Introduction
The insulin-like growth factors 1 and 2 (IGFs) were
initially discovered in the mid-1950s, as mediators of growth and
developmental processes (1). IGFs
have been shown to recapitulate numerous functions of insulin,
including increasing glucose metabolism and transport, inhibiting
lipolysis, and accelerating lipid, glycogen and protein syntheses
(2). The role of IGFs in
tumorigenesis has attracted a lot of attention since the early
1990s, when it was demonstrated that a lack of insulin-like growth
factor-1 receptor (IGF-1R) conferred resistance to viral and
cellular oncogene-induced transformation in R-cells (3). Later studies showed that enhanced
IGF-1R expression is a pre-requisite for cells about to undergo
proliferation or transformation (4). In addition, amplification or
overexpression of IGF-1R has been detected in numerous human
cancers, including breast, kidney, prostate, lung and stomach
cancers (5). Therefore, targeting
IGF-1R for a potential therapeutic strategy in human cancers has
gained considerable interest.
The discovery of RNA interference (RNAi) was
initially presented in 1998 (6),
and subsequently the technology was suggested to be a novel
therapeutic strategy for treating human diseases (7). Small interfering RNA (siRNA), a class
of double-stranded RNA ranging from 20–25 base pairs in length, can
recruit the RNA-induced silencing complex to target genes with high
sequence specificity, and consequently either suppress translation
or cause mRNA degradation (8).
Human cancer is a complex consequence of the interplay between
oncogenes that promote tumor cell growth or metastasis, and tumor
suppressor genes that do the opposite. Therefore, inhibition of
oncogene expression and elevation of tumor suppressor expression,
through siRNA-based therapeutics, may be a potential therapeutic
strategy for cancer treatment. Previous studies have shown that the
in vivo delivery of siRNAs targeting survivin, androgen
receptor, enhancer of zeste homolog 2, polo-like kinase 1, or
vascular endothelial growth factor, in prostate cancer, markedly
reduced tumor cell growth and metastasis (9). The tumor suppressor gene p53 is
frequently mutated to gain oncogenic activity in >50% human
cancers, and is down-regulated in other cancers by its negative
regulators, which include transforming protein E6 of human
papilloma virus and mouse double minute 2 homolog (MDM2) (10). RNAi-based inhibition of mutant p53
or MDM2 have previously been shown to effectively repress
tumorigenesis in human cancer cells and a mouse model (11,12).
A previous study demonstrated that IGF-1R is highly
expressed in gastric cancer (GC) and is associated with tumor size,
quantity of stroma, depth of wall invasion, lymph node metastasis,
TNM (Tumor size/lymph Node/Metastasis) stages and differentiation
status (13). The present study
aimed to further investigate whether RNAi-based IGF-1R depletion
could affect the tumorigenic and metastatic activity of the BGC823
GC cell line. These findings suggested a role for IGF-1R in
carcinogenesis; however, to the best of our knowledge, the role of
IGF-1R has not been investigated in gastric cancer. Therefore, the
objective of the present study was to elucidate the role of IGF-1R
in gastric cancer.
Materials and methods
Cell culture
The BGC823 GC cell line was purchased from the
Xiangya School of Medicine Central Experiment Laboratory (Changsha,
China). The cells were cultured in Dulbecco’s modified Eagle’s
medium (DMEM; Gibco Life Technologies, Carlsbad, CA, USA),
supplemented with 10% fetal bovine serum (FBS; Gibco Life
Technologies), 100 U/ml penicillin and 0.1 mg/ml streptomycin, at
37°C in a 5% CO2 humidified atmosphere.
Antibodies
The primary antibodies against IGF-1R and β-actin,
were purchased from Abcam (Cambridge, UK), and Sigma-Aldrich (St
Louis, MO, USA), respectively. Thegoat anti-mouse Immunoglobulin G
(IgG) and goat anti-rabbit IgG secondary antibodies were purchased
from Kirkegaard & Perry Laboratories, Inc. (Gaithesburg, MD,
USA).
MTT assay
BGC823 cell suspensions were seeded
(4×104 cells/well) in a 96 well plate. Following a 16 h
culture, the cells were infected with pSUPER-IGF-IR-siRNA2 or
pSUPER-control-siRNA2 (Genechem, Shanghai, China), or treated with
phosphate-buffered saline (PBS) for 0, 24, 48 or 72 h. The cells
were supplemented with 0.5 μg/μl MTT and cultured for a further 4
h, followed by treatment with dimethyl sulfoxide for 10 min. The
absorbance of the samples was measured at 570 nm using an iMark
platereader (Bio-Rad Laboratories, Hercules, CA, USA).
