Introduction
Ankylosing spondylitis (AS), which is the most
frequently occurring form of Spondyloarthritis (SpA), is a chronic
immune-mediated inflammatory disease characterized by inflammation
that predominantly affects the axial skeleton. Peripheral arthritis
and enthesopathy have been reported to be present in a large group
of AS patients (1). In addition,
specific organ involvement including anterior uveitis, psoriasis
and chronic inflammatory bowel disease, may simultaneously occur in
AS, accompanied by an increased risk of cardiovascular or pulmonary
complications (1,2). Chronic inflammation in the attachment
of tendons, ligaments and joint capsules to bone leads to
alterations in joint architecture, with new bone formations and
joint fusions (3). The unique
structural alterations in syndesmophyte formation and ankylosis of
the vertebrae are the primary causes of early severe disability in
patients with AS during disease progression. However, the
pathogenesis and contributing etiological factors of AS remain to
be elucidated.
Genetic studies have contributed the most
significant information to our understanding of AS. Familial
aggregation studies have revealed that heritability contributes a
substantial proportion of AS susceptibility (4). A strong genetic predisposition was
confirmed by the discovery of a remarkably high association between
AS and the human leukocyte antigen (HLA)-B27 in 1973 (5). A proportion of AS cases that do not
involve HLA-B27 have been reported, however, it is still regarded
as one of the most important factors for the development of AS with
a high association (>100), and is present in up to 90% of
patients in the majority of ethnic groups that present with the
disease (6). Genetic studies have
concluded that HLA-B27 in the major histocompatibility complex
(MHC) locus contributes to ~20.1% of AS heritability, with 4.3%
associated with loci other than HLA-B (7). It has a high degree of genetic
polymorphism, with up to 105 known subtypes, termed HLA-B*27:01 to
HLA-B*27:106, which are encoded by 132 alleles. The most common
subtypes associated with AS are HLA-B*27:05 (Caucasians),
HLA-B*27:04 (Chinese), and HLA-B*27:02 (Mediterranean) (8). However, two subtypes, HLA-B*27:06 and
HLA-B*27:09, do not appear to present any association with the
disease (9). In addition,
genome-wide association studies and those involving Immunochip
arrays, have further substantiated that the development of AS is
determined, to a large extent, by genes located outside of the MHC
locus. These involve loci of the interleukin (IL)-23/IL-17 axis,
including IL23R, IL12B, tyrosine kinase 2, signal transducer and
activator of transcription 3, IL6R, IL27, as well as several
aminopeptidase genes, including endoplasmic reticulum
aminopeptidase (ERAP) 1 and 2, leucyl and cystinyl aminopeptidase
(LNPEP) and aminopeptidase puromycin sensitive (NPEPPS) genes
(7,10). It has previously been suggested
that a strong association exists between ERAP1 and AS that is
restricted to HLA-B27-positive patients (11,12).
Previous studies suggest that the development of AS may be
associated with antigen processing and presentation (11,12).
Classical and non-classical forms of
HLA-B27
MHC Class I molecules are important for the
initiation and propagation of immune responses (13,14).
The classical heterotrimeric MHC class I molecule is composed of
three non-covalently bound individual polypeptides: A highly
polymorphic heavy chain (HC), β2-microglobulin (β2m) light chain
and an oligopeptide, typically 8 to 10 residues in length (13,14).
Assembly of a stable MHC molecule in the endoplasmic reticulum (ER)
is a necessary step prior to export to the cell surface. Following
synthesis and glycosylation, free HCs are initially stabilized by
chaperones (calreticulin and tapasin) until a conformation suitable
to bind β2m and a peptide with suitable length is achieved
(15). Nascent MHC class I
molecules typically bind antigen peptides and transport them to the
cell surface for presentation to the T-cell receptors (TCR) on T
lymphocytes (15).
In the absence of β2m, HCs will misfold and
ER-associated degradation may occur in the ER. However, HLA-B27
appears to exhibit a tendency to misfold and a predilection for
forming dimers or multimers (16).
HLA-B27 possesses three unpaired cysteine (C) residues at positions
67, 308 and 325, and four conserved cysteine residues at positions
101, 164, 203 and 259 (17). Three
distinct forms of dimeric MHC-I structures have been described
previously, including cell surface HLA-B27 homodimers,
intracellular and the exosomal fully-folded MHC-I dimers (18). The intracellular dimers are termed
as ER-resident and endosomal dimers, the latter of which may be
expressed on the cell surface. Previous studies have proposed that
the cysteine at position 67 (C67) is involved in intracellular and
cell surface dimer formation (15,19).
However, the structurally conserved C101 and C164 residues have
additionally been demonstrated to participate in the formation of a
pool of intracellular dimers (17).
In addition to the widely-accepted cell surface
HLA-B27 homodimers, ER-resident dimers and endosomal dimers, the
present review introduces the exosomal fully-folded MHC-I dimers.
Exosomes are small vesicles of different sizes that are formed by
the inward budding of endosomes to generate multivesicular bodies
(MVBs). A proportion of these MVBs will fuse with the plasma
membrane, releasing the internal vesicles to the extracellular
environment (20). An extensive
variety of cell types have been demonstrated to release exosomes
and various MHC-I dimers have been revealed to be expressed on the
surface of these vesicles (20).
The exosome-associated HLA-B27 dimers are fully-folded, and the
cysteine residues at position 325 in the cytoplasmic tail domain
participate in the formation of these structures (21). In addition, a novel population of
mixed-allele dimers comprising HLA-A2 and HLA-B27 has been
described in exosomes. Further details of the various forms of
HLA-B27 are described in Table
I.
 | Table I.Different forms of HLA-B27 and their
pathogenic roles in ankylosing spondylitis. |
Table I.
Different forms of HLA-B27 and their
pathogenic roles in ankylosing spondylitis.
| First author,
year | HLA-B27 form | Location | Structural
feature | Formation | Condition | Pathogenic
role/receptor | Refs. |
|---|
| Chen, 2013; | Classical | ER | Expressed at the
cell | Assembly of a
stable | Three
non-covalently | TCR, KIR3DL1, | 23 |
| Allen, 2004; | HLA-B27 |
| surface as
heterotrimeric | HLA-B27 molecule in
the ER | bound
individual | LILRB1, | 35 |
| Allen, 2001; |
|
| peptide-MHC
complexes | is necessary.
