Introduction
Intracerebral hemorrhage (ICH) is a common and
severe subtype of stroke associated with high rates of morbidity
and mortality (1). Resulting from
ruptured blood vessel(s) in the brain, ICH is defined as bleeding
into the brain parenchyma (2). A
series of pathophysiological processes occurs following an acute
ICH outbreak, including apoptosis and necrosis, breakdown of the
blood-brain barrier, edema formation, inflammation and
extracellular matrix remodeling (1). These complications may result in
severe neuronal injury. Thus, it is important to understand the
underlying mechanisms of ICH in order to control the subsequent
complications. Despite this, relatively few studies have focused on
ICH and its subsequent effects compared with studies on ischemic
stroke (3–5).
Characterized by elevated expression levels of
pro-inflammatory cytokines, including interferon-β (IFN-β), tumor
necrosis factor-α (TNF-α) and interleukin-6 (IL-6), inflammation
induced by ICH is hypothesized to contribute to neurodegeneration
(6,7). Glucocorticoids (GCs) exert
anti-inflammatory effects, and are therefore considered to be
effective therapeutic agents in acute and chronic inflammation,
including severe shock, asthma, inflammatory bowel disease,
rheumatoid arthritis and multiple sclerosis (8–10).
In addition, studies investigating their efficacy in the treatment
of ICH-induced inflammation have revealed positive results
(11–13). However, the underlying mechanisms
through which GCs exert their anti-inflammatory and
immunosuppressive functions in ICH remain to be elucidated
(14).
MicroRNAs (miRs) are endogenous 20–23 nucleotide
small noncoding RNAs that are expressed in various tissues and are
responsible for regulating the expression of various genes. miRs
affect numerous biological processes, including development,
autoimmune diseases, inflammation and proliferation (15–19).
miRs function by shortening mRNA half-life or inhibiting
translation via binding the 3′untranslated region of target genes
(20). miR-155 is encoded by the
B-cell integration (BIC) gene located on chromosome 21 (20,21).
Previous studies have suggested that miR-155, similar to other
miRs, is closely associated with the development of inflammatory
pathogenesis (22). Furthermore,
Liu et al (23) revealed
the involvement of miR-155 in rat ICH.
Previous studies have focused on the
miR-155-mediated signaling pathway, in order to elucidate its
precise underlying molecular mechanisms in various biological
processes (20,21). Identified targets of miR-155 to
date include Fas-associated death domain protein, Src homology
2-containing inositol 5-phosphatase 1 and the suppressor of
cytokine signaling-1 (SOCS-1) (20,24–26).
In microglia-mediated immune reactions, miR-155 promoted
inflammation via the downregulation of SOCS-1 protein (6). However, the underlying mechanisms by
which miR-155 functions in ICH-induced inflammatory pathogenesis
remain to be elucidated.
In the present study, a mouse ICH model was
established according to our previous paper (27,28)
and miR-155 was overexpressed in astrocytes to investigate the role
of miR-155 in ICH-induced inflammation, as well as the underlying
mechanism of the anti-inflammatory effects of GCs.
Materials and methods
Animals and ICH-induction
All experimental procedures were in compliance with
the National Institutes for Health Guide for the Care and Use of
Laboratory Animals (29) and
approved by the Institutional Animal Care and Use Committee at
Soochow University (Suzhou, China). Animals, including both mice
and rats, were housed under pathogen-free conditions with 10:14 h
light: Dark cycle, at room temperature and 75% humidity. Animals
had free access to sterile water and chow. Protocols for
ICH-induction were conducted as described previously (27,28).
Briefly, 30 adult male ICR mice weighing 25–30 g and 6 neonatal
Sprague-Dawley rats were obtained from Shanghai Laboratory Animal
Center (Shanghai, China). ICH was induced by collagenase injection,
as previously described (27).
Mice were anesthetized with 4% chloral hydrate (0.4 mg/g) purchased
from Sigma-Aldrich (St. Louis, MO, USA) and placed in a
stereotactic apparatus. A cranial burr hole (1-mm in diameter) was
drilled, through which type IV collagenase purchased from
Sigma-Aldrich was injected (0.075 units in 500 nl of saline) into
the left striatum (1 mm anterior and 2 mm lateral of the bregma,
3.5 mm in depth). Collagenase was delivered over 5 min. The needle
was kept in place for an additional 5 min to prevent any reflux.
The control (sham) mice received an equivalent volume of sterile
saline injected in an identical manner. Body temperature was
maintained at 37±0.5°C using a heat lamp throughout surgery and
recovery.
Experimental groups and drug
administration
Mice were randomly assigned to three groups: control
(Ctrl), Sham surgery (sham) and ICH, with 6 mice per group. To
investigate the effect of GCs, dexamethasone (30 mg/kg) was
administered via i.p. injection twice a day for 3 consecutive days,
beginning on the day of surgery. Animals were sacrificed one day
after the last dose, a total of three days following surgery.
Animals were sacrificed by an intraperitoneal injection of
pentobarbital (40 mg/kg, Sigma-Aldrich) and brain tissues were
collected for analysis. Dexamethasone was purchased from
Sigma-Aldrich and administered according to previously published
protocols (27,30).
RNA isolation
Total RNA was extracted from brain tissue samples or
cultured astrocytes using TRIzol® Reagent (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's instructions. Any remaining DNA was removed with the
DNA-free kit (Ambion; Thermo Fisher Scientific, Inc.) and RNA was
purified with an RNeasy kit (Qiagen, Inc., Valencia, CA, USA),
according to the manufacturer's instructions.
