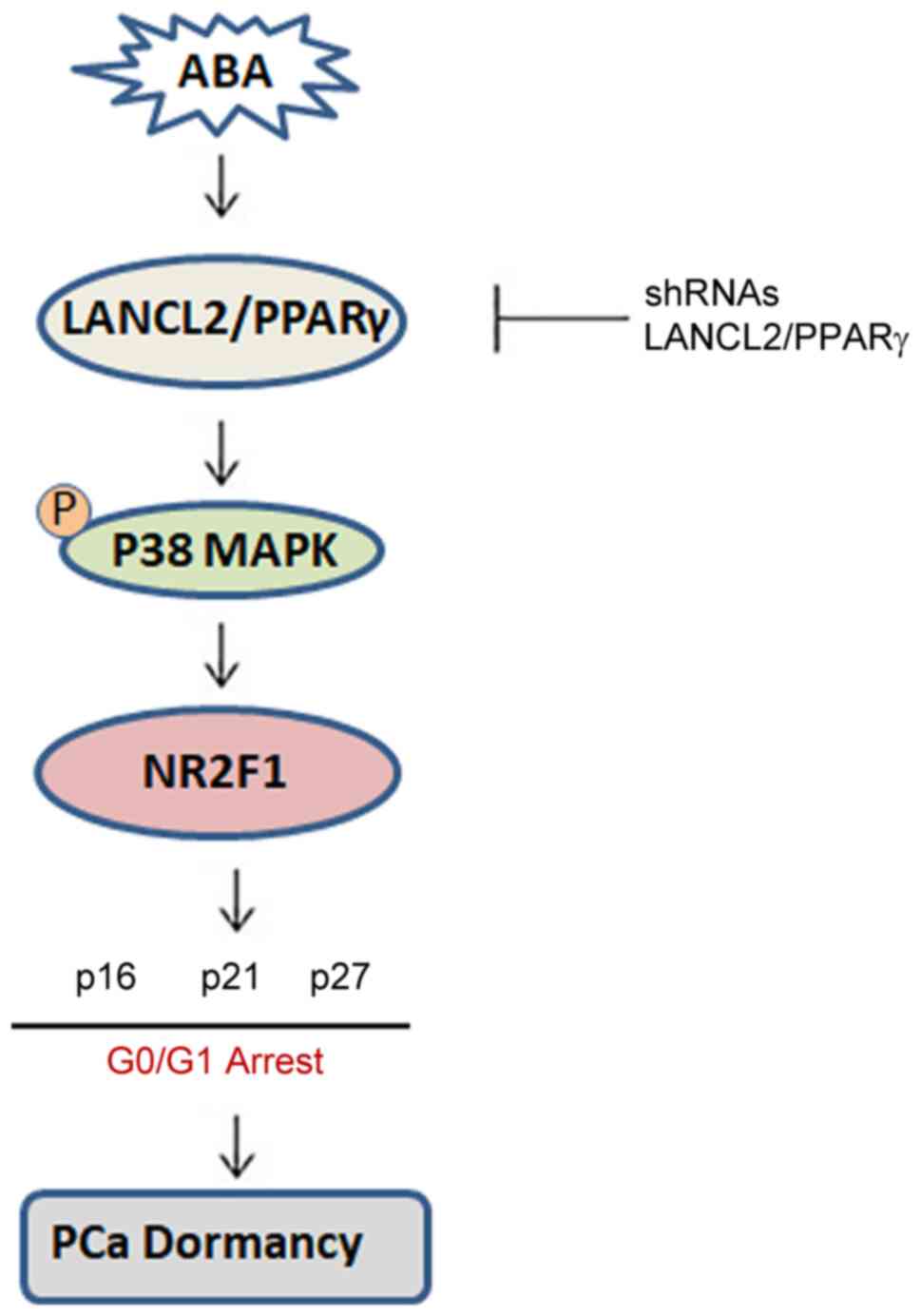
Together, these data suggest the following model: ABA signaling
through LANCL2 or PPARγ receptors activates signaling via p38MAPK,
NR2F1, p27, p16 and p21, which ultimately leads to dormancy
(Fig. 8). These results suggest
that interference with ABA signaling under the appropriate
conditions, or perhaps even from dietary sources such as fruits and
vegetables, may prove beneficial in treating men with prostate
cancer, by either keeping DTCs as dormant cells or by interfering
with ABA signaling together with chemotherapy to selectively kill
DTCs.