Following synthesis | polypeptides are
all | LILRB2, | 37 |
| Giles, 2012; |
|
| with β2m and
peptide | and glycosylation,
free HCs | required: a
highly | LILRA1 | 40 |
| Shaw, 2014 |
|
|
| are initially
stabilized by | polymorphic HC,
β2m |
| 57 |
|
|
|
|
| chaperones
(calreticulin and tapasin) | light, chain and
an |
|
|
|
|
|
|
| until a
conformation suitable to | oligopeptide,
typically |
|
|
|
|
|
|
| bind β2m and a
peptide is achieved | of 8 to 10
residues |
|
|
| Kollenberger,
2007; | Cell surface | Endosome | Formed by two
covalently | Recycling of
fully-folded | Acidic environment
of | KIR3DL1, | 36 |
| Allen, 2001; | HLA-B27 |
| bonded
β2m-dissociated | HLA-B27 cell
surface molecules | the endosome
and | KIR3DL2, | 37 |
| Giles, 2012; | homodimers |
| HCs | via the endocytic
pathway, the | the low affinity
binding | LILRB2, | 40 |
| Campbell,
2012; |
|
|
| β2m-dissociated HCs
form covalent | of β2m and
peptides | LILRA1 | 43 |
| Shaw, 2014 |
|
|
| homodimers by
cysteine residue at | with HC |
| 57 |
|
|
|
|
| C67 in the α1
domain, and are |
|
|
|
|
|
|
|
| re-express at the
cell surface |
|
|
|
| Lenart, 2012; | ER HLA-B27 | ER | The two
β2m-dissociated, | Form via C67-C67,
C101-C101 or | HLA-B27 exhibited
an enhanced | UPR | 17 |
| Colbert, 2009; | homodimers |
| partially unfolded
HCs form | C164-C164 disulfide
bonds. | tendency to misfold
and was |
| 48 |
| Turner, 2005 |
|
| covalent
homodimers, but |
| susceptible to
aggregation |
| 49 |
|
|
|
| do not transit out
of the ER |
|
|
|
|
| Raposo, 2013; | Redox-induced | Exosomes/ | Fully-folded
β2m- | Critically depend
on C325 in the | Lower levels of
glutathione inside | Intercellular | 20 |
| Lynch, 2009; | HLA-B27 | Apoptosing | associated
HLA-B27 | cytoplasmic tail
(or with C339 in | exosomes creating a
more oxidizing | communication | 21 |
| Campbell,
2012; | dimers | cells | dimers that are
detected | HLA-A alleles) | environment |
| 43 |
| Shaw, 2014 |
|
| on exosomes |
|
|
| 57 |
|
Luthra-Guptasarma, | HLA-B27 | ER | Misfolded
monomeric | Residues 169–181
(identical to a | β2m-free,
peptide-free HCs support | UPR/recognized | 58 |
| 2004 | with peptide |
| HLA-B27 with
the | known HLA-B27
ligand) loop | a helix-coil
transition facilitating | by receptors |
|
| binding cleft |
| molecule's own
peptide | around and occupy
the molecules | rotation of
backbone angles around |
|
|
|
| occupied |
| binding cleft
occupied | own peptide-binding
cleft | amino acid
167/168 |
|
|
| Dakwar, 2008; | HLA-B27 that | ER | Misfolded
HLA-B27 | Misfolding occurs
in the ER | B pocket in the
peptide- | UPR | 26 |
| Bowness, 2011; | have not yet |
| monomer that
folds | prior to β2m
association and | binding groove
conferred a |
| 39 |
| Rajagopalan,
2012 | folded
properly |
| improperly | peptide
optimization | slow folding
phenotype and |
| 46 |
|
|
|
|
|
| a tendency to
misfold |
|
|
Role of HLA-B27 in the pathogenesis of
AS
The mechanism of antigen processing and presentation
is presented in Fig. 1. The
majority of proteins are initially degraded in the cytoplasm by the
multi-unit proteasome complex, typically generating peptide
fragments of up to 25 amino acids in length. These antigen peptides
and their N-terminal extended precursors are subsequently
transported into the ER by a transporter associated with antigen
processing that preferentially transports peptides 8–16 residues in
length. Transporter, ATP-binding cassette subfamily B member (TAP)
is an adenosine triphospate-driven transporter welladapted for the
transfer of the precursor peptides that are continuously generated
by the proteasome or other cytosolic proteases (22). Subsequently, longer peptides will
be further cleaved to the length required for antigen presentation
by ERAP1 residing in the ER. ERAP1 efficiently cleaves the
precursors to oligopeptides 8 or 9 residues in length, which is
optimal for binding to HLA-B27. These peptide-MHC complexes will
subsequently enter the Golgi apparatus for the generation of mature
epitopes (23). However, previous
genetic studies have identified an association between various
aminopeptidase genes (NPEPPS, LNPEP and ERAP2) and AS (7). The NPEPPS protein localizes to the
cytoplasm and has previously been demonstrated to be involved in
processing proteasome-derived peptides prior to their transport to
the ER. LNPEP and ERAP2 are members of the ER aminopeptidase family
and have substantial sequence homology to ERAP1 (7,24).
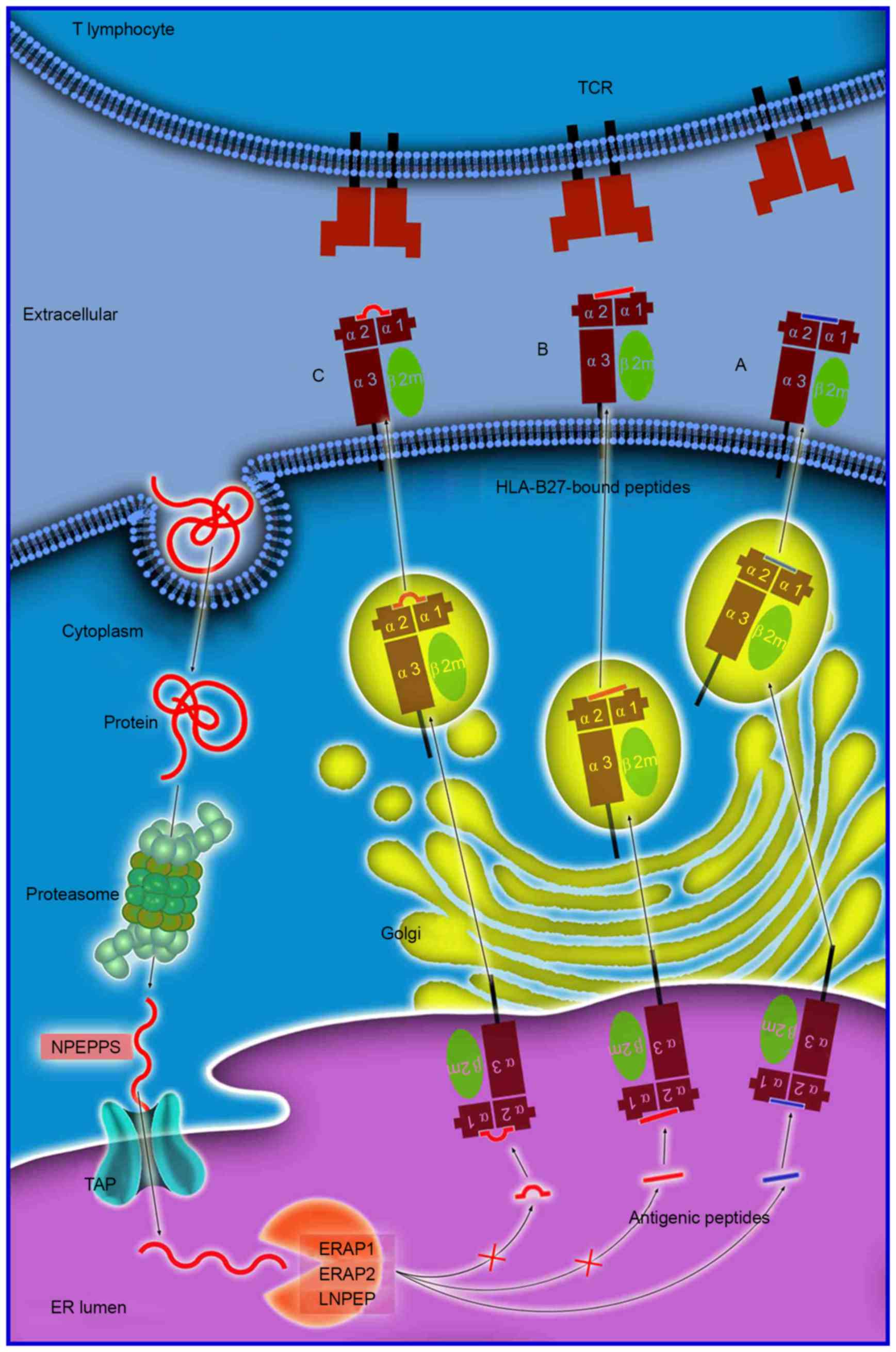 | Figure 1.Antigen processing and presentation
of peptides of various sizes. Antigen processing and presentation
is a sequenced process. Numerous proteins are initially degraded
into peptide fragments of up to 25 amino acids in length by the
multi-unit proteasome complex followed by NPEPPS. TAP
preferentially transports antigen peptides of 8–16 residues into
the ER. N-terminal extended precursors will be further cleaved by
ERAP1/ERAP2/LNPEP into oligopeptides of 8 or 9 residues, which is
the optimal length for binding to HLA-B27. The peptides
