Introduction
Traumatic brain injury (TBI) is a complex and
devastating clinical condition mediated at least in part by
pro-inflammatory cytokines that produce neuronal loss, axonal
destruction and demyelination during the secondary injury cascade
(1). In contrast to primary
injuries that occur at the time of impact, secondary pathological
processes develop while under supervised medical care and
profoundly affect patient recovery (2,3).
These processes trigger an increase in ATP release and the
activation of the microglia, which leads to neuronal cell death and
the release of inflammatory factors (4,5).
Identifying novel strategies to inhibit the upregulation of harmful
factors following increased ATP release and to reduce the release
of inflammatory factors, has provided a new window of opportunity
for the clinical treatment of TBI.
Neuronal cell death is the main cause of
neurological deficits following TBI, and glial cells have an
important role in triggering the processes that lead to apoptosis.
Microglia represent the most abundant time of normal central
nervous system (CNS) immune cell and are widely distributed,
accounting for ~20% of the total number of glial cells in the brain
and spinal cord. The role of the guardian of the actor, the inlet
to the role of the warning system, also taking into account the
protection and restoration (6).
Microglia activation is associated with a number of CNS diseases,
including Alzheimer’s, Parkinson’s disease, amyotrophic lateral
sclerosis and multiple sclerosis (7–11).
In certain pathological conditions, including brain trauma,
ischemia, inflammation and others, local cell damage induces the
release of large amounts of ATP and degradation products, and when
the concentrations of these substances increase locally, nearby
microglia are activated (12).
Microglial cells are sensitive to changes in the surrounding
environment and a variety of physiological and pathological factors
activate rapid microglia hyperplasia, induce an ‘activated’
amoeba-like morphology that includes increasing cell body size,
shortening of processes, and also trigger the release of white
blood cell interleukin-1β (IL-1β), interleukin-6, cyclooxygenase -2
and other inflammatory factors. These factors not only cause damage
to the neigh-boring neurons, but also convene a series of immune
cells into the CNS causing further damage.
Purinergic P2X7 receptors mediate, at least in part,
the biological actions of extracellular ATP (13). Sustained activation of P2X7 with
high concentrations of ATP induces the release of biologically
active IL-1β (14,15), a potent pro-inflammatory cytokine.
Notably, IL-1β exhibited a prolonged induction in multiple
pre-clinical models of TBI (16–21),
and increased IL-1β levels in the CSF and brain was positively
correlated with elevated intracranial pressure (ICP) and
unfavorable outcomes in TBI patients (22–24).
Furthermore, it has been previously demonstrated that genetic or
pharmacological inhibition of IL-1β attenuates cerebral edema and
secondary injury following TBI (25–28),
indicative of a deleterious role for IL-1β after head trauma.
Therefore, once the role of IL-1β in TBI has been fully elucidated,
it may provide opportunities for the development of novel
therapeutic strategies. As a robust inflammatory response that
clinically correlates with secondary neurovascular injury following
TBI, it was hypothesized that activation of P2X7 mediates
neurological demise following TBI via attenuation of IL-1β.
The gamma isotype of protein kinase C (PKCγ) is a
member of the classical PKC (CPKC) subfamily which is activated by
Ca2+ and diacylglycerol in the presence of
phosphatidylserine (29–31). PKCγ is expressed solely in the
brain and spinal cord and its localization is restricted to neurons
(32). Within the brain, PKCγ
levels are most abundant in the cerebellum, hippocampus and
cerebral cortex, where notable neuronal plasticity occurs (33,34).
Recently, Matsumoto et al demonstrated that the PKCγ
expression was significantly increased following TBI in rats
(35). Nevertheless, to the best
of our knowledge, no studies have examined the potential for BBG to
regulate the expression of PKCγ in neurons in an animal model.
The present study aimed to investigate the
hypothesis that the P2X7 inhibitor, BBG, induced neuroprotective
properties via reducing PKCγ and the levels of inflammatory
cytokine IL-1β following TBI.
Materials and methods
Animals
A total of 150 Sprague-Dawley rats (obtained from
Hebei United University Experimental Animal Center, Tangshan,
Hebei, China), weighing 280–320 g, were housed under a 12 h
light/dark cycle with regular food and water supply. All
experimental procedures were conducted in conformity with the
Institutional Guidelines for the Care and Use of Laboratory Animals
of Hebei United University, (Shijiazhuang, China) and all
procedures were performed in accordance with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals (NIH Publication no. 80–23, revised 1996).
Models of TBI
A rat model of TBI was established by using a
modified weight-drop device, as described previously by Marmarou
et al (36). Briefly, the
rats were anesthetized with sodium pentobarbital (Nembutal, 60
mg/kg) prior to the surgery. A midline incision was performed to
expose the skull between the bregma and lambda suture lines, and a
steel disc (10 mm in diameter and 3 mm thickness) was adhered to
the skull using dental acrylic. Following this, the rats were
placed on a foam mattress underneath a weight-drop device in which
a 450 g weight falls freely through a vertical tube from 1.5 m onto
the steel disk. The sham-operated animals underwent the same
surgical procedure without being exposed to percussion injury, but
no trauma was induced. Following surgery, the rats received
supporting oxygenation with 95% O2 for no longer than 2
min and were returned to their cages. All of the rats were housed
in individual cages and placed on heat pads (37°C) for 24 h to
maintain normal body temperature during the recovery period.
Group and drug administration
The rats were randomly assigned to the sham-operated
group (sham; n=30), TBI treated with BBG group (BBG; n=60) and TBI
received only equal volumes of 0.9% saline solution (vehicle;
n=60). BBG was dissolved in 2% sucrose water and stored at 4°C.