In conclusion, the present findings suggested that
ABA may induce cell cycle arrest in PCa. ABA does so by signaling
through LANCL2 and PPARγ, which activate p38MAPK and the CDK
inhibitors, p21, p16 and p27, and NR2F1 resulting in PCa cell
dormancy. Further investigation into how the ABA signaling pathway
results in dormancy may reveal novel opportunities for eradicating
dormant cancer cells and/or keeping them in a perpetual dormant
state, thus preventing PCa metastasis.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to express their gratitude to
Professor Amjad Javed (University of Alabama at Birmingham) for
generously providing the mice used for isolating primary bone
marrow and conducting the co-culture experiments with prostate
cancer cells.
Funding
This work was supported by the NIH/NCI (grant no.
3P01CA093900-018), the Prostate Cancer Foundation (2014 Challenge
Award) and the Department of Defense (grant no. PC140665).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KRP, YJ and RST conceived or designed the study. KRP
collected the data. KRP and RST analyzed and interpreted the data.
KRP drafted the article. YJ and RST critically revised the article.
KRP and RST confirm the authenticity of all the raw data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The University of Alabama at Birmingham
Institutional Animal Care and Use Committee (IACUC) approved the
present study (approval no. IACUC-21928).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Taichman RS, Loberg RD, Mehra R and Pienta
KJ: The evolving biology and treatment of prostate cancer. J Clin
Invest. 117:2351–2361. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wenzel M, Garcia CC, Hoeh B, Jorias C,
Humke C, Koll F, Tselis N, Rödel C, Graefen M, Tilki D, et al:
Real-world evidence of outcomes of oligometastatic
hormone-sensitive prostate cancer patients treated with
metastasis-directed therapy. Prostate. 83:1365–1372. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rogowski P, Trapp C, von Bestenbostel R,
Schmidt-Hegemann NS, Shi R, Ilhan H, Kretschmer A, Stief C,
Ganswindt U, Belka C and Li M: Outcomes of metastasis-directed
therapy of bone oligometastatic prostate cancer. Radiat Oncol.
16:1252021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bubendorf L, Schöpfer A, Wagner U, Sauter
G, Moch H, Willi N, Gasser TC and Mihatsch MJ: Metastatic patterns
of prostate cancer: An autopsy study of 1,589 patients. Hum Pathol.
31:578–583. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Berruti A, Dogliotti L, Bitossi R, Fasolis
G, Gorzegno G, Bellina M, Torta M, Porpiglia F, Fontana D and
Angeli A: Incidence of skeletal complications in patients with bone
metastatic prostate cancer and hormone refractory disease:
Predictive role of bone resorption and formation markers evaluated
at baseline. J Urol. 164:1248–1253. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sartor O and de Bono JS: Metastatic
prostate cancer. N Engl J Med. 378:645–657. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Uhr JW and Pantel K: Controversies in
clinical cancer dormancy. Proc Natl Acad Sci USA. 108:12396–12400.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ruppender NS, Morrissey C, Lange PH and
Vessella RL: Dormancy in solid tumors: Implications for prostate
cancer. Cancer Metastasis Rev. 32:501–509. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aguirre-Ghiso JA: Models, mechanisms and
clinical evidence for cancer dormancy. Nat Rev Cancer. 7:834–846.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Phan TG and Croucher PI: The dormant
cancer cell life cycle. Nat Rev Cancer. 20:398–411. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aguirre-Ghiso JA, Estrada Y, Liu D and
Ossowski L: ERK(MAPK) activity as a determinant of tumor growth and
dormancy; regulation by p38(SAPK). Cancer Res. 63:1684–1695.
2003.PubMed/NCBI
|
|
13
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bragado P, Estrada Y, Parikh F, Krause S,
Capobianco C, Farina HG, Schewe DM and Aguirre-Ghiso JA: TGF-β2
dictates disseminated tumour cell fate in target organs through
TGF-beta-RIII and p38α/β signalling. Nat Cell Biol. 15:1351–1361.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Prunier C, Baker D, Ten Dijke P and Ritsma
L: TGF-β family signaling pathways in cellular dormancy. Trends
Cancer. 5:66–78. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sosa MS, Avivar-Valderas A, Bragado P, Wen
HC and Aguirre-Ghiso JA: ERK1/2 and p38α/β signaling in tumor cell
quiescence: Opportunities to control dormant residual disease. Clin
Cancer Res. 17:5850–5857. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yumoto K, Eber MR, Wang J, Cackowski FC,
Decker AM, Lee E, Nobre AR, Aguirre-Ghiso JA, Jung Y and Taichman
RS: Axl is required for TGF-β2-induced dormancy of prostate cancer
cells in the bone marrow. Sci Rep. 6:365202016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kobayashi A, Okuda H, Xing F, Pandey PR,
Watabe M, Hirota S, Pai SK, Liu W, Fukuda K, Chambers C, et al:
Bone morphogenetic protein 7 in dormancy and metastasis of prostate
cancer stem-like cells in bone. J Exp Med. 208:2641–2655. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Le Page-Degivry MT, Bidard JN, Rouvier E,
Bulard C and Lazdunski M: Presence of abscisic acid, a
phytohormone, in the mammalian brain. Proc Natl Acad Sci USA.