Cell cycle analysis
The BGC823 cells infected with pSUPER-IGF-IR-siRNA2
or pSUPER-control-siRNA2, or treated with PBS, were fixed with 70%
ice-cold ethanol at 4°C overnight. The cells were then treated with
RNase A (Sigma-Aldrich, St. Louis, MO, USA) at 37°C for 30 min and
propidium iodide (PI; Sigma-Aldrich) at 4°C for 1 h. The DNA
content of the cells was assessed using a FACSCalibur flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and the data
were analyzed using Modfit 3.0 DNA software (BD Biosciences,
Franklin Lakes, NJ, USA).
PI staining for apoptosis analysis
The BGC823 cells treated with the indicated siRNAs,
or PBS, were incubated with 20 μg/ml RNase A and stained with 50
μg/ml PI solution. A total of 500 cells were randomly counted under
a microscope (Axio observer Z1; Zeiss Meditec, Jena, Germany). The
apoptotic rate was calculated using the following formula: Rate of
cell death = stained cells/total cells.
DNA laddering analysis
The cells infected with the siRNAs, or treated with
PBS, were treated with Tris-NaCl-EDTA buffer, containing proteinase
K, at 55°C overnight. Genomic DNA was isolated from the cells using
DNA purification kits (Life Technologies, Carlsbad, CA, USA) and
fractionated by 2% agarose gel electrophoresis.
Wound healing assay
The cells were infected with the specified siRNAs,
or treated with PBS for 48 h, prior to being seeded in a
fibronectin-coated 6 well plate and cultured until confluent. The
cells were then cultured in serum-free DMEM and a straight scratch
was made using a 200 μl pipette tip. The width of the scratch was
observed and recorded using a microscope, at 0, 6, 12, 24 and 36 h.
Cells of three independent wound areas were counted. A minimum of
three wells were counted per experiment.
Transwell cell invasion assay
Transwell chambers were sterilized by UV treatment,
and the inserts and PVDF membrane were coated with
Matrigel® matrix (Life Technologies). The cells
(1×105), in serum-free media, were plated in the upper
wells of the chambers, and media supplemented with 10% FBS was
added to the lower wells. Following a 24 h incubation, the cells on
the Matrigel® side of the chambers were removed using a
cotton swab. The inserts were fixed in methanol and stained using
0.2% crystal violet staining solution (Life Technologies).
Statistical analysis
The data are expressed as the means ± standard
deviation. Statistical differences were evaluated using an unpaired
student’s t test using SPSS version 15.0 (SPSS Inc., Chicago, IL,
USA) software. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
RNAi-mediated IGF-1R silencing inhibits
the growth of BGC823 gastric cancer cells
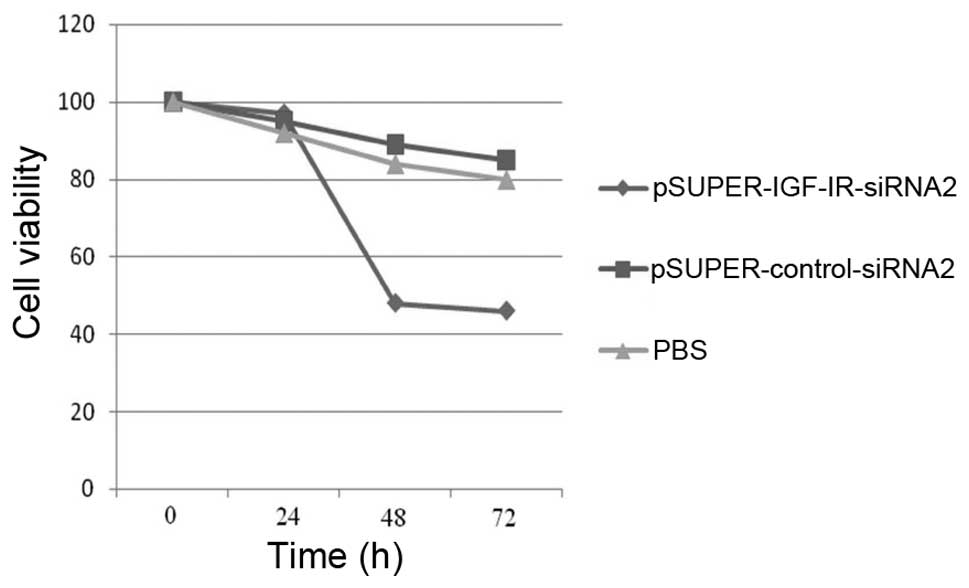
An MTT assay was performed to determine whether
silencing of IGF-1R expression influenced gastric cancer cell
growth. The cells were infected with pSUPER-IGF-IR-siRNA2 or
pSUPER-control-siRNA2, or treated with PBS, for 0, 24, 48 and 72 h.
