Introduction
Lung cancer is one of the most common malignant
tumor types worldwide, with increasing incidence and mortality
rates (1). Despite the development
of improved therapy modalities in the past two decades, the
five-year survival rate has remained <15% (2). Several studies have suggested that
conventional therapies may have reached a therapeutic plateau.
Therefore, the current challenge is to investigate new therapeutic
agents to treat lung cancer and other malignancies.
Previously, there has been growing interest in the
use of natural products as a new source of anticancer drugs
(3,4). Cucurbitacins are compounds originally
isolated from Cucurbitaceae plants (5). They are a group of diverse
triterpenoid molecules with a number of biological properties,
including cytotoxic, antitumor, hepatoprotective,
anti-inflammatory, antimicrobial, antihelminthic and cardiovascular
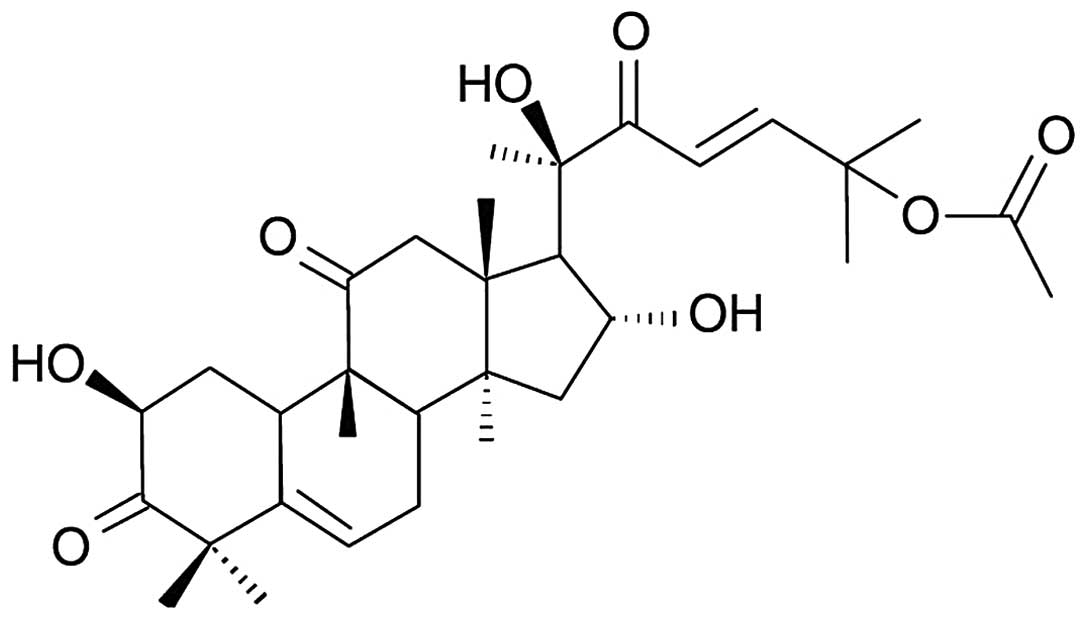
activities (6,7). Cucurbitacin B (CuB) (Fig. 1) is one of the most potent and
widely used cucurbitacins (5).
Accumulated evidence demonstrated that CuB induces apoptosis and
inhibits the growth of various human cancer cell lines (5,7,8).
It was reported that numerous components of herbal
medicine may induce apoptosis in lung cancer cells through the
mitochondrial pathway (9,10). In another study, CuB treatment
increased the protein levels of caspase-9 in the pancreatic cancer
cell line Panc-1 (11). However,
whether CuB is able to cause apoptosis of lung cancer cells in a
mitochondria-dependent manner remains elusive.
In the present study, the anticancer effect of CuB
on A549 lung cancer cells was investigated. The effect of CuB on
cell proliferation, cell cycle distribution, apoptosis, caspase
activity and cytochrome c release was examined. In addition,
the possible mechanisms underlying this effect were investigated by
screening a panel of proteins relevant to cell proliferation and
apoptosis pathways.
Materials and methods
Reagents and chemicals
Highly purified CuB was purchased from the National
Institute for the Control of Pharmaceutical and Biological Products
(Beijing, China). RPMI-1640 and trypsin were purchased from
Biological Industries (Kibutz Beit Haemek, Israel). Fetal bovine
serum (FBS) and 3-(N-Morpholino)propanesulfonic acid (MOPS) buffer
were purchased from Solarbio (Beijing Solarbio Science &
Technology, Beijing, China). MTT, dimethyl sulfoxide (DMSO),
propidium iodide (PI), Hoechst 33258 and rhodamine 123 were
purchased from Sigma-Aldrich (St. Louis, MO, USA). Annexin
V-fluorescein isothiocyanate (FITC) Apoptosis kit and bicinchoninic
acid (BCA) protein assay kit were purchased from Key Gene (Nanjing,
China). Mouse monoclonal antibodies specific to phosphorylated and
total signal transducer and activator of transcription 3 (STAT3),
cytochrome c, B-cell lymphoma 2 (Bcl-2), cyclin B1 and
β-actin and the horseradish peroxidase (HRP) conjugated goat anti
mouse immunoglobulin (Ig) G secondary antibody were purchased from
Santa Cruz Biotechnology, Inc., (Santa Cruz, CA, USA). Enchanced
Chemoluminescence Plus (ECL Plus) kit was purchased from Thermo
Fisher Scientific, Inc., (Thermo Scientific Pierce, Waltham, MA,
USA). All other chemicals were obtained from Sinopharm Chemical
Reagent Shenyang Co., Ltd. (Shenyang, China).
Cell culture
The human lung cancer cell line A549 was obtained
from the China Center for Type Culture Collection (Wuhan, China).
The cells were cultured in RPMI-1640 containing 10% fetal calf
serum at 37°C in 5% CO2. The culture medium was replaced
every day. The cells for the assays were detached using a solution
of 0.25% trypsin and 0.02% EDTA.
Cell viability assay
Viability was assessed by the MTT assay.
Approximately 5×103 cells were plated per well in
96-well plates and treated with different concentrations of CuB
(0.02, 0.1, 0.5, 2.5, 12.5 and 62.5 μmol/l) for 24, 48 and 72 h,
respectively. Corresponding DMSO and culture medium were used
either as a control or empty control. For quantitation of cell
viability, 25 μl MTT solution [(2 mg/ml in phosphate-buffered
saline (PBS)] was added to each well, and the plates were incubated
for an additional 4 h at 37°C. The medium was then removed and 150
μl DMSO was added to each well to solubilize the formazan crystals
formed in viable cells. Each solution was measured
spectrophotometrically at 570 nm (OD570) using an ELISA plate
reader (Model 550; Bio-Rad, Hercules, CA, USA). At least three
independent experiments were performed.