83:1155–118. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bruzzone S, Ameri P, Briatore L, Mannino
E, Basile G, Andraghetti G, Grozio A, Magnone M, Guida L, Scarfì S,
et al: The plant hormone abscisic acid increases in human plasma
after hyperglycemia and stimulates glucose consumption by
adipocytes and myoblasts. FASEB J. 26:1251–1260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bruzzone S, Bodrato N, Usai C, Guida L,
Moreschi I, Nano R, Antonioli B, Fruscione F, Magnone M, Scarfì S,
et al: Abscisic acid is an endogenous stimulator of insulin release
from human pancreatic islets with cyclic ADP ribose as second
messenger. J Biol Chem. 283:32188–32197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bruzzone S, Basile G, Mannino E, Sturla L,
Magnone M, Grozio A, Salis A, Fresia C, Vigliarolo T, Guida L, et
al: Autocrine abscisic acid mediates the UV-B-induced inflammatory
response in human granulocytes and keratinocytes. J Cell Physiol.
227:2502–2510. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bruzzone S, Moreschi I, Usai C, Guida L,
Damonte G, Salis A, Scarfì S, Millo E, De Flora A and Zocchi E:
Abscisic acid is an endogenous cytokine in human granulocytes with
cyclic ADP-ribose as second messenger. Proc Natl Acad Sci USA.
104:5759–5764. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Magnone M, Bruzzone S, Guida L, Damonte G,
Millo E, Scarfì S, Usai C, Sturla L, Palombo D, De Flora A and
Zocchi E: Abscisic acid released by human monocytes activates
monocytes and vascular smooth muscle cell responses involved in
atherogenesis. J Biol Chem. 284:17808–17818. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Magnone M, Sturla L, Jacchetti E, Scarfì
S, Bruzzone S, Usai C, Guida L, Salis A, Damonte G, De Flora A and
Zocchi E: Autocrine abscisic acid plays a key role in
quartz-induced macrophage activation. FASEB J. 26:1261–1271. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Scarfi S, Ferraris C, Fruscione F, Fresia
C, Guida L, Bruzzone S, Usai C, Parodi A, Millo E, Salis A, et al:
Cyclic ADP-ribose-mediated expansion and stimulation of human
mesenchymal stem cells by the plant hormone abscisic acid. Stem
Cells. 26:2855–2864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li HH, Hao RL, Wu SS, Guo PC, Chen CJ, Pan
LP and Ni H: Occurrence, function and potential medicinal
applications of the phytohormone abscisic acid in animals and
humans. Biochem Pharmacol. 82:701–712. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sakthivel P, Sharma N, Klahn P, Gereke M
and Bruder D: Abscisic Acid: A phytohormone and mammalian cytokine
as novel pharmacon with potential for future development into
clinical applications. Curr Med Chem. 23:1549–1570. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fresia C, Vigliarolo T, Guida L, Booz V,
Bruzzone S, Sturla L, Di Bona M, Pesce M, Usai C, De Flora A and
Zocchi E: G-protein coupling and nuclear translocation of the human
abscisic acid receptor LANCL2. Sci Rep. 6:266582016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bassaganya-Riera J, Guri AJ, Lu P, Climent
M, Carbo A, Sobral BW, Horne WT, Lewis SN, Bevan DR and Hontecillas
R: Abscisic acid regulates inflammation via ligand-binding
domain-independent activation of peroxisome proliferator-activated
receptor gamma. J Biol Chem. 286:2504–2516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Leber A, Hontecillas R, Zoccoli-Rodriguez
V and Bassaganya-Riera J: Activation of LANCL2 by BT-11 Ameliorates
IBD by supporting regulatory T cell stability through
immunometabolic mechanisms. Inflamm Bowel Dis. 24:1978–1991. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Koeffler HP: Peroxisome
proliferator-activated receptor gamma and cancers. Clin Cancer Res.
