Introduction
It is estimated that >285 million people
worldwide have diabetes mellitus, and that this figure will reach
439 million by 2030 (1). Diabetes
mellitus is a leading cause of end-stage renal disease and chronic
kidney failure that has become a worldwide problem (2–4).
Growing evidence suggests that tubulointerstitial fibrosis is the
final common pathway of almost all forms of chronic progressive
renal disease, including diabetic nephropathy (DN) (5–7).
Clinical evidence has confirmed that early tubular injury,
including fibrosis, has been reported in patients with diabetes
mellitus (8–10). Renal interstitial fibrosis involves
expansion of interstitial fibroblasts, myofibroblast activation and
extracellular matrix (ECM) accumulation, leading to the loss of
normal kidney function and, ultimately, renal failure (11). The specific therapeutic options to
inhibit the progression of DN are not available in the clinic. A
major reason for this relentless progression is potentially
associated with the incomplete understanding of the pathogenic
mechanisms of DN, which is fundamental for the development of more
effective preventive or therapeutic strategies.
Transforming growth factor (TGF)-β1, a strong
profibrotic cytokine, has been consistently implicated in the
pathogenesis of ECM accumulation in DN (12,13).
Factors that are associated with the pathogenesis of DN can
increase renal TGF-β1 expression in vivo in experimentally
induced-diabetes, and in diabetic humans (14,15).
A previous study reported that chronic treatment of
db/db mice with a neutralizing anti-TGF-β1 antibody
successfully prevented mesangial matrix expansion and renal
insufficiency, indicating that the TGF-β1 system is important in
the development of DN (13). In
addition, in cell culture experiments, cultured proximal tubular
cells exposed to media containing increasing concentrations of
D-glucose synthesize more TGF-β1 than control cells cultured in
normal-glucose medium (16,17).
Furthermore, in vivo study demonstrated that repeated
administration of a neutralizing anti-TGF-β1 antibody ameliorates
certain early changes observed in the kidneys of streptozotocin
(STZ)-induced diabetic mice, including increased mRNA levels of
collagen and fibronectin, and renal and glomerular hypertrophy
(18). The Smad protein family is
an important signaling pathway by which TGF-β1 activates the
transcription of several well-established TGF-β1-induced genes with
various functions. TGF-β1 receptor activation triggers
phosphorylation of receptor-regulated Smad2 and 3, which bind to
Smad4 and accumulate in the nucleus, where they activate
transcription (19,20).
Zinc is an essential element. Intracellular zinc is
associated with proteins, primarily via complex interactions with
cysteines, acting as an integral component of numerous
metalloenzymes, structural proteins and transcription factors
(21,22). Zinc deficiency (ZnD) is associated
with multiple disorders. In particular, hyperzincuria and low
intestinal absorption of zinc in diabetic patients suggests that
they are more susceptible to ZnD, which may result from
hyperglycemia, impaired intestinal absorption or increased urinary
zinc loss (23–25). In addition, low-dietary zinc intake
and low levels of serum zinc are associated with a high prevalence
of cardiovascular diseases, diabetes and glucose intolerance, and
low zinc status may to contribute to diabetes-associated renal
injury (26,27). Furthermore, a recent clinical study
demonstrated that advancing DN, indicated by decreased glomerular
filtration rate, and increasing microalbuminuria is associated with
lower serum zinc levels (27).
Indeed, numerous studies have indicated that zinc supplementation
inhibits fibrosis, including myocardial, liver, perivascular and
cystic fibrosis (28–30). A previous report demonstrated that
zinc is involved in high glucose-induced epithelial-mesenchymal
transition (EMT) in normal rat tubular epithelial cells by
modulating TGF-β1 and reactive oxygen species production, and
phosphoinositide 3-kinase and mitogen-activated protein kinase
activation (31). However, the
effect of zinc on DN, particularly in renal interstitial fibrosis,
in vivo has not been previously studied.
As there are few reports that have investigated the
effects of ZnD in renal interstitial fibrosis, it is necessary to
understand the matrix synthesis and degradation, and the mechanisms
by which ZnD influences the functional and pathological
consequences of DN in mouse models of DN.
Materials and methods
Animals
C57BL/6 J mice (4 weeks old; ~13±2 g) were purchased
from the Experimental Animal Center, China Medical University
(Shenyang, China). C57BL/6 J mice were administered with STZ (n=16;
150 mg/kg; Sigma-Aldrich; Merck Millipore, Darmstadt, Germany)
diluted in 0.1 M citrate buffer (pH 4.5) or citrate buffer alone
(n=8) by intraperitoneal injection, as described previously
(32). Mice serving as vehicle
controls (nondiabetic mice) were administered with the same volume
of citrate buffer. Mice were weighed and their blood glucose levels
were measured using an Accu-chek glucometer (Roche Diagnostics,
Basel, Switzerland) and only diabetic animals with blood glucose
>16 mmol/l were considered diabetic. When diabetic mice were
diagnosed on day 3 following STZ treatment, the mice were then
randomized into two groups, receiving treatment with zinc-deficient
(n=8) or zinc-adequate (n=8) diets for 5 weeks prior to euthanasia
at 12 weeks. The zinc-deficient mice received egg white-based diet
(0.85 ppm; cat. no. AIN-76A; Research Diets Company, New Brunswick,
NJ, USA) and were provided with deionized water; the zinc-adequate
mice (30 ppm) and had ad libitum access to water. The
zinc-adequate mice received a diet with normal zinc levels produced
by the Experimental Animal Center of China Medical University. The
mice were housed in cages in a controlled environment (22–25°C; 50%
humidity; and 12-h light/dark cycle). All animal experimental
procedures were approved by the Experimental Animal Ethical
Committee of China Medical University, in accordance with the
criteria described in the National Institutes of Health Guide for
the Care and Use of Laboratory Animals. At the time of sacrifice,
24-h urines were collected in metabolic cages. Urine albumin levels
were determined using the murine microalbuminuria ELISA kit (cat.
no. 1011; Exocell, Inc., Philadelphia, PA, USA). The mice were
terminally anesthetized with sodium pentobarbital (4%; Merck
Millipore) and perfused with isotonic saline, followed by 4%
paraformaldehyde in 0.1 M PBS (pH 7.4). The kidney was removed and
postfixed with 4% paraformaldehyde overnight at 4°C for use in
immunofluorescence. To obtain tissues for western blot analysis,
the mice anaesthetized with 4% sodium pentobarbital and
subsequently decapitated.
