Introduction
Ovarian cancer is one of the most common types of
carcinoma in females, usually associated with a high mortality as
signs and symptoms are frequently absent (1). The exact causes of ovarian cancer
remain to be elucidated. Genetic factors have been identified as
important in certain ovarian cancer patients, such as mutations in
the BRCA1 and BRCA2 genes, which are also risk
factors for breast cancer (2).
Mutations in BRCA1 confer a high risk of ovarian cancer and
can reduce lifespan (3). In
addition, marker genes of ovarian cancer can be classified into
several types: Presentation (4,5),
recurrence (6), inheritance
(7), prognosis (8,9) and
therapy targets (10). Although a
large number of markers have been observed to be associated with
ovarian carcinoma, the incidence of and the associations and
interactions between these markers have not been extensively
investigated.
In the present study, all markers associated with
ovarian cancer were determined by analyzing the whole transcriptome
of ovarian cancer cell lines. Tumor samples were selected at two
stages (presentation and recurrence) from two patients as the focus
of the present study. RNAs were extracted from tumor cell lines and
sequenced with Illumina GAII. By aligning RNA-seq data to marker
sequences, differentially expressed markers (DEMs) were filtered
out in different stages, and the interactions between them were
assessed.
Materials and methods
Data sources and processing
A total of four ovarian carcinoma cell lines at
different tumor stages: PEO1 (first recurrence, 22 month post
chemotherapy; Patient ID, 3), PEO4 (second recurrence, 10 months
post first recurrence; Patient ID, 3), PEO23 (recurrence, 7 months
post chemotherapy; Patient ID, 2), and PEO14 (presentation, prior
to chemotherapy; Patient ID, 2) (11), were grown in vitro. Original
tissue materials were collected in a study which was approved by
the Cambridge Local Research Ethics Committee (LREC), all patients
gave written informed consent prior to participation (12). and mRNA of the cells was extracted
and then sequenced on Illumina GAII genome analyzer (Illumina Inc.,
San Diego, CA, USA). Each sample was subject to three experimental
repeats for statistical calculation. The raw sequence data were
downloaded from the European Molecular Biology Laboratory, European
Bioinformatics Institute database (http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-691/).
High-quality data were obtained by filtering raw reads with the
fastx-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) (13) (Table
I). The quality of the data was controlled using FastQC
(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
(14).
 | Table IData description. |
Table I
Data description.
| Sample | Read length
(bp) | Patient ID | Stage | Raw bases | Bases after
filtering |
|---|
| PEO1 | 42*2 | 2 | First
recurrence | 1.3 G | 1.2 G |
| PEO1 | 42*2 | 2 | First
recurrence | 1.4 G | 1.2 G |
| PEO1 | 42*2 | 2 | First
recurrence | 1.7 G | 1.5 G |
| PEO4 | 42*2 | 2 | Second
recurrence | 1.6 G | 1.3 G |
| PEO4 | 42*2 | 2 | Second
recurrence | 1.5 G | 1.3 G |
| PEO4 | 42*2 | 2 | Second
recurrence | 1.4 G | 1.3 G |
| PEO14 | 43*2 | 3 | Presentation | 1.2 G | 1.1 G |
| PEO14 | 43*2 | 3 | Presentation | 933.6 M | 860 M |
| PEO14 | 43*2 | 3 | Presentation | 1.3 G | 1.2 G |
| PEO23 | 42*2 | 3 | Recurrence | 1.8 G | 1.6 G |
| PEO23 | 42*2 | 3 | Recurrence | 1.7 G | 1.5 G |
| PEO23 | 43*2 | 3 | Recurrence | 701.8 M | 670 M |
Marker selection
All the markers of ovarian carcinoma were selected
from the Nucleotide database in National Center for Biotechnology
Information (http://www.ncbi.nlm.nih.gov/). Markers were selected
according to the following criteria: The nucleic acid sequence
existed, the sequence type was RNA and there were supporting
documents (listed in the Nucleotide database with their
corresponding marker). Normal human genes were downloaded from
University of California Santa Cruz (UCSC) Genome Bioinformatics
(http://genome.ucsc.edu/, version hg19). Gene
family information was downloaded from the HUGO Gene Nomenclature
database (http://www.genenames.org/).