Following the brain injuries, BBG was immediately administered to
the rats of the BBG group following TBI as a tail vein injection
(50 mg/kg body weight). All of the tests were blinded, and the
animal codes were revealed only at the end of the behavioral and
histological analyses.
Immunofluorescence
The brain tissues were fixed in 4% paraformaldehyde
for 24 h, and placed into 30% sucrose solution with 0.1 mol/l
phosphate-buffered saline (PBS; pH 7.4) until sinking to the
bottom. The tissues were divided 200 µm apart from each
section from anterior to posterior hippo-campus (bregma -1.90 to
-3.00 mm) from TBI rats, and then embedded in OCT. Frozen sections
(15 µm) were sliced with a frozen slicer, treated with 0.4%
Triton X-100 for 10 min and blocked in normal donkey serum for 1 h.
For double labeling, the frozen sections were incubated with a
mixture of rabbit anti-microtubule-associated protein 1 PKCγ
polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA; diluted 1:100) and mouse anti-neuron-specific nuclear
protein (NeuN) polyclonal antibody (Santa Cruz Biotechnology, Inc.;
diluted 1:100) overnight at 4°C. The next day, the sections were
incubated with a mixture of fluorescein-conjugated anti-rabbit IgG
and anti-mouse IgG (Santa Cruz Biotechnology, Inc.; diluted,
1:1,000) for 2 h at 37°C in the dark. All cell nuclei were
counterstained by 4′,6-diamidino-2-phenylindole (DAPI). The images
were captured in a laser scanning confocal microscope (Olympus
FV1000). Primary antibodies were replaced with PBS in the negative
control group.
Western blot analysis
Briefly, rats were anesthetized and underwent
intracardiac perfusion with 0.1 mol/l PBS (pH 7.4). The cortex of
the brains were rapidly isolated, the total proteins were extracted
and the protein concentration was determined by the BCA reagent
(Solarbio, Beijing, China) method. Samples were subjected to sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Separated proteins on the gel were transferred onto PVDF membranes
(Roche Diagnostics, Mannheim, Germany). The blots were blocked with
5% fat-free dry milk for 1 h at room temperature. Following
blocking, the membrane was incubated with the indicated primary
antibodies overnight at 4°C, including rabbit anti-PKCγ polyclonal
antibodies (Santa Cruz Biotechnology, Inc.; diluted 1:500), rabbit
anti-P2X7 polyclonal antibodies (Santa Cruz Biotechnology, Inc.;
diluted 1:500), rabbit anti-IL-1β polyclonal antibody (Santa Cruz
Biotechnology, Inc.; diluted 1:500), mouse anti-β-actin monoclonal
antibody (Santa Cruz Biotechnology, Inc.; diluted 1:500). Next, the
membrane was incubated with horseradish peroxidase conjugated
anti-rabbit IgG and anti-mouse IgG (Cell Signaling Technology,
Inc., Danvers, MA, USA; diluted 1:5,000) for 2 h at room
temperature. Following incubation with a properly titrated
secondary antibody, the immunoblot on the membrane was visible
following development with an enhanced chemiluminescence (ECL)
detection system and the densitometric signals were quantified
using an imaging program. Immunoreactive bands of all protein
expression were normalized to the intensity of the corresponding
bands for β-actin. The western blotting results were analyzed with
National Institutes of Health Image 1.41 software (Bethesda, MD,
USA).
Evaluation of brain edema
Brain edema was examined by analysis of brain water
content as described previously (37). The rat brains were separated and
weighed immediately with a chemical balance to obtain the wet
weight (WW). Following drying in a desiccating oven for 24 h at
100°C, the dry tissues were weighed again to obtain the constant
dry weight (DW). The percentage of water in the tissues was
calculated according to the formula: % brain water = [(WW−DW)/(WW)
× 100].
Recovery of motor function
The neurobehavioral status of the rats was
determined using a set of ten tasks, collectively termed the
neurological severity score (NSS), which examines reflexes,
alertness, coordination and motor abilities. One point is awarded
for failure to perform a particular task, thus, a score of 10
reflects maximal impairment, whereas a normal rat scores 0
(38). Post-injury, NSS was
evaluated at 12 and 24 h. Each animal was assessed by an observer
who was blinded to the animal treatment. The difference between the
initial NSS and that at any later time was calculated for each rat,
and this value (ΔNSS) reflects the spontaneous or treatment-induced
recovery of motor function.
Statistical analysis
All data are presented as the mean ± SD. SPSS 16.0
(SPSS, Inc., Chicago, IL, USA) was used for statistical analysis of
the data. Statistical analysis was performed using analysis of
variance (ANOVA) and followed by the Student-Newman-Keuls post hoc
tests. P<0.05 was considered to indicate a statistically
significant difference.