9:1–9. 2003.PubMed/NCBI
|
|
34
|
Hisatake JI, Ikezoe T, Carey M, Holden S,
Tomoyasu S and Koeffler HP: Down-Regulation of prostate-specific
antigen expression by ligands for peroxisome proliferator-activated
receptor gamma in human prostate cancer. Cancer Res. 60:5494–5498.
2000.PubMed/NCBI
|
|
35
|
Sikka S, Chen L, Sethi G and Kumar AP:
Targeting PPARγ signaling cascade for the prevention and treatment
of prostate cancer. PPAR Res. 2012:9680402012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fizazi K and Navone NM: Preclinical models
of prostate cancer. Bull Cancer. 92:129–141. 2005.(In French).
PubMed/NCBI
|
|
37
|
Rosol TJ, Tannehill-Gregg SH, LeRoy BE,
Mandl S and Contag CH: Animal models of bone metastasis. Cancer. 97
(Suppl 3):S748–S57. 2003. View Article : Google Scholar
|
|
38
|
Chung LW, Kao C, Sikes RA and Zhau HE:
Human prostate cancer progression models and therapeutic
intervention. Hinyokika Kiyo. 43:815–820. 1997.PubMed/NCBI
|
|
39
|
Wang Y, Herroon MK, Zielske SP, Ellis L,
Podgorski I, Taichman RS and Cackowski FC: Use of FVB Myc-CaP cells
as an immune competent, androgen receptor positive, mouse model of
prostate cancer bone metastasis. J Bone Oncol. 30:1003862021.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Westbrook TF, Martin ES, Schlabach MR,
Leng Y, Liang AC, Feng B, Zhao JJ, Roberts TM, Mandel G, Hannon GJ,
et al: A genetic screen for candidate tumor suppressors identifies
REST. Cell. 121:837–848. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lin L, Chamberlain L, Pak ML, Nagarajan A,
Gupta R, Zhu LJ, Wright CM, Fong KM, Wajapeyee N and Green MR: A
large-scale RNAi-based mouse tumorigenesis screen identifies new
lung cancer tumor suppressors that repress FGFR signaling. Cancer
Discov. 4:1168–1181. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oktem G, Bilir A, Uslu R, Inan SV, Demiray
SB, Atmaca H, Ayla S, Sercan O and Uysal A: Expression profiling of
stem cell signaling alters with spheroid formation in
CD133high/CD44high prostate cancer stem cells. Oncol Lett.
7:2103–2109. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jung Y, Cackowski FC, Yumoto K, Decker AM,
Wang Y, Hotchkin M, Lee E, Buttitta L and Taichman RS: Abscisic
acid regulates dormancy of prostate cancer disseminated tumor cells
in the bone marrow. Neoplasia. 23:102–111. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sharma S, Xing F, Liu Y, Wu K, Said N,
Pochampally R, Shiozawa Y, Lin HK, Balaji KC and Watabe K: Secreted
protein acidic and rich in cysteine (SPARC) mediates metastatic
dormancy of prostate cancer in bone. J Biol Chem. 291:19351–19363.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rebello RJ, Oing C, Knudsen KE, Loeb S,
Johnson DC, Reiter RE, Gillessen S, Van der Kwast T and Bristow RG:
Prostate cancer. Nat Rev Dis Primers. 7:92021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Roudier MP, Corey E, True LD, Hiagno CS,
Ott SM and Vessell RL: Histological, immunophenotypic and
histomorphometric characterization of prostate cancer bone
metastases. Cancer Treat Res. 118:311–339. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mehra R, Kumar-Sinha C, Shankar S, Lonigro
RJ, Jing X, Philips NE, Siddiqui J, Han B, Cao X, Smith DC, et al:
Characterization of bone metastases from rapid autopsies of
prostate cancer patients. Clin Cancer Res. 17:3924–3932. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Viale PH: The American Cancer Society's
facts & figures: 2020 edition. J Adv Pract Oncol. 11:135–136.
2020.PubMed/NCBI
|
|
50
|
Ku SY, Gleave ME and Beltran H: Towards
precision oncology in advanced prostate cancer. Nat Rev Urol.
16:645–654. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Patel VG and Oh WK: The evolving landscape
of immunotherapy in advanced prostate cancer. Immunotherapy.
