Introduction
Alzheimer’s disease (AD) is the most common cause of
dementia worldwide and is characterized by pathological β-amyloid
(Aβ) deposition and neurodegeneration (1). Although the precise mechanisms
underlying neurodegeneration in AD remain to be elucidated, the
role of the complex interaction between genetic and environmental
factors has been supported in previous studies (2-4). In
addition, emerging evidence has indicated the critical involvement
of the ectopic expression of miRNA in the pathogenesis of AD
(5).
MicroRNAs (miRNAs), a class of endogenous, small
(~22 nt), noncoding RNAs, mediate post-transcriptional regulation
of protein-coding genes by binding to the 3′ untranslated region
(3′UTR) of target genes. This leads to translational inhibition or
mRNA degradation, reducing the production of hundreds of proteins
(6). There is evidence indicating
that miRNAs expressed in the brain are involved in neuronal
development, survival and apoptosis (7). β-site amyloid precursor
protein-cleaving enzyme1 (BACE1), a rate-limiting enzyme involved
in the production of Aβ, is important in the pathogenesis of AD
(8). Several down-regulated miRNAs
have been demonstrated in the brain, including the miR-107
(9) and miR-29 families (5), which regulate the expression of BACE1
and consequently affect the production of Aβ. Of the presently
identified miRNAs, ~70% are expressed in the brain, however, the
pathological implications of dysregulated miRNA expression in AD
remain to be fully elucidated (10).
The present study aimed to investigate the
neuroprotective role of miR-29c in AD. The results of the present
study may be therapeutically beneficial for the treatment of
AD.
Materials and methods
Blood samples
A total of 60 samples of peripheral blood were
collected from the median cubital veins of patients with AD and
from normal age-matched individuals (>65 years old) Haikou
People’s Hospital (Hainan, China). The patients with AD were aged
between 65 and 86 years (male, n=17; female n=13), and the normal
control individuals were aged between 65 and 74 years (male, n=19;
female, n=11). Individuals with significant illness, including
diabetes, heart disease, stroke or cancer, were excluded from the
present study. All the samples were collected according to the
legislation and ethical boards of Haikou People’s Hospital. Written
informed consent was obtained from the patients. The samples were
stored at −80°C until use.
Cell culture and treatment
Primary hippocampal neurons were obtained from the
hippocampi of SAMR1 mice at embryonic day 15. Briefly, the
hippocampi were mechanically dissociated and treated with 0.2%
trypsin (Sigma-Aldrich, St. Louis, MO, USA) for 15 min at 37°C in
phosphate-buffered saline (PBS). The hippocampal cells were
collected by centrifugation at 118 × g for 5 min, and then
2×105 cells/ml were washed in Dulbecco’s modified
Eagle’s medium (DMEM; Invitrogen Life Technologies, Carlsbad, CA,
USA), containing 10% fetal bovine serum (FBS; Invitrogen Life
Technologies), and resuspended in DMEM supplemented with 10% FBS
and 100 kU/l penicillin and streptomycin (Sigma-Aldrich). The cells
were cultured in a humidified atmosphere of 95% air and 5%
CO2. Ectopic expression of miR-29c in cells was
introduced by transfection with miR-29c mimics or inhibitors using
Lipofectamine 2000 (Invitrogen Life Technologies).
Animals and treatment
The SAMR1 and SAMP8 mice were obtained from the
Animal Center of Beijing University Medical Department (Beijing,
China) and housed in standard conditions (12 h light/dark; 25±1°C;
50% humidity). Male 8-month-old SAMP8 (n=6) and SAMR1 (n=6) mice
were used. The anesthetized (40 mg/kg 1% pentobarbital sodium;
Sigma-Aldrich) SAMP8 mice were positioned in a stereotaxic
apparatus (model 68016; RWD Life Science, Shenzhen, China), and 1.5
μl PBS, containing either 0.5 nmol miR-29c mimic or a
scrambled control (Intvitrogen Life Technologies, Carlsbad, CA,
USA), was injected for 8 min into the third ventricle. The control
SAMR1 mice received an equal volume of the vehicle (PBS only). The
present study was approved by the ethics committee of Southern
Medical University (Guangzhou, China), in compliance with the
National Institute of Health Guide for the Care and Use of
Laboratory Animals (11).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was extracted from the indicated
tissues or cells using RNA Extraction reagent, according to the
manufacturer’s instructions (CWBio, Beijing, China). The mRNA
expression of BACE1 was detected using a SYBR green qPCR assay
(CWBio). RevertAid First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Waltham, MA, USA) was used to reverse transcribe the
RNA. The expression of β-actin was used as an endogenous control.
The specific primers used were as follows: BACE1, forward
5′-AATTCGAAATGGCCCAAGCCCTGCCCT-3′ and reverse
5′-AGGGATCCGGGCCTCCTCACTTCAGCAG-3′, and β-actin, forward
5′-CATTAAGGAGAAGCTGTGCT-3′ and reverse 5′-GTTGAAGGTAGTTTCGTGGA-3′.