Detection of miR-155 by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
For quantification of miR-155 levels, RT-qPCR was
performed on total RNA. miR-155 expression levels were determined
using a Hairpin-it™ miRs qPCR kit (Shanghai GenePharma Co., Ltd.,
Shanghai, China). In brief, the assay has two steps: Stem-loop RT
reaction and qPCR detection. Stem-loop RT primers bind to the 3′end
of miR molecules and are transcribed with reverse transcriptase.
Briefly, the total RNA was reverse transcribed using the M-MLV
reverse transcriptase system (Promega Corporation, Madison, WI,
USA). The reaction was performed at 42°C for 1 h and terminated by
deactivation of the enzyme at 70°C for 10 min. The RT product was
quantified by qPCR, using miR-specific forward and reverse primers
as follows: Forward, 5′-GTGCTGCAAACCAGGAAGG-3′ and reverse,
5′-CTGGTTGAATCATTGAAGATGG-3′, and a carboxyfluorescein dye-labeled
reporter probe. Thermal cycling conditions were as follows:
Pre-denaturation at 95°C for 5 min, denaturation at 95°C for 10
sec, annealing at 58°C for 15 sec, and extension at 72°C for 20
sec. Gene expression levels were standardized to a housekeeping
gene (U6) and expressed as a fold of control. To normalize RNA
content, the U6 small nuclear RNA served as the internal control.
The relative miR-155 expression levels of each group were
calculated using the ΔΔCq method (31).
Cytokine detection was performed by RT-qPCR using an
ABI 7300 RT-PCR instrument (Applied Biosystems; Thermo Fisher
Scientific, Inc.) using a SYBR Green-based RT-PCR kit
[SYBR® Premix Ex Taq™ (Tli RNaseH Plus); Takara Bio,
Inc., Otsu, Japan]. The primers used were as follows: IFN-β
(32) forward, 5′
AAGAGTTACACTGCCTTTGCCATC3′ and reverse, 5′CACTGTCTGCTG
GTGGAGTTCATC3′; TNF-α forward, 5′TGAACTTCGGGGTGA TTG GTC3′ and
reverse, 5′GCCTTGTCCCTTGAAGAGAAC3′; IL-6 forward,
5′-TACTTCACAAGTCCGGAG-3′ and reverse, 5′-TCCAGAAGACCAGAGCAG-3′;
β-actin forward, 5′-ACATCTGCTGGAAGGTCCAC-3′ and reverse,
5′-GGTACCACCATGTACCCAGG-3′. All reactions were performed three
times for each group.
Western blot analysis of SOCS-1 protein
expression levels
Protein extract prepared from tissue samples or
cultured astrocytes were examined by western blot analysis. Protein
was extracted using the ProteoExtract® Transmembrane
Protein Extraction kit (Merck Millipore, Darmstadt, Germany)
according to the manufacturer's protocol, and determination of
protein concentration was performed using a bicinchoninic acid
protein assay kit (Pierce; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Equal amounts of protein
(15 µg) were subjected to 10% SDS-PAGE (running at 60 V for
1 h through the stacking gel, and at 120 V for 1 h through the
resolving gel). Western blot analysis was performed using a mouse
antibody against SOCS1 (1:1,000 dilution, catalog no. 04-002, EMD
Millipore, Billerica, MA, USA) (27). Briefly, proteins were blotted onto
a polyvinylidene difluoride membrane (Roche Applied Science,
Penzburg, Germany) for 90 min at 350 mA. Then the membranes were
blocked with 0.1% Tris-buffered saline (TBS)-Tween buffer
containing 5% skim milk either overnight at 4°C or for 2 h at room
temperature. The membrane was subsequently incubated with
anti-SOCS1 antibody overnight at 4°C. Following the incubation, the
membrane was washed with 0.1% TBS-Tween buffer and probed with goat
anti-mouse HRP-conjugated IgG secondary antibody (1:2,000 dilution;
catalog no. sc-2005; Santa Cruz Biotechnology, Inc., Dallas, TX
USA) for 1 h at room temperature. Rabbit antibody against GADPH
(1:5,000 dilution; catalog no. KC-5G4; Kangcheng, Inc., Shanghai,
China) was used as the loading control. Protein bands were
visualized using enhanced chemiluminescence (Vazyme Biotech Co.,
Ltd., Nanjing, China) (27,33).
SOCS-1 expression levels were quantified by densitometry in
arbitrary units using ImageJ 1.44p software (National Institutes of
Health, Bethesda, MD, USA), and normalized to GAPDH.
Astrocyte isolation and culture
Astrocytes were isolated from mixed glial cultures
as described previously (34).
Briefly, brains harvested from 1- to 3-day-old Sprague Dawley rat
pups were minced, filtered and cultured in Dulbecco's modified
Eagle's medium (DMEM)/F-12 (Invitrogen; Thermo Fisher Scientific,
Inc.). At 100% confluence (1 week post-plating), the mixed glial
cultures were shaken at 200 rpm overnight at 37°C on a rotary
shaker to separate the microglia and oligodendrocytes from the
adherent astrocytes. The media, microglia and oligodendrocytes were
removed; astrocytes were trypsinized and subcultured in DMEM/F-12
supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, 100
mg/ml streptomycin, 100 mg/ml L-glutamine and 15 mM
4-(2-hydroxyethyl)-1-piperazineethanesulphonic aciction id (all
Sigma-Aldrich). Astrocytes were maintained at 37°C in 5%
CO2 and 95% air for 3 days.