11:903–912. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Watson PA, Arora VK and Sawyers CL:
Emerging mechanisms of resistance to androgen receptor inhibitors
in prostate cancer. Nat Rev Cancer. 15:701–711. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Takezawa D, Komatsu K and Sakata Y: ABA in
bryophytes: How a universal growth regulator in life became a plant
hormone? J Plant Res. 124:437–453. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lievens L, Pollier J, Goossens A, Beyaert
R and Staal J: Abscisic acid as pathogen effector and immune
regulator. Front Plant Sci. 8:5872017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sano N and Marion-Poll A: ABA metabolism
and homeostasis in seed dormancy and germination. Int J Mol Sci.
22:50692021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chaqour J, Lee S, Ravichandra A and
Chaqour B: Abscisic acid-an anti-angiogenic phytohormone that
modulates the phenotypical plasticity of endothelial cells and
macrophages. J Cell Sci. 131:jcs2104922018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Baliño P, Gómez-Cadenas A, López-Malo D,
Romero FJ and Muriach M: Is there a role for abscisic acid, a
proven anti-inflammatory agent, in the treatment of ischemic
retinopathies? Antioxidants (Basel). 8:1042019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sturla L, Fresia C, Guida L, Bruzzone S,
Scarfì S, Usai C, Fruscione F, Magnone M, Millo E, Basile G, et al:
LANCL2 is necessary for abscisic acid binding and signaling in
human granulocytes and in rat insulinoma cells. J Biol Chem.
284:28045–28057. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cao LQ, Shao ZL, Liang HH, Zhang DW, Yang
XW, Jiang XF and Xue P: Activation of peroxisome
proliferator-activated receptor-γ (PPARγ) inhibits hepatoma cell
growth via downregulation of SEPT2 expression. Cancer Lett.
359:127–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bolden A, Bernard L, Jones D, Akinyeke T
and Stewart LV: The PPAR gamma agonist troglitazone regulates Erk
1/2 phosphorylation via a PPARγ-Independent, MEK-dependent pathway
in human prostate cancer cells. PPAR Res. 2012:9290522012.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Cho SJ, Kook MC, Lee JH, Shin JY, Park J,
Bae YK, Choi IJ, Ryu KW and Kim YW: Peroxisome
proliferator-activated receptor γ upregulates galectin-9 and
predicts prognosis in intestinal-type gastric cancer. Int J Cancer.
136:810–820. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Grommes C, Landreth GE and Heneka MT:
Antineoplastic effects of peroxisome proliferator-activated
receptor gamma agonists. Lancet Oncol. 5:419–429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ogino S, Shima K, Baba Y, Nosho K, Irahara
N, Kure S, Chen L, Toyoda S, Kirkner GJ, Wang YL, et al: Colorectal
cancer expression of peroxisome proliferator-activated receptor
gamma (PPARG, PPARgamma) is associated with good prognosis.
Gastroenterology. 136:1242–1250. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang C, Fu M, D'Amico M, Albanese C, Zhou
JN, Brownlee M, Lisanti MP, Chatterjee VK, Lazar MA and Pestell RG:
Inhibition of cellular proliferation through IkappaB
kinase-independent and peroxisome proliferator-activated receptor
gamma-dependent repression of cyclin D1. Mol Cell Biol.
21:3057–3070. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Elnemr A, Ohta T, Iwata K, Ninomia I,
Fushida S, Nishimura G, Kitagawa H, Kayahara M, Yamamoto M, Terada
T and Miwa K: PPARgamma ligand (thiazolidinedione) induces growth
arrest and differentiation markers of human pancreatic cancer
cells. Int J Oncol. 17:1157–1164. 2000.PubMed/NCBI
|
|
66
|
Itami A, Watanabe G, Shimada Y, Hashimoto
Y, Kawamura J, Kato M, Hosotani R and Imamura M: Ligands for
peroxisome proliferator-activated receptor gamma inhibit growth of
pancreatic cancers both in vitro and in vivo. Int J Cancer.
94:370–376. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jabbar ZR and Sahib HB: The effects of
abscisic acid on angiogenesis in both ex vivo and in vivo assays.