Zinc analysis
The zinc concentration in the plasma and kidney was
measured by atomic absorption spectrophotometry (AAS) at the
Experimental Center, China Medical University as described
previously (33). Total zinc in
the tissues, including free zinc and protein-bound zinc, were
measured and expressed as µg/g of dry tissue. The results are
expressed as mg/g of wet weight and as averages of at least two
determinations per sample.
Immunofluorescence staining
Serial paraffin sections (5 µm) of kidney tissues
were prepared, dewaxed in xylene, and rehydrated using gradient
alcohol solutions. Subsequently, the cryostat sections were
pre-incubated with 5% normal donkey serum (1:20; Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA) for 1 h and
incubated overnight at room temperature with anti-lectin (1:100;
polyclonal antibody; cat. no. HPA000646; Sigma-Aldrich; Merck
Millipore), anti-collagen-I (1:100; cat. no. sc-59772; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and anti-α-smooth muscle
actin (α-SMA; 1:100; cat. no. sc-324317; Santa Cruz Biotechnology,
Inc.). Secondary antibodies used included fluorescein
isothiocyanate (FITC)-conjugated donkey anti-rabbit (FITC-DAR)
immunoglobulin G (IgG; cat. no. 705 07 003) and Texas
Red-conjugated donkey anti-goat (Texas Red-DAG) IgG (cat. no. 711
542 152). They were purchased from Jackson ImmunoResearch
Laboratories, Inc. Following rinsing with PBS, the sections were
incubated for 2 h with DAR-FITC (1:50) and Texas Red-DAM (1:50) at
room temperature. The sections were mounted and examined using a
confocal laser scanning microscope (SP2; Leica Microsystems GmbH,
Wetzlar, Germany).
Western blot analysis
Kidney tissues were homogenized in cold
radioimmunoprecipitation assay lysis buffer, incubated on ice for 1
h, centrifuged at 12,000 × g for 20 min at 4°C and then the
supernatants were transferred to a clean tube. Protein
concentrations were quantified using a Bio-Rad protein assay kit
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Total protein from
each sample (50 µg) was subjected to SDS-PAGE using 10% gradient
Tris/glycine gels. Then, the proteins were transferred to
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). Following blocking with 5% fat-free milk for 1 h, the blots
were incubated with the following primary antibodies at 4°C
overnight: Type I collagen (1:800; cat. no. sc-59772; Santa Cruz
Biotechnology, Inc.); rabbit polyclonal anti-vimentin (1:800; cat.