Transfection of miRs
miR-155 and its mutant constructs were synthesized
by Integrated DNA Technologies, Inc., Coralville, IA, USA. The
sequences of anti-miR-155 antisense inhibitor oligonucleotides
(AMOs) used in the present study are exact antisense copies of
mature miR sequences, and the nucleotides in the AMOs contain
2′-O-methyl modifications at every base and a 3′C3 amino linker
(31). Oligo transfection was
performed according to an established protocol (35). Briefly, wild type (WT) cells were
transfected using PolyFect transfection reagent (Qiagen, Inc.) for
6 h. Transfection complexes were prepared according to the
manufacturer's instructions, and 2′OMe-miR-155 or control oligo
2′OMe-enhanced green fluorescent protein was added directly to the
complexes to a final oligonucleotide concentration of 30, 50 or 100
nmol/l. The transfection efficiency was low at 30 or 50 nmol/l.
However, when transfected at 100 nmol/l, transfection efficiency
reached ≥80%. Therefore, only cells receiving 100 nmol/l were
analyzed. The transfection medium was replaced 6 h subsequent to
transfection with regular culture medium, and cells were cultured
for 3 days prior to analysis.
Statistical analysis
Data are presented as the mean ± standard error.
Comparisons were made between groups using the Student's
t-test using SigmaPlot version 10 software (Systat Software,
Inc., San Jose, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-155 is involved in ICH pathogenesis
in mice in vivo
To evaluate the role of miR-155 in ICH-mediated
inflammation, a mouse model of ICH was used, and the expression of
miR-155 was determined by RT-qPCR three days subsequent to ICH
treatment. As presented in Fig.
1A, the expression level of miR-155 in the ICH group was
significantly increased compared with the control (P= 0.030) and
sham groups (P=0.038). Furthermore, to identify the signaling
pathway mediated by miR-155 in the ICH model, western blotting was
performed to examine the SOCS-1 protein expression levels in the
three groups. In contrast to miR-155, the expression level of
SOCS-1 protein was significantly decreased in the ICH group
compared with the control mice (P=0.021; Fig. 1B). In addition, significantly
increased mRNA expression levels of the pro-inflammatory cytokines,
IFN-β, IL-6 and TNF-α were observed following ICH treatment
compared with control groups (P=0.005,0.002 and P<0.001,
respectively; Fig. 1C). These data
indicate that miR-155 and SOCS-1 are involved in the inflammatory
pathogenesis induced by ICH. Furthermore, miR-155 and SOCS-1 are
antagonistic in this process.
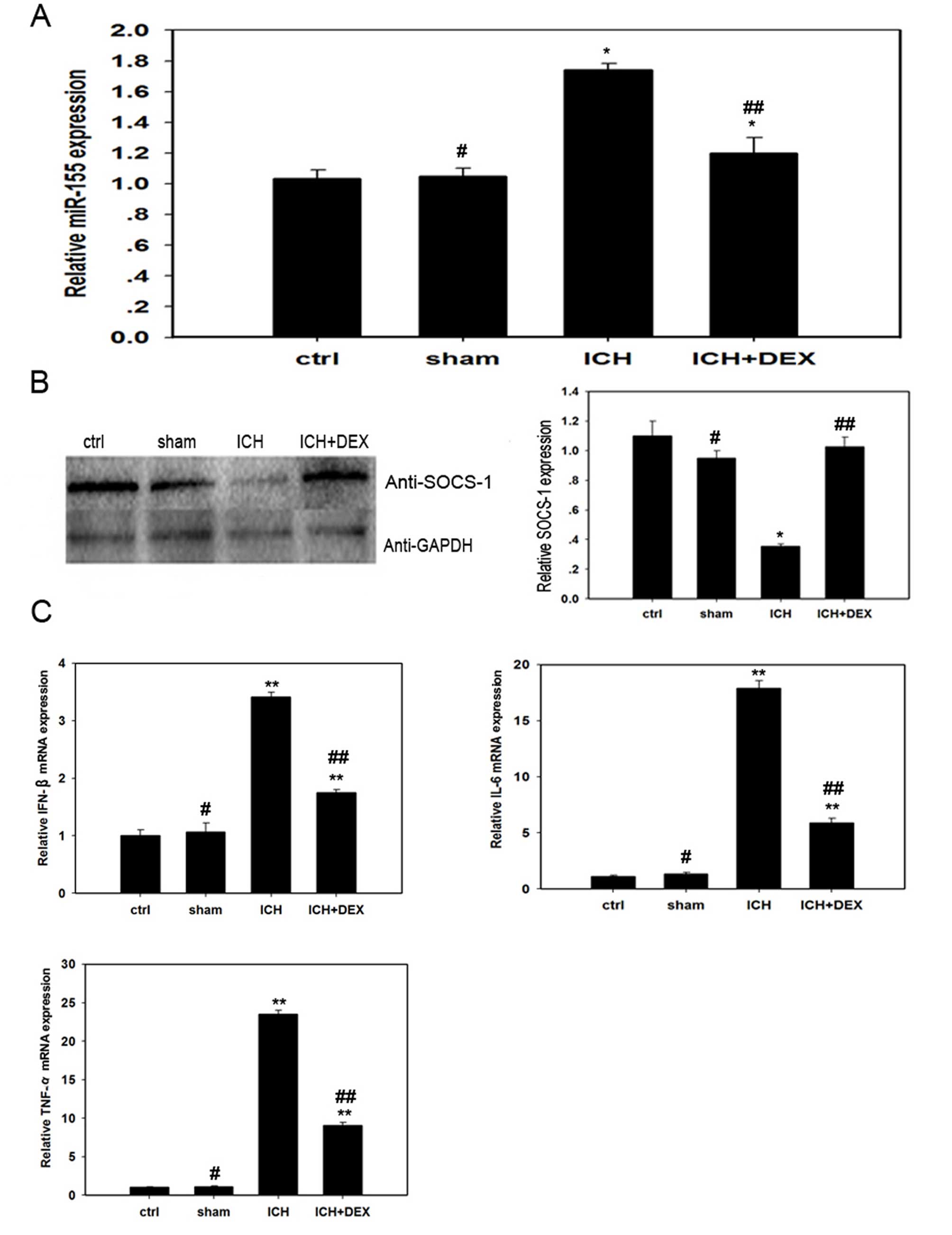 | Figure 1miR-155/SOCS-1 are involved in
ICH-induced inflammation and dexamethasone reduces inflammation by
inhibiting miR-155 expression in vivo. The expression levels
of (A) miR-155, (B) SOCS-1 and (C) pro-inflammatory cytokines were
determined under various treatment conditions. Mice were divided
randomly into four groups (n=36 per group): Ctrl group, without any
treatment; ICH group, injected with collagenase for ICH induction:
Sham group, injected with the equivalent volume of saline; and
ICH+DEX group, treated with dexamethasone following ICH induction.