A MiScript SYBR-Green PCR kit (Ribobio, Co., Ltd., Guangzhou,
China) was used for qPCR to detect the expression levels of
miR-29c. The specific primer sets for miRNA-29c and U6 were
purchased from GeneCopoeia (Rockville, MD, USA). The expression of
U6 was used as an endogenous control. A total of 2 μl cDNA
(50 ng/μl) was used to analyze the expression levels on a
CFX96 Real-Time system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). The PCR conditions were as follows: 95°C for 5 min, followed
by 40 cycles at 95°C for 30 sec, 58°C for 30 sec and 72°C for 30
sec, and finally 72°C for 10 min. The data were then analyzed using
the 2−ΔΔCT method (12).
ELISA determination of Aβ
Mouse Aβ immunoassay kits (Invitrogen Life
Technologies) were used to determine the levels of Aβ in the
primary cultured hippocampal cells and the brain tissues of the
mice, according to the manufacturer’s instructions. Briefly, the
media in the cultured cells or the supernatants of the indicated
tissues were used to measure the total protein quantities of each
sample. The samples and the Aβ antibody were incubated overnight at
4°C, prior to incubation with horseradish peroxidase (HRP)-labeled
anti-rabbit antibody for 30 min at room temperature. The wells were
then developed using 100 μl tetramethylbenzidine reagent in
the dark, and the absorbance was measured at 450 nm (Synergy™ Mx;
BioTek Instrument, Inc., Winooski, VT, USA).
Western blotting
The total protein was extracted from the indicated
cells or tissues using cold radioimmunoprecipitation lysis buffer
(CWBio). A bicinchoninic acid protein assay kit (Pierce
Biotechnology, Rockford, IL, USA) was used to determine the protein
concentrations. The protein (60 μg) was subsequently
separated on a 10% SDS-PAGE gel (Wuhan Boster Biological
Technology, Ltd., Wuhan, China) and transferred onto a
nitrocellulose membrane (Wuhan Boster Biological Technology, Ltd.).
The membrane was blocked in 5% nonfat dried milk in PBS for 4 h,
and was incubated with the following primary antibodies overnight
at 4°C: Rabbit anti-protein kinase A (PKA; 1:1,00; cat. no. 4781;
Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit
anti-BACE (1:500; cat. no. 5606; Cell Signaling Technology, Inc.),
rabbit anti-cAMP response element binding protein (CREB; 1:500;
cat. no. 4820; Cell Signaling Technology, Inc.), rabbit
anti-phosphorlyated (p) CREB (1:500; cat. no. 9198; Cell Signaling
Technology, Inc.) and mouse anti-β-actin (1:3,000; cat. no. BM0627;
Wuhan Boster Biological Technology, Ltd.). The membranes were
washed with Tris-buffered saline containing 0.1% Tween and
incubated with HRP-labeled goat anti-rabbit (cat. no. A12004-1;
1:2,000) and goat anti-mouse (cat. no. A12003-1; 1:3,000) secondary
antibodies (Epigentek, Farmingdale, NY, USA) for 2 h at room
temperature. Enhanced chemiluminescence reagent (Wuhan Boster
Biological Technology, Ltd.) was used to detect the signal on the
membrane. The expression data were analyzed by densitometry using
Image-Pro plus software 6.0 (Media Cybernetics, Inc., Rockville,
MD, USA) and normalized against that of the internal control.
Dual luciferase reporter assay
Wild-type (wt) and mutant (mut) 3′-UTRs of BACE1
were constructed, which were inserted into the dual luciferase
reporter vector (Promega Corporation, Madison, WI, USA). Briefly,
to generate the wt-BACE1-3′-UTR, the 3′-UTR was amplified and
cloned into the XbaI (Thermo Fisher Scientific) site of the
pGL3-control vector (Promega Corporation), downstream of the
luciferase gene. The mut-BACE1-3′-UTR was generated from
wt-BACE1-3′-UTR by site-directed mutagenesis, by Genecopeoia
(Guangzhou, China). For the luciferase assay, 100,000 cells were
cultured to ~70% confluence in 24-well plates. Following culture,
the cells were co-transfected with the miR-29c mimic and either the
wt or mut 3′-UTR of the BACE1 dual luciferase reporter vector.
Following incubation for 5 h with the transfection reagent/DNA
complex, the medium was replaced with fresh medium, containing 10%
FBS. At 48 h post-transfection, a Dual Luciferase Reporter Gene
Assay kit (BioVision, Milpitas, CA, USA) was used to determine the
luciferase activities in each group on a luminometer (Elecsys 2010;
Roche Diagnostics, Basel, Switzerland). The activity of Renilla
luciferase was normalized against that of firefly luciferase.
Cell proliferation assay
For all the groups, 5,000-6,000 cells/well were
seeded into a 96-well plate. Following treatment, the plates were
incubated for 0, 12, 24, 48 or 72 h at 37°C with 5% CO2.
To assess cell proliferation, an MTT assay was performed, according
to the manufacturer’s instructions. MTT reagent (20 μl; 5
mg/ml; Wuhan Boster Biological Technology, Ltd.) in 200 μl
FBS-free medium was added to each well and incubated for 4 h at
37°C. The medium was then removed and 100 μl dimethyl
sulfoxide (Wuhan Boster Biological Technology, Ltd.) was added. The
absorbance was detected at 490 nm using a microplate reader
(Elecsys 2010). The assay was repeated three times in triplicate
wells.