Flow cytometry for cell cycle
analysis
The cells were seeded into six-well plates at a
concentration of 5×105/well and allowed to attach in
culture overnight, then treated with either 0.1 or 1 μmol/l CuB for
24 h and harvested. For cell cycle analysis, the cells were fixed
in 70% ice cold ethanol at 4°C overnight. The cells were then
washed with PBS, treated with RNase and stained with PI (100 μg/ml)
in the dark for 30 min at room temperature. The samples were
analyzed by a FACScan flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA). At least three independent experiments were
performed.
Flow cytometric analysis of
apoptosis
For Annexin V/PI apoptosis analysis,
5×105 cells were plated per well in six-well plates and
treated with either 0.1 or 1 μmol/l CuB for 24 h. Following
harvesting, the cells were resuspended in cold PBS and stained
using an Annexin V-FITC Apoptosis kit according to the
manufacturer’s instructions. The cells were analyzed using a
FACScan flow cytometer. At least three independent experiments were
performed.
Fluorescence microscopy
The cells (5×105) were grown on
coverslips placed into six-well plates. Following treatment with
different concentrations of CuB (0.1 μmol/l, 1 μmol/l) or an equal
concentration of DMSO for 24 h, the cells were washed twice with
cold PBS, fixed with cold methanol and acetic acid (3/1, v/v) at
4°C overnight and stained with Hoechst 33258 for 30 min in the
dark, washed again in PBS and finally mounted in mounting medium
(80% glycerol in PBS). Morphological changes were analyzed under an
E800 fluorescence microscope (Nikon, Tokyo, Japan).
Transmission electron microscopy
The cells treated with 1 μmol/l CuB for 24 h were
harvested and fixed with 3% glutaraldehyde overnight. Following
removal of the primary fixative, the cells were washed three times
with MOPS buffer, post-fixed in 1% osmium tetroxide
(OsO4), dehydrated in an ethanol series and embedded in
epoxy resin. Ultra-thin sections were double-stained with lead
citrate/uranyl acetate prior to examination using a JEM-100CX
transmission electron microscope (Japan Electron Optics Laboratory
Co., Ltd, Tokyo, Japan).
Caspase-3 and caspase-9 activity
assay
The activity of caspase-3 and caspase-9 was detected
by chromogenic substrate assay. The cells were seeded in 75
cm2-culture flasks and cultured for 24 h. The cells were
then treated with 1 μmol/l CuB for 24 h and harvested. An equal
volume of culture medium was added to the control group. The
protein was then extracted with lysis buffer and quantified with a
BCA protein assay kit. A total of 200 μg protein was prepared,
diluted to a volume of 50 μl, 50 μl of 2× reaction buffer was added
with 5 μl substrate of caspase-3 or caspase-9. A total of 50 μl
lysis buffer and 50 μl 2× reaction buffer was added to the control
group. Following incubation for 4 h at 37°C, the absorbance was
measured at 405 nm. At least three independent experiments were
performed.
Detection of the mitochondrial membrane
potential (Δψm)
The A549 cells (5×105/well) were seeded
into six-well plates and then treated with 0.1 μmol/l or 1 μmol/l
CuB for 24 h. An equal amount of culture medium was added as the
control. The cells were harvested and incubated with 10 mg/ml
rhodamine 123 for 30 min at 37°C. The cells were resuspended with
PBS and analyzed using a FACScan flow cytometer. At least three
independent experiments were performed.
Western blot analysis
The cells were seeded in culture flasks and then
incubated with 0.1 μmol/l or 1 μmol/l CuB for 24 h. An equal amount
of RPMI-1640 was added to the control group. Total extracted
protein, cytoplasmic protein or mitochondrial protein were analyzed
separately. The proteins were separated by SDS-PAGE and transferred
to polyvinylidene fluoride membranes. The membranes were blocked
with 5% non-fat milk and incubated overnight at 4°C with antibodies
against phosphorylated and total STAT3, cytochrome c, Bcl-2,
cyclin B1 and β-actin (1:1,000). Following incubation with
peroxidase-conjugated anti-mouse IgG (1:10,000) at room temperature
for 1 h, the proteins were visualized using an ECL Plus kit and
detected using a ChemiDoc-It BioImaging system (UVP Inc., Upland,
CA, USA).
Statistical analysis
Values are expressed as the mean ± standard
deviation. The statistical correlation of data was examined for
significance by analysis of variance and Student’s t-test.
P<0.05 was considered to indicate a statistically significant
difference. These analyses were performed using SPSS 13.0 software
(SPSS, Inc., Chicago, IL, USA).
Results
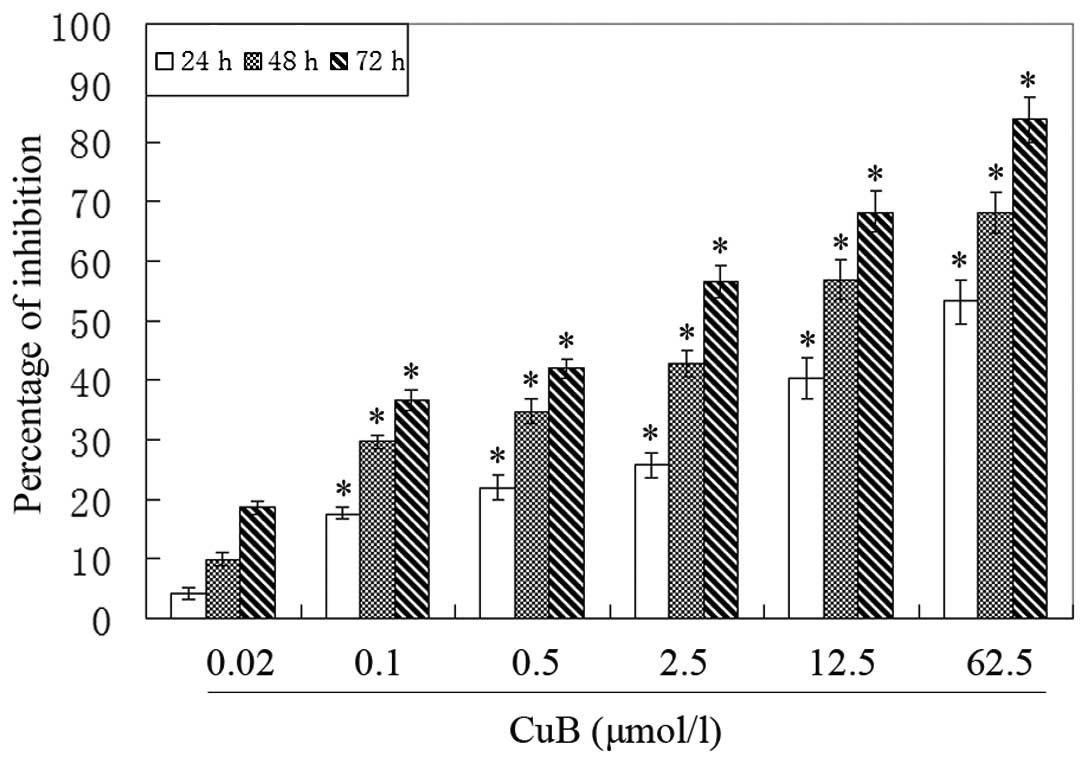
CuB inhibits A549 cell proliferation
To evaluate the effect of CuB on the proliferation
of A549 cells, an MTT assay was performed. It was observed that the
growth of A549 cells was suppressed in a dose- and time-dependent
manner (Fig. 2).