Results
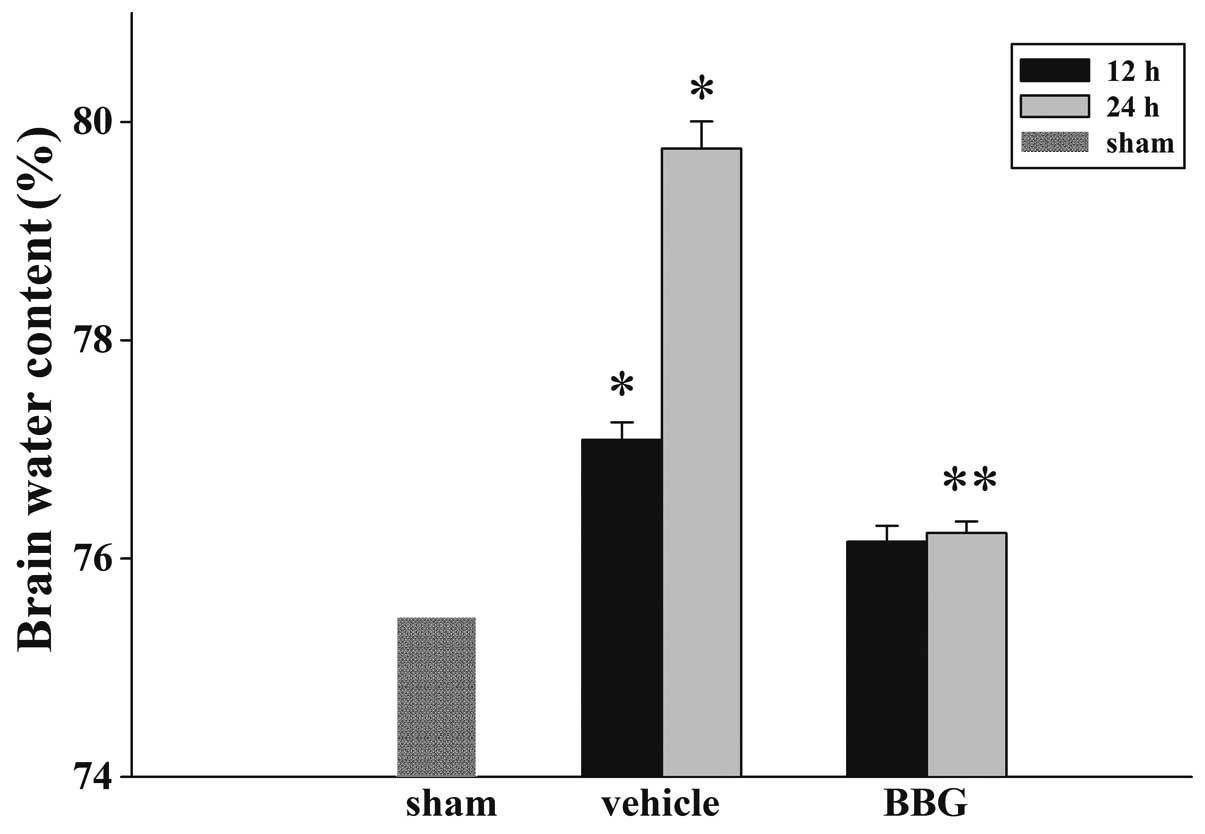
Treatment of BBG attenuates TBI-induced
cerebral edema
The wet-dry weight method was used to examine brain
edema. As demonstrated in Fig. 1,
BBG post-injury administration attenuated cerebral edema following
TBI. In the vehicle group, the brain water content was
significantly increased compared with the sham group at 12 and 24 h
following injury. The tissue water content in the BBG treatment
group was significantly reduced at 24 h compared with the vehicle
group.
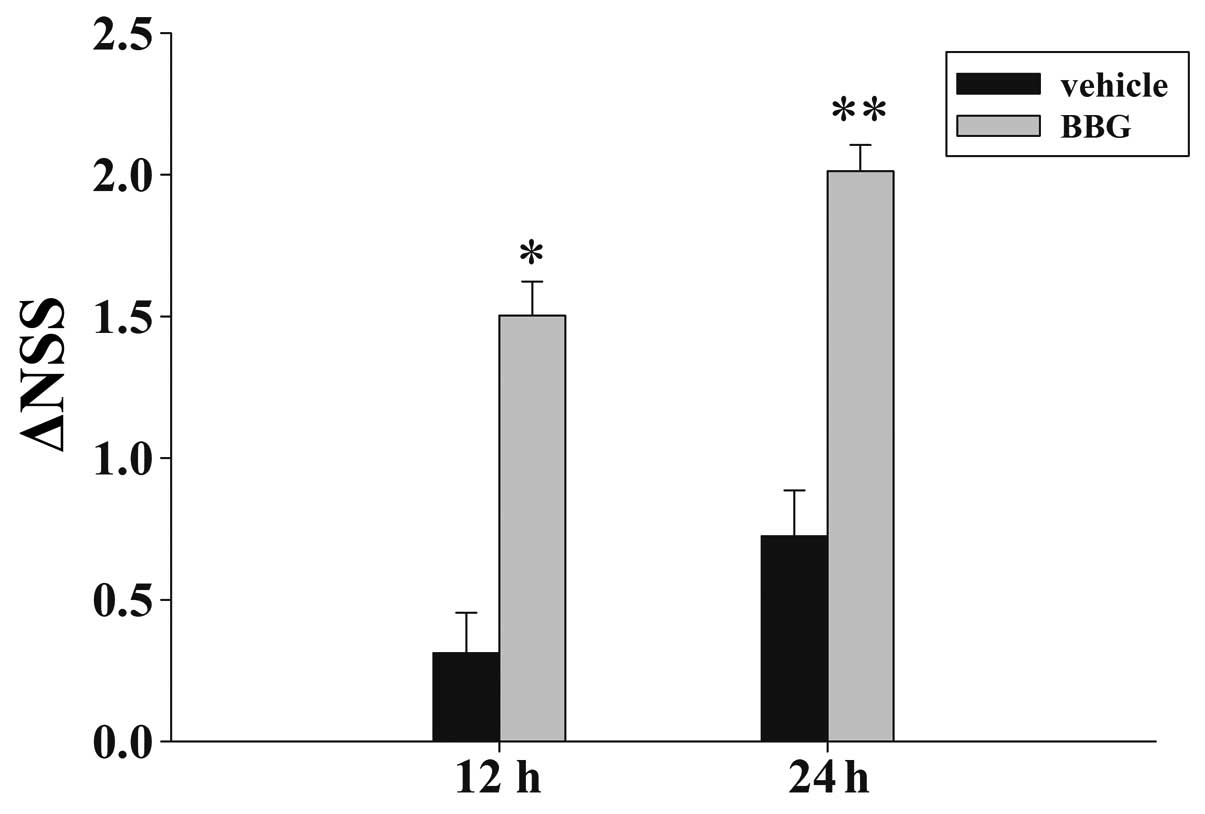
Treatment of BBG attenuates TBI-induced
motor deficits
Fig. 2 depicts the
temporal changes in functional recovery of the rat, expressed as
ΔNSS. It is evident that post-injury administration of BBG improved
the motor function recovery of the trauma rats at 12 and 24 h
following TBI.