subsequently (A) enter the Golgi apparatus for generation of mature
epitopes. However, various longer peptides may bind to HLA-B27,
where they reside in the peptide groove of the HLA-B27 with (B) a
protruding C-terminus, or (C) a bulge in the center. These
HLA-B27-bound peptides may be highly immunogenic and may stimulate
an extremely biased T-cell response repertoire. NPEPPS,
aminopeptidase puromycin sensitive; TAP, transporter, ATP-binding
cassette subfamily B member; HLA, human leukocyte antigen; ER,
endoplasmic reticulum; ERAP1/2, endoplasmic reticulum
aminopeptidase 1/2; LNPEP, leucyl cystinyl aminopeptidase; TCR,
T-cell receptor. |
Structurally unique peptide-MHC
complexes
The process of antigen peptide processing and
presentation to receptors on immune cells may occur via a variety
of pathways. The ‘arthritogenic’ peptide hypothesis suggests that
the HLA-B27-specific autoimmune response may be directly initiated
for structurally unique peptide-MHC complexes, depending on the
amino-acid composition of the antigen peptides (6). The theory of ‘molecular mimicry’
suggests that a cross-reactive peptide derived from an infecting
bacterial pathogen stimulates T cells, which subsequently respond
to an HLA-B27 associated ‘self-peptide’, or to peptides derived
from HLA-B27 directly (25).
Numerous self and foreign antigen peptides have previously been
investigated and sequenced, however, there is no conclusive
evidence demonstrating that any of these peptides are indeed
cross-reactive or self-peptides (26). Furthermore, Taurog et al
(27) demonstrated that disease
manifestations arose in HLA-B27/Huβ2m-transgenic rats in the
absence of functional cluster of differentiation (CD)8+ T cells.
Generating a model of AS disease pathogenesis, whereby HLA-B27
presents a peptide to CD8+ T cells, presents a significant
challenge.
The length of antigen peptides
The length of antigen peptides is an important
consideration in the process of antigen presenting. Extended
peptides which are generated via aberrant peptide processing may
bind HLA-B27, and have been demonstrated to reside in the peptide
groove of MHC class I molecules, with either a protruding
C-terminus (28), or a middle
bulge (Fig. 1) (29). These HLA-B27-bound peptides may be
highly immunogenic and may stimulate an extremely biased TCR
repertoire. When they are presented to the TCR on T cells, a
HLA-B27-specific, T-cell-mediated impaired peptide presentation is
initiated, which leads to differences in immunodominance
hierarchies for the presentation of a variety of aberrant epitopes,
which is important in the pathogenesis of AS (23).
It was previously demonstrated that three antigens
of the Epstein-Barr virus with overlapping sequences of different
lengths, including the 9-mer (56LPQGQLTAY64), 11-mer
(54EPLPQGQLTAY64) and 13-mer (52LPEPLPQGQLTAY64) peptides, all
bound efficiently to HLA-B*35:01. By contrast, the cytotoxic T cell
(CTL) response in individuals expressing HLA-B*35:01 was directed
exclusively toward the 11-mer peptide (30). Conversely, individuals with
HLA-B*35:03 demonstrated no significant CTL response to these
peptides; however, individuals expressing HLA-B*35:08 exhibited a
CTL response targeted to the 13-mer peptide (30). It was therefore hypothesized that
the CTL response to infecting pathogens may target antigen peptides
of >9 amino acids in length. It is therefore formally possible
that HLA-B27 may exhibit a similar role in the pathogenesis of AS,
by targeting unusually long peptides with subtype specificity.
For numerous variants of ERAP1, when the function of
peptide processing is suppressed or disordered, the modification of
peptide fragments may be deregulated and lead to an increase in the
number of extended peptides or abnormal peptides. It has previously
been demonstrated that mice lacking the ERAP1 enzyme exhibit
disrupted presentation of peptide-MHC-I molecules, which leads to a
marked shift in the hierarchy of immunodominance (31). This is followed by a 100-fold
increase in the immune response to various ERAP1-sensitive viral
peptides, a reduced or absent immune response to those that are
ERAP1-dependent, and an unaltered response to ERAP1-independent
peptides (31). Furthermore,
ERAP-deficient cells present numerous unstable and structurally
unique peptide-MHC complexes, which may elicit potent
CD8+ T cell and B cell responses (32). An additional factor that may
influence structural features of antigen peptides is TAP, as it
serves a role in altering antigen peptide selection and
transportation, which is the basis for further processing (33). The increase in the frequency of
longer peptides for dysfunctional ERAP1 and TAP may increase the
antigen peptide precursors and the frequency of downstream
abnormalities, thus resulting in a greater incidence of AS.
Therefore, it may be feasible to ameliorate CTL-mediated autoimmune
assaults by altering epitope generation via the administration of
selective proteasome inhibitors (34).