Interaction network of markers
pathways
Pathways enrichment analysis for all the selected
markers was performed by the Kyoto Encyclopedia of Genes and
Genomes (KEGG) database (15),
then the enrichment pathways were linked together to form an
interaction network.
Marker expression evaluation and DEMs
detection
Bioinformatics protocols were applied to analyze the
sequencing data. Firstly, sequencing data were mapped to marker
sequences using bowtie (16) with
a maximum of two mismatches in the read, at most three mismatches
in a whole read, and only the best alignment reported of each read,
while the rest parameters were left as default. Secondly,
expression levels of markers were measured in reads per kilobase of
transcript per million mapped reads (RPKM) (17). Only markers with an average RPKM
value of three experimental duplicates ≥1 were defined as expressed
markers. Thirdly, the DEMs of the four samples were detected
referring to the method developed by Audic and Claverie (18). The P-values were adjusted using the
Benjamin and Hochberg correction (19) in the R programming language.
According to the criteria, expressed markers with fold change ≥1.5
and false discovery rate-adjusted P-value ≤0.01 were defined as
differentially-expressed. Spearman’s coefficient was calculated to
demonstrate the correlation between experimental duplicates
(20).
Results
Expressed marker analysis
In the present study, a total of 563 markers were
selected, all of which were documented to be associated with
ovarian cancer. Therefore, it was expected that they would be
expressed in the samples. The expression levels of the markers were
validated by RPKM, which is a quantitative value with widespread
use in RNA-seq analysis (21). As
a result, 484 markers (~85.97%) were detected to be expressed in
one or more samples. It was found that the RPKM values of the
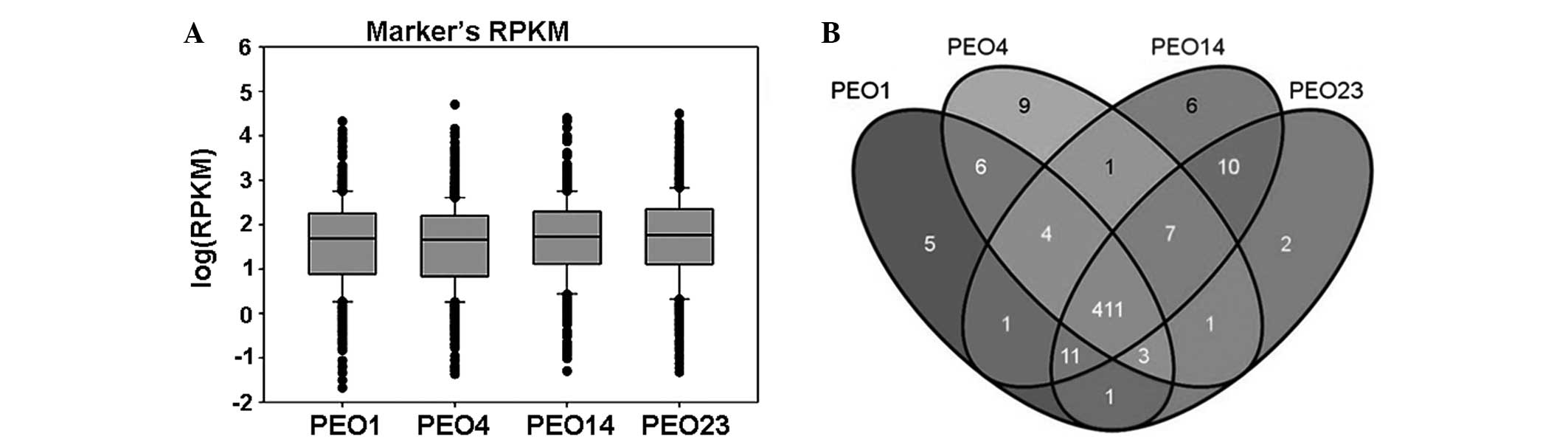
markers were similar in the four samples (Fig. 1A). The majority of the markers were
expressed in all four samples (411 of 484; ~84.92%), with each
sample also exhibiting specifically-expressed genes (Fig. 1B).