no. sc-5565; Santa Cruz Biotechnology, Inc.); goat polyclonal
anti-α-SMA (1:800; cat. no. sc-324317; Santa Cruz Biotechnology,
Inc.); anti-TGF-β1 (1:1,000; cat. no. 3711; Cell Signaling
Technology, Inc., Danvers, MA, USA); anti-Smad2 (1:1,000; Cell
Signaling Technology, Inc.); anti-Smad3 (1:1,000; Cell Signaling
Technology, Inc.); and mouse monoclonal anti-β-actin (1:2,000;
sc-8432; Santa Cruz Biotechnology, Inc.). The membranes were then
incubated with horseradish peroxidase-conjugated secondary
antibodies, including rabbit anti-goat IgG (1:1,000; cat. no.
sc-2922; Santa Cruz Biotechnology, Inc.) and rabbit anti-mouse IgG
(1:1,000; cat. no. sc-358920; Santa Cruz Biotechnology, Inc.) for 2
h at 4°C. Subsequently, the membranes were visualized using an
enhanced chemiluminescence kit (Santa Cruz Biotechnology, Inc.)
using the ChemiDoc™ XRS system with Quantity One software (version
4.6; Bio-Rad Laboratories, Inc.) and the G-BOX EF Chemi HR16 gel
imaging system (Syngene, Frederick MD, USA). Following development,
the band intensities were quantified using Image Pro Plus 6.0
analysis software (Media Cybernetics, Inc., Rockville, MD,
USA).
Statistical analysis
Results from at least three independent experiments
were expressed as the mean ± standard error. Statistical analysis
of the data for multiple groups was performed by one-way analysis
of variance and the Tukey's multiple comparison test was used.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ZnD enhances renal injury in an
STZ-induced diabetic model
The effect of ZnD on DN was determined using an
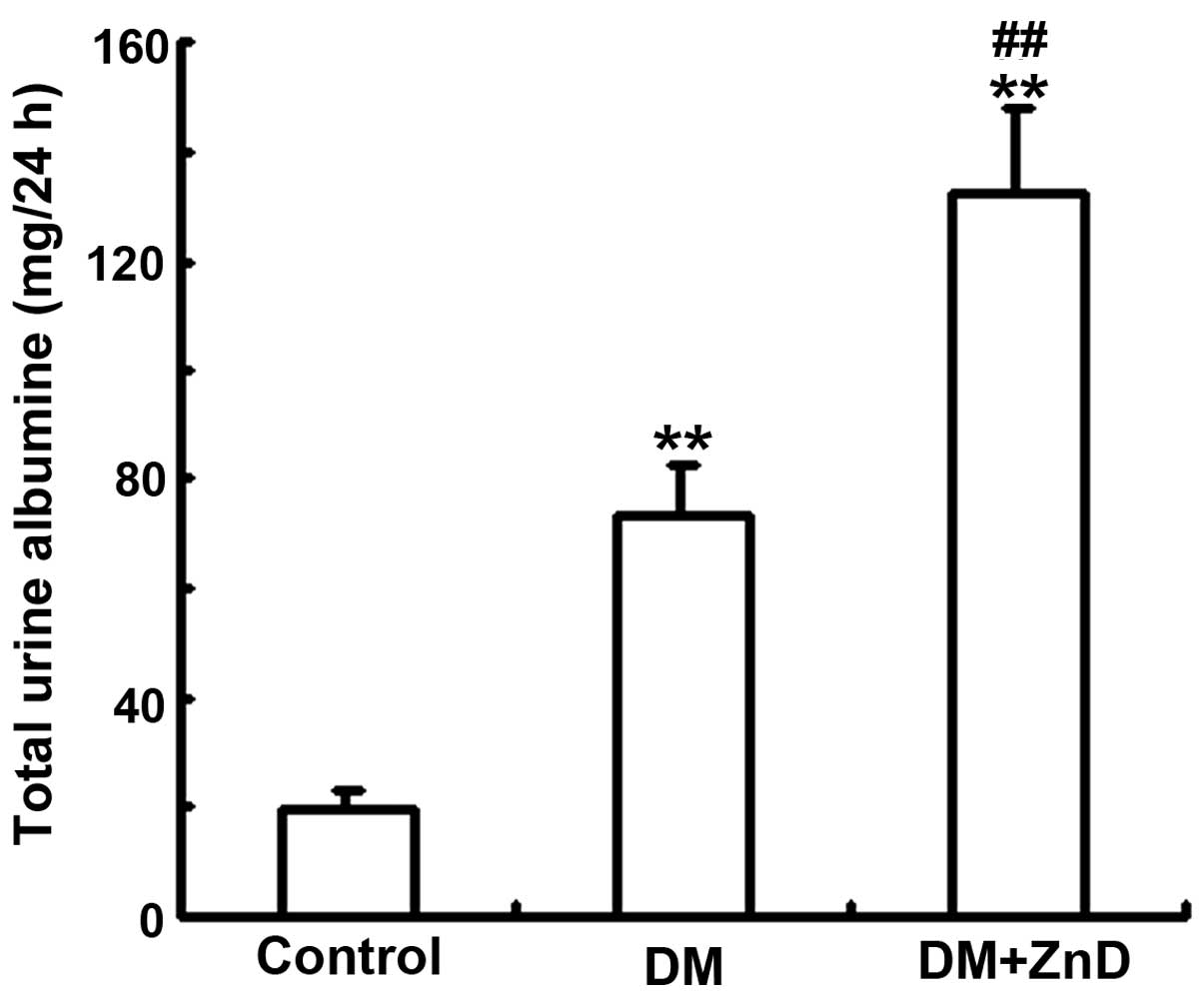
in vivo model of DN. Urinary albumin excretion over 24 h was
significantly increased in the diabetic mice (113.6+12.53 mg/24 h)
compared with the non-diabetic control (27.63±6.31 mg/24 h;
P<0.01), and this increase was significantly enhanced by ZnD
(156.2+16.27 mg/24 h) compared with the diabetic mice (P<0.01;
Fig. 1).
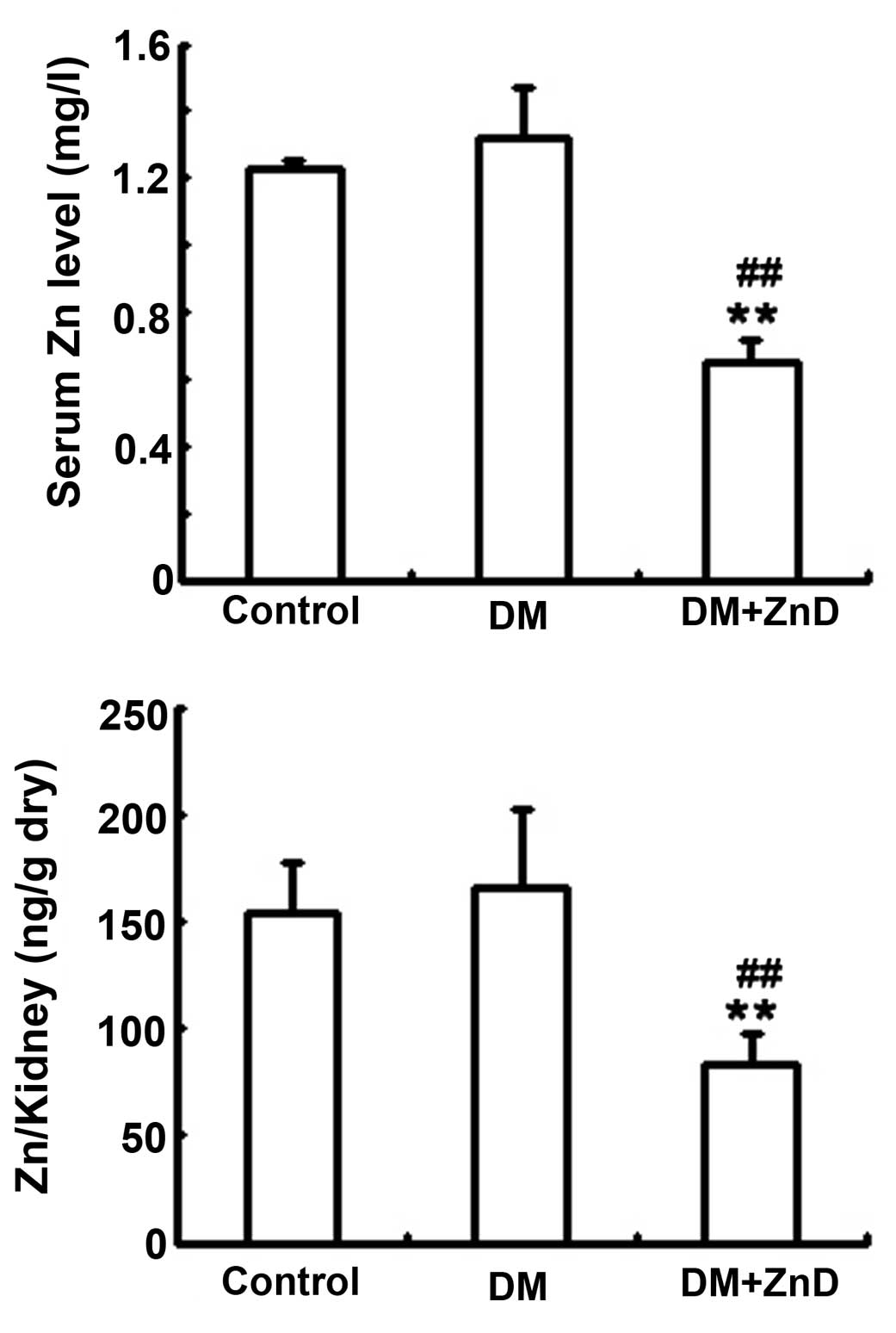
Effect of ZnD on the concentration of
zinc ions
In an in vivo experiment, AAS technology was
used to examine the concentration of zinc ions in the mouse kidney
and plasma. As reported previously (34), diabetic mice fed ZnD diet exhibited
significantly decreased levels of zinc in the kidney and plasma
compared with the control and diabetic mice (all P<0.01),
indicating that the diet successfully induced ZnD (Fig. 2).