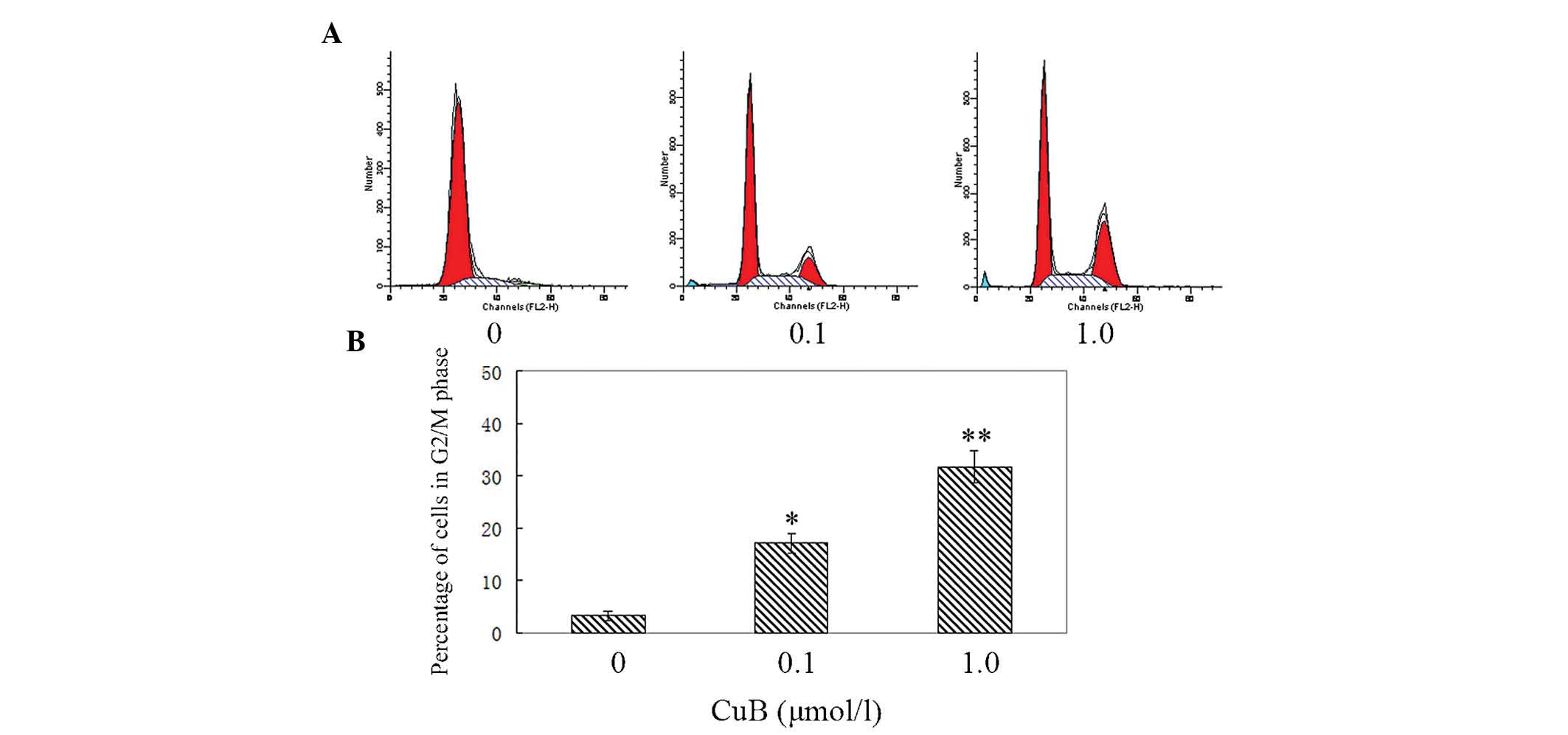
CuB induces G2/M cell cycle arrest in
A549 cells
To further investigate the inhibitory effect of CuB
on cell growth, the cell cycle distribution in A549 cells was
examined by flow cytometry. The cells in the experimental groups
were treated with 0.1 μmol/l and 1 μmol/l CuB. As shown in Fig. 3, when the dose of CuB was
increased, the percentage of cells in the G2/M phase increased
markedly. In the control group, the percentage of cells in G2/M
phase was 3.26±1.02% and in the CuB high-dose group, the G2/M
percentage was 31.78±3.69%. The results demonstrated that CuB
induced G2/M arrest significantly.