Y-maze test
The Y-maze apparatus (model RD1102-YM-M; Mobiledatum
Co., Ltd., Shanghai, China) was constructed from wood, painted in
black, and had three arms at 120° angles. Each arm was 50 cm long,
15 cm high, 5 cm wide. The mice were initially placed at the end of
one arm and allowed to move freely for 10 min. The series of arm
entries was recorded using a video camera (model HDR-PJ790E; Sony,
Tokyo, Japan). Spontaneous alternation was defined as: Successive
entries into the three arms in overlapping triplet sets. The
alternation percentages were determined as the ratios of actual
alternations to maximum alternations, multiplied by 100. The
percentages of alternation behaviors were recorded by an observer,
in a blinded manner, followed by statistical analysis.
Statistical analysis
Statistical analyses were performed using Graphpad
Prism 5 software (Graphpad Software, Inc., San Diego, CA, USA) and
the data are expressed as the mean ± standard deviation. An
unpaired two-tailed Student’s t-test was used to analyze the
differences between the samples. P<0.05 was considered to
indicate a statistically significant difference.
Results
Levels of miR-29c are negatively
correlated with protein levels of BACE1 in the blood of patients
with AD
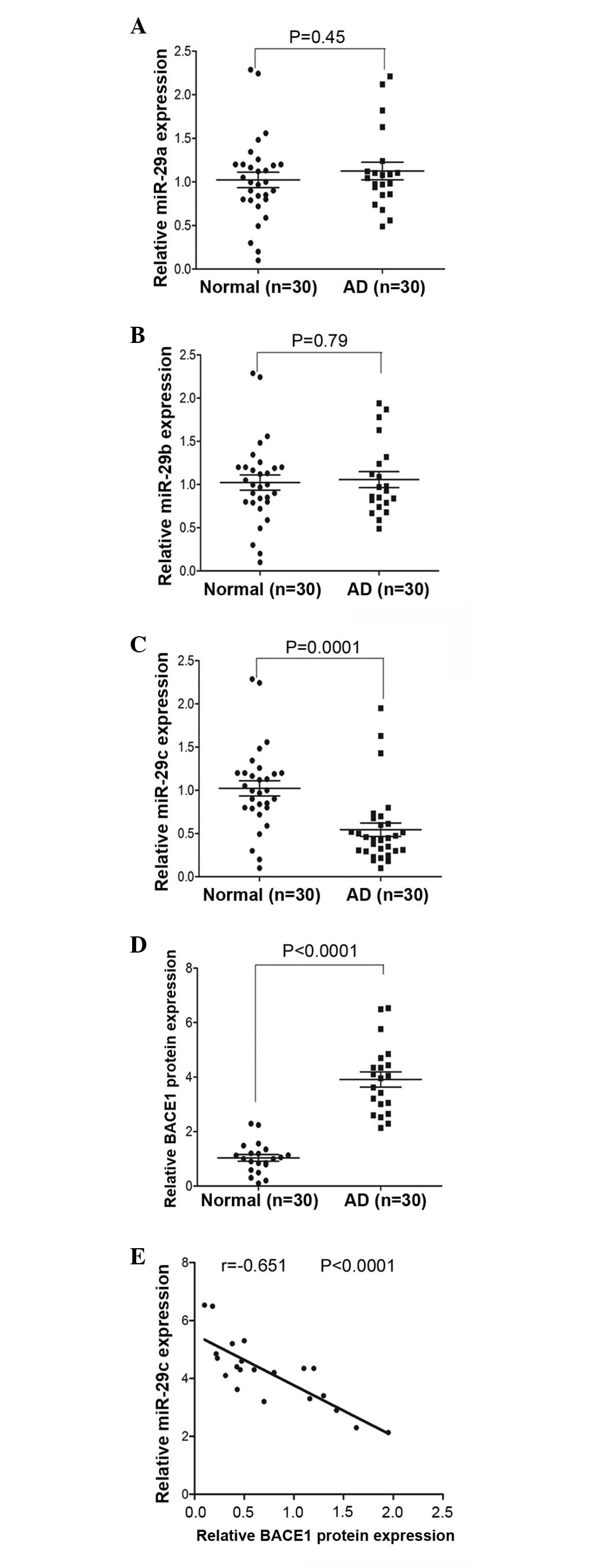
The expression levels of miR-29 were detected using
SYBR green qPCR analysis. In the sample of 30 patients with AD and
30 normal individuals, no significant differences were observed in
the expression levels of miR-29a and miR-29b (Fig. 1A and B), however, the results
demonstrated that miR-29c was significantly decreased in 27 (90%)
of the peripheral blood samples from the patients with AD, compared
with the paired normal blood samples (Fig. 1C). However. In addition, an ELISA
was used to measure the protein expression levels of BACE1. As
shown in Fig. 1D, the protein
expression levels of BACE1 were significantly increased in the
peripheral blood from patients with AD, compared with the normal
control individuals. In addition, the expression of miR-29c was
negatively correlated with the protein expression of BACE1 in the
peripheral blood of the patients with AD (Fig. 1E).