Knockdown of IGF-1R expression for 24 h did not affect cell growth,
as compared with the control siRNA and PBS groups. However, IGF-1R
ablation for 48 to 72 h, significantly reduced cell growth by 62.15
and 59.82%, respectively (Table I
and Fig. 1). This observation
indicates that the greatest effects of IGF-1R expression knockdown
on cell growth inhibition were achieved 48 h post-infection.
 | Table ISilencing of insulin-like growth
factor-1 receptor (IGF-1R) affects BGC823 gastric cancer cell
proliferation, as determined by an MTT assay (n=3). |
Table I
Silencing of insulin-like growth
factor-1 receptor (IGF-1R) affects BGC823 gastric cancer cell
proliferation, as determined by an MTT assay (n=3).
| Inhibition (%) |
|---|
|
|
|---|
| 0 h | 24 h | 48 h | 72 h |
|---|
|
pSUPER-IGF-IR-siRNA2 | 7.62±0.43 | 8.84±0.36 | 62.15±0.98 | 59.82±0.68 |
|
pSUPER-Control-siRNA | 7.39±0.26 | 8.28±0.20 | 8.07±0.33 | 7.95±0.25 |
| PBS | 7.68±0.32 | 7.96±0.16 | 8.13±0.29 | 7.85±0.26 |
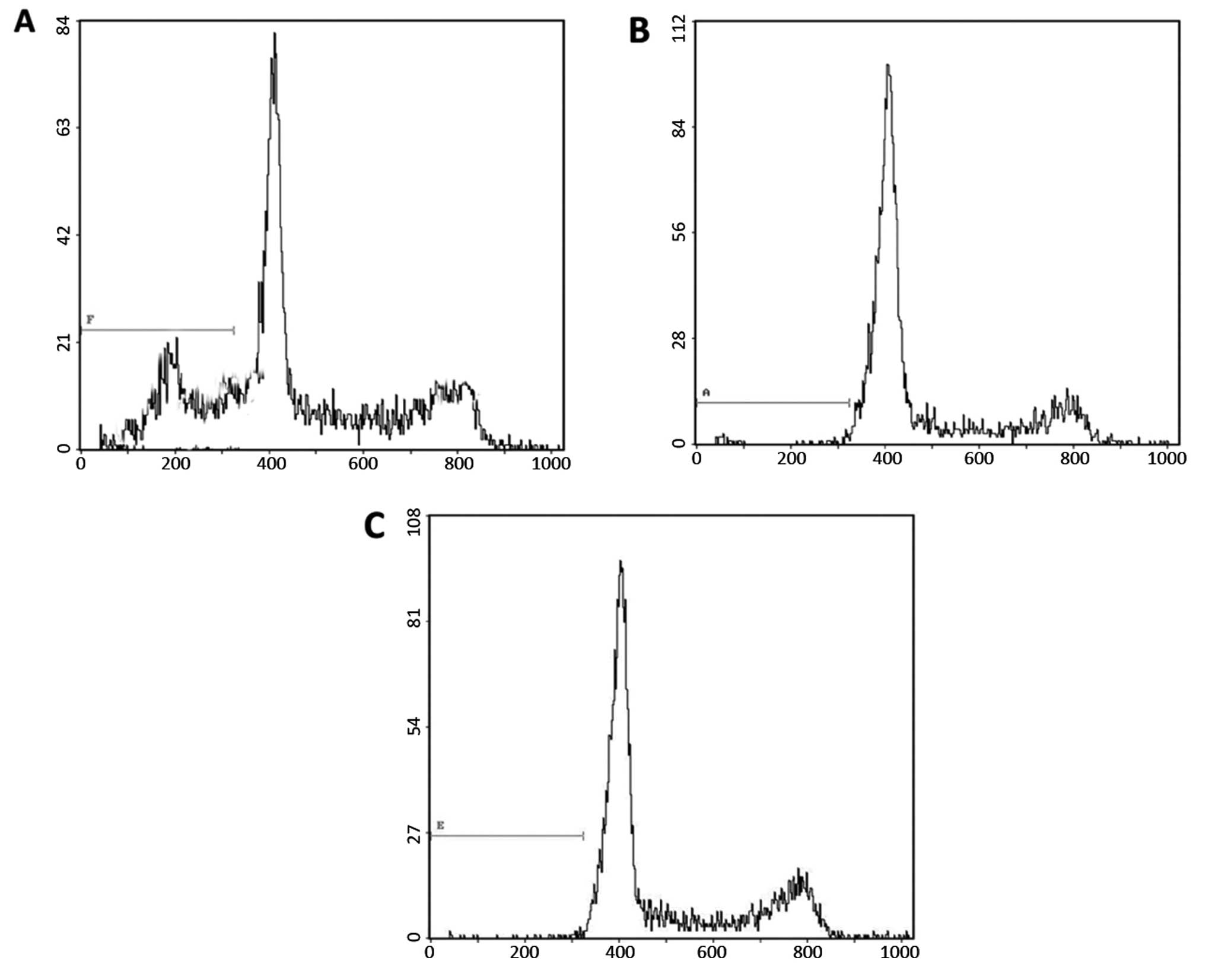
RNAi-mediated IGF-1R silencing leads to
G1 arrest in BGC823 cells
To determine whether silencing of IGF-1R expression
affected the progression of the cell cycle a flow cytometric
analysis was performed. The cells were fixed with ethanol 48 h
post-infection, stained with PI and subjected to
fluorescence-activated cell sorting. The percentages of the
IGF-1R-depleted cells in the G0/G1, S and
G2/M phases were 59.42, 37.83 and 3.71%, respectively
(Table II and Fig. 2). The control cells, either
transfected with control siRNA or treated with PBS, had a
significantly lower G0/G1 cell population and
higher S population (Table II and
Fig. 2). These data demonstrate
that knockdown of IGF-1R expression results in G1 cell
cycle arrest, and a subsequent reduction in the S phase population,
in gastric cancer cells.