Certain genes in the same gene family have similar
roles in metabolic processes, therefore it was inferred that they
may be expressed synchronously in the process of transcription. To
verify this hypothesis, all the known gene families found in UCSC
were selected, and the 484 markers were categorized into 94 gene
families. By comparing the RPKM values of these markers using
cluster analysis (Fig. 2), it was
identified that the three experimental replicates of each cell line
can be clustered together. Samples of the two patients could be
distinguished. However, samples of PEO14 and PEO23 could not be
separated by the cluster analysis. This inferred that the
differences between the two individuals had a greater effect on the
expression of the marker than the differences between the tumor
stages.
Pathway interaction network
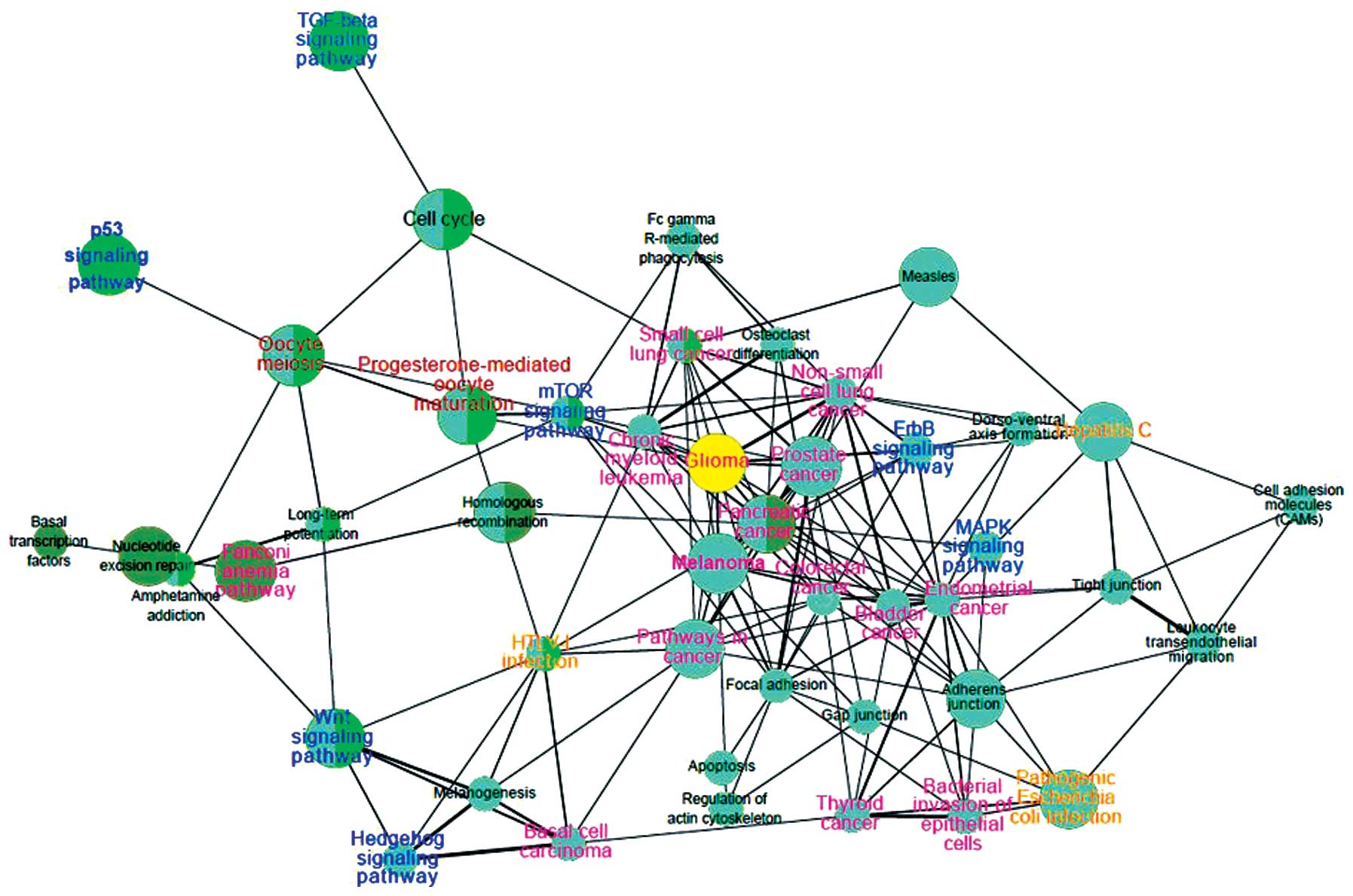
A KEGG pathway interaction network was developed
(Fig. 3). In the 484 expressed
markers, 257 markers were detected to be enriched in 44 pathways,
and 72 markers appeared in pathways associated with cancer
(Fig. 3, pink). AKT2
(22,23), a putative oncogene, was enriched in
the majority of the cancer-associated pathways in the network,
including small cell lung cancer, non-small cell lung cancer,
thyroid cancer and bladder cancer. Similarly, E2F2 and
BRCA2 also appeared in several cancer-associated pathways.