Reverse transcription-quantitative polymerase chain reaction was
performed to determine miR-155, IFN-β, IL-6 and TNF-α mRNA
expression levels, while western blotting determined SOCS-1 protein
expression levels. Representative data is presented, as the mean ±
standard error, from one of at least three experiments.
*P<0.05 vs. ctrl; **P<0.01 vs. ctrl
(for ICH group) or P<0.01 vs. ICH group (for ICH+DEX group);
#P>0.05 vs. ctrl; and ##P<0.05 compared
with ICH group, as determined by Student's t-test using
SigmaPlot version 10.0 software. miR, microRNA; SOCS-1, suppressor
of cytokine signaling-1; ICH, intracerebral hemorrhage; ctrl,
control; DEX, dexamethasone; IFN-β, interferon-β; IL-6,
interleukin-6; TNF-α, tumor necrosis factor-α. |
Dexamethasone inhibits ICH-induced
inflammation by targeting miR-155 and SOCS-1
To elucidate the underlying functional mechanism of
GCs in ICH-induced inflammation, 30 mg/kg dexamethasone was
administered to mice following ICH induction. A total of three days
subsequent to administration, miR-155 expression was determined by
RT-qPCR. As presented in Fig. 1A,
the level of miR-155 expression in mice treated with dexamethasone
(ICH+DEX group) was significantly reduced compared with mice that
did not receive dexamethasone (ICH group; P=0.042). In addition,
ICH-mediated inhibition of the protein expression levels of SOCS-1
was reversed following dexamethasone administration (P=0.022;
Fig. 1B). Furthermore, the mRNA
expression levels of the cytokines IFN-β, IL-6 and TNF-α were
markedly decreased in the ICH+DEX group compared with the ICH group
(P=0.037, 0.024 and 0.018, respectively; Fig. 1C).
MiR-155 inhibits the expression of SOCS-1
protein in astrocytes in vitro
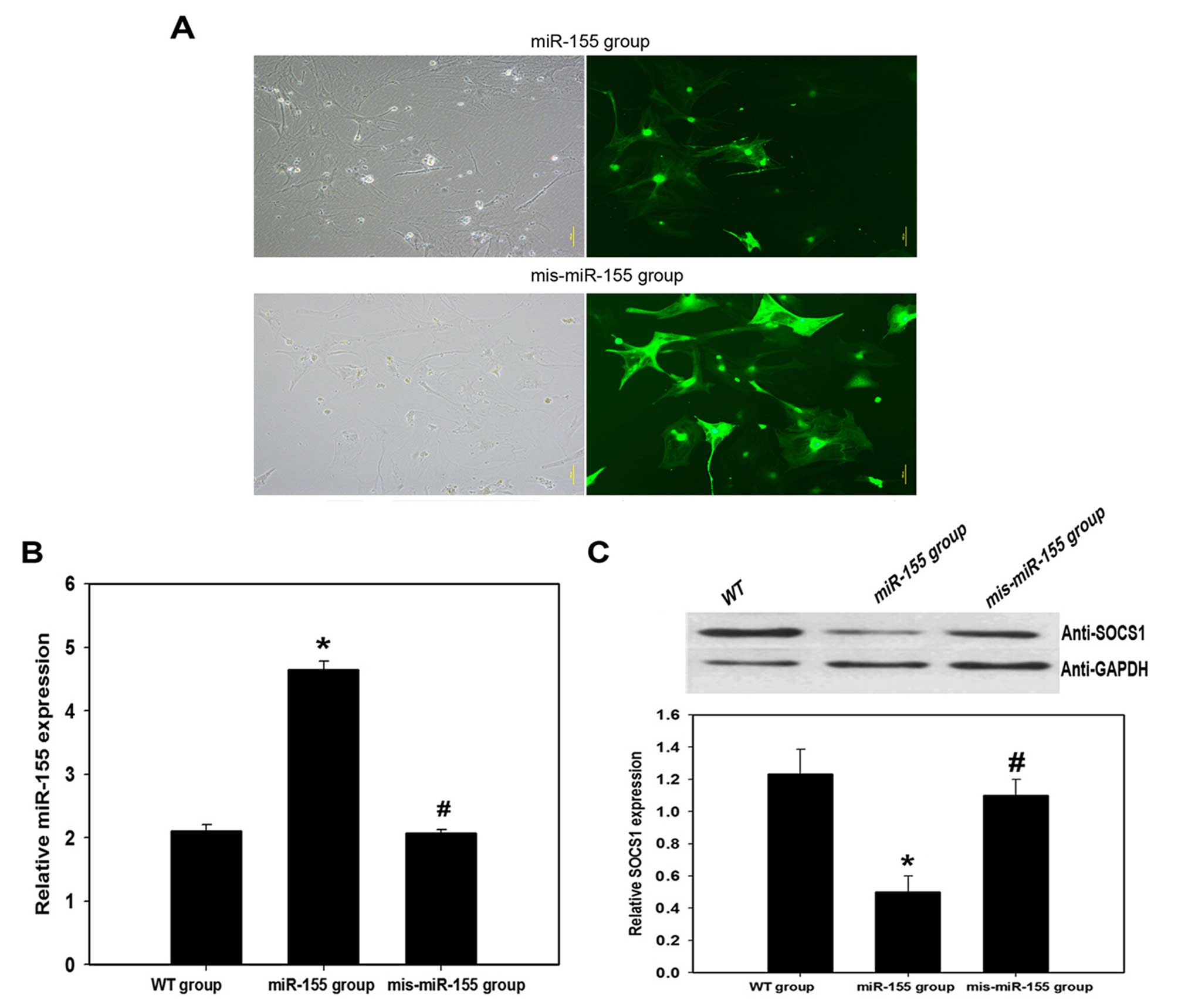
As astrocytes are the primary effector cells in the
inflammatory pathogenesis triggered by ICH, experiments were
performed in vitro using astrocytes to attempt to clarify
the association between miR-155 and SOCS-1 in the ICH model. WT
astrocytes were transfected with miR-155 oligonucleotides to