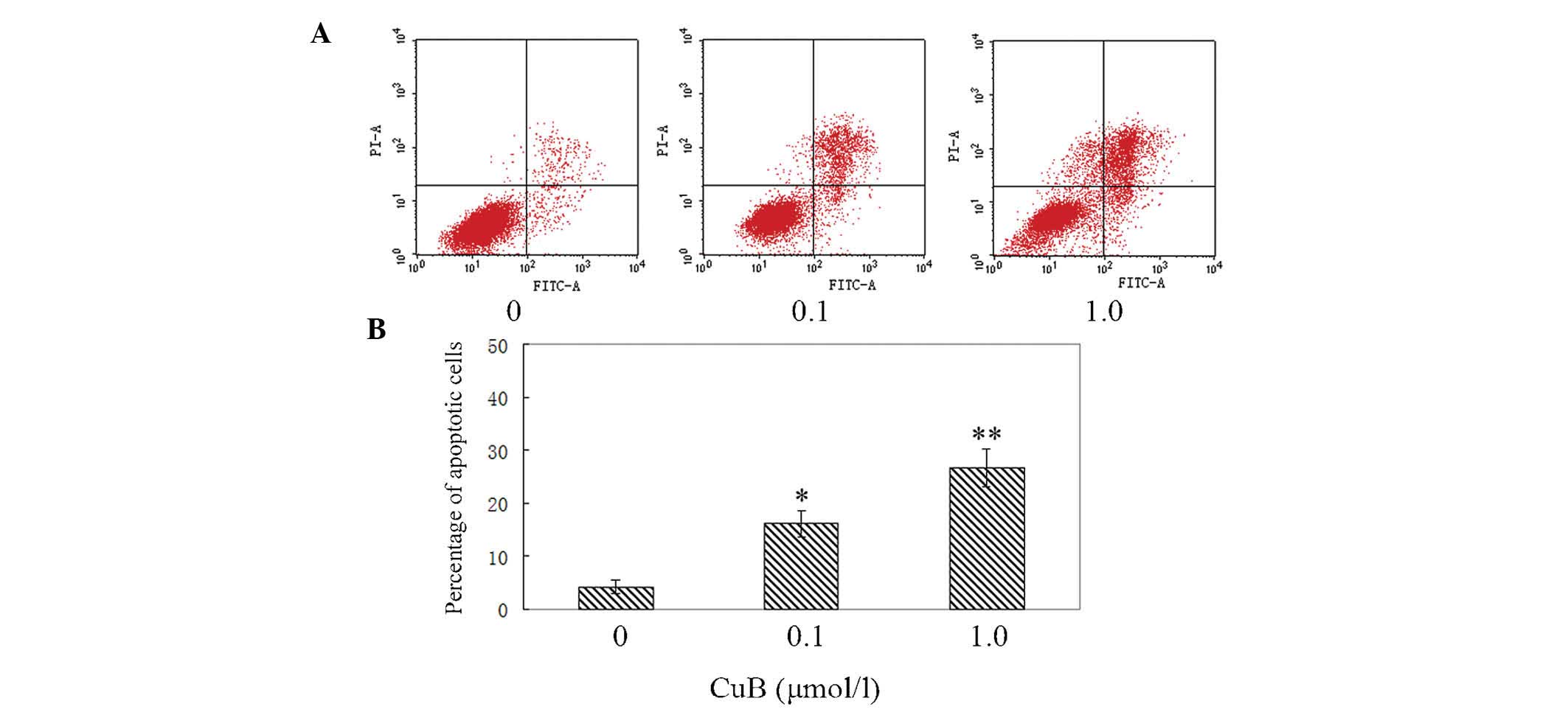
CuB induces apoptosis of A549 cells
AnnexinV/PI analysis was employed to examine the
effect of CuB on apoptosis in A549 cells. It was identified that
CuB induced apoptosis of A549 cells evidently. The percentages of
early and late apoptotic cells were significantly increased
compared with the control group. The proportion of apoptotic cells
in the treated cells was increased in a dose-dependent manner
(Fig. 4).
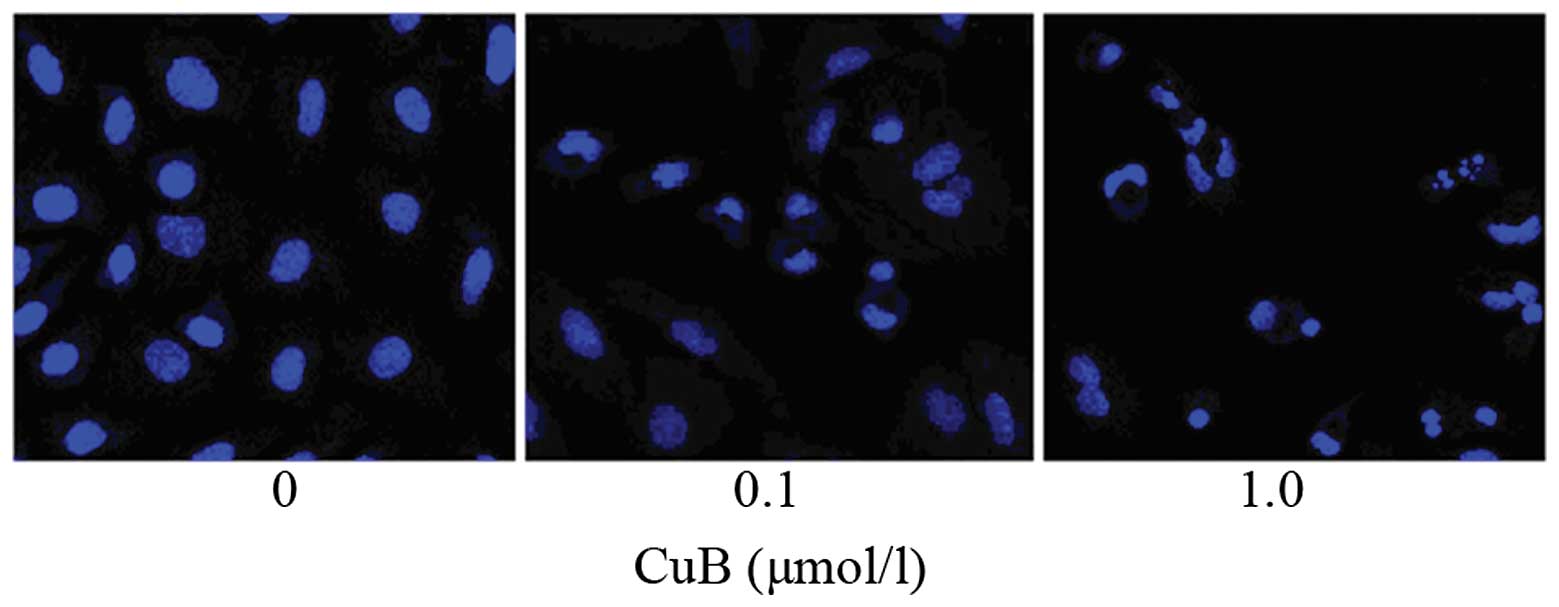
Following treatment with CuB for 24 h, the A549
cells exhibited typical morphological hallmarks of apoptosis,
including condensation of chromatin, karyopyknosis and nuclear
fragmentation, which was observed using Hoechst 33258 staining
(Fig. 5). As the dose of CuB
increased, the rate of cell apoptosis increased.