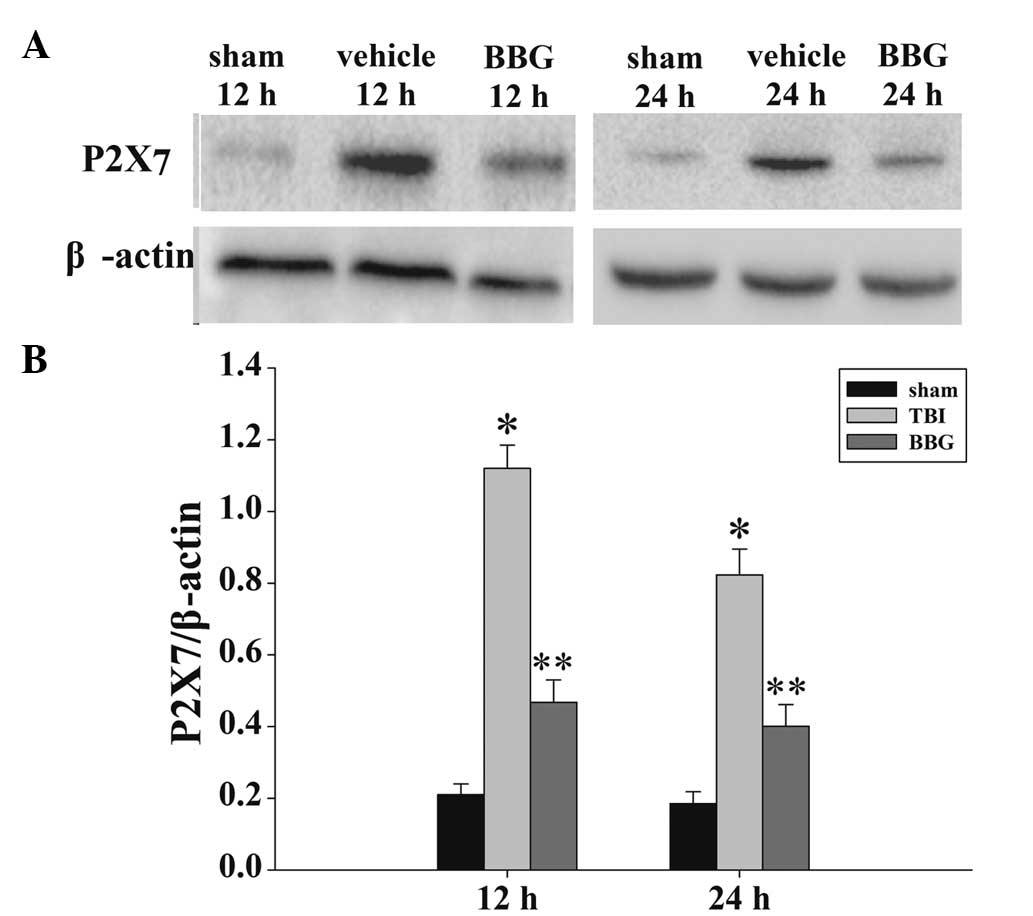
Treatment of BBG inhibits P2X7 expression
in the cortex following TBI
The expression levels of P2X7 in the rat cortex at
12 and 24 h were measured by western blot analysis (Fig. 3A). As demonstrated in Fig. 3B, the P2X7 levels in the sham rat
cortex at the two time points following injury were consistently
presented in a low background. In the vehicle group, all measured
P2X7 levels exhibited significant increases following injury.
Administration of BBG produced significant reductions in the
injury-induced upregulation of P2X7.