| Table IISilencing of insulin-like growth
factor-1 receptor (IGF-1R) affects BGC823 gastric cancer cell cycle
progression (n=3). |
Table II
Silencing of insulin-like growth
factor-1 receptor (IGF-1R) affects BGC823 gastric cancer cell cycle
progression (n=3).
| Cell cycle phases
(%) |
|---|
|
|
|---|
|
G1/G0 | S | G2/M |
|---|
|
pSUPER-IGF-IR-siRNA2 | 59.42±4.01 | 37.83±2.13 | 3.71±0.18 |
|
pSUPER-control-siRNA | 19.72±1.61 | 50.33±4.91 | 9.09±0.62 |
| PBS | 14.33±2.15 | 55.21±3.81 | 9.64±0.71 |
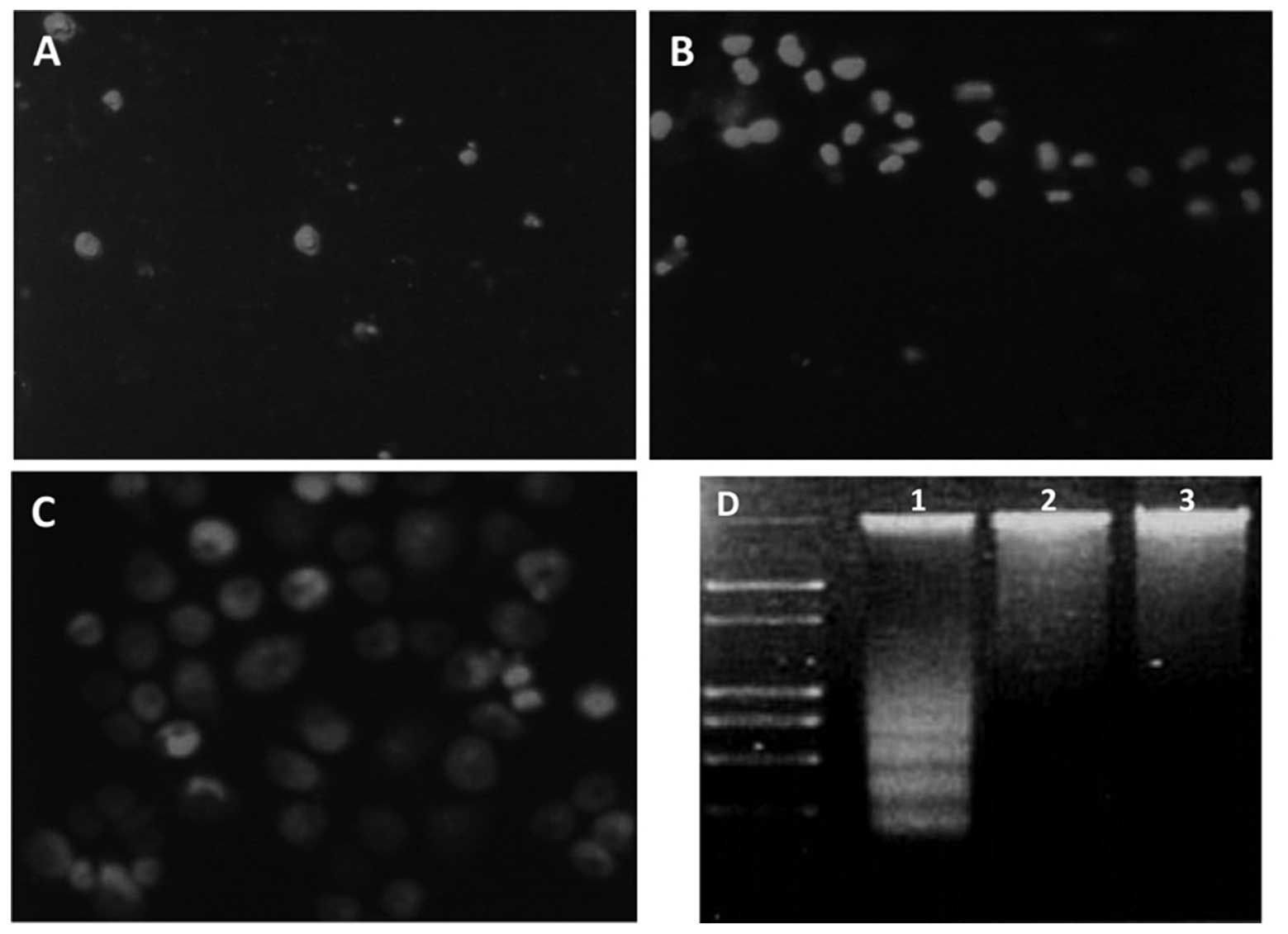
RNAi-mediated IGF-1R silencing promotes
BGC823 cell apoptosis
An accumulated sub-G1 population was
observed in response to IGF-1R expression knockdown, suggesting an
increased number of apoptotic cells (Fig. 2). To further confirm whether IGF-1R
ablation triggered apoptosis in gastric cancer cells, PI staining
and microscopic analysis were performed. Living cells with an
intact cytomembrane are resistant to PI staining; however,
apoptotic cells are easily stained. The percentages of living cells
in the IGF-1R siRNA, control siRNA and PBS groups were 8.43, 49.72
and 47.53%, respectively (Fig. 3).