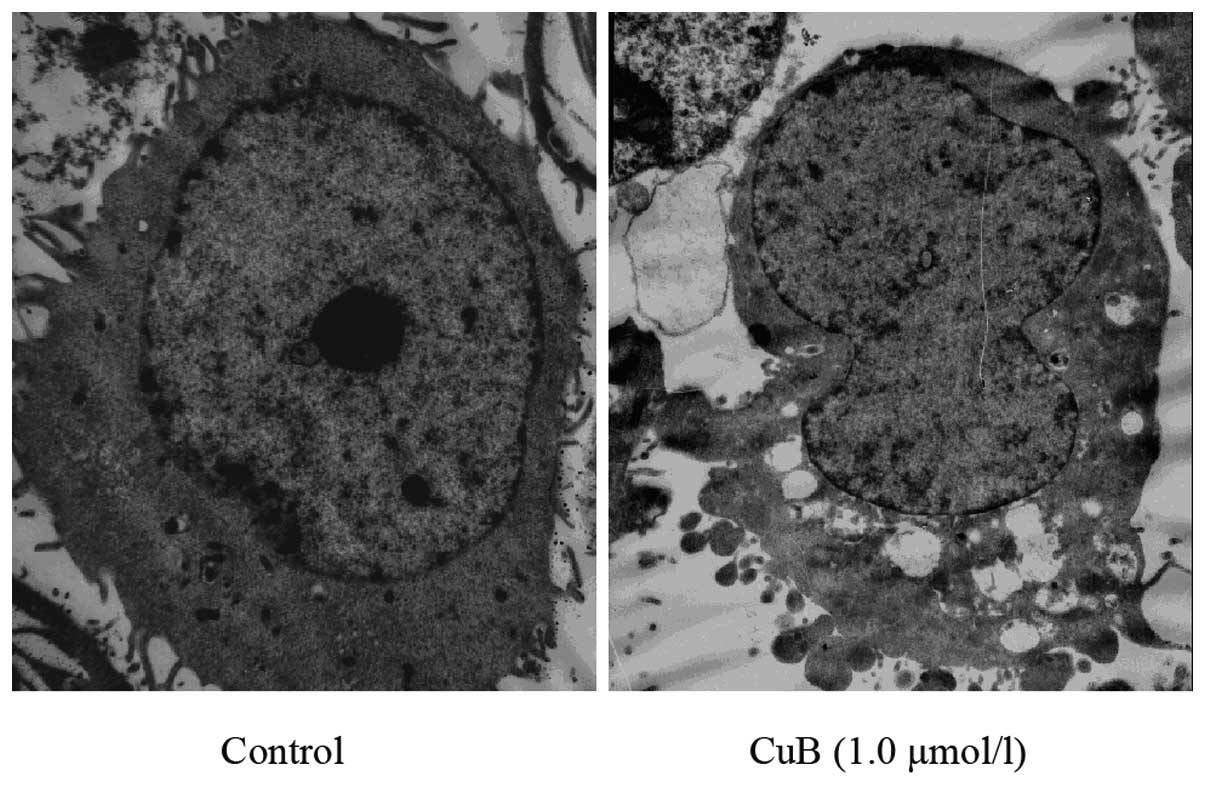
Ultrastructural changes caused by CuB in A549 cells
were also examined. Transmission electron microscopic analysis
demonstrated morphological alterations in the A549 cells treated
with CuB. The cells treated with 1.0 μmol/l CuB for 24 h lost their
pseudopodia and exhibited evidence of cell shrinking,
intracytoplasmic vacuoles, chromatin condensation and mitochondrial
swelling (Fig. 6).
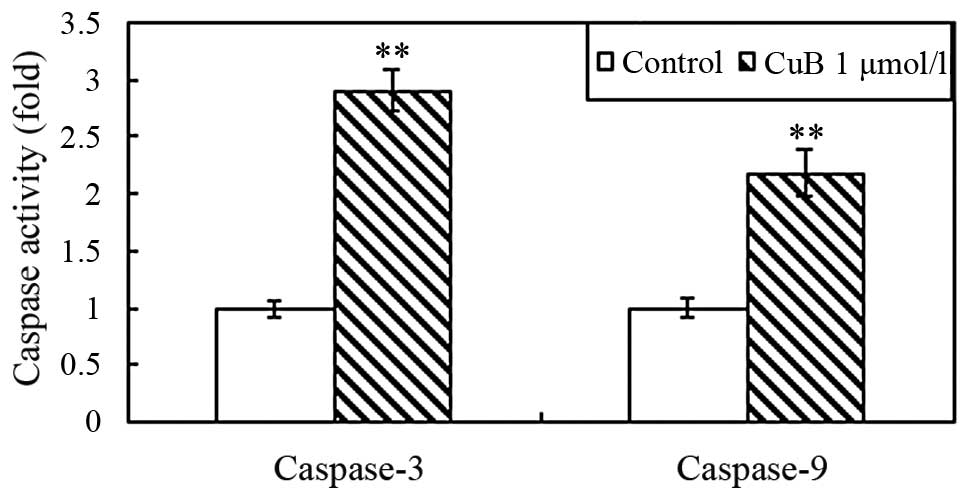
CuB induces caspase-3 and -9
activation
To further investigate the effect of CuB on
apoptosis, the activation of caspase-3 and caspase-9 in A549 cells
was examined using caspase activity assay kits. The proteolytic
activity of both caspase-3 and caspase-9 was significantly
increased following treatment with CuB for 24 h (Fig. 7).