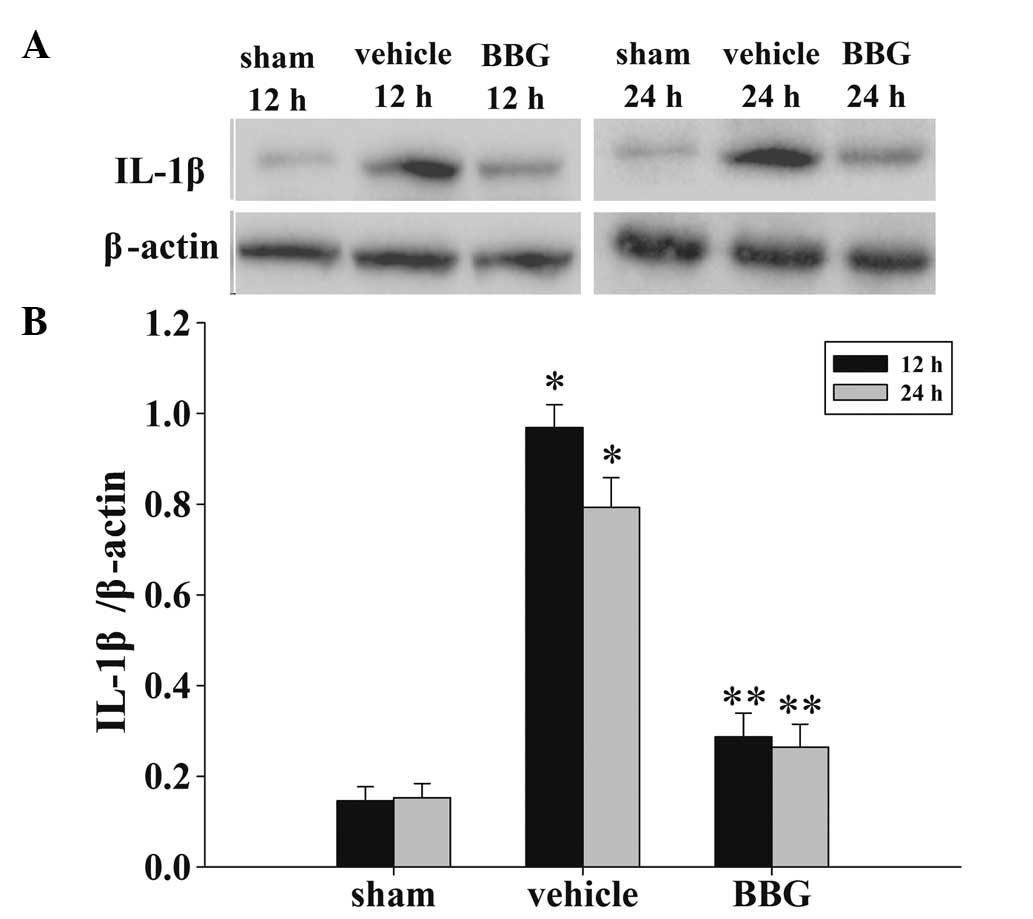
Treatment of BBG suppresses inflammatory
cytokine levels in the cortex following TBI
The levels of IL-1β in the cortex at 12 and 24 h
were measured by western blot analysis(Fig. 4A). As demonstrated in Fig. 4B, the IL-1β levels in the sham rat
cortex at the two time points following injury were in a low
background. The IL-1β expression levels were significantly
increased in the vehicle group. By contrast, in the BBG group,
treatment with BBG produced marked reductions in the injury-induced
upregulation of IL-1β expression.
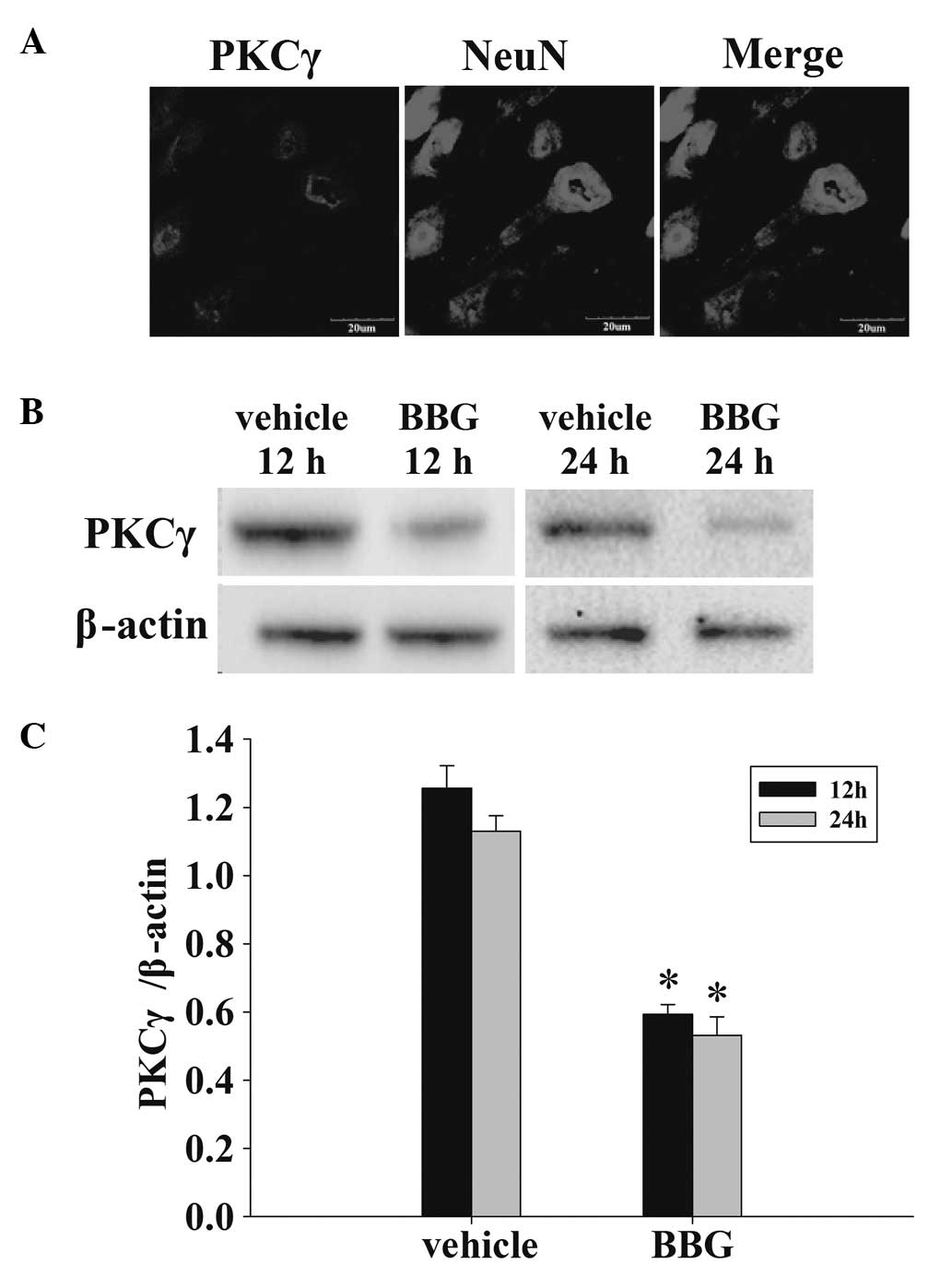
Treatment of BBG attenuates PKCγ in
neurons following TBI
The co-localization of NeuN and PKCγ was examined
with immunofluorescent staining at 24 h. As demonstrated in
Fig. 5A, the vast majority of PKCγ
expression following TBI was localized to neurons. Following this,
whether BBG attenuated the expression of PKCγ was determined by
western blot analysis (Fig. 5B).