These results indicate that IGF-1R ablation was capable of inducing
apoptosis. A DNA laddering analysis was also conducted and IGF-1R
expression knockdown dramatically enhanced the formation of a DNA
ladder, which could barely be seen in the control cells (Fig. 3D). These results demonstrate that
silencing of IGF-1R expression induces gastric cancer cell
apoptosis.
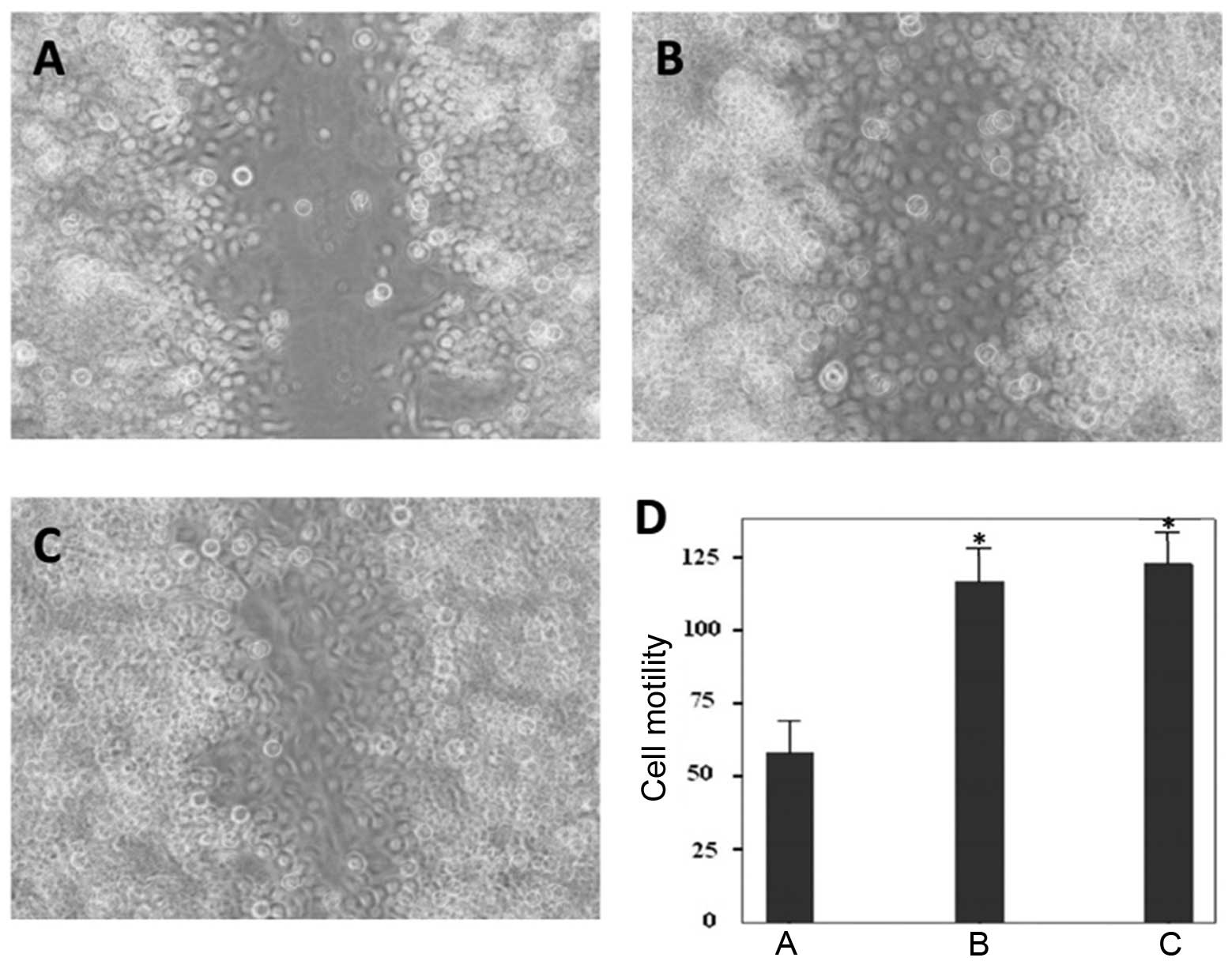
RNAi-mediated IGF-1R silencing inhibits
migration of BGC823 cells
Wound-healing assays were conducted to assess the
migratory ability of BGC823 cells in response to IGF-1R ablation.