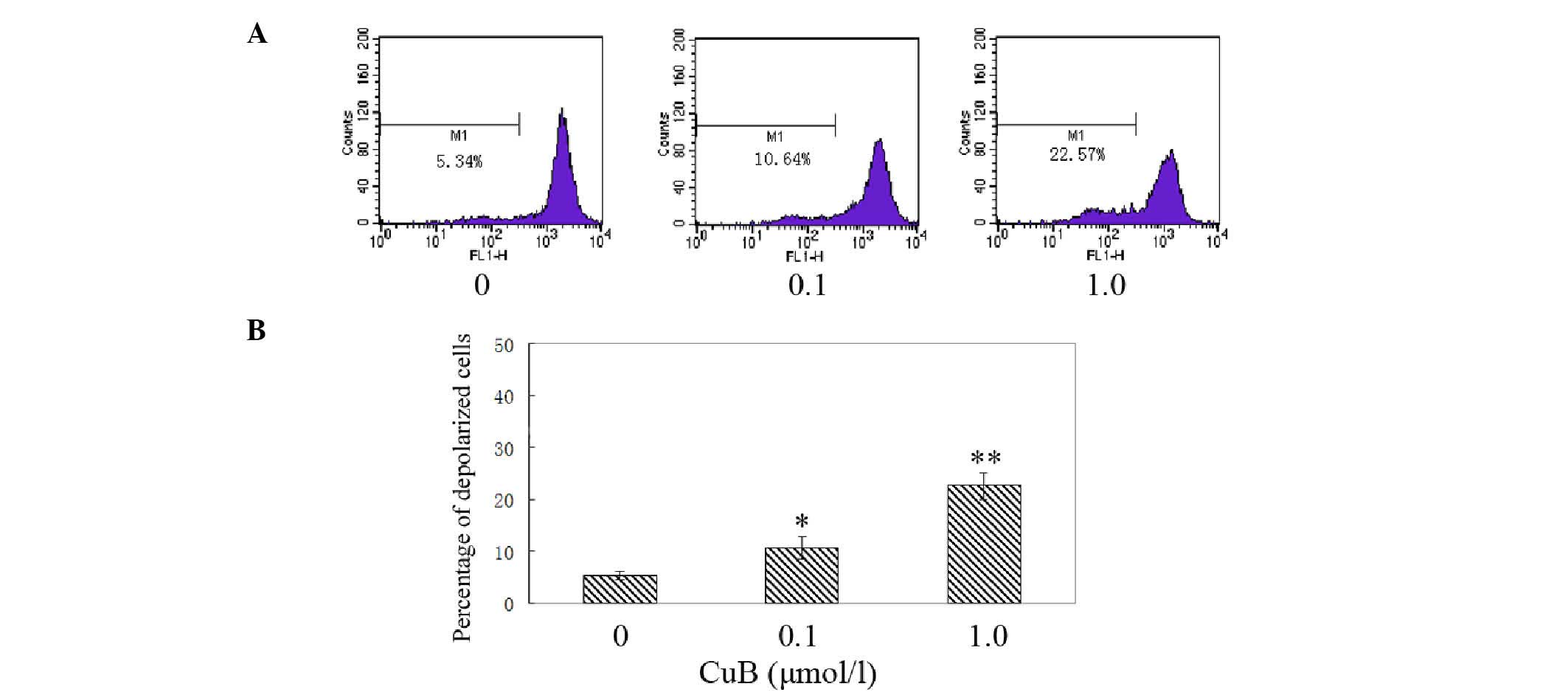
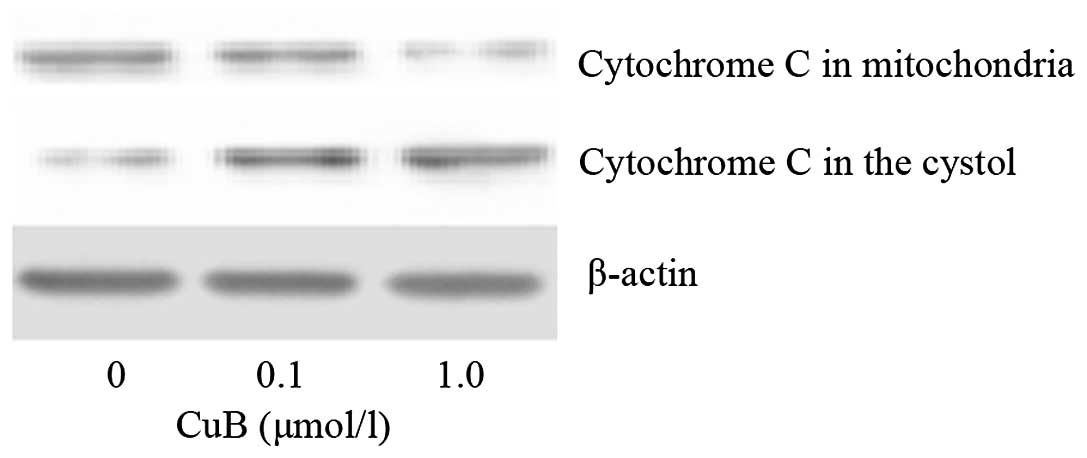
CuB induces disruption of the Δψm and
cytochrome c release
The effect of CuB on Δψm following CuB treatment was
examined by rhodamine 123 staining and flow cytometric analysis.
The integral under the Δψm curve decreased and shifted toward the
left; therefore, the proportion of depolarized cells increased in a
dose-dependent manner following CuB treatment (Fig. 8). The results indicated that
cucurbitacin B treatment induced significant disruption of the Δψm.
As cytochrome c release may be a limiting factor in
caspase-9 activation and represents a control coordinating step in
apoptosis, the ability of CuB to trigger cytochrome c
release was examined in A549 cells. As demonstrated in Fig. 9, CuB treatment induced the release
of mitochondrial cytochrome c into the cytosol.
CuB downregulates the protein expression
of phosphorylated (p)-STAT3, cyclinB1 and Bcl-2
To further examine the mechanisms of the effect of
CuB on proliferation and apoptosis in A549 cells, a panel of
proteins which are closely associated with cell growth and
apoptosis were detected. CuB suppressed p-STAT3 in a dose-dependent
manner, while it had no effect on the levels of total STAT3.
Furthermore, it was identified that CuB treatment decreased the
protein levels of cyclinB1 and Bcl-2 as well, which are downstream
targets of STAT3 and are associated with cell growth and apoptosis.
The results indicated that CuB affects proliferation and apoptosis
through inhibiting STAT3 activation and subsequently decreased the
levels of cyclin B1 and Bcl-2 protein expression (Fig. 10).