With the exception of cancer, pathways associated with infection
also appeared in the network with 35 markers. In the network, the
p53 signaling pathway was linked with oocyte meiosis and
progesterone-mediated oocyte maturation. The mTOR signaling pathway
was also found to be associated with the progesterone-mediated
oocyte maturation.
DEMs analysis
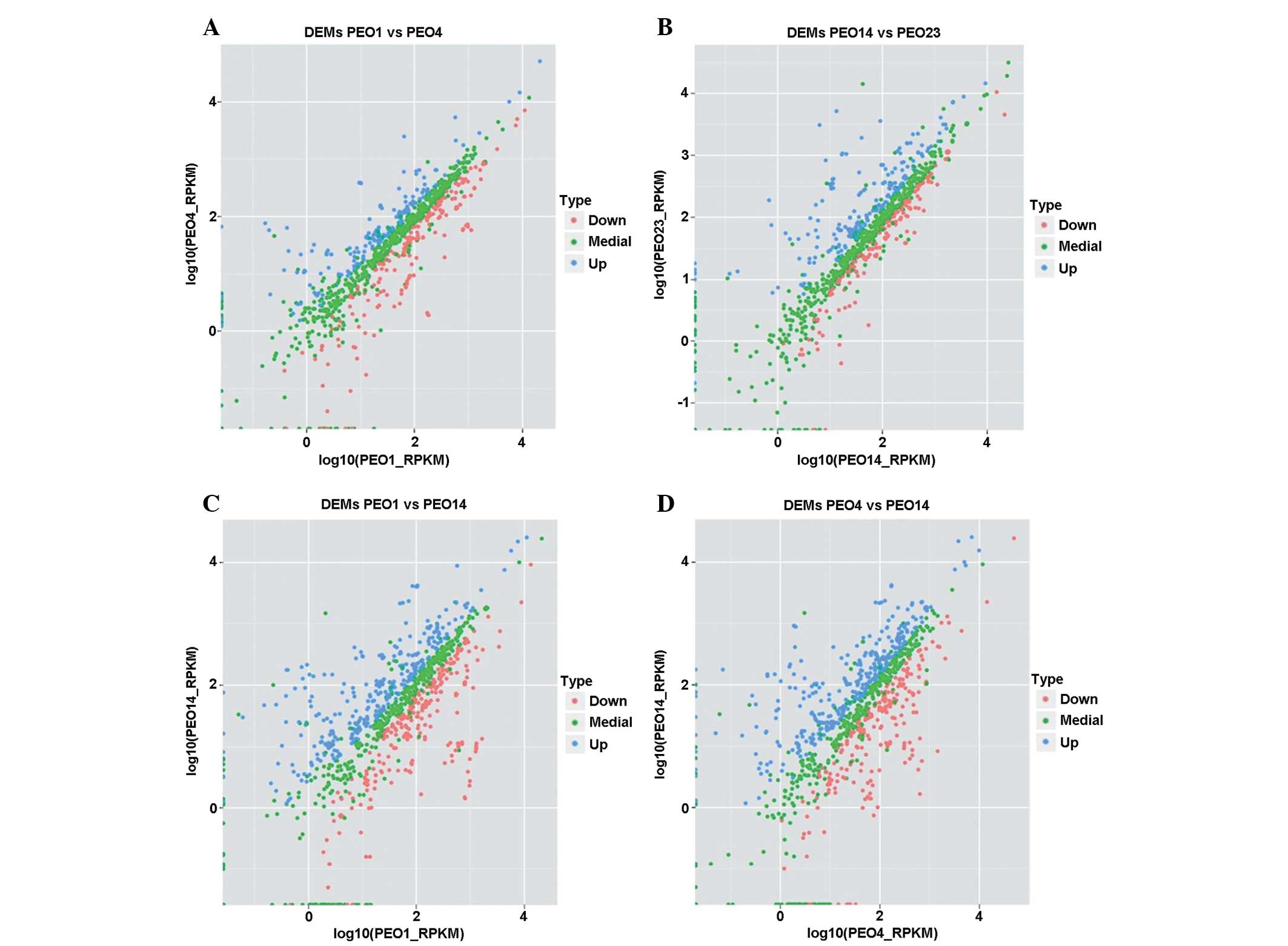
By comparing the changes of expression levels of
markers from the presentation stage to the recurrence stage, it was
found that certain markers had roles in recurrence in ovarian
cancer. In the present study, the expression levels of markers
between different patients with different tumor stages were
compared. More DEMs were detected between two different tumor
stages of the same patient, compared with between patients
(Fig. 4). DEMs were detected in
four pairs of comparisons, PEO14 vs. PEO23, PEO14 vs. PEO1, PEO14
vs. PEO4, and PEO4 vs. PEO1. As a result, 85 markers were found to
be differentially-expressed.
Discussion
Markers associated with ovarian cancer were analyzed
in the present study. To verify which markers were expressed in
tumor cells, RNA-seq data analysis was applied. Of a total of 563
markers that were selected, 484 markers (~85.97%) were expressed in
human ovarian tumor cells, which gave clear evidence that these are
markers of ovarian cancer from the respect of the whole
transcriptome. Differences between individuals are often omitted,
but the present result demonstrated that expression levels of
markers were more likely to be different between individuals than
between tumors of different stages. In addition, more DEMs were
detected between two different tumor stages of the same patient
compared with between patients. Thus, in a future study, more