IGF-1R ablation markedly suppressed cell motility at 36 h
post-infection. The control siRNA or PBS treated cells migrated to
heal the wound areas, whereas the IGF-1R-depleted cells were
incapable of migrating (Fig.
4A–C). Furthermore, the cells of the wound areas were counted,
and the number of cells were significantly lower in the IGF-1R
depletion group, as compared with the control groups (Fig. 4D). These results indicate that
suppression of IGF-1R expression inhibits gastric cancer cell
motility.
RNAi-mediated IGF-1R silencing suppresses
invasion of BGC823 cells
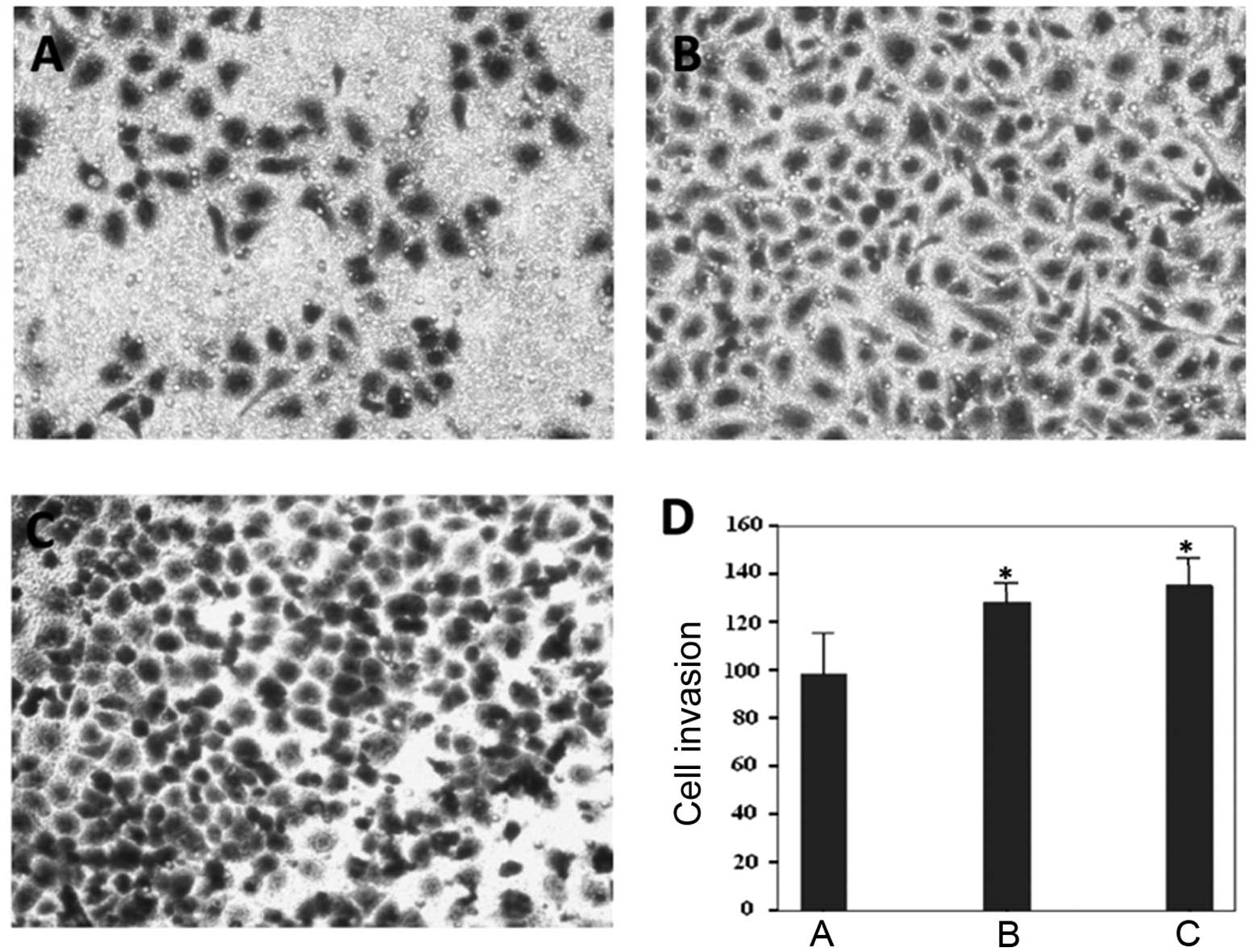
An in vitro Transwell assay was used to
determine whether IGF-1R ablation would affect gastric cancer cell
invasion (Fig. 5A–C). The average
number of invasive IGF-1R-depleted cells was 98.33 (Fig. 5D), which was significantly reduced,
as compared with the control siRNA (128.33) and PBS groups (135.33)
(Fig. 5D). These results suggest
that IGF-1R depletion may also prevent gastric cancer cell
invasion.