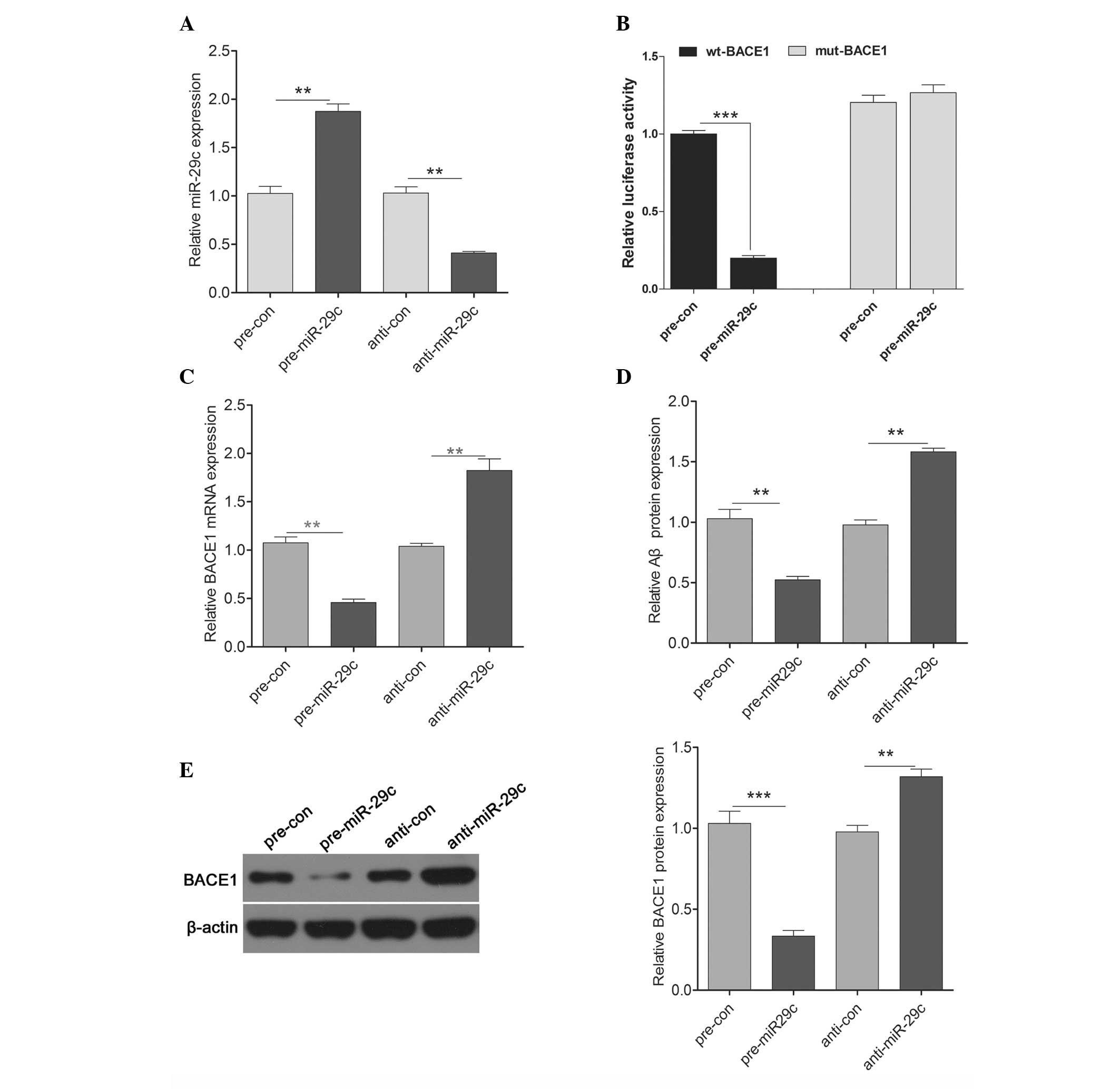
miR-29c regulates the expression of BACE1
by directly targeting its 3′UTR
To investigate whether miR-29c targets the 3′UTR of
BACE1, the present study cloned the 3′UTR of BACE1 downstream to a
luciferase reporter gene (wt-BACE1). The mutant version
(mut-BACE1), with binding site mutagenesis, was also constructed.
To determine whether miR-29c regulated the expression of BACE1 at
the transcriptional or translational level, the wt-BACE1 vector and
either the pre-miR-29c mimic or scramble control (anti-miR-29c)were
co-transfected into primary cultured hippocampal cells. As shown in
Fig. 2A, the transfection
efficiency was satisfactory for further investigation. The
luciferase activity of the pre-miR-29c transfected cells was
significantly reduced, compared with the scramble control cells
(Fig. 2B). Additionally, The
results demonstrated that the overexpression of miR-29c
significantly reduced the mRNA and protein expression levels of
BACE1, whereas downregulation of miR-29c increased the mRNA and
protein expression levels of BACE1 (Fig. 2C). In addition, the protein
expression of Aβ in the culture medium was determined. The protein
expression of Aβ was decreased by the upregulation of miR-29c and
increased by downregulation of miR-29c (Fig. 2D). These results suggested that
miR-29c regulated the expression of BACE1 (Fig. 2E) at the transcriptional level by
directly targeting its 3′UTR.