As demonstrated in Fig. 5C, at 12
and 24 h following TBI, administration of BBG significantly
attenuated the PKCγ protein expression in rat cortex compared with
the vehicle group.
Discussion
TBI most commonly results in chronic neurological
abnormalities, including cognitive deficits, emotional disturbances
and motor impairments, and represents a severe international health
concern. Microglia activation and secretion of inflammatory factors
in the post-traumatic brain promotes clinical deterioration and
worsens long-term outcomes, at least in part, by inflammatory
cytokine production, infiltration of immune cells into the CNS, and
by increasing the manifestation of neurological impairments,
including headaches, anxiety, depression, sleep disturbances,
cognitive dysfunction and appetite loss (39,40).
Therefore, elucidation of the cellular mechanisms of neurological
injury may permit the development of efficacious therapeutics to
improve patient outcomes after TBI.
The biological actions of ATP are mediated, at least
in part, by the activation of either metabotropic P2Y receptors or
ionotropic P2X receptors (13).
Among the purine receptor family members, P2X7 is a low-affinity
receptor that preferentially responds to sustained elevations in
ATP, including those which occur following trauma, which suggests
that P2X7 possesses the optimal biophysical properties for
mediating the detrimental actions of ATP after brain injury.
Observations from a study by Wang et al demonstrated that
spinal cord injury was associated with prolonged purinergic
receptor activation, which results in excitotoxicity-based neuronal
degeneration. Furthermore, P2X7R antagonists inhibited this
process, reducing the histological extent and functional
consequences of acute spinal cord injury (41). The present study used BBG on a rat
model of TBI, and demonstrated that BBG inhibition of P2X7 reduced
secondary brain injury and improved the functional outcomes
following moderate TBI in mice. Therefore, it was hypothesized that
the inhibitor also has an markedly important role in the CNS
recovery following TBI. Similar neuroprotective effects of BBG for
TBI have been reported by Kimbler et al, that demonstrated
that BBG reduced post-traumatic cerebral edema with an extended
therapeutic window. Their study focussed on the effects of P2X7
expression of aquaporin aqp4, including reducing edema and
increased intracranial pressure following TBI (42). These results are consistent with
the earlier evidence, and the data of the present study also
reported that BBG significantly reduced the expression of PKCγ in
neurons and the levels of the inflammatory factor IL-1β.
IL-1β is an important pro-inflammatory cytokine that
mediates a variety of host defense responses to tissue injury and
exogenous antigens. Accumulative evidence demonstrates the presence
of P2X7 receptors on microglia and the activation of these
receptors by ATP triggers microglia release of IL-1β (43). Blockade of P2X7R has accordingly
been demonstrated to attenuate microglial activation and
inflammation in spinal cord injury (6). In the present study, it was observed
that administration of the P2X7R inhibitor BBG significantly
reduced the expression of IL-1β without any evident toxicity. These
results are consistent with the previous findings of Peng et
al (44) and it is therefore
conceivable to hypothesize that the mechanism underlying the
neuroprotective effect of BBG on TBI may be associated with the
attenuation of the levels of inflammatory cytokine IL-1β.
PKCγ is predominantly expressed in the CNS.
Staurosporine, a PKCγ inhibitor, has been demonstrated to reduce
ischemic cell death in vivo (45). Hypothermia and substances that
diminish PKC translocation to cell membranes are known to be
neuroprotective (46). In the
present study, it was identified that PKCγ expression in the
injured rat hippocampus of brain was suppressed by BBG. It may
therefore be one of the mechanisms that underlies the
neuroprotective effects of BBG following TBI.
Of note, BBG is a derivative of a commonly used blue
food color (FD&C blue no. 1), which crosses the blood-brain
barrier. Systemic administration of BBG may thus comprise a readily
feasible approach by which to treat TBI in humans. However, in the
present study, several considerations must be further considered.
Although BBG is considered as a highly selective P2X7 antagonist,
it also inhibits both P2X2 and P2X5, albeit less potently than that
of P2X7 (47). In the present
study, while BBG demonstrated protective and potential therapeutic
effects in a rat model of TBI, the possibility that off-target
effects on receptors other than P2X7 mediated the beneficial
actions of BBG may not be excluded. Therefore, further studies are
required to identify more specific inhibitors of P2X7, or to
develop a P2X7(−/−) rat.
In conclusion, the present study demonstrated that
BBG was able to attenuate secondary brain edema and improve
cognitive function following TBI. The upregulation of PKCγ and
IL-1β was also attenuated by post-injury treatment of BBG in rats.