overexpress miR-155 (miR-155 group) or with mis-miR-155
oligonucleotides represented as scramble miR as a control
(mis-miR-155 group). Transfection efficiency was >80% and no
clear morphology impairment was observed following transfection
(Fig. 2A). The three populations
of astrocytes were collected and assayed for miR-155 expression by
RT-qPCR (Fig. 2B), and for SOCS-1
expression by western blotting (Fig.
2C). Overexpression of miR-155 led to a significant inhibition
in SOCS-1 protein expression levels compared with the control (WT
group; P=0.027). This indicates that SOCS-1 is a target of miR-155
in astrocytes, and that miR-155 may promote ICH-triggered
inflammatory pathogenesis by downregulating SOCS-1 expression.
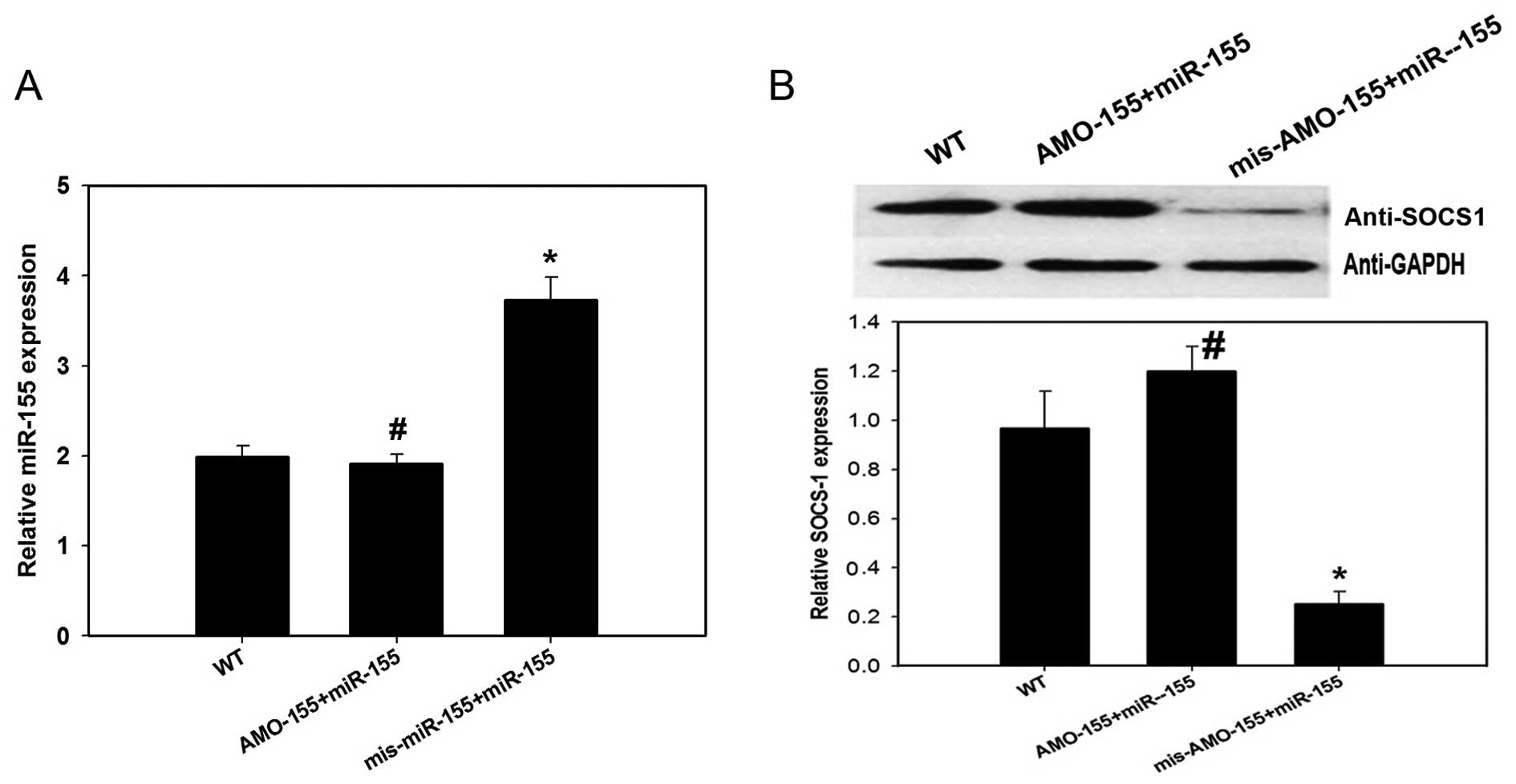
To confirm the association between miR-155 and
SOCS-1, anti-miR-155 antisense inhibitor oligonucleotides (AMO-155)
were synthesized to antagonize the expression of miR-155, and
mis-AMO-155 oligonucleotides represented as scramble miR served as
a control. AMO-155 and miR-155 oligonucleotides were transfected
simultaneously into certain astrocytes (AMO-155+miR-155 group) to
cancel out the miR-155 expression, while a separate group were
transfected with mis-AMO-155 together with miR-155 oligonucleotides
to examine the overexpression of miR-155 (mis-AMO-155+miR-155
group). As presented in Fig. 3A,
expression levels of miR-155 were increased in the
mis-AMO-155+miR-155 group (P=0.030) although not in the
AMO-155+miR-155 group (P=0.724), compared to WT cells. As presented
in Fig. 3B, expression levels of
SOCS-1 protein were significantly inhibited in the
mis-AMO-155+miR-155 group compared with the WT group (P=0.013);
however, this effect was abrogated in the AMO-155+miR-155 group
(P=0.373). These data suggest that miR-155 functions upstream and
inhibits the expression of SOCS-1 in astrocytes. As astrocytes are
important effector cells in ICH, the results of the in vitro
experiments suggest that miR-155 contributes to ICH-induced
inflammatory pathogenesis by inhibiting SOCS-1 expression.