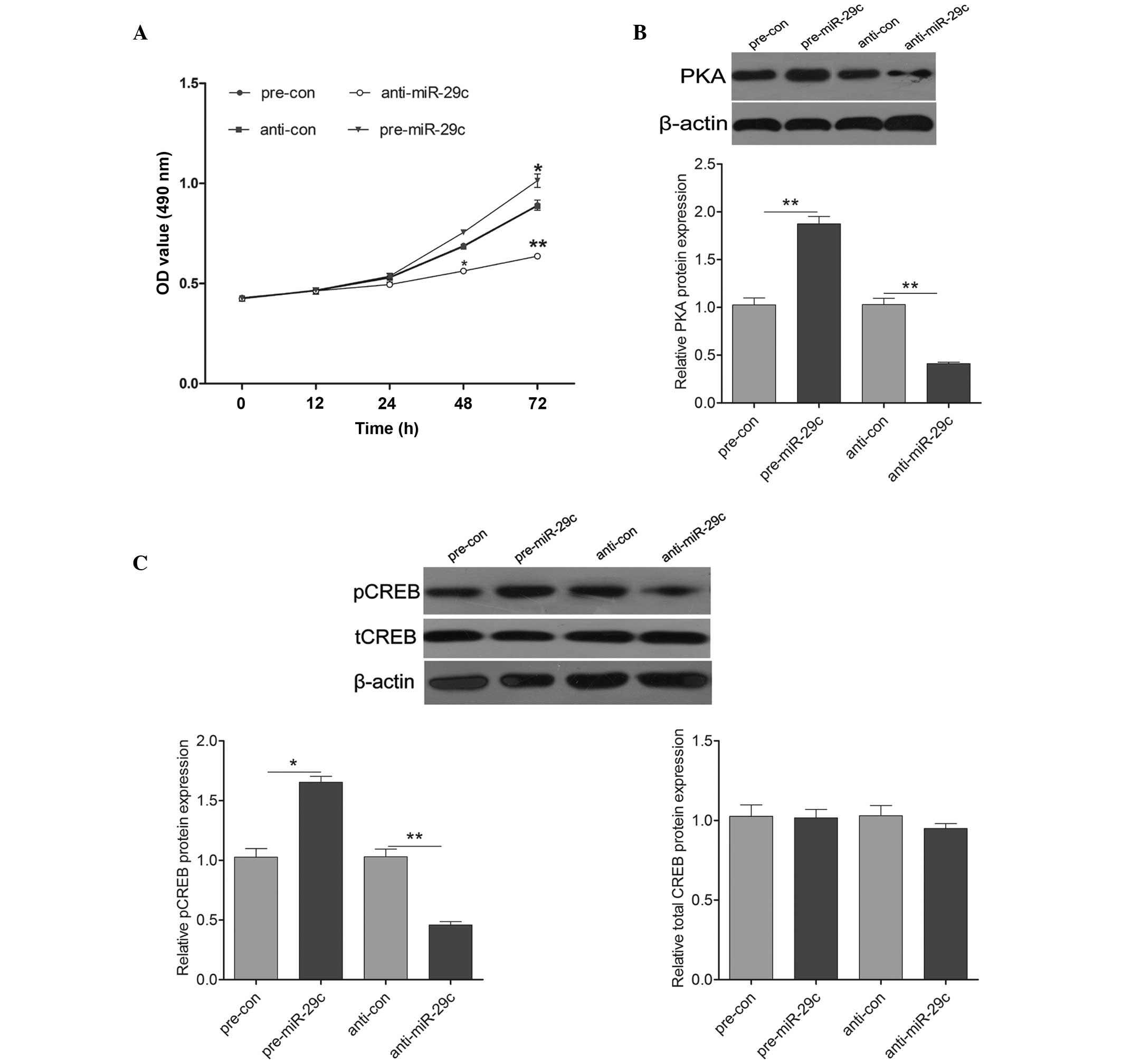
miR-29c promotes cell proliferation via
PKA signaling
To determine whether miR-29c regulated hippocampal
neuron proliferation, an MTT assay was performed by transfecting
either pre-miR-29c or anti-miR-29c into the primary cultured
hippocampal cells. Pre-miR-29c transfection exhibited a significant
promotion of cell proliferation, compared with the control cells,
whereas transfection with anti-miR-29c led to a significant
inhibition on cell proliferation (Fig.
3A). To further investigate the molecular mechanism underlying
miR-29c induction on cell growth, the expression levels of
molecules associated with PKA signaling were detected. As shown in
Fig. 3B, the protein expression of
PKA was increased by the upregulation of miR-29c and decreased by
the downregulation of miR-29c. Additionally, the expression of
pCREB was induced significantly following transfection with
pre-miR-29c, and was reduced following transfection with
anti-miR-29c (Fig. 3C). These
results suggested that miR-29c promoted hippocampal neuron cell
proliferation by activating PKA signaling.
Effects of miR-29c on SAMP8 mice
The present study subsequently examined the effects
of miR-29c in vivo, by injecting miR-29c mimics into the
hippocampi of SAMP8 mice. The expression levels of miR-29c in the
SAMP8 mice were significantly lower compared with those in the
SAMR1 mice. When the miR-29c mimic was injected into the hippocampi
of SAMP8 mice, the expression of miR-29c was significantly
increased compared with the vehicle-injected control mice (Fig. 4A). Similar to the effects in
vitro, the upregulation of miR-29c significantly decreased the
protein expression levels of BACE1 and Aβ in vivo (Fig. 4B and C). The present study also
examined the learning and memory behaviors of the SAMP8 mice 2
months following transfection with miR-29c. As shown in Fig. 4D, overexpression of miR-29c in the
hippocampus promoted the learning and memory behaviors of the SAMP8
mice. Furthermore, PAK signaling was also examined, and the protein
expression levels of PKA and pCREB were induced significantly by
the upregulation of miR-29c (Fig. 4E
and F).