These findings emphasize that BBG administration immediately
following TBI may be neuroprotective, and that this effect may be
associated with the attenuation of PKCγ and IL-1β expression. The
present study provides a viable therapeutic window for the
development of novel clinical treatment strategies for TBI.
Acknowledgments
This study was supported by grants from the Natural
Science Foundation of Hebei, Hebei, China (grant nos. H2012401071
and NO.H2014105079).
Abbreviations:
|
TBI
|
traumatic brain injury
|
|
BBG
|
brilliant blue G
|
|
PKCγ
|
protein kinase Cγ
|
|
NeuN
|
neuron-specific nuclear protein
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
NSS
|
neurological severity score
|
|
IL-1
|
interleukin-1
|
References
|
1
|
Nortje J and Menon DK: Traumatic brain
injury: physiology, mechanisms, and outcome. Curr Opin Neurol.
17:711–718. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bramlett HM and Dietrich WD: Progressive
damage after brain and spinal cord injury: pathomechanisms and
treatment strategies. Prog Brain Res. 161:125–141. 2007.PubMed/NCBI
|
|
3
|
Song SX, Gao JL, Wang KJ, et al:
Attenuation of brain edema and spatial learning deficits by the
inhibition of NADPH oxidase activity using apocynin following
diffuse traumatic brain injury in rats. Mol Med Rep. 7:327–331.
2013.
|
|
4
|
Levin HS, Eisenberg HM, Gary HE, et al:
Intracranial hypertension in relation to memory functioning during
the first year after severe head injury. Neurosurgery. 28:196–199.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saul TG and Ducker TB: Effect of
intracranial pressure monitoring and aggressive treatment on
mortality in severe head injury. J Neurosurg. 56:498–503. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chessell IP, Hatcher JP, Bountra C, et al:
Disruption of the P2X7 purinoceptor gene abolishes chronic
inflammatory and neuropathic pain. Pain. 114:386–396. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Labasi JM, Petrushova N, Donovan C, et al:
Absence of the P2X7 receptor alters leukocyte function and
attenuates an inflammatory response. J Immunol. 168:6436–6445.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Le Feuvre R, Brough D and Rothwell N:
Extracellular ATP and P2X7 receptors in neurodegeneration. Eur J
Pharmacol. 447:261–269. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Solle M, Labasi J, Perregaux DG, et al:
Altered cytokine production in mice lacking P2X(7) receptors. J
Biol Chem. 276:125–132. 2001. View Article : Google Scholar
|
|
10
|
Parvathenani LK, Tertyshnikova S, Greco
CR, Roberts SB, Robertson B and Posmantur R: P2X7 mediates
superoxide production in primary microglia and is up-regulated in a
transgenic mouse model of Alzheimer’s disease. J Biol Chem.
278:13309–13317. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sperlágh B, Vizi ES, Wirkner K and Illes
P: P2X7 receptors in the nervous system. Prog Neurobiol.
78:327–346. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sanz JM and Di Virgilio F: Kinetics and
mechanism of ATP-dependent IL-1beta release from microglial cells.
J Immunol. 164:4893–4898. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Surprenant A, Rassendren F, Kawashima E,
North R and Buell G: The cytolytic P2Z receptor for extracellular
ATP identified as a P2X receptor (P2X7). Science. 272:735–738.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Calogero S, Grassi F, Aguzzi A, et al: The
lack of chromosomal protein Hmg1 does not disrupt cell growth but
causes lethal hypoglycaemia in newborn mice. Nat Genet. 22:276–280.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takenouchi T, Sugama S, Iwamaru Y,
Hashimoto M and Kitani H: Modulation of the ATP-lnduced release and
processing of IL-1beta in microglial cells. Crit Rev Immunol.
29:335–345. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Folkersma H, Brevé JJ, Tilders FJ, Cherian
L, Robertson CS and Vandertop WP: Cerebral microdialysis of
interleukin (IL)-1beta and IL-6: extraction efficiency and
production in the acute phase after severe traumatic brain injury
in rats. Acta Neurochir (Wien). 150:1277–1284. 2008. View Article : Google Scholar
|
|
17
|
Herx LM, Rivest S and Yong VW: Central
nervous system-initiated inflammation and neurotrophism in trauma:
IL-1β is required for the production of ciliary neurotrophic
factor. J Immunol. 165:2232–2239. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kamm K, Vanderkolk W, Lawrence C, Jonker M
and Davis AT: The effect of traumatic brain injury upon the
concentration and expression of interleukin-1beta and
interleukin-10 in the rat. J Trauma Acute Care Surg. 60:152–157.
2006. View Article : Google Scholar
|
|
19
|
Kinoshita K, Chatzipanteli K, Vitarbo E,
Truettner JS, Alonso OF and Dietrich WD: Interleukin-1beta
messenger ribonucleic acid and protein levels after
fluid-percussion brain injury in Rats: importance of injury
severity and brain temperature. Neurosurgery. 51:195–203. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Laird MD, Sukumari-Ramesh S, Swift AE,
Meiler SE, Vender JR and Dhandapani KM: Curcumin attenuates
cerebral edema following traumatic brain injury in mice: a possible
role for aquaporin-4? J Neurochem. 113:637–648. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Taupin V, Toulmond S, Serrano A, Benavides
J and Zavala F: Increase in IL-6, IL-1 and TNF levels in rat brain
following traumatic lesion: Influence of pre-and post-traumatic
treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine
ligand. J Neuroimmunol. 42:177–185. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shiozaki T, Hayakata T, Tasaki O, et al:
Cerebrospinal fluid concentrations of anti-inflammatory mediators
in earlyphase severe traumatic brain injury. Shock. 23:406–410.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hayakata T, Shiozaki T, Tasaki O, et al:
Changes in CSF S100B and cytokine concentrations in early-phase
severe traumatic brain injury. Shock. 22:102–107. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chiaretti A, Genovese O, Aloe L, et al:
Interleukin 1beta and interleukin 6 relationship with paediatric
head trauma severity and outcome. Childs Nerv Syst. 21:185–193.