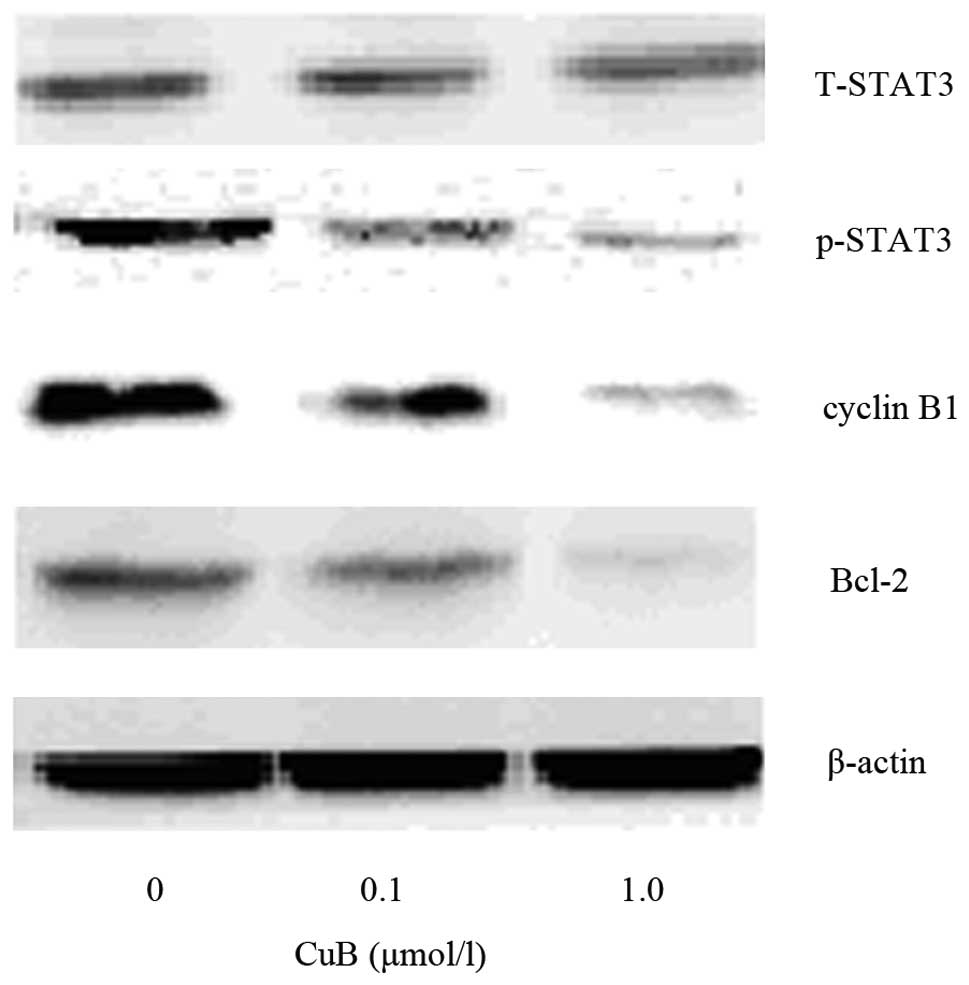 | Figure 10Effect of CuB on the expression of
cyclin B1, p-STAT3, T-STAT3 and Bcl-2 by western blot analysis.
A549 cells were treated with CuB (0, 0.1 and 1.0 μmol/l) for 24 h.
The proteins were extracted, then cyclin B1, p-STAT3, T-STAT3,
Bcl-2 and β-actin expression were analyzed by western blot. CuB,
cucurbitacin B; P/T-STAT3, phosphorylated/total signal transducer
and activator of transcription 3; Bcl-2, B-cell lymphoma 2. |
Discussion
Cucurbitacin B is a compound originally isolated
from Cucurbitaceae plants and has hepatoprotective biological
properties. Accumulating evidence has indicated that CuB inhibits
proliferation and induces apoptosis in several human cancer cell
lines (5,11–13).
In the present study, it was identified that CuB may induce
apoptosis in the lung cancer cell line A549. In addition, CuB
inhibited the proliferation rate of A549 cells in a dose- and
time-dependent manner. Further study revealed that CuB treatment
caused G2/M cell cycle arrest, elevated caspase-3 and caspase-9
activity, ΔΨm disruption and cytochrome c release.
Examination of potential target protein expression revealed that
CuB inhibited STAT3 phosphorylation, and downregulated cyclin B1
and Bcl-2 expression.
The induction of cell cycle arrest and apoptosis are
common mechanisms proposed for the cytotoxic effects of anticancer
drugs extracted from medicinal plants (14). In the present study the potential
mechanism by which CuB inhibits cell proliferation was examined.
Flow cytometry results demonstrated that CuB arrested cell cycle
progression at the G2/M check point with a decreased G0/G1 ratio,
thus inhibiting the cell proliferation rate. Accordingly, the
expression of cyclin B1 was also decreased. Cyclin B1 is a
regulatory protein involved in mitosis and may form a complex with
cyclin-dependent kinase 1 (cdk1) (15). Cyclin B1-Cdk1 is involved in the
early events of mitosis, including chromosome condensation, nuclear
envelope breakdown and spindle pole assembly. Previous reports
demonstrated that CuB was able to inhibit G2/M transition in breast
cancer cells, laryngeal cancer cells and colon adenocarcinoma
cells, which was in accordance with the results of the present
study (12,16,17).
CuB was reported to induce apoptosis in various
cancer cell lines, including laryngeal, pancreatic, colon and
hepatocellular carcinoma (5,11,13,16).
Using flow cytometry, fluorescence microscopy and transmission
electron microscopy, it was demonstrated that CuB treatment induced
lung cancer cell apoptosis in a dose-dependent manner.
Mitochondrial dysfunction has been demonstrated to participate in
the induction of apoptosis and has been suggested to be central to
the apoptotic pathway. Further analysis of mitochondrial function
revealed a disruption of ΔΨm and cytochrome c release. In
addition, the levels of caspase-3 and caspase-9 activity were also
upregulated, coupled with downregulation of the anti-apoptotic
Bcl-2 protein. The Bcl-2 protein regulates apoptosis by controlling
mitochondrial permeability (18).
Bcl-2 resides in the outer mitochondrial wall and inhibits
cytochrome c release (19).
Upon release from the mitochondria, cytochrome c binds to
apoptotic protease activating factor 1 and forms an activation
complex with caspase-9 (20,21).
Caspase-9 activates caspase-3 by proteolytic cleavage and caspase-3
then cleaves vital cellular proteins or other caspases (22). Based on the present results, CuB
may activate the apoptosis cascade through initiating cytochrome
c release and downregulating Bcl-2 protein expression.
Previous studies demonstrated that CuB suppressed
the activation of STAT3, regulated STAT3 downstream genes and
consequently inhibited tumor growth in several types of cancer
(11,13). Accordingly, a decrease of STAT3
phosphorylation following CuB treatment was identified. The STAT
family proteins have been demonstrated to have important roles in
tumorigenesis (23–25). Recent in vitro and in
vivo studies have revealed that several strategies to target
STAT3 signaling have been proposed as cancer therapies (26,27).
In addition, Bcl-2 was a downstream target gene of STAT3 signaling.
The results indicated that CuB may downregulate Bcl-2 and induce
apoptosis through inhibition of the STAT3 pathway.