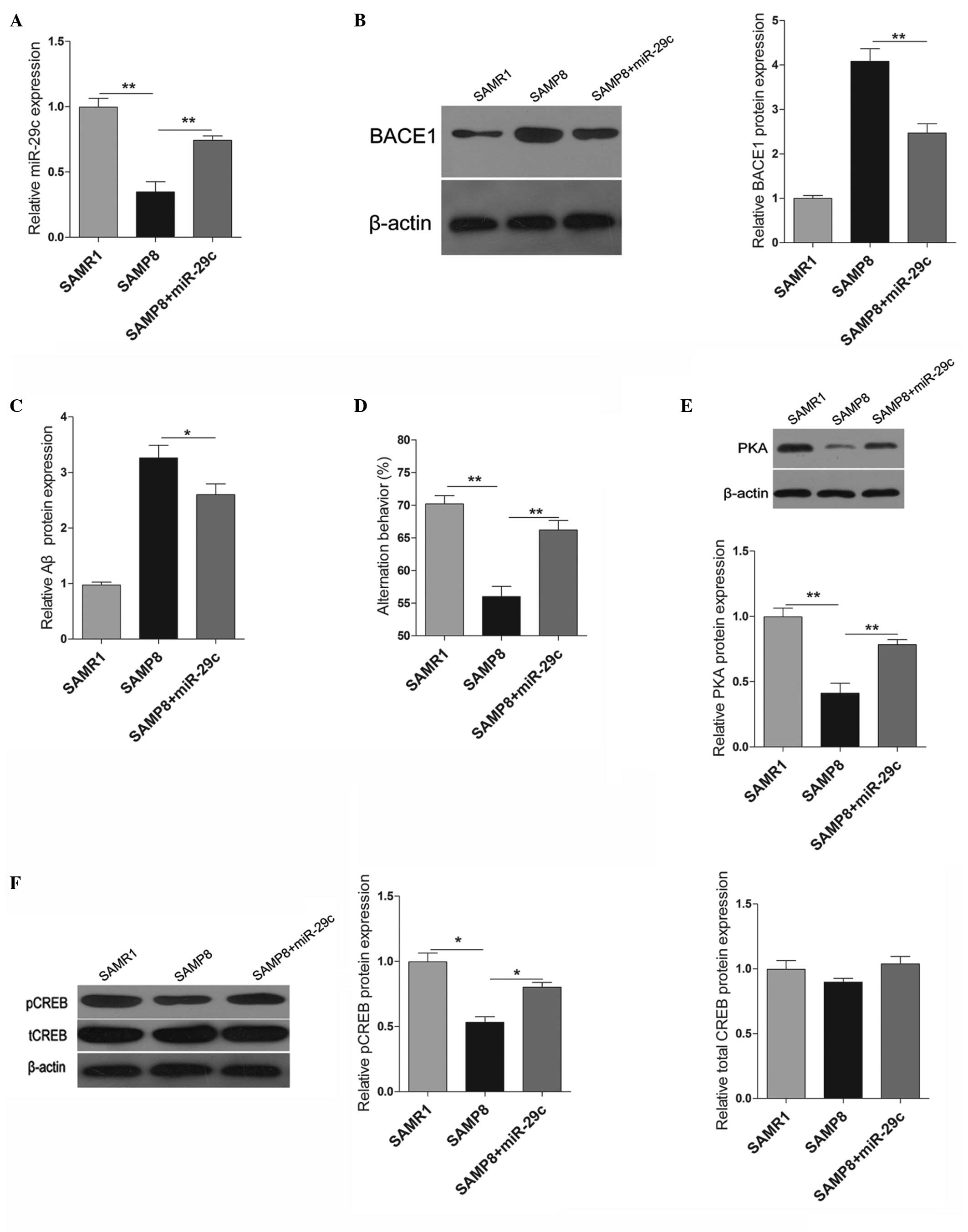 | Figure 4Effects of miR-29c in SAMP8 mice. (A)
Expression levels of miR-29c in the hippocampi of SAMR1, SAMP8 and
miR-29c-transfected-SAMP8 mice. (B) Protein expression levels of
BACE1 in the hippocampi of SAMR1, SAMP8 and
miR-29c-transfected-SAMP8 mice, and quantification of western
blotting. (C) An ELISA was used to determine the protein expression
of β-amylase in the hippocampi. (D) A Y-maze test was performed to
measure the learning and memory behaviors of the mice. (E) Western
blotting and quantification of the protein expression levels of
PKA. (F) Western blotting and quantification of the protein
expression levels of pCREB and tCREB. The data are expressed as the
mean ± standard deviation (*P<0.05 and
**P<0.01, vs. control). PKA, protein kinase A; CREB,
cAMP response element-binding protein; pCREB, phosphorylated CREB;
tCREB, total CREB; miR, microRNA; BACE1, β-site amyloid precursor
protein-cleaving enzyme 1. |
Discussion
Current cerebrospinal fluid (CSF) and peripheral
blood-based biomarkers, including Aβ, BACE1 (13) and tau, can support the clinical
diagnosis of AD with a sensitivity and specificity of ~90%
(14,15). Previous studies have focused on the
involvement of miRNAs in AD. In a previous study, miRNAs with a
differential expression in either the hippocampus or CSF from
patients with AD and age-matched healthy control individuals were
identified, the results of which suggested that low levels of
miR-146a in the CSF were associated with AD (16). In the present study, miR-29c was
significantly decreased in the peripheral blood of patients with AD
compared with the age-matched control individuals, and was
negatively correlated with an increased expression of BACE1. It was
previously reported, using a microarray, containing 328 miRNAs,
that reduced expression of the miR29a/b cluster is inversely
correlated with the protein expression of BACE1 in the anterior
temporal cortex of AD (5). The
present study revealed no differential expression of miR-29a/b,
however, differential expression of miR-29c was observed. A
reasonable explanation is that the expression levels of miRNAs
differs in different tissue samples. A previous study also
demonstrated that miR-29c is reduced in the peripheral blood of
patients with AD (17).