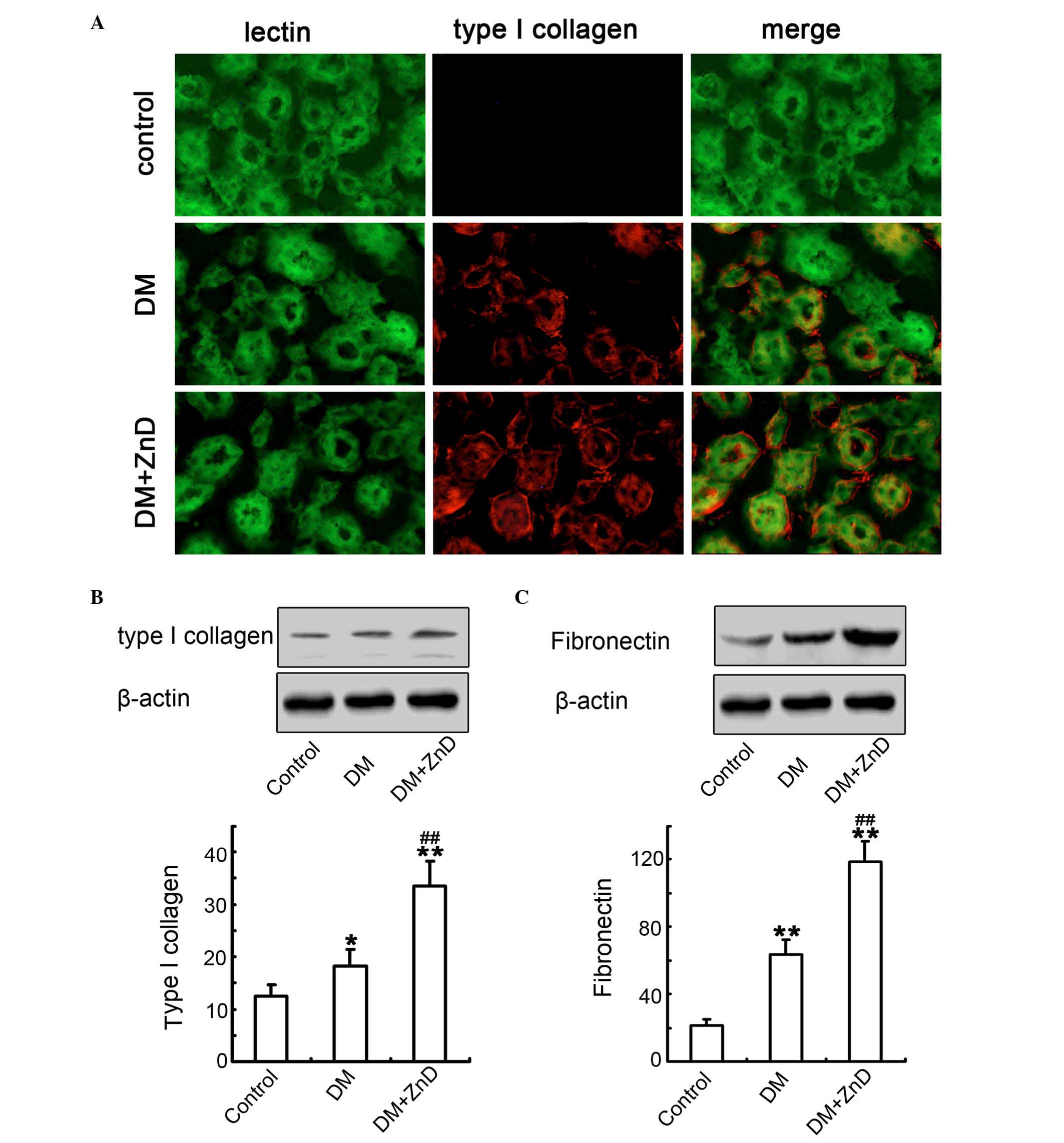
ZnD improves ECM gene expression and
increases renal interstitial fibrosis in diabetic mice
Increased ECM protein expression is a key
characteristic of renal fibrosis in DN (35,36).
Double-immunofluorescence staining for lectin (a tubular marker)
and type I collagen was performed to analyze the distribution and
localization of type I collagen protein in the renal interstitium.
Diabetes increased the expression of type I collagen localized to
the interstitial areas of diabetic kidneys compared with control
mice, and ZnD enhanced this response (Fig. 3A). Furthermore, western blot
analysis also demonstrated that the expression of type I collagen
(Fig. 3B) and fibronectin
(Fig. 3C) in ZnD-diabetic kidneys
was significantly increased compared with control and diabetic
mice. These observations were consistent with the results of
immunofluorescence staining. It was clear that ZnD increases
interstitial matrix production and enhances renal fibrosis in the
model of DN in vivo.