Cell surface HLA-B27 dimers and their
receptors
As the low binding affinity of β2m and peptides with
HLA-B27 HCs by hydrogen bonding, as well as HCs may form covalent
homodimers via the α1 domain of C67, the ‘cell surface HLA-B27
homodimers’ hypothesis proposes that formation of disulphide bonds
between the cysteine residue at C67 in the peptide binding groove
of two separate HC molecules generates homodimers without the
participation of β2m (23). It has
been previously demonstrated that HLA-B27 homodimers may bind to
immunoreceptors expressed on natural killer (NK) cells,
myelomonotic cells or lymphocytes [killer cell immunoglobulin-like
receptors (KIR) and leucocyte immunoglobulin-like receptors
(LILR)], and therefore may be important in the pathogenesis of
autoimmune disorders (37). A
previous study revealed that patients with SpA exhibit an increased
number of NK and CD4+ T cells (38). These cells express the killer cell
immunoglobulin-like receptor, three Ig domains and long cytoplasmic
tail 2 (KIR3DL2) receptor, which recognizes cell surface HLA-B27
homodimers (38,39). Interestingly, LILRB2 and KIR3DL2
bind to HLA-B27 dimers with a stronger affinity than HLA-B27, as
well as additional conventional HLA-class I heterotrimers, and the
binding of KIR3DL2 has been demonstrated to promote the survival
and differentiation of inflammatory leukocytes in SpA (40,41).
Enhanced proliferation and survival of KIR3DL2+ CD4+ T cells and
increased IL-17 production have been observed in AS patients upon
stimulation with antigen presenting cells that express HLA-B27
homodimers (38). In addition, the
majority of IL-17-producing KIR3DL2+ CD4+ T cells were reported to
produce tumor necrosis factor (TNF)-α, and were enriched with the
production of interferon (IFN)-γ, when compared with KIR3DL2-Th17
cells. KIR3DL2-expressing CD4+ T cells account for the majority of
peripheral blood CD4+ T cell IL-23 receptor expression, and produce
increased IL-17 in the presence of IL-23 (39). In addition to KIR3DL2, the
interaction between these cell surface HLA-B27 dimers and their
receptors require further investigation.
ER-resident and cell surface dimers are not
associated
The cysteine at position 67 is known to be involved
in cell surface and intracellular dimer formation (42). In addition, the structurally
conserved cysteines at positions 101 and 164 have been demonstrated
to be involved in the formation of ER-resident dimers (17). ER-resident dimers form via C67-C67
or C164-C164 disulfide bonds, however, they do not transit out of
the ER (43). Cell surface dimers
form following the recycling of fully-folded HLA-B27 cell surface
molecules via the endocytic pathway, prior to re-expression as
dimers mediated by C67-C67 interactions (25). It is important to note that
intracellular dimers and cell surface dimers may represent two
different mechanisms or hypotheses (Fig. 2). One potential mechanism may be
that the structurally unique peptide-MHC complexes are unstable,
which may lead to dissociation of heterotrimeric HLA-B27 from the
cell surface and the formation of cell surface homodimers.
Furthermore, the formation of these dimers from different HLA-B27
subtypes may contribute to the differential association of these
alleles with AS. It has been reported that HLA-B*27:05, which is
strongly associated with AS, forms a greater number of HLA-B27
dimers for KIR3DL2 compared with HLA-B*27:09 which is not
associated with AS (33).
Increased proportions of peripheral blood NK and CD4+ T cells that
express KIR3DL2 have been demonstrated to be present in AS patients
with HLA-B*27:05+, compared with healthy HLA-B*27:05+, HLA-B*27:09+
or HLA-B27- controls (44). By
contrast, it is possible that HLA-B27 homodimers may affect prior
to their expression on the cell surface (45). An additional subtype of MHC-I
molecules, HLA-G, has been demonstrated to form homodimers in
endosomes with a fully-folded β2m-associated form (21). This non-classical HLA-G subtype is
considered to be the ligand for KIR2DL4 (CD158d). Unlike the other
KIRs that are expressed on the surface of NK cells, KIR2DL4 resides
in endosomes (46). It remains to
be verified whether additional potential receptors are present in
endosomes that recognize endosomal HLA-B27 dimers, and whether they
demonstrate pathogenic roles in AS.
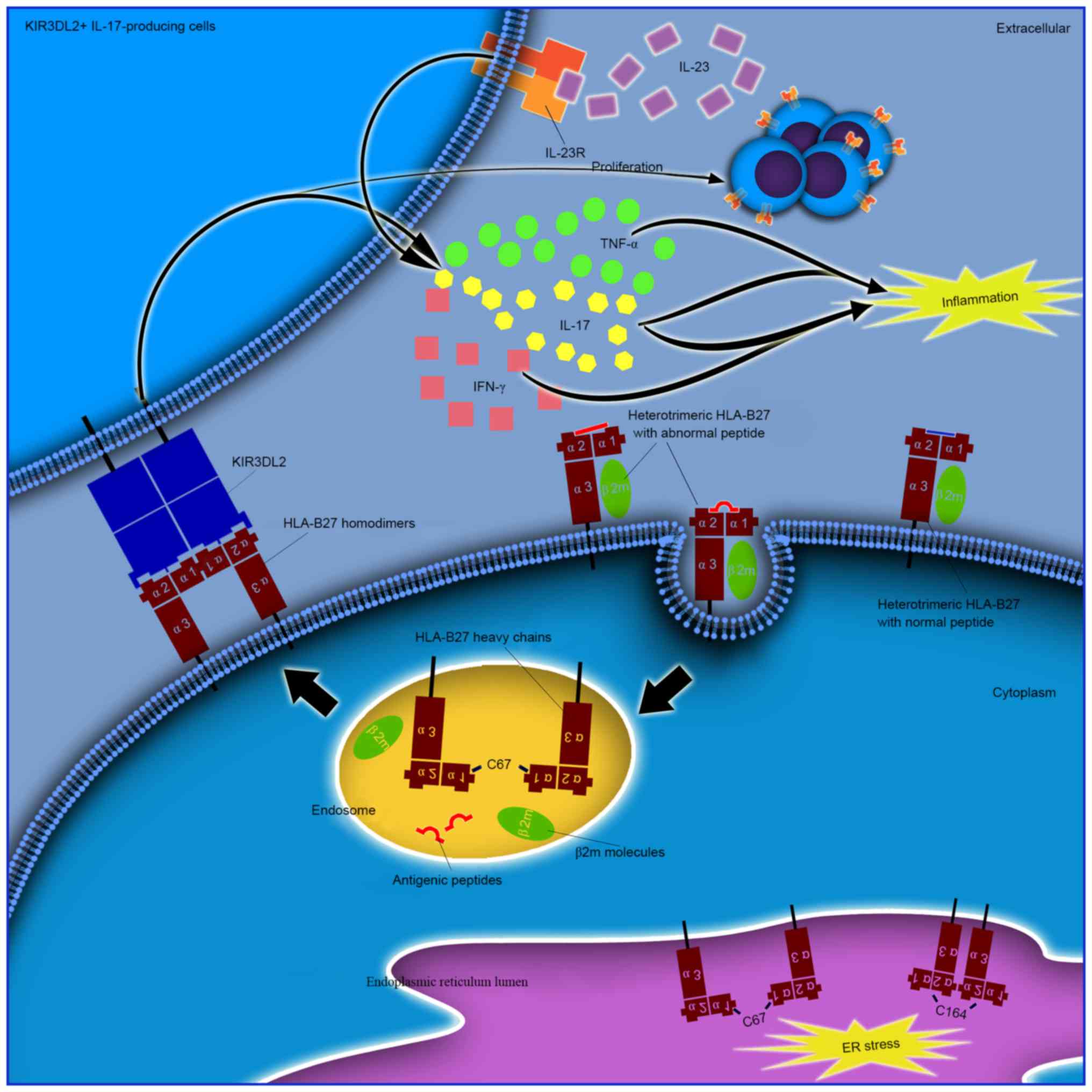 | Figure 2.Different pathogenic roles of
ER-resident and cell surface HLA-B27 dimers. ER resident dimers may
result in ER stress as a cellular response and lead to activation
of the unfolded protein response. Cell surface dimers are reported
to form following the recycling of fully-folded HLA-B27 cell
surface molecules via the endocytic pathway, and re-express as
dimers for presentation to immunoreceptors, including KIR and LILR.