Increased expression of BACE1 has been previously
observed in the brain, CSF and peripheral blood of patients with AD
(18,19), suggesting that increased expression
of BACE1 is an important risk factor for AD. The present study also
revealed, using an ELISA, that the expression of BACE1 was
increased in the peripheral blood of patients with AD. In a
previous study, miR-29c, which is highly expressed in APPswe/PSΔE9
mice, can lower the protein expression of BACE1 in vitro and
in transgenic miR-29c mice (20).
In primary cultured hippocampal neurons obtained from SAMR1 mice,
the present study demonstrated that miR-29c regulated the
expression of BACE1 at the transcriptional level by directly
targeting its 3′UTR. Furthermore, the upregulation of miR-29c also
reduced the protein expression of Aβ by regulating BACE1. The
present study provided evidence to suggest that low expression
levels of miR-29c may have contributed to the deposition of Aβ by
regulating BACE1 in the hippocampalneurons, which the downregulated
miR-29c exhibited in the peripheral blood of patients with AD.
Therefore, combined with previous studies, miR-29c may be a
peripheral biomarker for AD.
Patients with AD are characterized by learning and
memory deficit, which is promoted by neurogenesis (21). The hippocampus is an important
region of neurogenesis in adulthood (22). Therefore, promoting the survival
and proliferation of hippocampal neurons is critical for the
treatment of AD. The present study demonstrated that upregulation
of miR-29c increased the proliferation of primary cultured
hippocampal neurons in vitro, whereas downregulation of
miR-29c impaired cell growth. PKA/CREB signaling has been
implicated in neuroprotection (23). It has been reported that induction
of pCREB via PKA is important in N-methyl-D-aspartate
receptor-mediated neuroprotection (24). Caffeine intake has been associated
with a lower incidence of AD in humans, and it has been revealed
that caffeine shifts the balance between neurodegeneration and
neuronal survival towards the stimulation of pro-survival cascades
and the inhibition of pro-apoptotic pathways in the striatum by
increasing PKA and pCREB (25). In
addition, several lines of evidence indicate that PKA/CREB are
involved in Aβ-trigged disruption of synaptic plasticity in AD
(26), and the overexpression of
BACE1 reduces the activity of PKA and pCREB, contributing to the
memory and cognitive deficits typical of AD (27). Therefore, activation of the
PKA/CREB pathway is beneficial against AD. The present study
demonstrated that upregulation of miR-29c induced the expression
levels of PKA and CREB in primary cultured hippocampal neurons
in vitro, and this molecular mechanism was subsequently
confirmed in vivo. Notably, significant reductions in BACE1
and Aβ were demonstrated in the hippocampi of SAMP8 mice following
the overexpression of miR-29c for 2 months. Furthermore, treatment
with miR-29c resulted in a significant increase in expression
levels of PKA and pCREB in the hippocampi of the SAMP8 mice. The
upregulation of miR-29c also improved the learning and memory
outcomes, which were determined using a Y-maze test, in the SAMP8
mice.
In conclusion, the results of the present study
suggested that miR-29c, as a peripheral AD biomarker, promoted
learning and memory behaviors in SAMP8 mice, which was associated
with a decrease in the production of Aβ by targeting BACE1 and
increasing the activity of PKA/CREB involved in neuro-protection.
Therefore, miR-29c may be a promising potential therapeutic target
against AD.
References
|
1
|
Maciotta S, Meregalli M and Torrente Y:
The involvement of microRNAs in neurodegenerative diseases. Front
Cell Neurosci. 7:2652013. View Article : Google Scholar
|
|
2
|
Rao AT, Degnan AJ and Levy LM: Genetics of
Alzheimer disease. AJNR Am J Neuroradiol. 35:457–458. 2014.
View Article : Google Scholar
|
|
3
|
Davinelli S, Calabrese V, Zella D and
Scapagnini G: Epigenetic nutraceutical diets in Alzheimer’s
disease. J Nutr Health Aging. 18:800–805. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wirth M, Villeneuve S, La Joie R, Marks SM
and Jagust WJ: Gene-environment interactions: Lifetime cognitive
activity, APOE genotype, and β-amyloid burden. J Neurosci.