Discussion
Based on the results of the present study, miR-155
may be required for the development of ICH-induced inflammation in
mice. Furthermore, the results of the present study revealed that
miR-155 downregulates the expression levels of SOCS-1 protein, but
increases the mRNA expression levels of the pro-inflammatory
cytokines IFN-β, IL-6 and TNF-α in vivo. In addition, the
association between miR-155 and SOCS-1 was demonstrated by
transfection of astrocytes in vitro. This signaling pathway
may be responsible for the effect of GCs on ICH-induced
inflammation, as miR-155 expression was significantly suppressed,
while SOCS-1 protein expression levels were significantly
increased, when dexamethasone was administered to ICH mice.
Previous studies have revealed that miR-155 is involved in
biological processes, including inflammatory pathogenesis (6,21,22);
however, the role of miR-155 in ICH-induced inflammation and its
specific signaling pathway remains to be elucidated. Although GCs
have been demonstrated to be effective in controlling various
inflammatory disorders (9,36,37),
the exact underlying mechanism of their anti-inflammatory effects
remains unclear. The results of the present study provide, to the
best of our knowledge, the first evidence of the involvement of
miR-155 in ICH-induced inflammatory pathogenesis and the signaling
pathway of GCs in this process. These results thus contribute to
our understanding of the underlying mechanisms involved in
ICH-induced inflammation.
The production of pro-inflammatory cytokines,
including IFN-β, IL-6 and TNF-α, is an important downstream effect
of ICH (1). Enhanced expression
levels of IFN-β, IL-6 and TNF-α mRNAs, accompanied by increased
expression of miR-155 and reduced expression of SOCS-1 protein
levels, indicates a critical role for the miR-155/SOCS-1 signaling
cascade in ICH-induced inflammation. Previous studies have
attributed the increased production of pro-inflammatory cytokines
following ICH to the activation of Toll-like receptors (TLRs),
particularly TLR2 and TLR4 (38).
The classical signaling pathway triggered by TLRs results in the
activation of nuclear factor (NF)-κB and mitogen-activated protein
kinases (JNK and p38), which leads to the transcription of
pro-inflammatory cytokine genes (39–41).
As miR-155 is transcribed from the BIC gene, the activation of
which is under the control of NF-κB, miR-155/SOCS-1 may function
downstream of TLR signaling following ICH. This has previously been
demonstrated in macrophage-mediated innate immune reactions
(42). Nevertheless, additional
studies are required to clarify the association between TLR
signaling and the miR-155/SOCS-1 signaling cascade following
ICH.
The significant decrease in the mRNA expression
levels of IFN-β, IL-6 and TNF-α, combined with the decrease in
miR-155 expression and enhanced expression levels of SOCS-1 protein
following dexamethasone administration, suggests that GCs,
including dexamethasone, function through the miR-155/SOCS-1
signaling pathway to attenuate ICH-induced inflammation. This
underlying functional mechanism of dexamethasone is consistent with
that of 1,25-dihydroxyvitamin D, which inhibits TLR-mediated
inflammation by targeting the miR-155/SOCS-1 signaling pathway in
macrophages (42). Whether other
regulatory mechanisms underlie GC function in ICH requires further
investigation.
The expression levels of miR-155 and the
pro-inflammatory cytokines in the ICH+DEX group, although inhibited
compared with the ICH group, remained significantly increased
compared with the control group. This suggests that GCs may produce
adverse effects on brain pathology. Potential adverse effects
caused by GCs have been reported previously, for instance on the
hypothalamus-pituitary-adrenal axis (43,44).
Due to the beneficial effects of GCs, future studies are required
to investigate their underlying functional mechanisms.
In conclusion, the results of the present study
demonstrate an essential role of the miR-155/SOCS-1 signaling
cascade in ICH-induced inflammation, and furthermore reveal that
GCs may relieve this inflammation by targeting the miR-155/SOCS-1
signaling pathway. Targeting the miR-155/SOCS-1 signaling pathway
may provide a novel therapeutic strategy for the treatment of
ICH-triggered inflammation.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81302617, 81172897,
81571848 and 81172898) and the Priority Academic Program
Development of Jiangsu Higher Education Institutions.