Asian Pac J Cancer Prev. 23:4193–4203. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhao HF, Zhou XM, Wang J, Chen FF, Wu CP,
Diao PY, Cai LR, Chen L, Xu YW, Liu J, et al: Identification of
prognostic values defined by copy number variation, mRNA and
protein expression of LANCL2 and EGFR in glioblastoma patients. J
Transl Med. 19:3722021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Yousefnia S, Momenzadeh S, Seyed Forootan
F, Ghaedi K and Nasr Esfahani MH: The influence of peroxisome
proliferator-activated receptor γ (PPARγ) ligands on cancer cell
tumorigenicity. Gene. 649:14–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Spinelli S, Begani G, Guida L, Magnone M,
Galante D, D'Arrigo C, Scotti C, Iamele L, De Jonge H, Zocchi E and
Sturla L: LANCL1 binds abscisic acid and stimulates glucose
transport and mitochondrial respiration in muscle cells via the
AMPK/PGC-1α/Sirt1 pathway. Mol Metab. 53:1012632021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Biagioni B, Tomei L, Valleriani C,
Liccioli G, Barni S, Sarti L, Citera F, Giovannini M and Mori F:
Allergy to Gibberellin-Regulated Proteins (Peamaclein) in Children.
Int Arch Allergy Immunol. 182:1194–1199. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Nakagawa M, Hanada M, Inomata N and Amano
H: A case of a gibberellin-regulated protein-positive patient
allergic to various fruits. Eur J Dermatol. 31:88–90. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Inuo C, Okazaki F, Shiraki R, Tanaka Y,
Momma K, Kondo Y and Narita H: Generalized allergic reaction in
response to exercise due to strawberry gibberellin-regulated
protein: a case report. Allergy Asthma Clin Immunol. 18:492022.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mo J, Kang M, Ye JX, Chen JB, Zhang HB and
Qing C: Gibberellin derivative GA-13315 sensitizes
multidrug-resistant cancer cells by antagonizing ABCB1 while
agonizes ABCC1. Cancer Chemother Pharmacol. 78:51–61. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Egbewande FA, Sadowski MC, Levrier C,
Tousignant KD, White JM, Coster MJ, Nelson CC and Davis RA:
Identification of gibberellic acid derivatives that deregulate
cholesterol metabolism in prostate cancer cells. J Nat Prod.
81:838–845. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Mukherjee A, Gaurav AK, Singh S, Yadav S,
Bhowmick S, Abeysinghe S and Verma JP: The bioactive potential of
phytohormones: A review. Biotechnol Rep (Amst). 35:e007482022.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Shuai HW, Meng YJ, Luo XF, Chen F, Qi Y,
Yang WY and Shu K: The roles of auxin in seed dormancy and
germination. Yi Chuan. 38:314–322. 2016.PubMed/NCBI
|
|
78
|
Sosa MS, Parikh F, Maia AG, Estrada Y,
Bosch A, Bragado P, Ekpin E, George A, Zheng Y, Lam HM, et al:
NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven
quiescence programmes. Nat Commun. 6:61702015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chambard JC, Lefloch R, Pouysségur J and
Lenormand P: ERK implication in cell cycle regulation. Biochim
Biophys Acta. 1773:1299–1310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yu C, Shiozawa Y, Taichman RS, McCauley
LK, Pienta K and Keller E: Prostate cancer and parasitism of the
bone hematopoietic stem cell niche. Crit Rev Eukaryot Gene Expr.
22:131–148. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Morrissey C, Vessella RL, Lange PH and Lam
HM: The biology and clinical implications of prostate cancer
dormancy and metastasis. J Mol Med (Berl). 94:259–265. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Cackowski FC and Taichman RS: Minimal
residual disease in prostate cancer. Adv Exp Med Biol. 1100:47–53.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Shiozawa Y, Pedersen EA, Patel LR, Ziegler
AM, Havens AM, Jung Y, Wang J, Zalucha S, Loberg RD, Pienta KJ and
Taichman RS: GAS6/AXL axis regulates prostate cancer invasion,
proliferation, and survival in the bone marrow niche. Neoplasia.
12:116–127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Jung Y, Shiozawa Y, Wang J, McGregor N,
Dai J, Park SI, Berry JE, Havens AM, Joseph J, Kim JK, et al:
Prevalence of prostate cancer metastases after intravenous
inoculation provides clues into the molecular basis of dormancy in
the bone marrow microenvironment. Neoplasia. 14:429–439. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Jung Y, Decker AM, Wang J, Lee E, Kana LA,
Yumoto K, Cackowski FC, Rhee J, Carmeliet P, Buttitta L, et al:
Endogenous GAS6 and Mer receptor signaling regulate prostate cancer
stem cells in bone marrow. Oncotarget. 7:25698–25711. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Cackowski FC and Taichman RS: Parallels
between hematopoietic stem cell and prostate cancer disseminated
tumor cell regulation. Bone. 119:82–86. 2019. View Article : Google Scholar : PubMed/NCBI
|