Discussion
The aim of the present study was to determine
whether IGF-1R may be a potential therapeutic target for gastric
cancer. RNAi technology was used to silence endogenous IGF-1R
expression, and the physiological outcomes were examined in BGC823
gastric cancer cells. Ablation of IGF-1R resulted in repressed cell
growth, accumulated G1-phase population, enhanced
apoptosis, and impaired abilities of cell motility and invasion.
These results suggested a tumor-inhibitory function of
IGF-1R-targeting siRNA; however, it remains to be elucidated
whether siRNA-mediated IGF-1R ablation is able to inhibit gastric
tumor growth in vivo. Therefore, delivery of
IGF-1R-targeting siRNA in a mouse model of gastric cancer or
RNAi-based xenografts may be a promising area of future study.
Overexpression of IGF-1R has been regarded as a
typical hallmark of numerous human cancers, and various tumor
suppressors, including p53 family proteins (14,15),
breast cancer 1 (16,17), Von Hippel-Lindau protein (18), Wilms’ tumor 1 (19,20),
have been shown to negatively regulate IGF-1R transcription
(4). These results imply that
IGF-1R may be a therapeutic target for human cancers. RNAi-based
IGF-1R repression has previously been applied to sensitize
drug-resistant cancer cells harboring mutated p53 (21). Furthermore, it has previously been
demonstrated that administering a half maximal inhibitory
concentration dose of Adriamycin®, with siRNA targeting
IGF-1R, considerably reduced cell growth in liver cancer cells
(21). However, it remains unknown
whether IGF-1R depletion may be detrimental to cell proliferation
and invasion in gastric cancer. A seminal study previously
demonstrated that IGF-1R prompts tumor growth and survival in
gastric cancer, and a dominant-negative fragment of IGF-1R may
suppress IGF-1R-induced tumorigenicity and trigger apoptosis
(22). Considering the results of
the present study, developing small molecules, including lead
compounds, short peptides or siRNAs, that target IGF-1R may be a
potential and promising strategy for gastric cancer therapy.
Mechanistically, IGF ligands, including IGF1 and
IGF2, can be either generated from remote sites and delivered
through circulation, or locally produced (23). These ligands specifically bind to
IGF-1R leading to stimulation of its intrinsic tyrosine kinase
activity, consequently eliciting intracellular signaling that
controls cell proliferation and survival (24). Two pathways are highly responsive
following the interaction of IGF1/IGF2 and IGF-1R. The
PI3K-AKT-mTOR pathway may be activated and substantially promotes
global protein synthesis, which supports enhanced cell
proliferation. In addition, IGF1/IGF2-IGF-1R binding also triggers
the RAF-MAPK cascade which is involved in numerous cellular
processes, including proliferation, differentiation, mitosis, cell
survival, and apoptosis (25).
Therefore, abrogation of IGF1/IGF2-IGF-1R signaling by RNAi-based
technology may impair both PI3K-AKT-mTOR and RAF-MAPK pathways,
thus suppressing tumor cell growth.
In conclusion, the present study is the first, to
the best of our knowledge, to demonstrate that siRNA-mediated
IGF-1R expression silencing markedly reduces the malignant
properties of gastric cancer cells. These results suggest that
targeting IGF-1R by siRNA may be an effective strategy for gastric
cancer treatment and that RNAi-based therapy for gastric cancer
requires further comprehensive exploration.
Acknowledgements
The authors of the present study would like to
acknowledge Biomedworld for the help in editing the manuscript. The
present study was supported by the Natural Science Foundation of
Central South University (no. 2012QNZT135).
References
|
1
|
Salmon WD Jr and Daughaday WH: A
hormonally controlled serum factor which stimulates sulfate
incorporation by cartilage in vitro. J Lab Clin Med. 49:825–836.
1957.
|
|
2
|
Maki RG: Small is beautiful: insulin-like
growth factors and their role in growth, development, and cancer. J
Clin Oncol. 28:4985–4995. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sell C, Rubini M, Rubin R, et al: Simian
virus 40 large tumor antigen is unable to transform mouse embryonic
fibroblasts lacking type 1 insulin-like growth factor receptor.