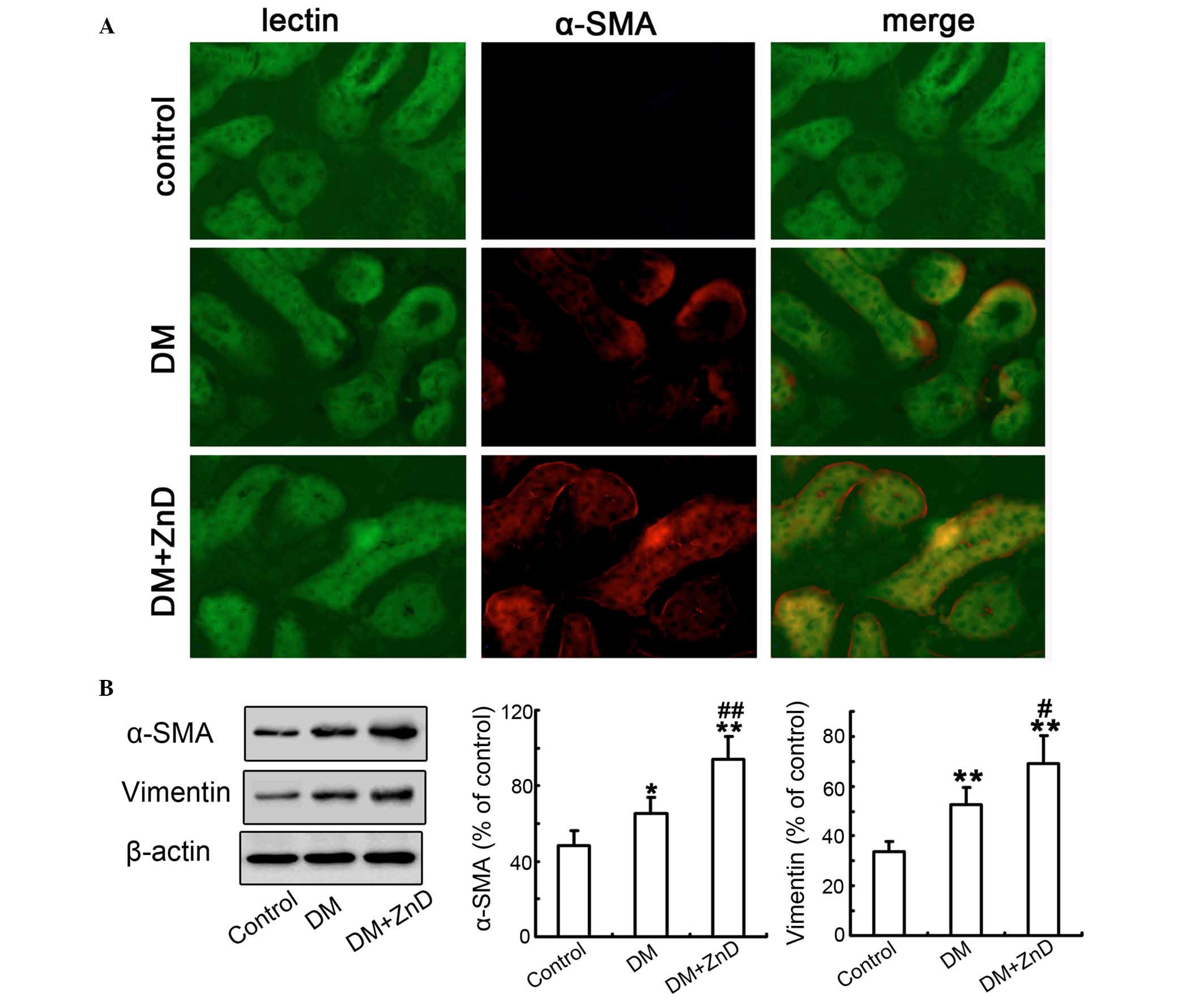
ZnD enhances the activation of renal
fibroblasts in the kidneys of diabetic mice
Subsequently, the effects of ZnD on the activation
of renal fibroblasts were examined. Double-immunofluorescence
staining for lectin and α-SMA demonstrated that α-SMA expression
was in the interstitial areas of diabetic kidneys compared with
non-diabetic controls, and that ZnD enhanced α-SMA expression in
the kidneys of diabetic mice compared with the diabetic controls
(Fig. 4A). Consistently, western
blot analyses of kidney tissues demonstrated that the expression
levels of α-SMA and vimentin were significantly increased in the
kidneys of diabetic mice compared with non-diabetic controls
(P<0.05 and P<0.01, respectively), and the levels were
significantly enhanced by the administration of ZnD compared with
the levels in diabetic mice (P<0.01 and P<0.05, respectively;
Fig. 4B). Collectively, these data
suggest that ZnD is mechanistically involved in the activation of
fibroblasts in DN in vivo.
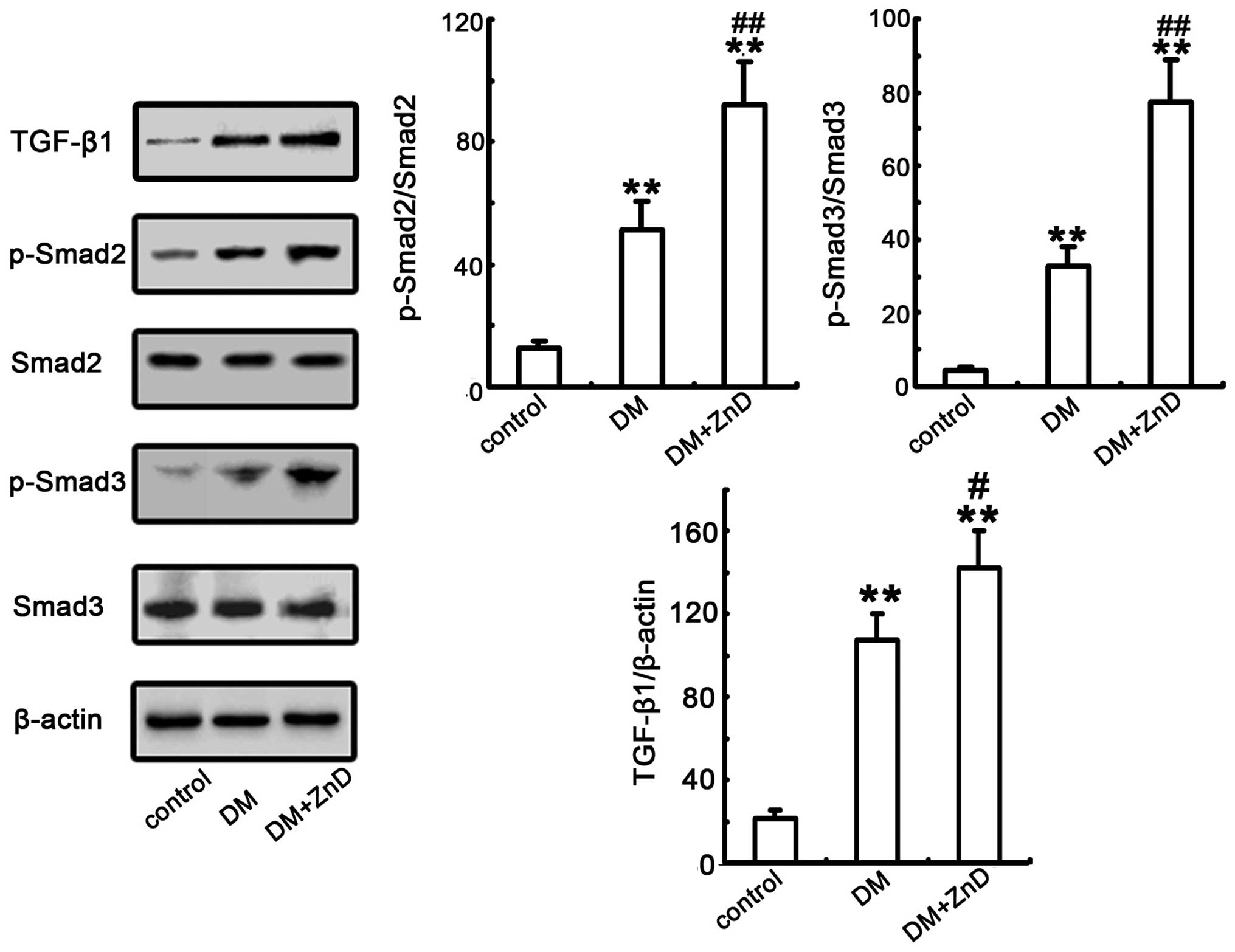
ZnD enhances the activation of TGF-β1
and Smad2/3 expression in the kidneys of diabetic mice
TGF-β1, and the TGF-β/Smad signaling pathway, has
been previously reported to serve a critical role in kidney
fibrosis (19,37). Thus, the effect of ZnD on the
expression of the TGF-β/Smad signaling pathway was examined. As
indicated in Fig. 5, the protein
expression level of TGF-β1 and phosphorylation of Smad2/3 were
significantly increased in diabetic mice compared with the control
group (all P<0.01). Additionally, TGF-β1 expression and Smad2/3
phosphorylation were significantly increased in ZnD-diabetic mice
compared with diabetic mice (P<0.05, P<0.01 and P<0.01,
respectively; Fig. 5). Considering
the above findings, these data clearly indicate that ZnD is
involved in mediating the expression or activation of several
important proteins known to be relevant in the TGF-β/Smad signaling
pathway that in stimulated in interstitial fibroblasts to regulate
ECM accumulation in vivo.