34:8612–8617. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hébert SS, Horré K, Nicolaï L, et al: Loss
of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease
correlates with increased BACE1/beta-secretase expression. Proc
Natl Acad Sci USA. 105:6415–6420. 2008. View Article : Google Scholar
|
|
6
|
Selbach M, Schwanhäusser B, Thierfelder N,
et al: Widespread changes in protein synthesis induced by
microRNAs. Nature. 455:58–63. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fineberg SK, Kosik KS and Davidson BL:
MicroRNAs potentiate neural development. Neuron. 64:303–309. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hampel H, Shen Y, Walsh DM, et al:
Biological markers of amyloid beta-related mechanisms in
Alzheimer’s disease. Exp Neurol. 223:334–346. 2010. View Article : Google Scholar :
|
|
9
|
Wang WX, Rajeev BW, Stromberg AJ, et al:
The expression of microRNA miR-107 decreases early in Alzheimer’s
disease and may accelerate disease progression through regulation
of beta-site amyloid precursor protein-cleaving enzyme 1. J
Neurosci. 28:1213–1223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kocerha J, Kauppinen S and Wahlestedt C:
microRNAs in CNS disorders. Neuromolecular Med. 11:162–172. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals. Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press; Washington DC, US: 2011
|
|
12
|
Livak KF and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
13
|
Decourt B, Walker A, Gonzales A, et al:
Can platelet BACE1 levels be used as a biomarker for Alzheimer’s
disease? Proof-of-concept study. Platelets. 24:235–238. 2013.
View Article : Google Scholar
|
|
14
|
Shaw LM, Vanderstichele H, Knapik-Czajka
M, et al: Alzheimer’s Disease Neuroimaging Initiative:
Cerebrospinal fluid biomarker signature in Alzheimer’s disease
neuroimaging initiative subjects. Ann Neurol. 65:403–413. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bibl M, Esselmann H, Lewczuk P, et al:
Combined Analysis of CSF Tau, Aβ42, Aβ1-42% and AβCombined Analysis
of CSF Tau, Aβ42, Aβ1-42% and Aβ1-40% in Alzheimer’s Disease,
Dementia with Lewy Bodies and Parkinson’s Disease Dementia. Int J
Alzheimers Dis pii. 7615712010.
|
|
16
|
Müller M, Kuiperij HB, Claassen JA,
Küsters B and Verbeek MM: MicroRNAs in Alzheimer’s disease:
Differential expression in hippocampus and cell-free cerebrospinal
fluid. Neurobiol Aging. 35:152–158. 2014. View Article : Google Scholar
|
|
17
|
Bettens K, Brouwers N, Engelborghs S, et
al: APP and BACE1 miRNA genetic variability has no major role in
risk for Alzheimer disease. Hum Mutat. 30:1207–1213. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marques SC, Lemos R, Ferreiro E, et al:
Epigenetic regulation of BACE1 in Alzheimer’s disease patients and
in transgenic mice. Neuroscience. 220:256–266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cattabeni F, Colciaghi F and Di Luca M:
Platelets provide human tissue to unravel pathogenic mechanisms of
Alzheimer disease. Prog Neuropsychopharmacol Biol Psychiatry.
28:763–770. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zong Y, Wang H, Dong W, et al: miR-29c
regulates BACE1 protein expression. Brain Res. 1395:108–115. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cogswell JP, Ward J, Taylor IA, et al:
Identification of miRNA changes in Alzheimer’s disease brain and
CSF yields putative biomarkers and insights into disease pathways.
J Alzheimers Dis. 14:27–41. 2008.PubMed/NCBI
|
|
22
|
Moon M, Cha MY and Mook-Jung I: Impaired
hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD
mice. J Alzheimers Dis. 41:233–241. 2014.PubMed/NCBI
|
|
23
|
Shi YQ, Huang TW, Chen LM, et al:
Ginsenoside Rg1 attenuates amyloid-beta content, regulates PKA/CREB
activity and improves cognitive performance in SAMP8 mice. J
Alzheimers Dis. 19:977–989. 2010.
|
|
24
|
Valera E, Sánchez-Martín FJ,
Ferrer-Montiel AV, Messeguer A and Merino JM: NMDA-induced
neuroprotection in hippo-campal neurons is mediated through the
protein kinase A and CREB (cAMP-response element-binding protein)
pathway. Neurochem Int. 53:148–154. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zeitlin R, Patel S, Burgess S, Arendash GW
and Echeverria V: Caffeine induces beneficial changes in PKA
signaling and JNK and ERK activities in the striatum and cortex of
Alzheimer’s transgenic mice. Brain Res. 1417:127–136. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sierksma AS, Rutten K, Sydlik S, et al:
Chronic phosphodiesterase type 2 inhibition improves memory in the
APPswe/PS1dE9 mouse model of Alzheimer’s disease.
Neuropharmacology. 64:124–136. 2013. View Article : Google Scholar
|
|
27
|
Chen Y, Huang X, Zhang YW, et al:
Alzheimer’s β-secretase (BACE1) regulates the cAMP/PKA/CREB pathway
independently of β-amyloid. J Neurosci. 32:11390–11395. 2012.
View Article : Google Scholar : PubMed/NCBI
|