References
|
1
|
Magistris FB, Bazak SB and Martin J:
Intracerebral hemorrhage: Pathophysiology, diagnosis and
management. Clinical Review. 10:14–22. 2013.
|
|
2
|
Derex L and Nighoghossian N: Intracerebral
haemorrhage after thrombolysis for acute ischaemic stroke: An
update. J Neurol Neurosurg Psychiatry. 79:1093–1099. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang C, Ji B, Cheng B, Chen J and Bai B:
Neuroprotection of microRNA in neurological disorders (Review).
Biomed Rep. 2:611–619. 2014.PubMed/NCBI
|
|
4
|
Jickling GC, Ander BP, Zhan X, Noblett D,
Stamova B and Liu D: microRNA expression in peripheral blood cells
following acute ischemic stroke and their predicted gene targets.
PLoS One. 9:e992832014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tan JR, Koo YX, Kaur P, Liu F, Armugam A,
Wong PT and Jeyaseelan K: microRNAs in stroke pathogenesis. Curr
Mol Med. 11:76–92. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cardoso AL, Guedes JR, Pereira de Almeida
L and Pedroso de Lima MC: miR-155 modulates microglia-mediated
immune response by down-regulating SOCS-1 and promoting cytokine
and nitric oxide production. Immunology. 135:73–88. 2012.
View Article : Google Scholar :
|
|
7
|
Lansberg MG, Albers GW and Wijman CA:
Symptomatic intracerebral hemorrhage following thrombolytic therapy
for acute ischemic stroke: A review of the risk factors.
Cerebrovasc Dis. 24:1–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Coutinho AE and Chapman KE: The
anti-inflammatory and immunosuppressive effects of glucocorticoids,
recent developments and mechanistic insights. Mol Cell Endocrinol.
335:2–13. 2011. View Article : Google Scholar :
|
|
9
|
Zheng Y, Xiong S, Jiang P, Liu R, Liu X,
Qian J, Zheng X and Chu Y: Glucocorticoids inhibit
lipopolysaccharide-mediated inflammatory response by downregulating
microRNA-155: A novel anti-inflammation mechanism. Free Radic Biol
Med. 52:1307–1317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Spies CM, Strehl C, van der Goes MC,
Bijlsma JW and Buttgereit F: Glucocorticoids. Best Pract Res Clin
Rheumatol. 25:891–900. 2011. View Article : Google Scholar
|
|
11
|
McMaster A and Ray DW: Modelling the
glucocorticoid receptor and producing therapeutic agents with
anti-inflammatory effects but reduced side-effects. Exp Physiol.
92:299–309. 2007. View Article : Google Scholar
|
|
12
|
Kreitzer N and Adeoye O: An update on
surgical and medical management strategies for intracerebral
hemorrhage. Semin Neurol. 33:462–467. 2013. View Article : Google Scholar
|
|
13
|
Gu YT, Zhang H and Xue YX: Dexamethasone
treatment modulates aquaporin-4 expression after intracerebral
hemorrhage in rats. Neurosci Lett. 413:126–131. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vandevyver S, Dejager L, Tuckermann J and
Libert C: New insights into the anti-inflammatory mechanisms of
glucocorticoids: An emerging role for
glucocorticoid-receptor-mediated transactivation. Endocrinology.
154:993–1007. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jing J, Wu J, Liu W, Xiong S, Ma W, Zhang
J, Wang W, Gui JF and Mei J: Sex-biased miRNAs in gonad and their
potential roles for testis development in yellow catfish. PLoS One.
9:e1079462014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin F, Yao L, Xiao J, Liu D and Ni Z:
MiR-206 functions as a tumor suppressor and directly targets K-Ras
in human oral squamous cell carcinoma. Onco Targets Ther.
7:1583–1591. 2014.PubMed/NCBI
|
|
17
|
Liu H, Yang Y, Zhang L, Liang R, Ge RS,
Zhang Y, Zhang Q, Xiang Q, Huang Y and Su Z: Basic fibroblast
growth factor promotes stem Leydig cell development and inhibits
LH-stimulated androgen production by regulating microRNA
expression. J Steroid Biochem Mol Biol. 144:483–491. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ramirez-Salazar EG, Salinas-Silva LC,
Vázquez-Manríquez ME, Gayosso-Gómez LV, Negrete-Garcia MC,
Ramírez-Rodriguez SL, Chávez R, Zenteno E, Santillán P,
Kelly-García J and Ortiz-Quintero B: Analysis of microRNA
expression signatures in malignant pleural mesothelioma, pleural
inflammation and atypical mesothelial hyperplasia reveals common
predictive tumorigenesis-related targets. Exp Mol Pathol.
97:375–385. 2014. View Article : Google Scholar
|
|
19
|
Lu C, Huang X, Zhang X, Roensch K, Cao Q,
Nakayama KI, Blazar BR, Zeng Y and Zhou X: miR-221 and miR-155
regulate human dendritic cell development, apoptosis, and IL-12
production through targeting of p27kip1, KPC1, and SOCS-1. Blood.
117:4293–4303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu
H, Liu MF and Wang ED: MicroRNA-155 functions as an OncomiR in
breast cancer by targeting the suppressor of cytokine signaling 1
gene. Cancer Res. 70:3119–3127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yao R, Ma YL, Liang W, Li HH, Ma ZJ, Yu X
and Liao YH: MicroRNA-155 modulates Treg and Th17 cells
differentiation and Th17 cell function by targeting SOCS1. PLoS
One. 7:e460822012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kanwal N, John P and Bhatti A:
MicroRNA-155 as a therapeutic target for inflammatory diseases.