In conclusion, the present study revealed that CuB
inhibited proliferation in lung cancer cells, with cell cycle
inhibition and cyclin B1 downregulation. CuB also induced lung
cancer cell apoptosis through cytochrome c release, Bcl-2
downregulation and STAT3 pathway inhibition. CuB may serve as a
potentially useful therapeutic strategy for patients with lung
cancer.
Acknowledgements
The study was supported by Outstanding Scientific
Fund of Shengjing Hospital (grant no. 201205).
References
|
1
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Schiller JH, Harrington D, Belani CP, et
al: Comparison of four chemotherapy regimens for advanced
non-small-cell lung cancer. N Engl J Med. 346:92–98. 2002.
View Article : Google Scholar
|
|
3
|
Zhang Y, Zhang GB, Xu XM, et al:
Suppression of growth of A549 lung cancer cells by waltonitone and
its mechanisms of action. Oncol Rep. 28:1029–1035. 2012.PubMed/NCBI
|
|
4
|
Reyes-Zurita FJ, Rufino-Palomares EE,
Lupiañez JA and Cascante M: Maslinic acid, a natural triterpene
from Olea europaea L., induces apoptosis in HT29 human
colon-cancer cells via the mitochondrial apoptotic pathway. Cancer
Lett. 273:44–54. 2009.PubMed/NCBI
|
|
5
|
Liu T, Zhang M, Zhang H, et al: Combined
antitumor activity of cucurbitacin B and docetaxel in laryngeal
cancer. Eur J Pharmacol. 587:78–84. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jayaprakasam B, Seeram NP and Nair MG:
Anticancer and antiinflammatory activities of cucurbitacins from
Cucurbita andreana. Cancer Lett. 189:11–16. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kongtun S, Jiratchariyakul W, Kummalue T,
et al: Cytotoxic properties of root extract and fruit juice of
Trichosanthes cucumerina. Planta Med. 75:839–842. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang M, Zhang H, Sun C, et al: Targeted
constitutive activation of signal transducer and activator of
transcription 3 in human hepatocellular carcinoma cells by
cucurbitacin B. Cancer Chemother Pharmacol. 63:635–642. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsu HF, Houng JY, Kuo CF, Tsao N and Wu
YC: Glossogin, a novel phenylpropanoid from Glossogyne
tenuifolia, induced apoptosis in A549 lung cancer cells. Food
Chem Toxicol. 46:3785–3791. 2008.PubMed/NCBI
|
|
10
|
Magesh V, Lee JC, Ahn KS, et al: Ocimum
sanctum induces apoptosis in A549 lung cancer cells and
suppresses the in vivo growth of Lewis lung carcinoma cells.
Phytother Res. 23:1385–1391. 2009. View
Article : Google Scholar
|
|
11
|
Thoennissen NH, Iwanski GB, Doan NB, et
al: Cucurbitacin B induces apoptosis by inhibition of the JAK/STAT
pathway and potentiates antiproliferative effects of gemcitabine on
pancreatic cancer cells. Cancer Res. 69:5876–5884. 2009. View Article : Google Scholar
|
|
12
|
Liu T, Zhang M, Zhang H, Sun C and Deng Y:
Inhibitory effects of cucurbitacin B on laryngeal squamous cell
carcinoma. Eur Arch Otorhinolaryngol. 265:1225–1232. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chan KT, Meng FY, Li Q, et al:
Cucurbitacin B induces apoptosis and S phase cell cycle arrest in
BEL-7402 human hepatocellular carcinoma cells and is effective via
oral administration. Cancer Lett. 294:118–124. 2010. View Article : Google Scholar
|
|
14
|
Xu X, Zhang Y, Qu D, Jiang T and Li S:
Osthole induces G2/M arrest and apoptosis in lung cancer A549 cells
by modulating PI3K/Akt pathway. J Exp Clin Cancer Res. 30:332011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jackman M, Lindon C, Nigg EA and Pines J:
Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat
Cell Biol. 5:143–148. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yasuda S, Yogosawa S, Izutani Y, et al:
Cucurbitacin B induces G2 arrest and apoptosis via a reactive
oxygen species-dependent mechanism in human colon adenocarcinoma
SW480 cells. Mol Nutr Food Res. 54:559–565. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang L, Wu S, Zhang Q, Liu F and Wu P:
23,24-Dihydrocucurbitacin B induces G2/M cell-cycle arrest and
mitochondria-dependent apoptosis in human breast cancer cells
(Bcap37). Cancer Lett. 256:267–278. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Braun F, de Carne Trecesson S,
Bertin-Ciftci J and Juin P: Protect and serve: Bcl-2 proteins as
guardians and rulers of cancer cell survival. Cell Cycle.
12:2937–2947. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang J, Liu X, Bhalla K, et al: Prevention
of apoptosis by Bcl-2: release of cytochrome C from mitochondria
blocked. Science. 275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu Y, Benedict MA, Ding L and Nuñez G:
Role of cytochrome C and dATP/ATP hydrolysis in Apaf-1-mediated
caspase-9 activation and apoptosis. EMBO J. 18:3586–3595. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Malladi S, Challa-Malladi M, Fearnhead HO
and Bratton SB: The Apaf-1*procaspase-9 apoptosome
complex functions as a proteolytic-based molecular timer. EMBO J.
28:1916–1925. 2009.
|
|
22
|
Inoue S, Browne G, Melino G and Cohen GM:
Ordering of caspases in cells undergoing apoptosis by the intrinsic
pathway. Cell Death Differ. 16:1053–1061. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Niu G, Wright KL, Huang M, et al:
Constitutive Stat3 activity up-regulates VEGF expression and tumor
angiogenesis. Oncogene. 21:2000–2008. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Diaz N, Minton S, Cox C, et al: Activation
of stat3 in primary tumors from high-risk breast cancer patients is
associated with elevated levels of activated SRC and survivin
expression. Clin Cancer Res. 12:20–28. 2006. View Article : Google Scholar
|
|
25
|
Yeh HH, Chang WT, Lu KC, et al:
Upregulation of tissue factor by activated Stat3 contributes to
malignant pleural effusion generation via enhancing tumor
metastasis and vascular permeability in lung adenocarcinoma. PLoS
One. 8:e752872013. View Article : Google Scholar
|
|
26
|
Chan KS, Sano S, Kiguchi K, et al:
Disruption of Stat3 reveals a critical role in both the initiation
and the promotion stages of epithelial carcinogenesis. J Clin
Invest. 114:720–728. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu H and Jove R: The STATs of cancer - new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|