Enhanced proliferation and survival of KIR3DL2+
CD4+ T cells and increased IL-17 production in patients
with AS following stimulation with antigen presenting cells
expressing HLA-B27 homodimers has been previously demonstrated. The
majority of these cells have been reported to produce TNF-α and
IFN-γ. IL-17 has been demonstrated to synergize with TNF-α or IFN-γ
to promote the release of inflammatory mediators and influence bone
metabolism, thus demonstrating its important role in the
pathogenesis of AS. ER, endoplasmic reticulum; HLA, human leukocyte
antigen; IFN-γ, interferon-γ; IL-17, interleukin-17; KIR, killer
cell immunoglobulin-like receptor; KIR3DL2, killer cell
immunoglobulin-like receptor three domains long cytoplasmic tail 2;
LILR, leucocyte immunoglobulin-like receptor; TNF-α, tumor necrosis
factor-α; AS, ankylosing spondylitis. |
Misfolded forms with or without the unfolded
protein response (UPR)
The majority of the disease-associated forms of
HLA-B27 molecules (HLA-B*27:05, HLA-B*27:04 and HLA-B*27:02)
demonstrate a reduced rate of folding when compared with
HLA-B*27:06 and HLA-B*27:09 and the majority of other MHC-I
molecules, which are not associated with AS (38,47).
The increase in duration for the disease-associated HLA-B27
molecules to assemble appears to subsequently lead to the
accumulation of misfolded HLA-B27 molecules in the ER; a proportion
of which may be in the form of dimers. The accumulation of
misfolded or unfolded proteins may perturb ER function and result
in ER stress and activation of the UPR, in an attempt to rescue or
dispose of the burden of misfolded proteins (48,49).
Furthermore, IL-23p19 (the unique subunit of the active IL-23
cytokine) was revealed to be synergistically upregulated by
lipopolysaccharide (LPS) in macrophages undergoing a UPR in an
HLA-B27 transgenic rat model of spondyloarthropathy (50). Furthermore, IL-23p19 was increased
in the colon of these rats, which was concurrent with the
development of intestinal inflammation. IL-17 exhibited robust
upregulation in a similar temporal pattern, with an expansion of
IL-17-expressing CD4+ T cells (50). However, increased production of
IL-23 in response to LPS without induction of significant UPR has
been reported in AS macrophages (51). Ciccia et al (52) reported that HLA-B27 misfolding
occurs in the gut of patients with AS, and is accompanied by
activation of autophagy rather than a UPR. However, Neerinckx et
al (53) failed to demonstrate
any significant increase in the expression of synovium
autophagy-associated genes by reverse transcription-polymerase
chain reaction, and no significant overexpression of IL-23p19 was
observed when compared with disease and healthy controls. IL-23p19
has been previously demonstrated, to be overexpressed in the
inflamed tissues of patients with AS (such as the gut and
zygapophysial joints) as determined by immunohistochemical analysis
(54,55). IL-23p19 may exhibit a
tissue-specific role in the gut and/or in the lymph nodes, by
priming specific subsets of IL-23-responsive proinflammatory cells.
A previous study investigated the hypothesis that ERAP1-mediated
HLA-B27 misfolding increases ER stress and induces an
IL-23-dependent, pro-inflammatory immune response (56). It was demonstrated that
disease-associated polymorphisms in the ERAP1 and HLA-B27 genes do
not alter ER-stress levels in AS (56). Therefore, it remains to be
elucidated whether ER-resident misfolded HLA-B27 molecules are
associated with the UPR and exhibit pathogenic roles in AS.
Exosomal fully-folded MHC I dimers
The redox-induced dimers in exosomes are
fully-folded and are independent of the cysteine residues at
positions 67 and 308. However these dimers are critically dependent
on cysteine 325 in the cytoplasmic tail (21). It has been suggested that they are
redox-induced due to the relative absence of the reducing agent
glutathione in exosomes, which contrasts with the low millimolar
levels normally observed in the cell cytoplasm (43). Considering that exosomes will be
released as extracellular vesicles, they may represent an important
mode of intercellular communication (20,57).
Therefore, the exosomal fully-folded MHC I dimers may transfer
signals to the resident cells in entheses to induce inflammation,
which may lead to alterations in joint architecture and new bone
formation.
Additional HLA-B27 hypotheses
β2m-free, peptide-free HCs support a helix-coil
transition facilitating rotation of backbone angles around amino
acid 167/168, thus leading to the residues 169–181 (identical to a
known HLA-B27 ligand) to loop around and occupy the molecule's own
peptide binding cleft. This ‘auto-display’, that occurs within or
between HLA-B27 molecules, may induce an autoimmune disease and be
important in the pathogenesis of AS (58).
Upon dissociation of the HLA-B27 dimers, β2m may
accumulate and become trapped in the synovia, where they may bind
to collagen and form amyloid deposits or interact with synovial
fibroblasts, thereby inducing the synthesis and secretion of
proteins involved in tissue destruction, finally resulting in AS
(59). This is termed the ‘β2m
deposition’ hypothesis. It was further hypothesized that β2m
expression levels may be associated with AS pathogenesis, as
spondylitis was successfully induced in a novel HLA-B27/β2m
transgenic rat model expressing increased levels of β2m (60). This lead to the proposed ‘β2m
over-expression’ hypothesis (60).
Discussion
According to the results of previous studies
discussed in the present review, it is possible that the onset of
AS may result from aberrant peptide presentation (11,12),
misfolded HLA-B27 molecules (16),
HLA-B27 dimers (17,19) or β2m accumulation and deposition
(6,23) (Fig.
3). Previous studies have demonstrated the involvement of the
IL-23/IL-17 axis in the pathogenesis of AS (61,62).
However, aberrant recognition and cytokine dysregulation may not be
two independent procedures. Aberrant recognition by specific
immunoreceptors may occur in upstream pathways, which may
subsequently contribute to downstream cytokine dysregulation,
particularly in the IL-23/IL-17 axis.