Rheumatol Int. 33:557–560. 2013. View Article : Google Scholar
|
|
23
|
Liu DZ, Tian Y, Ander BP, Xu H, Stamova
BS, Zhan X, Turner RJ, Jickling G and Sharp FR: Brain and blood
microRNA expression profiling of ischemic stroke, intracerebral
hemorrhage, and kainate seizures. J Cereb Blood Flow Metab.
30:92–101. 2010. View Article : Google Scholar
|
|
24
|
Zhu GF, Yang LX, Guo RW, Liu H, Shi YK,
Wang H, Ye JS, Yang ZH and Liang X: miR-155 inhibits oxidized
low-density lipoprotein-induced apoptosis of RAW264.7 cells. Mol
Cell Biochem. 382:253–261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xue H, Hua LM, Guo M and Luo JM: SHIP1 is
targeted by miR-155 in acute myeloid leukemia. Oncol Rep.
32:2253–2259. 2014.PubMed/NCBI
|
|
26
|
O'Connell RM, Chaudhuri AA, Rao DS and
Baltimore D: Inositol phosphatase SHIP1 is a primary target of
miR-155. Proc Natl Acad Sci USA. 106:7113–7118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chang P, Dong W, Zhang M, Wang Z, Wang Y,
Wang T, Gao Y, Meng H, Luo B, Luo C, et al: Anti-necroptosis
chemical necrostatin-1 can also suppress apoptotic and autophagic
pathway to exert neuroprotective effect in mice intracerebral
hemorrhage model. J Mol Neurosci. 52:242–249. 2014. View Article : Google Scholar
|
|
28
|
Wang T, Huang Y, Zhang M, Wang L, Wang Y,
Zhang L, Dong W, Chang P, Wang Z, Chen X and Tao L: [Gly14]-Humanin
offers neuroprotection through glycogen synthase kinase-3β
inhibition in a mouse model of intracerebral hemorrhage. Behav
Brain Res. 247:132–139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
McGlone JJ and Swanson J: Update on the
guide for the care and use of agricultural animals in research and
teaching. J Dairy Sci. 93:122010.
|
|
30
|
Wang T, Zhang L, Zhang M, Bao H, Liu W,
Wang Y, Wang L, Dai D, Chang P, Dong W, et al: [Gly14]-Humanin
reduces histopathology and improves functional outcome after
traumatic brain injury in mice. Neuroscience. 231:70–81. 2013.
View Article : Google Scholar
|
|
31
|
Luo X, Xiao J, Lin H, Li B, Lu Y, Yang B
and Wang Z: Transcriptional activation by stimulating protein 1 and
post-transcriptional repression by muscle-specific microRNAs of
IKs-encoding genes and potential implications in regional
heterogeneity of their expressions. J Cell Physiol. 212:358–367.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Roth-Cross JK, Martínez-Sobrido L, Scott
EP, García-Sastre A and Weiss SR: Inhibition of the alpha/beta
interferon response by mouse hepatitis virus at multiple levels. J
Virol. 81:7189–7199. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fang X, Hu H, Xie J, Zhu H, Zhang D, Mo W,
Zhang R and Yu M: An involvement of neurokinin-1 receptor in
FcεRI-mediated RBL-2H3 mast cell activation. Inflamm Res.
61:1257–1263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tallant EA and Higson JT: Angiotensin II
activates distinct signal transduction pathways in astrocytes
isolated from neonatal rat brain. Glia. 19:333–342. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Caplen NJ, Parrish S, Imani F, Fire A and
Morgan RA: Specific inhibition of gene expression by small
double-stranded RNAs in invertebrate and vertebrate systems. Proc
Natl Acad Sci USA. 98:9742–9747. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van der Velden VH: Glucocorticoids:
Mechanisms of action and anti-inflammatory potential in asthma.
Mediators Inflamm. 7:229–237. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schleimer RP: An overview of
glucocorticoid anti-inflammatory actions. Eur J Clin Pharmacol.
45(Suppl 1): S3–S7; discussion S43–S44. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang YC, Zhou Y, Fang H, Lin S, Wang PF,
Xiong RP, Chen J, Xiong XY, Lv FL, Liang QL and Yang QW: Toll-like
receptor 2/4 heterodimer mediates inflammatory injury in
intracerebral hemorrhage. Ann Neurol. 75:876–889. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Arroyo DS, Soria JA, Gaviglio EA,
Rodriguez-Galan MC and Iribarren P: Toll-like receptors are key
players in neurodegeneration. Int Immunopharmacol. 11:1415–1421.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kong Y and Le Y: Toll-like receptors in
inflammation of the central nervous system. Int Immunopharmacol.
11:1407–1414. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Eberle ME and Dalpke AH: Dectin-1
stimulation induces suppressor of cytokine signaling 1, thereby
modulating TLR signaling and T cell responses. J Immunol.
188:5644–5654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen Y, Liu W, Sun T, Huang Y, Wang Y, Deb
DK, Yoon D, Kong J, Thadhani R and Li YC: 1,25-Dihydroxyvitamin D
promotes negative feedback regulation of TLR signaling via
targeting microRNA-155-SOCS1 in macrophages. J Immunol.
190:3687–3695. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li ZQ, Liang GB, Xue YX and Liu YH:
Effects of combination treatment of dexamethasone and melatonin on
brain injury in intracerebral hemorrhage model in rats. Brain Res.
1264:98–103. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lema PP, Girard C and Vachon P: Evaluation
of dexamethasone for the treatment of intracerebral hemorrhage
using a collagenase-induced intracerebral hematoma model in rats. J
Vet Pharmacol Ther. 27:321–328. 2004. View Article : Google Scholar : PubMed/NCBI
|