Discussion
Several studies in animal models and certain
clinical studies have demonstrated that zinc deficiency may be
associated with fibrosis in chronic inflammatory diseases,
including in liver, myocardial and cystic fibrosis (38–41).
Although renal interstitial fibrosis was previously demonstrated in
rat and mouse models of progressive DN, and in kidneys from
patients with long-standing type 1 diabetes, the majority of DN
research has generally focused on the glomerular lesions in this
condition (42). However, it is
increasingly appreciated that tubular injury has an important role
in the progression of DN resulting in several of the molecular and
cellular changes. Thus, the present study was undertaken to define
the effect and underlying mechanism of ZnD on renal interstitial
fibrosis in DN. The results in the present study demonstrated that
ZnD enhances diabetic renal interstitial fibrosis, as indicated by
an increase in levels of type I collagen, fibronectin, α-SMA and
vimentin, which is may occur via the TGF-β/Smad2/3 pathway. To the
best of our knowledge, these results are the first to demonstrate
the effect of ZnD on the pathogenic mechanisms of renal
interstitial fibrosis during the development of DN.
Clinical studies have previously demonstrated that
patients with diabetes with low serum zinc levels have a high risk
of developing kidney injury compared with those with normal serum
zinc levels, and zinc supplementation may potentially be used
clinically to prevent diabetes-induced complications in multiple
organs (43,44). A previous study confirmed that the
protective effects of zinc supplementation on renal pathological
changes, fibrosis and oxidative damage were more significant
compared with the effects on 24 h urinary protein increase,
partially because urinary protein level is a reflection of both
tubular and glomerular function (43).
Increased ECM protein synthesis and/or decreased ECM
degradation ultimately contributes to the development of
diabetes-associated tubulointerstitial fibrosis (19,45).
The activation of interstitial fibroblasts to become α-SMA-positive
myofibroblasts is recognized as a crucial step in the development
of chronic kidney disease, including DN. In addition to activated
resident interstitial fibroblasts, myofibroblasts may also be
derived from tubular epithelial cells via EMT, which is considered
to be primarily responsible for interstitial matrix accumulation
and deposition (33,46). The present study observed that STZ
induction of diabetes increased the number of α-SMA-positive renal
fibroblasts, and ZnD significantly increased the activation of
kidney fibroblasts, parallel with a substantial increase in ECM
synthesis, which is in accordance with a previous report (47). Collectively, previous studies and
the current study have demonstrated that ZnD induced an increase in
renal tubular collagen synthesis and may be also responsible for
collagen upregulation and renal tubular injury during diabetes
in vivo.
TGF-β1, a strong profibrotic cytokine, and the
TGF-β/Smad pathway has been consistently implicated to be critical
during the pathogenic ECM accumulation in DN (3,5,19). A
previous study has demonstrated that TGF-β1 upregulation was
induced by high glucose in the p38 mitogen-activated protein kinase
signaling pathway (48). In
addition, there is evidence that high glucose-induced TGF-β1
production is involved in the extracellular matrix metabolism in DN
(12,49). Furthermore, a previous study
demonstrated that a ZnD lead to TGF-β1 induction during
neurogenesis, impairs neuronal precursor cell proliferation and
induces apoptosis via regulating p53-dependent molecular mechanisms
(50). Another study indicated
that TGF-β1 has stimulatory and inhibitory effects on
osteoclast-like cell formation in mouse marrow cultures, and that
zinc can inhibit the stimulatory effect of TGF-β1 (51). A recent study reported that zinc
can inhibit human lens epithelial cell migration and proliferation
by decreasing the expression of TGF-β1 and increasing the
expression of tumor necrosis factor-α, and subsequently induce
apoptosis/necrosis (52). In the
present study, expression of TGF-β1 and phosphorylation of Smad2/3
were increased in diabetic mice, and further increased by ZnD,
suggesting that ZnD can aggravate renal tubular interstitial
fibrosis in DN, and may affect key fibrotic factors and signaling
pathways.
In conclusion, the present study provides novel
evidence regarding the association between ZnD and renal
interstitial fibrosis in STZ-induced diabetic mice. ZnD enhanced
albuminuria and ECM protein expression associated with diabetic
renal interstitial fibrosis through activation of renal
interstitial fibroblasts and regulation of the expression of
fibrosis-associated factors expressed by fibroblasts, which may
occur via the TGF-β/Smad2/3 pathway. These findings suggest that
suboptimal zinc status induces renal tubulointerstitial injury
associated with the development of DN. These findings indicate that
suboptimal zinc status induces renal tubulointerstitial injury
associated with the development of DN and suggest that zinc
supplementation may benefit in the medication and prevention of the
disease.
Acknowledgements
This work was supported by the National Grand
Fundamental Research 973 Program of China (grant no. 2012CB722405),
the Natural Science Foundation of China (grant no. 81170561), the
Liaoning Province Science and Technology Plan Project (grant nos.
2012408002 and 2013225090) and the Postdoctoral Science Foundation
of China (grant no. 2014MM551144).