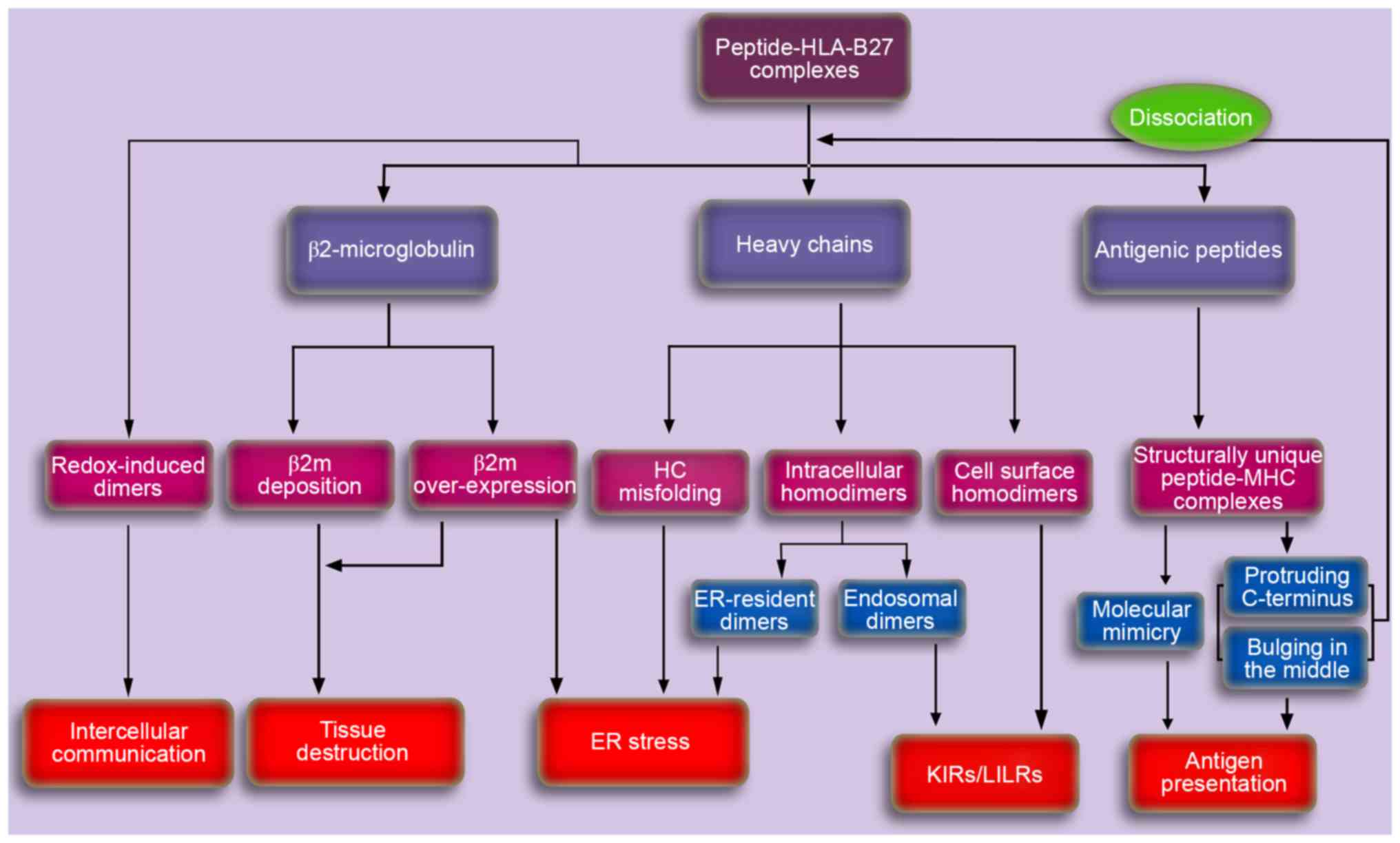 | Figure 3.A schematic depicting the potential
pathogenesis of AS caused by HLA-B27. Aberrant processing and
presentation of structurally unique peptides were initially
proposed to explain the potential pathogenesis of AS. Cell surface
HLA-B27 dimers may be recognized by various immunoreceptors and may
be important in the pathogenesis of autoimmune disorders.
Accumulation of proteins in the ER, including ER-resident dimers,
misfolded HCs and β2m may result in the ER stress response, thereby
activating the unfolded protein response, which is associated with
cytokine dysregulation. In addition, the accumulating β2m in
synovia for dissociation and/or overexpression, may induce the
synthesis and secretion of proteins involved in tissue destruction,
thus leading to AS. The exosomal fully-folded HLA-B27 dimers may be
important in the pathogenesis of AS via intercellular
communication. AS, ankylosing spondylitis; HLA, human leukocyte
antigen; HC, heavy chain; KIR, killer cell immunoglobulin-like
receptor; LILR, leucocyte immunoglobulin-like receptor; ER,
endoplasmic reticulum; MHC, major histocompatibility complex; β2m,
β2microglobulin. |
Currently, three major mechanistic hypotheses exist
to describe the association between HLA-B27 and AS. Firstly,
aberrant peptide processing and presentation may be involved in the
pathogenesis of AS due to the interaction between HLA-B27 and ERAP1
(11,12). However, the molecular mechanisms
underlying this process remain to be fully elucidated. Secondly,
misfolded HLA-B27 molecules in the ER may trigger ER stress and
provoke the UPR (16). It has been
previously demonstrated that this is followed by the subsequent
upregulation of various cytokines, particularly IL-23 and IL-17,
accompanied by the development of immune dysregulation. However,
macrophages from AS patients exhibited greater IL-23 production in
response to LPS, and no significant UPR induction was observed. The
induction of the UPR is dependent on the magnitude and duration of
ER stress, as well as the type of cells that are affected. Further
studies are required to determine whether cells process misfolded
monomers, intracellular HLA-B27 homodimers or β2m in the ER
differently, and whether they may be associated with the UPR and
further cytokine dysregulation. In addition, further studies are
required to reassess the cellular source of IL-17 in the primary
target tissues of AS, including γδ T cells, mast cells, neutrophils
or innate lymphoid cells that have been implicated in previous
studies (3). Furthermore, cell
surface HLA-B27 dimers may be important in AS pathogenesis, due to
their role in binding to receptors on immune cells (17,19).
The recognition of HLA-B27 dimers by KIR3DL2 is reportedly
associated with KIR3DL2+ IL-17-producing CD4+
T cells, IL-23 receptor expression and the production of IL-17,
TNF-α and IFN-γ. Future studies investigating the intrinsic
association between the pathogenic role of HLA-B27 and the
IL-23/IL-17 axis may provide novel insights into understanding the
molecular mechanisms involved in the development and progression of
AS.
Acknowledgements
The present review was supported by grant-in-aid for
scientific research from the National Natural Science Foundation of
China (grant no. 81171686) and the Natural Science Foundation of
Shanghai (grant no. 14140903802).
References
|
1
|
Dougados M and Baeten D:
Spondyloarthritis. Lancet. 377:2127–2137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cho H, Kim T, Kim TH, Lee S and Lee KH:
Spinal mobility, vertebral squaring, pulmonary function, pain,
fatigue, and quality of life in patients with ankylosing
spondylitis. Ann Rehabil Med. 37:675–682. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Braun J and Sieper J: Ankylosing
spondylitis. Lancet. 369:1379–1390. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Végvári A, Szabó Z, Szántó S, Glant TT,
Mikecz K and Szekanecz Z: The genetic background of ankylosing
spondylitis. Joint Bone Spine. 76:623–628. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brewerton DA, Hart FD, Nicholls A, Caffrey
M, James DC and Sturrock RD: Ankylosing spondylitis and HL-A 27.