samples from various ovarian cancer patients should be sequenced to
prevent interindividual differences affecting the data.
In the expressed markers, the majority of the
markers were enriched in pathways associated with cancer (72
markers), signaling (129 markers) and infection (35 markers).
AKT2, a putative oncogene (24), was enriched in the majority of the
cancer-associated pathways in the network and was first found to be
aberrantly expressed in human ovarian cancer (25). Similarly, E2F2 and BRCA2 were also
enriched in several cancer-associated pathways. The existence of
pathways associated with infection in the pathway network once
again supported the view that external infections, such as bacteria
and viruses may be associated with oncogenes in the induction of
the occurrence of carcinomas (26–28).
Signaling pathways also have important roles in cancer progression
(29), and there were 129 detected
markers in this type of pathway. Inactivation of p53 is implicated
in tumor progression and numerous activated oncogenes included in
the p53 signaling pathway have been elaborately investigated
(30,31). In the network determined in teh
present study, the p53 signaling pathway was linked with oocyte
meiosis and progesterone-mediated oocyte maturation (http://www.genome.jp/kegg/pathway/hsa/hsa04115.html).
As has been established, the oocyte is the area where ovarian
carcinoma formation is initiated, and unbalanced estrogen
metabolism is hypothesized to be one of the causes of ovarian
carcinoma (32). Thus, markers
enriched in this pathway may have a role in the oncogenesis of
ovarian cancer. The mTOR signaling pathway was also found to be
associated with progesterone-mediated oocyte maturation. The
deregulation of mTOR, a downstream effector of the AKT pathway, has
been reported to have effects on tumor progression (33).