Glossary
Abbreviations
Abbreviations:
|
FITC
|
fluorescein isothiocyanate
|
|
TGF-β1
|
transforming growth factor-β1
|
|
ECM
|
extracellular matrix
|
|
DN
|
diabetic nephropathy
|
|
ZnD
|
zinc deficiency
|
References
|
1
|
Chen L, Magliano DJ and Zimmet PZ: The
worldwide epidemiology of type 2 diabetes mellitus-present and
future perspectives. Nat Rev Endocrinol. 8:228–236. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kelly DJ, Gilbert RE, Cox AJ, Soulis T,
Jerums G and Cooper ME: Aminoguanidine ameliorates overexpression
of prosclerotic growth factors and collagen deposition in
experimental diabetic nephropathy. J Am Soc Nephrol. 12:2098–2107.
2001.PubMed/NCBI
|
|
3
|
Schmid H, Boucherot A, Yasuda Y, Henger A,
Brunner B, Eichinger F, Nitsche A, Kiss E, Bleich M, Gröne HJ, et
al: Modular activation of nuclear factor-kappaB transcriptional
programs in human diabetic nephropathy. Diabetes. 55:2993–3003.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seckeler MD and Hoke TR: The worldwide
epidemiology of acute rheumatic fever and rheumatic heart disease.
Clin Epidemiol. 3:67–84. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Navarro JF, Mora C, Muros M and Garcia J:
Urinary tumour necrosis factor-alpha excretion independently
correlates with clinical markers of glomerular and
tubulointerstitial injury in type 2 diabetic patients. Nephrol Dial
Transplant. 21:3428–3434. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oldfield MD, Bach LA, Forbes JM,
Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T,
Jerums G and Cooper ME: Advanced glycation end products cause
epithelial-myofibroblast transdifferentiation via the receptor for
advanced glycation end products (RAGE). J Clin Invest.
108:1853–1863. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gilbert RE and Cooper ME: The
tubulointerstitium in progressive diabetic kidney disease: More
than an aftermath of glomerular injury? Kidney Int. 56:1627–1637.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fioretto P and Mauer M: Histopathology of
diabetic nephropathy. Semin Nephrol. 27:195–207. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lam S, van der Geest RN, Verhagen NA, van
Nieuwenhoven FA, Blom IE, Aten J, Goldschmeding R, Daha MR and van
Kooten C: Connective tissue growth factor and igf-I are produced by
human renal fibroblasts and cooperate in the induction of collagen
production by high glucose. Diabetes. 52:2975–2983. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wada T, Furuichi K, Sakai N, Iwata Y,
Yoshimoto K, Shimizu M, Takeda SI, Takasawa K, Yoshimura M, Kida H,
et al: Up-regulation of monocyte chemoattractant protein-1 in
tubulointerstitial lesions of human diabetic nephropathy. Kidney
Int. 58:1492–1499. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang C, Shen S, Ma Q, Gill A, Pollock CA
and Chen XM: KCa3.1 mediates activation of fibroblasts in diabetic
renal interstitial fibrosis. Nephrol Dial Transplant. 29:313–324.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simonson MS: Phenotypic transitions and
fibrosis in diabetic nephropathy. Kidney Int. 71:846–854. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sharma K and Ziyadeh FN: Hyperglycemia and
diabetic kidney disease. The case for transforming growth
factor-beta as a key mediator. Diabetes. 44:1139–1146. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pankewycz OG, Guan JX, Bolton WK, Gomez A
and Benedict JF: Renal TGF-beta regulation in spontaneously
diabetic NOD mice with correlations in mesangial cells. Kidney Int.
46:748–758. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han DC, Isono M, Hoffman BB and Ziyadeh
FN: High glucose stimulates proliferation and collagen type I
synthesis in renal cortical fibroblasts: Mediation by autocrine
activation of TGF-beta. J Am Soc Nephrol. 10:1891–1899.
1999.PubMed/NCBI
|
|
17
|
Hong SW, Isono M, Chen S, Iglesias-De La
Cruz MC, Han DC and Ziyadeh FN: Increased glomerular and tubular
expression of transforming growth factor-beta1, its type II
receptor and activation of the Smad signaling pathway in the db/db
mouse. Am J Pathol. 158:1653–1663. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sharma K, Jin Y, Guo J and Ziyadeh FN:
Neutralization of TGF-beta by anti-TGF-beta antibody attenuates
kidney hypertrophy and the enhanced extracellular matrix gene
expression in STZ-induced diabetic mice. Diabetes. 45:522–530.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kato M, Yuan H, Xu ZG, Lanting L, Li SL,
Wang M, Hu MC, Reddy MA and Natarajan R: Role of the Akt/FoxO3a
pathway in TGF-beta1-mediated mesangial cell dysfunction: A novel
mechanism related to diabetic kidney disease. J Am Soc Nephrol.
17:3325–3335. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang M, Fraser D and Phillips A: ERK, p38
and Smad signaling pathways differentially regulate transforming
growth factor-beta1 autoinduction in proximal tubular epithelial
cells. Am J Pathol. 169:1282–1293. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tomat AL, Costa MA, Girgulsky LC, Veiras
L, Weisstaub AR, Inserra F, Balaszczuk AM and Arranz CT: Zinc
deficiency during growth: Influence on renal function and
morphology. Life Sci. 80:1292–1302. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hagmeyer S, Haderspeck JC and Grabrucker
AM: Behavioral impairments in animal models for zinc deficiency.
Front Behav Neurosci. 8:4432015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Salgueiro MJ, Krebs N, Zubillaga MB, Weill
R, Postaire E, Lysionek AE, Caro RA, De Paoli T, Hager A and Boccio
J: Zinc and diabetes mellitus: Is there a need of zinc
supplementation in diabetes mellitus patients? Biol Trace Elem Res.
81:215–228. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Batista MN, Cuppari L, de Fátima Campos
Pedrosa L, Almeida Md, de Almeida JB, de Medeiros AC and Canziani
ME: Effect of end-stage renal disease and diabetes on zinc and
copper status. Biol Trace Elem Res. 112:1–12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ozcelik D, Tuncdemir M, Ozturk M and Uzun
H: Evaluation of trace elements and oxidative stress levels in the
liver and kidney of streptozotocin-induced experimental diabetic
rat model. Gen Physiol Biophys. 30:356–363. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Parham M, Amini M, Aminorroaya A and
Heidarian E: Effect of zinc supplementation on microalbuminuria in
patients with type 2 diabetes: A double blind, randomized,
placebo-controlled, cross-over trial. Rev Diabet Stud. 5:102–109.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Al-Timimi DJ, Sulieman DM and Hussen KR:
Zinc status in type 2 diabetic patients: Relation to the
progression of diabetic nephropathy. J Clin Diagn Res. 8:CC04–CC08.