Lancet. 1:904–907. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chatzikyriakidou A, Voulgari PV and Drosos
AA: What is the role of HLA-B27 in spondyloarthropathies? Autoimmun
Rev. 10:464–468. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
International Genetics of Ankylosing
Spondylitis Consortium (IGAS), ; Cortes A, Hadler J, Pointon JP,
Robinson PC, Karaderi T, Leo P, Cremin K, Pryce K, Harris J, et al:
Identification of multiple risk variants for ankylosing spondylitis
through high-density genotyping of immune-related loci. Nat Genet.
45:730–738. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sheehan NJ: HLA-B27: What's new?
Rheumatology (Oxford). 49:621–631. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khan MA: Polymorphism of HLA-B27: 105
subtypes currently known. Curr Rheumatol Rep. 15:3622013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brown MA: Progress in the genetics of
ankylosing spondylitis. Brief Funct Genomics. 10:249–257. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Warde N: Spondyloarthropathies: HLA-B27
and ERAP1 contribute to ankylosing spondylitis via aberrant peptide
processing and presentation. Nat Rev Rheumatol. 7:4982011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Evans DM, Spencer CC, Pointon JJ, Su Z,
Harvey D, Kochan G, Oppermann U, Dilthey A, Pirinen M, Stone MA, et
al: Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis
implicates peptide handling in the mechanism for HLA-B27 in disease
susceptibility. Nat Genet. 43:761–767. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nguyen TT, Chang SC, Evnouchidou I, York
IA, Zikos C, Rock KL, Goldberg AL, Stratikos E and Stern LJ:
Structural basis for antigenic peptide precursor processing by the
endoplasmic reticulum aminopeptidase ERAP1. Nat Struct Mol Biol.
18:604–613. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yewdell JW: DRiPs solidify: Progress in
understanding endogenous MHC class I antigen processing. Trends
Immunol. 32:548–558. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Madden DR: The three-dimensional structure
of peptide-MHC complexes. Annu Rev Immunol. 13:587–622. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Colbert RA, Tran TM and Layh-Schmitt G:
HLA-B27 misfolding and ankylosing spondylitis. Mol Immunol.
57:44–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lenart I, Guiliano DB, Burn G, Campbell
EC, Morley KD, Fussell H, Powis SJ and Antoniou AN: The MHC Class I
heavy chain structurally conserved cysteines 101 and 164
participate in HLA-B27 dimer formation. Antioxid Redox Signal.
16:33–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alvarez-Navarro C and López de Castro JA:
ERAP1 structure, function and pathogenetic role in ankylosing
spondylitis and other MHC-associated diseases. Mol Immunol.
57:12–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Colbert RA: The immunobiology of HLA-B27:
Variations on a theme. Curr Mol Med. 4:21–30. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Raposo G and Stoorvogel W: Extracellular
vesicles: Exosomes, microvesicles, and friends. J Cell Biol.
200:373–383. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lynch S, Santos SG, Campbell EC, Nimmo AM,
Botting C, Prescott A, Antoniou AN and Powis SJ: Novel MHC class I
structures on exosomes. J Immunol. 183:1884–1891. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lorente E, Infantes S, Abia D, Barnea E,
Beer I, García R, Lasala F, Jiménez M, Mir C, Morreale A, et al: A
viral, transporter associated with antigen processing
(TAP)-independent, high affinity ligand with alternative
interactions endogenously presented by the nonclassical human
leukocyte antigen E class I molecule. J Biol Chem. 287:34895–34903.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen B, Li D and Xu W: Association of
ankylosing spondylitis with HLA-B27 and ERAP1: Pathogenic role of
antigenic peptide. Med Hypotheses. 80:36–38. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lévy F, Burri L, Morel S, Peitrequin AL,
Lévy N, Bachi A, Hellman U, Van den Eynde BJ and Servis C: The
final N-terminal trimming of a subaminoterminal proline-containing
HLA class I-restricted antigenic peptide in the cytosol is mediated
by two peptidases. J Immunol. 169:4161–4171. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Antoniou AN, Lenart I and Guiliano DB:
Pathogenicity of misfolded and dimeric HLA-B27 molecules. Int J
Rheumatol. 2011:4868562011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dakwar E, Reddy J, Vale FL and Uribe JS: A
review of the pathogenesis of ankylosing spondylitis. Neurosurg
Focus. 24:E22008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Taurog JD, Dorris ML, Satumtira N, Tran
TM, Sharma R, Dressel R, van den Brandt J and Reichardt HM:
Spondylarthritis in HLA-B27/human beta2-microglobulin-transgenic
rats is not prevented by lack of CD8. Arthritis Rheum.
60:1977–1984. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Collins EJ, Garboczi DN and Wiley DC:
Three-dimensional structure of a peptide extending from one end of
a class I MHC binding site. Nature. 371:626–629. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Probst-Kepper M, Hecht HJ, Herrmann H,
Janke V, Ocklenburg F, Klempnauer J, van den Eynde BJ and Weiss S:
Conformational restraints and flexibility of 14-meric peptides in
complex with HLA-B*3501. J Immunol. 173:5610–5616. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Green KJ, Miles JJ, Tellam J, van Zuylen
WJ, Connolly G and Burrows SR: Potent T cell response to a class
I-binding 13-mer viral epitope and the influence of HLA
micropolymorphism in controlling epitope length. Eur J Immunol.
34:2510–2519. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
York IA, Brehm MA, Zendzian S, Towne CF
and Rock KL: Endoplasmic reticulum aminopeptidase 1 (ERAP1) trims
MHC class I-present edpeptides in vivo and plays an important role
in immunodominance. Proc Natl Acad Sci USA. 103:9202–9207. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hammer GE, Gonzalez F, James E, Nolla H
and Shastri N: In the absence of aminopeptidase ERAAP, MHC class I
molecules present many unstable and highly immunogenic peptides.
Nat Immunol. 8:101–108. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lorente E, García R, Mir C, Barriga A,
Lemonnier FA, Ramos M and López D: Role of metalloproteases in
vaccinia virus epitope processing for transporter associated with
antigen processing (TAP)-independent human leukocyte antigen
(HLA)-B7 class I antigen presentation. J Biol Chem. 287:9990–10000.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schwarz K, De Giuli R, Schmidtke G, Kostka
S, van den Broek M, Kim KB, Crews CM, Kraft R and Groettrup M: The
selective proteasome inhibitors lactacystin and epoxomicin can be
used to either up- or down-regulate antigen presentation at
nontoxic doses. J Immunol. 164:6147–6157. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Allen RL and Trowsdale J: Recognition of
classical and heavy chain forms of HLA-B27 by leukocyte receptors.