2005. View Article : Google Scholar
|
|
25
|
Clausen F, Hånell A, Björk M, et al:
Neutralization of interleukin-1beta modifies the inflammatory
response and improves histological and cognitive outcome following
traumatic brain injury in mice. Eur J Neurosci. 30:385–396. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jones NC, Prior MJ, Burden Teh E, Marsden
CA, Morris PG and Murphy S: Antagonism of the interleukin-1
receptor following traumatic brain injury in the mouse reduces the
number of nitric oxide synthase-2-positive cells and improves
anatomical and functional outcomes. Eur J Neurosci. 22:72–78. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tehranian R, Andell-Jonsson S, Beni SM, et
al: Improved recovery and delayed cytokine induction after closed
head injury in mice with central overexpression of the secreted
isoform of the interleukin-1 receptor antagonist. J Neurotrauma.
19:939–951. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Toulmond S and Rothwell NJ: Interleukin-1
receptor antagonist inhibits neuronal damage caused by fluid
percussion injury in the rat. Brain Res. 671:261–266. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kishimoto A, Mikawa K, Hashimoto K, et al:
Limited proteolysis of protein kinase C subspecies by
calcium-dependent neutral protease (calpain). J Biol Chem.
264:4088–4092. 1989.PubMed/NCBI
|
|
30
|
Nishizuka Y: Protein kinase C and lipid
signaling for sustained cellular responses. FASEB J. 9:484–496.
1995.PubMed/NCBI
|
|
31
|
Ohno S: The distinct biological potential
of PKC isotypes. Protein Kinase C. Parker PJ and Deckker LV: RG
Landes; Texas, USA: pp. 178–188. 1997
|
|
32
|
Saito N, Kikkawa U, Nishizuka Y and Tanaka
C: Distribution of protein kinase C-like immunoreactive neurons in
rat brain. J Neurosci. 8:369–382. 1988.PubMed/NCBI
|
|
33
|
Huang FL, Yoshida Y, Nakabayashi H, Young
WS III and Huang KP: Immunocytochemical localization of protein
kinase C isozymes in rat brain. J Neurosci. 8:4734–4744.
1988.PubMed/NCBI
|
|
34
|
Tanaka C and Saito N: Localization of
subspecies of protein kinase C in the mammalian central nervous
system. Neurochem Int. 21:499–512. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Matsumoto S, Murozono M, Nagaoka D, et al:
Isoflurane inhibits protein kinase Cgamma and calcium/calmodulin
dependent protein kinase II-alpha translocation to synaptic
membranes in ischemic mice brains. Neurochem Res. 33:2302–2309.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Marmarou A, Foda MA, van den Brink W,
Campbell J, Kita H and Demetriadou K: A new model of diffuse brain
injury in rats: Part I: Pathophysiology and biomechanics. J
Neurosurg. 80:291–300. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tang J, Liu J, Zhou C, et al: Mmp-9
deficiency enhances collagenase-induced intracerebral hemorrhage
and brain injury in mutant mice. J Cerebral Blood Flow Metab.
24:1133–1145. 2004. View Article : Google Scholar
|
|
38
|
Beni-Adani L, Gozes I, Cohen Y, et al: A
peptide derived from activity-dependent neuroprotective protein
(ADNP) ameliorates injury response in closed head injury in mice. J
Pharmacol Exp Ther. 296:57–63. 2001.
|
|
39
|
Whelan-Goodinson R, Ponsford J, Johnston L
and Grant F: Psychiatric disorders following traumatic brain
injury: their nature and frequency. J Head Trauma Rehabil.
24:324–332. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rogers JM and Read CA: Psychiatric
comorbidity following traumatic brain injury. Brain Inj.
21:1321–1333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang X, Arcuino G, Takano T, et al: P2X7
receptor inhibition improves recovery after spinal cord injury. Nat
Med. 10:821–827. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kimbler DE, Shields J, Yanasak N, Vender
JR and Dhandapani KM: Activation of P2X7 promotes cerebral edema
and neurological injury after traumatic brain injury in mice. PloS
One. 7:e412292012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Elssner A, Duncan M, Gavrilin M and Wewers
MD: A novel P2X7 receptor activator, the human cathelicidin-derived
peptide LL37, induces IL-1beta processing and release. J Immunol.
172:4987–4994. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Peng W, Cotrina ML, Han X, et al: Systemic
administration of an antagonist of the ATP-sensitive receptor P2X7
improves recovery after spinal cord injury. Proce Natl Acad Sci
USA. 106:12489–12493. 2009. View Article : Google Scholar
|
|
45
|
Iwashita A, Muramatsu Y, Yamazaki T, et
al: Neuroprotective efficacy of the peroxisome
proliferator-activated receptor delta-selective agonists in vitro
and in vivo. J Pharmacol Exp Ther. 320:1087–1096. 2007. View Article : Google Scholar
|
|
46
|
Karpiak SE, Mahadik SP and Wakade CG:
Ganglioside reduction of ischemic injury. Crit Rev Neurobiol.
5:221–237. 1990.PubMed/NCBI
|
|
47
|
Jiang LH, Mackenzie AB, North RA and
Surprenant A: Brilliant blue G selectively blocks ATP-gated rat
P2X7 receptors. Mol Pharmacol. 58:82–88. 2000.PubMed/NCBI
|