All the markers that were selected are associated
with the presentation, development and diagnosis of ovarian cancer,
according to the findings from associated studies. In total, 85
markers were abnormally expressed in tumor cells from patients who
had recurrences. Of the 33 upregulated markers, TMP
(34), MAL2 (35), ERCC (36), CD (37), KLK (38) and SCARA3 (39) were also observed to be
over-expressed in previous studies. The CD9 gene encodes a
member of trans-membrane 4 superfamily-tetraspanin family (40), which regulates cell surface
glycoprotein function in differentiation and signal transduction.
Notably, gene expression is involved in the suppression of cancer
cell motility and metastasis (41). Although a study found that
downregulation of CD9 may be associated with the process of
ovarian tumor dissemination (42),
the present study demonstrated that CD9 had a high
expression level (RPKM) in all four samples (1290.7 in PEO14,
2141.98 in PEO1, 2283.54 in PEO4 and 4084.01 in PEO23 with a
significant increase at the recurrence stage). AKTIP may
have a role in apoptosis; however, at present no studies have ruled
out its association with the risk of developing ovarian cancer
(43). In the present study,
AKTIP was overexpressed in the PEO23 cell line, with an ~2
fold increase at the recurrence stage compared with that of the
PEO14 cell line. The Hox genes are a large gene family,
including numerous genes located on different chromosomes in the
human genome (44). In the present
study, four Hox genes were found to be expressed in ovarian
cancer cells, HOXA4, HOXA5, HOXD1 and
HOXD3, all of which were downregulated in the recurrence
stage compared with the presentation stage. In a further study, the
HOXA4 gene is over-expressed in human ovarian cancer when
compared with benign tumors (45).
The inappropriate expression of the HOXA5 gene disrupts
normal growth and differentiation programs (46). In the present study, HOXA5
was expressed in all four ovarian cancer samples with a
downregulated expression level at the recurrence stage. However,
there have been no previous studies, to the best of our knowledge,
regarding the contribution of HODX1 and HODX3 genes
to ovarian cancer, while HODX9 and HODX11 genes were
reported to be significantly increased in ovarian tumor cells
(47). Although all the marker
genes that were selected can be connected to the progression,
invasion or high risk of ovarian carcinoma, there have been no
previous studies regarding genetic associations with its
recurrence. The DEMs that were identified may be connected with the
recurrence of human ovarian cancer by regulating the expression
levels of the genes, but the exact regulatory mechanism requires
further investigation.
In conclusion, DEMs of ovarian cancer samples at
different stages and the associations between them can be clearly
investigated by examining the RNA-seq data. These markers may
provide novel prospects for further studies on ovarian cancer.
References
|
1
|
Moss EL, Hollingworth J and Reynolds TM:
The role of CA125 in clinical practice. J Clin Pathol. 58:308–312.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lakhani SR, Manek S, Penault-Llorca F, et
al: Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clin
Cancer Res. 10:2473–2481. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rafnar T, Gudbjartsson DF, Sulem P, et al:
Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet.
43:1104–1107. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ducros E, Mirshahi S, Azzazene D, et al:
Endothelial protein C receptor expressed by ovarian cancer cells as
a possible biomarker of cancer onset. Int J Oncol. 41:433–440.
2012.PubMed/NCBI
|
|
5
|
Fung FK, Chan DW, Liu VW, Leung TH, Cheung
AN and Ngan HY: Increased expression of PITX2 transcription factor
contributes to ovarian cancer progression. PLoS One. 7:e370762012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ishibashi M, Nakayama K, Yeasmin S, et al:
A BTB/POZ gene, NAC-1, a tumor recurrence-associated gene, as a
potential target for taxol resistance in ovarian cancer. Clin
Cancer Res. 14:3149–3155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rajkumar T, Soumittra N, Nancy NK,
Swaminathan R, Sridevi V and Shanta V: BRCA1, BRCA2 and CHEK2 (1100
del C) germline mutations in hereditary breast and ovarian cancer
families in south india. Asian Pac J Cancer Prev. 4:203–208.