Curr Mol Med. 4:59–65. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kollnberger S, Chan A, Sun MY, Chen LY,
Wright C, di Gleria K, McMichael A and Bowness P: Interaction of
HLA-B27 homodimers with KIR3DL1 and KIR3DL2, unlike HLA-B27
heterotrimers, is independent of the sequence of bound peptide. Eur
J Immunol. 37:1313–1322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Allen RL, Raine T, Haude A, Trowsdale J
and Wilson MJ: Leukocyte receptor complex-encoded immunomodulatory
receptors show differing specificity for alternative HLA-B27
structures. J Immunol. 167:5543–5547. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chan AT, Kollnberger SD, Wedderburn LR and
Bowness P: Expansion and enhanced survival of natural killer cells
expressing the killer immunoglobulin-like receptor KIR3DL2 in
spondylarthritis. Arthritis Rheum. 52:3586–3595. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bowness P, Ridley A, Shaw J, Chan AT,
Wong-Baeza I, Fleming M, Cummings F, McMichael A and Kollnberger S:
Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers
are increased in ankylosing spondylitis. J Immunol. 186:2672–2680.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Giles J, Shaw J, Piper C, Wong-Baeza I,
McHugh K, Ridley A, Li D, Lenart I, Antoniou AN, DiGleria K, et al:
HLA-B27 homodimers and free H chains are stronger ligands for
leukocyte Ig-like receptor B2 than classical HLA class I. J
Immunol. 188:6184–6193. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wong-Baeza I, Ridley A, Shaw J, Hatano H,
Rysnik O, McHugh K, Piper C, Brackenbridge S, Fernandes R, Chan A,
et al: KIR3DL2 binds to HLA-B27 dimers and free H chains more
strongly than other HLA class I and promotes the expansion of T
cells in ankylosing spondylitis. J Immunol. 190:3216–3224. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dangoria NS, DeLay ML, Kingsbury DJ, Mear
JP, Uchanska-Ziegler B, Ziegler A and Colbert RA: HLA-B27
misfolding is associated with aberrant intermolecular disulfide
bond formation (dimerization) in the endoplasmic reticulum. J Biol
Chem. 277:23459–23468. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Campbell EC, Antoniou AN and Powis SJ: The
multi-faceted nature of HLA class I dimer molecules. Immunology.
136:380–384. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cauli A, Shaw J, Giles J, Hatano H, Rysnik
O, Payeli S, McHugh K, Dessole G, Porru G, Desogus E, et al: The
arthritis-associated HLA-B*27:05 allele forms more cell surface B27
dimer and free heavy chain ligands for KIR3DL2 than HLA-B*27:09.
Rheumatology (Oxford). 52:1952–1962. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kuśnierczyk P and Majorczyk E: Pas de
quatre: An interaction of HLA-B*27:05 and KIR3DL2 homodimers in
spondyloarthropathies. Rheumatology (Oxford). 52:1931–1912. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rajagopalan S and Long EO: KIR2DL4
(CD158d): An activation receptor for HLA-G. Front Immunol.
3:2582012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Antoniou AN, Ford S, Taurog JD, Butcher GW
and Powis SJ: Formation of HLA-B27 homodimers and their
relationship to assembly kinetics. J Biol Chem. 279:8895–8902.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Colbert RA, DeLay ML, Layh-Schmitt G and
Sowders DP: HLA-B27 misfolding and spondyloarthropathies. Prion.
3:15–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Turner MJ, Sowders DP, DeLay ML, Mohapatra
R, Bai S, Smith JA, Brandewie JR, Taurog JD and Colbert RA: HLA-B27
misfolding in transgenic rats is associated with activation of the
unfolded protein response. J Immunol. 175:2438–2348. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
DeLay ML, Turner MJ, Klenk EI, Smith JA,
Sowders DP and Colbert RA: HLA-B27 misfolding and the unfolded
protein response augment interleukin-23 production and are
associated with Th17 activation in transgenic rats. Arthritis
Rheum. 60:2633–2643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zeng L, Lindstrom MJ and Smith JA:
Ankylosing spondylitis macrophage production of higher levels of
interleukin-23 in response to lipopolysaccharide without induction
of a significant unfolded protein response. Arthritis Rheum.
63:3807–3817. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ciccia F, Accardo-Palumbo A, Rizzo A,
Guggino G, Raimondo S, Giardina A, Cannizzaro A, Colbert RA,
Alessandro R and Triolo G: Evidence that autophagy, but not the
unfolded protein response, regulates the expression of IL-23 in the
gut of patients with ankylosing spondylitis and subclinical gut
inflammation. Ann Rheum Dis. 73:1566–1574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Neerinckx B, Carter S and Lories R: IL-23
expression and activation of autophagy in synovium and PBMCs of
HLA-B27 positive patients with ankylosing spondylitis. Response to:
‘Evidence that autophagy, but not the unfolded protein response,
regulates the expression of IL-23 in the gut of patients with
ankylosing spondylitis and subclinical gut inflammation’ by Ciccia.
Ann Rheum Dis. 73:e682014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ciccia F, Bombardieri M, Principato A,
Giardina A, Tripodo C, Porcasi R, Peralta S, Franco V, Giardina E,
Craxi A, et al: Overexpression of interleukin-23, but not
interleukin-17, as an immunologic signature of subclinical
intestinal inflammation in ankylosing spondylitis. Arthritis Rheum.
60:955–965. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Appel H, Maier R, Bleil J, Hempfing A,
Loddenkemper C, Schlichting U, Syrbe U and Sieper J: In situ
analysis of interleukin-23- and interleukin-12-positive cells in
the spine of patients with ankylosing spondylitis. Arthritis Rheum.
65:1522–1529. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kenna TJ, Lau MC, Keith P, Ciccia F,
Costello ME, Bradbury L, Low PL, Agrawal N, Triolo G, Alessandro R,
et al: Disease-associated polymorphisms in ERAP1 do not alter
endoplasmic reticulum stress in patients with ankylosing
spondylitis. Genes Immun. 16:35–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shaw J, Hatano H and Kollnberger S: The
biochemistry and immunology of non-canonical forms of HLA-B27. Mol
Immunol. 57:52–58. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Luthra-Guptasarma M and Singh B: HLA-B27
lacking associated beta2-microglobulin rearranges to auto-display
or cross-display residues 169–181: A novel molecular mechanism for
spondyloarthropathies. FEBS Lett. 575:1–8. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Uchanska-Ziegler B and Ziegler A:
Ankylosing spondylitis: A beta2m-deposition disease? Trends
Immunol. 24:73–76. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tran TM, Dorris ML, Satumtira N,
Richardson JA, Hammer RE, Shang J and Taurog JD: Additional human
beta2-microglobulin curbs HLA-B27 misfolding and promotes arthritis
and spondylitis without colitis in male HLA-B27-transgenic rats.
Arthritis Rheum. 54:1317–1327. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yeremenko N, Paramarta JE and Baeten D:
The interleukin-23/interleukin-17 immune axis as a promising new
target in the treatment of spondyloarthritis. Curr Opin Rheumatol.
26:361–370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jethwa H and Bowness P: The interleukin
(IL)-23/IL-17 axis in ankylosing spondylitis: New advances and
potentials for treatment. Clin Exp Immunol. 183:30–36. 2016.
View Article : Google Scholar : PubMed/NCBI
|