Proc Natl Acad Sci USA. 90:11217–11221. 1993. View Article : Google Scholar
|
|
4
|
Werner H: Tumor suppressors govern
insulin-like growth factor signaling pathways: implications in
metabolism and cancer. Oncogene. 31:2703–2714. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Seccareccia E and Brodt P: The role of the
insulin-like growth factor-I receptor in malignancy: an update.
Growth Horm IGF Res. 22:193–199. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fire A, Xu S, Montgomery MK, et al: Potent
and specific genetic interference by double-stranded RNA in
Caenorhabditis elegans. Nature. 391:806–811. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deng Y, Wang CC, Choi KW, et al:
Therapeutic potentials of gene silencing by RNA interference:
principles, challenges, and new strategies. Gene. 538:217–227.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Castel SE and Martienssen RA: RNA
interference in the nucleus: roles for small RNAs in transcription,
epigenetics and beyond. Nat Rev Genet. 14:100–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guo J, Evans JC and O’Driscoll CM:
Delivering RNAi therapeutics with non-viral technology: a promising
strategy for prostate cancer? Trends Mol Med. 19:250–261. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Levine AJ and Oren M: The first 30 years
of p53: growing ever more complex. Nat Rev Cancer. 9:749–758.
2009.PubMed/NCBI
|
|
11
|
Lane DP, Cheok CF and Lain S: p53-based
cancer therapy. Cold Spring Harb Perspect Biol.
2:a0012222010.PubMed/NCBI
|
|
12
|
Oren M and Rotter V: Mutant p53
gain-of-function in cancer. Cold Spring Harb Perspect Biol.
2:a0011072010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ge J, Chen Z, Wu S, et al: Expression
levels of insulin-like growth factor-1 and multidrug
resistance-associated protein-1 indicate poor prognosis in patients
with gastric cancer. Digestion. 80:148–158. 2009. View Article : Google Scholar
|
|
14
|
Werner H, Karnieli E, Rauscher FJ and
LeRoith D: Wild-type and mutant p53 differentially regulate
transcription of the insulin-like growth factor I receptor gene.
Proc Natl Acad Sci USA. 93:8318–8323. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nahor I, Abramovitch S, Engeland K and
Werner H: The p53-family members p63 and p73 inhibit insulin-like
growth factor-I receptor gene expression in colon cancer cells.
Growth Horm IGF Res. 15:388–396. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maor S, Yosepovich A, Papa MZ, et al:
Elevated insulin-like growth factor-I receptor (IGF-IR) levels in
primary breast tumors associated with BRCA1 mutations. Cancer Lett.
257:236–243. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maor SB, Abramovitch S, Erdos MR, Brody LC
and Werner H: BRCA1 suppresses insulin-like growth factor-I
receptor promoter activity: potential interaction between BRCA1 and
Sp1. Mol Genet Metab. 69:130–136. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yuen JS, Cockman ME, Sullivan M, et al:
The VHL tumor suppressor inhibits expression of the IGF1R and its
loss induces IGF1R upregulation in human clear cell renal
carcinoma. Oncogene. 26:6499–6508. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Werner H, Re GG, Drummond IA, et al:
Increased expression of the insulin-like growth factor I receptor
gene, IGF1R, in Wilms tumor is correlated with modulation of IGF1R
promoter activity by the WT1 Wilms tumor gene product. Proc Natl
Acad Sci USA. 90:5828–5832. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Werner H, Shen-Orr Z, Rauscher FJ III, et
al: Inhibition of cellular proliferation by the Wilms’ tumor
suppressor WT1 is associated with suppression of insulin-like
growth factor I receptor gene expression. Mol Cell Biol.
15:3516–3522. 1995.
|
|
21
|
Niu J, Xu Z, Li XN and Han Z:
siRNA-mediated type 1 insulin-like growth factor receptor silencing
induces chemosensitization of a human liver cancer cell line with
mutant P53. Cell Biol Int. 31:156–164. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Min Y, Adachi Y, Yamamoto H, et al:
Insulin-like growth factor I receptor blockade enhances
chemotherapy and radiation responses and inhibits tumour growth in
human gastric cancer xenografts. Gut. 54:591–600. 2005. View Article : Google Scholar
|
|
23
|
Tian D and Kreeger PK: Analysis of the
quantitative balance between insulin-like growth factor (IGF)-1
ligand, receptor, and binding protein levels to predict cell
sensitivity and therapeutic efficacy. BMC Syst Biol. 8:982014.
View Article : Google Scholar
|
|
24
|
Denley A, Cosgrove LJ, Booker GW, Wallace
JC and Forbes BE: Molecular interactions of the IGF system.
Cytokine Growth Factor Rev. 16:421–439. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pollak MN, Schernhammer ES and Hankinson
SE: Insulin-like growth factors and neoplasia. Nat Rev Cancer.
4:505–518. 2004. View
Article : Google Scholar : PubMed/NCBI
|