2014.PubMed/NCBI
|
|
28
|
Takahashi M, Saito H, Higashimoto M and
Hibi T: Possible inhibitory effect of oral zinc supplementation on
hepatic fibrosis through downregulation of TIMP-1: A pilot study.
Hepatol Res. 37:405–409. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gandhi MS, Deshmukh PA, Kamalov G, Zhao T,
Zhao W, Whaley JT, Tichy JR, Bhattacharya SK, Ahokas RA, Sun Y, et
al: Causes and consequences of zinc dyshomeostasis in rats with
chronic aldosteronism. J Cardiovasc Pharmacol. 52:245–252. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Van Biervliet S, Velde S Vande, Van
Biervliet JP and Robberecht E: The effect of zinc supplements in
cystic fibrosis patients. Ann Nutr Metab. 52:152–156. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang X, Wang J, Fan Y, Yang L, Wang L and
Ma J: Zinc supplementation attenuates high glucose-induced
epithelial-to-mesenchymal transition of peritoneal mesothelial
cells. Biol Trace Elem Res. 150:229–235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tesch GH and Allen TJ: Rodent models of
streptozotocin-induced diabetic nephropathy. Nephrology (Carlton).
12:261–266. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang X, Liang D, Chi ZH, Chu Q, Zhao C,
Ma RZ, Zhao Y and Li H: Effect of zinc on high glucose-induced
epithelial-to-mesenchymal transition in renal tubular epithelial
cells. Int J Mol Med. 35:1747–1754. 2015.PubMed/NCBI
|
|
34
|
Pories WJ, DeWys WD, Flynn A, Mansour EG
and Strain WH: Implications of the inhibition of animal tumors by
dietary zinc deficiency. Adv Exp Med Biol. 91:243–257. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang S, Denichilo M, Brubaker C and
Hirschberg R: Connective tissue growth factor in tubulointerstitial
injury of diabetic nephropathy. Kidney Int. 60:96–105. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kelly DJ, Chanty A, Gow RM, Zhang Y and
Gilbert RE: Protein kinase Cbeta inhibition attenuates osteopontin
expression, macrophage recruitment and tubulointerstitial injury in
advanced experimental diabetic nephropathy. J Am Soc Nephrol.
16:1654–1660. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gilbert RE, Cox A, Wu LL, Allen TJ,
Hulthen UL, Jerums G and Cooper ME: Expression of transforming
growth factor-beta1 and type IV collagen in the renal
tubulointerstitium in experimental diabetes: Effects of ACE
inhibition. Diabetes. 47:414–422. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nawar O, Akridge RE, Hassan E, el Gazar R,
Doughty BL and Kemp WM: The effect of zinc deficiency on granuloma
formation, liver fibrosis and antibody responses in experimental
schistosomiasis. Am J Trop Med Hyg. 47:383–389. 1992.PubMed/NCBI
|
|
39
|
Gomot MJ, Faure P, Roussel AM, Coudray C,
Osman M and Favier A: Effect of acute zinc deficiency on insulin
receptor binding in rat adipocytes. Biol Trace Elem Res.
32:331–335. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zalewski PD: Zinc metabolism in the
airway: Basic mechanisms and drug targets. Curr Opin Pharmacol.
6:237–243. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Szuster-Ciesielska A, Plewka K, Daniluk J
and Kandefer-Szerszeń M: Zinc supplementation attenuates ethanol-
and acetaldehyde-induced liver stellate cell activation by
inhibiting reactive oxygen species (ROS) production and by
influencing intracellular signaling. Biochem Pharmacol. 78:301–314.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Burns WC, Twigg SM, Forbes JM, Pete J,
Tikellis C, Thallas-Bonke V, Thomas MC, Cooper ME and Kantharidis
P: Connective tissue growth factor plays an important role in
advanced glycation end product-induced tubular
epithelial-to-mesenchymal transition: Implications for diabetic
renal disease. J Am Soc Nephrol. 17:2484–2494. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tang Y, Yang Q, Lu J, Zhang X, Suen D, Tan
Y, Jin L, Xiao J, Xie R, Rane M, et al: Zinc supplementation
partially prevents renal pathological changes in diabetic rats. J
Nutr Biochem. 21:237–246. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Özcelik D, Naziroglu M, Tuncdemir M, Celik
Ö, Öztürk M and Flores-Arce MF: Zinc supplementation attenuates
metallothionein and oxidative stress changes in kidney of
streptozotocin-induced diabetic rats. Biol Trace Elem Res.
150:342–349. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hills CE, Al-Rasheed N, Willars GB and
Brunskill NJ: C-peptide reverses TGF-beta1-induced changes in renal
proximal tubular cells: Implications for treatment of diabetic
nephropathy. Am J Physiol Renal Physiol. 296:F614–F621. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu Y: New insights into
epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212–222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wolf G and Ziyadeh FN: Molecular
mechanisms of diabetic renal hypertrophy. Kidney Int. 56:393–405.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lv ZM, Wang Q, Wan Q, Lin JG, Hu MS, Liu
YX and Wang R: The role of the p38 MAPK signaling pathway in high
glucose-induced epithelial-mesenchymal transition of cultured human
renal tubular epithelial cells. PLoS One. 6:e228062011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mason RM and Wahab NA: Extracellular
matrix metabolism in diabetic nephropathy. J Am Soc Nephrol.
14:1358–1373. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Corniola RS, Tassabehji NM, Hare J, Sharma
G and Levenson CW: Zinc deficiency impairs neuronal precursor cell
proliferation and induces apoptosis via p53-mediated mechanisms.
Brain Res. 1237:52–61. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yamaguchi M and Kishi S: Differential
effects of transforming growth factor-beta on osteoclast-like cell
formation in mouse marrow culture: Relation to the effect of
zinc-chelating dipeptides. Peptides. 16:1483–1488. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Du Y, Guo D, Wu Q, Liu D and Bi H: Zinc
chloride inhibits human lens epithelial cell migration and
proliferation involved in TGF-β1 and TNF-α signaling pathways in
HLE B-3 cells. Biol Trace Elem Res. 159:425–433. 2014. View Article : Google Scholar : PubMed/NCBI
|