2003.PubMed/NCBI
|
|
8
|
Ghadersohi A, Odunsi K, Zhang S, et al:
Prostate-derived Ets transcription factor as a favorable prognostic
marker in ovarian cancer patients. Int J Cancer. 123:1376–1384.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tassi RA, Calza S, Ravaggi A, et al:
Mammaglobin B is an independent prognostic marker in epithelial
ovarian cancer and its expression is associated with reduced risk
of disease recurrence. BMC Cancer. 9:2532009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rahman M, Nakayama K, Rahman M, et al:
Fatty acid synthase expression associated with NAC1 is a potential
therapeutic target in ovarian clear cell carcinomas. Brit J Cancer.
107:300–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ng CK, Cooke SL, Howe K, et al: The role
of tandem duplicator phenotype in tumour evolution in high-grade
serous ovarian cancer. J Pathol. 226:703–712. 2012. View Article : Google Scholar
|
|
12
|
Ahmed AA, Mills AD, Ibrahim AE, et al: The
extracellular matrix protein TGFBI induces microtubule
stabilization and sensitizes ovarian cancers to paclitaxel. Cancer
Cell. 12:514–527. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Krueger F, Kreck B, Franke A and Andrews
SR: DNA methylome analysis using short bisulfite sequencing data.
Nat Methods. 9:145–151. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Madougou S, Santcroos M, Benabdelkader A,
et al: Provenance for distributed biomedical workflow execution.
Stud Health Technol Inform. 175:91–100. 2012.PubMed/NCBI
|
|
15
|
Mao X, Cai T, Olyarchuk JG and Wei L:
Automated genome annotation and pathway identification using the
KEGG orthology (KO) as a controlled vocabulary. Bioinformatics.
21:3787–3793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Langmead B, Trapnell C, Pop M and Salzberg
SL: Ultrafast and memory-efficient alignment of short DNA sequences
to the human genome. Genome Biol. 10:R252009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mortazavi A, Williams BA, Mccue K,
Schaeffer L and Wold B: Mapping and quantifying mammalian
transcriptomes by RNA-seq. Nat Methods. 5:621–628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Audic S and Claverie JM: The significance
of digital gene expression profiles. Genome Res. 7:986–995.
1997.PubMed/NCBI
|
|
19
|
Benjamini Y and Yekutieli D: The control
of the false discovery rate in multiple testing under dependency.
Ann Stat. 29:1165–1188. 2001.
|
|
20
|
Harr B and Schlötterer C: Comparison of
algorithms for the analysis of affymetrix microarray data as
evaluated by co-expression of genes in known operons. Nucleic Acids
Res. 34:e82006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Adamidi C, Wang Y, Gruen D, et al: De novo
assembly and validation of planaria transcriptome by massive
parallel sequencing and shotgun proteomics. Genome Res.
21:1193–1200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Virtakoivu R, Pellinen T, Rantala JK,
Perälä M and Ivaska J: Distinct roles of AKT isoforms in regulating
β1-integrin activity, migration and invasion in prostate cancer.
Mol Biol Cell. 23:3357–3369. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dobashi Y, Kimura M, Matsubara H, Endo S,
Inazawa J and Ooi A: Molecular alterations in AKT and its protein
activation in human lung carcinomas. Hum Pathol. 43:2229–2240.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bellacosa A, De Feo D, Godwin AK, et al:
Molecular alterations of the AKT2 oncogene in ovarian and breast
carcinomas. Int J Cancer. 64:280–285. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Altomare DA, Wang HQ, Skele KL, et al: AKT
and mTOR phosphorylation is frequently detected in ovarian cancer
and can be targeted to disrupt ovarian tumor cell growth. Oncogene.
23:5853–5857. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chu CM: Natural history of chronic
hepatitis B virus infection in adults with emphasis on the
occurrence of cirrhosis and hepatocellular carcinoma. J Gastroen
Hepatol. 15:E25–E30. 2000. View Article : Google Scholar
|
|
27
|
Parsonnet J, Friedman GD, Vandersteen DP,
et al: Helicobacter pylori infection and the risk of gastric
carcinoma. N Engl J Med. 325:1127–1131. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sobhani I, Walker F, Roudot-Thoraval F, et
al: Anal carcinoma: incidence and effect of cumulative infections.
Aids. 18:1561–1569. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dreesen O and Brivanlou AH: Signaling
pathways in cancer and embryonic stem cells. Stem Cell Rev. 3:7–17.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Osman I, Drobnjak M, Fazzari M, et al:
Inactivation of the p53 pathway in prostate cancer: impact on tumor
progression. Clin Cancer Res. 5:2082–2088. 1999.PubMed/NCBI
|
|
31
|
Puzio-Kuter AM, Castillo-Martin M, Kinkade
CW, et al: Inactivation of p53 and Pten promotes invasive bladder
cancer. Genes Dev. 23:675–680. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ho SM: Estrogen, progesterone and
epithelial ovarian cancer. Reprod Biol Endocrinol. 1:732003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pópulo H, Lopes JM and Soares P: The mTOR
signalling pathway in human cancer. Int J Mol Sci. 13:1886–1918.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guerrero K, Wang Z, Bachvarova M, et al: A
novel genome-based approach correlates TMPRSS3 overexpression in
ovarian cancer with DNA hypomethylation. Gynecol Oncol.
125:720–726. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Byrne J, Maleki S, Hardy JR, et al: MAL2
and tumor protein D52 (TPD52) are frequently overexpressed in
ovarian carcinoma, but differentially associated with histological
subtype and patient outcome. BMC cancer. 10:4972010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo Y, Fu P, Zhu H, et al: Correlations
among ERCC1, XPB, UBE2I, EGF, TAL2 and ILF3 revealed by gene
signatures of histological subtypes of patients with epithelial
ovarian cancer. Oncol Rep. 27:286–292. 2012.
|
|
37
|
Hwang JR, Jo K, Lee Y, Sung BJ, Park YW
and Lee JH: Upregulation of CD9 in ovarian cancer is related to the
induction of TNF-α gene expression and constitutive NF-κB
activation. Carcinogenesis. 33:77–83. 2012. View Article : Google Scholar
|
|
38
|
Seiz L, Dorn J, Kotzsch M, et al: Stromal
cell-associated expression of kallikrein-related peptidase 6 (KLK6)
indicates poor prognosis of ovarian cancer patients. Biol Chem.
393:391–401. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bock AJ, Nymoen DA, Brenne K, Kærn J and
Davidson B: SCARA3 mRNA is overexpressed in ovarian carcinoma
compared with breast carcinoma effusions. Hum Pathol. 43:669–674.
2012. View Article : Google Scholar
|
|
40
|
Chuan Y, Pang ST, Bergh A, Norstedt G and
Pousette A: Androgens induce CD-9 in human prostate tissue. Int J
Androl. 28:291–296. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cook GA, Wilkinson DA, Crossno JT Jr,
Raghow R and Jennings LK: The tetraspanin CD9 influences the
adhesion, spreading and pericellular fibronectin matrix assembly of
chinese hamster ovary cells on human plasma fibronectin. Exp Cell
Res. 251:356–371. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Furuya M, Kato H, Nishimura N, et al:
Down-regulation of CD9 in human ovarian carcinoma cell might
contribute to peritoneal dissemination: morphologic alteration and
reduced expression of β1 integrin subsets. Cancer Res.
65:2617–2625. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Notaridou M, Quaye L, Dafou D, et al:
Common alleles in candidate susceptibility genes associated with
risk and development of epithelial ovarian cancer. Int J Cancer.
128:2063–2074. 2011. View Article : Google Scholar :
|
|
44
|
Acampora D, D’esposito M, Faiella A, et
al: The human hox gene family. Nucleic Acids Res. 17:10385–10402.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ota T, Klausen C, Salamanca MC, Woo HL,
Leung PC and Auersperg N: Expression and function of HOXA genes in
normal and neoplastic ovarian epithelial cells. Differentiation.
77:162–171. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gendronneau G, Boucherat O, Aubin J,
Lemieux M and Jeannotte L: The loss of Hoxa5 function causes
estrous acyclicity and ovarian epithelial inclusion cysts.
Endocrinology. 153:1484–1497. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Morgan R, Plowright L, Harrington KJ,
Michael A and Pandha HS: Targeting HOX and PBX transcription
factors in ovarian cancer. BMC cancer. 10:892010. View Article : Google Scholar : PubMed/NCBI
|