Introduction
Myocardial infarction (MI) is a leading cause of
congestive heart failure. The left ventricle (LV) undergoes
molecular, cellular and extracellular matrix alterations following
MI that can significantly affect myocardial size, shape and
function (1). Ventricular
remodeling due to persistent hypoxia and ischemia following MI has
been associated with the development of heart failure (2). Therefore, elucidating the mechanisms
underlying post-MI ventricular remodeling may yield novel
therapeutic targets for the treatment of patients with cardiac
disorders.
The nuclear hormone receptor family comprises of
transcriptional regulators that have been implicated in numerous
physiological and pathophysiological functions, such as cellular
metabolism, proliferation and apoptosis, as well as tumorigenesis
and angiogenesis. Several nuclear hormone receptors, such as liver
X receptors α and β, and the estrogen, androgen and retinoic acid
receptors, have been implicated in the physiological regulation of
cardiovascular function (3–8). The
farnesoid X receptor (FXR; NR1H4) is another member of the nuclear
receptor superfamily (9–12). FXR expression is localized
primarily in the liver, intestine, kidneys and adrenal glands,
where it regulates the homeostasis and metabolism of cholesterol,
bile acid and lipids (9). FXR is
also expressed in the heart and adipose tissue, and in the
vasculature (11,13,14).
Previous studies have suggested that FXR, apart from its
implication in metabolism, may serve an important role in
cardiovascular physiology and pathology (15,16).
FXR has been identified as a novel apoptotic mediator and has been
revealed to contribute to myocardial ischemia/reperfusion injury
(13). However, the mechanisms
underlying the implication of FXR in pathophysiological processes
following MI have yet to be elucidated. Therefore, the aim of the
present study was to investigate the effects of FXR knockdown on LV
remodeling and dysfunction following MI.
Materials and methods
Ethics statement
The present study was conducted according to the
Guide for the Care and Use of Laboratory Animals published by the
US National Institutes of Health (NIH Publication No. 85–23,
revised 1996) and was approved by the Ethics Review Board for
Animal Studies of Shanghai Jiaotong University School of Medicine
(approval no. SYKX-2008-0050; Shanghai, China).
Animals
FXR−/− and wild type (WT) male mice were
obtained from The Jackson Laboratory (Bar Harbor, ME, USA).
FXR−/− mice and control WT C57BL/6 mice (age, 8–12
weeks; weight, 20–25 g) were housed in a specific pathogen-free
barrier facility at 24°C, 55% humidity and under 12/12 h light/dark
cycles, with free access to food and water.
Mouse model of MI
The MI model was generated by an experienced
investigator who was blind to the experiment. Mice were
anesthetized with 2% isoflurane prior to treatment with 0.5%
bupivacaine. The left anterior descending coronary artery (LAD) was
ligated as previously described (17). Sham-operated mice (n=6) underwent
open chest surgery without coronary artery ligation. A total of 6
WT and 6 FXR−/− mice that underwent MI (n=6) were
sacrificed on days 7 and 28 following MI. All possible measures
were taken to minimize suffering.
Ultrasonic echocardiography (UCG)
assessment
Cardiac function was evaluated using transthoracic
UCG prior to surgery and 1 day, 1 week and 4 weeks following MI in
a blind manner. UCG was performed using the Vevo® 2100
ultrasound system (VisualSonics, Inc., Toronto, ON, Canada)
equipped with a 30 MHz transducer.
LV dimensions were examined digitally in M-mode
tracings and averaged from three consecutive cardiac cycles. LV
end-systolic diameter (LVESD) and end-diastolic diameter (LVEDD),
interventricular septal thickness in diastole and LV posterior wall
thickness were assessed. The percentage of LV fractional shortening
(%FS) was calculated as follows: % FS = (LVEDD-LVESD)/LVEDD × 100
(%).
Assessment of infarct size
2,3,5-triphenyltetrazolium chloride (TTC) was used
for assessment of the infarction size. Briefly, 28 days after
surgery the hearts from randomly selected mice were removed, frozen
at −20°C and cut into 1-mm thick sections perpendicular to the long
axis of the heart. The hearts of these mice were harvested to
assess the infarct size only. It was observed that the infarct area
was not stained, and the non-infarct area was dyed red. The slice
was photographed with a digital camera following TTC staining.
Infarct size on day 28 post-MI was evaluated, as described by
Pfeffer et al (18,19), and calculated as a percentage of
the whole LV size, using ImageJ version k1.45 software (National
Institutes of Health, Bethesda, MD, USA).
Histological examination
The hearts of left ventricular myocardium were
arrested by intravenous administration of 2 mol/l KCl (Shanghai
Pharmaceuticals Holding Co., Ltd., Shanghai, China) following
isoflurane anesthesia and abdominal aorta bleeding. The samples
were immediately fixed with 4% paraformaldehyde at room temperature
for 24 h and dehydrated, paraffin embedded and sliced into 4 µm
sections, on weeks 1 and 4 following MI, to be used for terminal
deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay,
hematoxylin and eosin (H&E) staining, Massons trichrome
staining for interstitial fibrosis and α-smooth muscle actin
(α-SMA) staining. Paraffin-embedded tissue sections were stained
with a Massons trichrome Staining kit (cat. no. G1006; Wuhan
Goodbio Technology Co., Ltd., Wuhan, China). Collagen volume
fraction (CVF) was quantified and calculated as an area of Massons
trichrome-stained connective tissue divided by total area of the
image as previously described (17,18)
using Image Pro Plus software (Media Cybernetics, Inc., Rockville,
MD, USA). Immunohistochemical staining of α-SMA as a marker of
myocardial fibrosis after myocardial infarction was also performed.
Paraffin-embedded tissue sections were stained with an α-SMA
primary antibody (1:2,000; cat. no. GB13063; Wuhan Goodbio
Technology Co., Ltd., Wuhan, China) overnight at 4°C, washed three
times with PBS and incubated with HRP-conjugated secondary goat
anti mouse IgG (1:10,000; cat. no. K5007; Wuhan Goodbio Technology
Co., Ltd., Wuhan, China) for 50 min at room temperature. Samples
were counterstained with hematoxylin and incubated at room
temperature for 3 min. Stained sections were observed under a
confocal laser scanning microscope. LV diameter was measured in two
perpendicular axes and averaged for each animal (18). LVEDD was measured as previously
described (19). Myocyte size was
measured using cross sections midway between the base and apex of
the LV with hematoxylin and eosin (H&E) staining.
Western blot analysis
FXR−/− and WT C57BL/6 mice were
sacrificed on days 3, 7 and 28 post-MI for the collection of heart
samples. Proteins extracted from heart tissue were analyzed using
western blot analysis. The harvested ventricular tissue was
homogenized and lysed using lysis buffer containing 1 protease
inhibitor cocktail tablet per 10 ml of lysis reagents (Roche
Diagnostics, Indianapolis, IN, USA). Protein concentration was
determined with a bicinchoninic acid (BCA) protein assay kit
(Beyotime Institute of Biotechnology, Shanghai, China). For western
blot analysis an equal amount of protein (60 mg) was loaded in each
well and subjected to 12% sodium dodecyl-sulfate polyacrylamide gel
electrophoresis. Separated proteins were then transferred from the
gel to nitrocellulose membranes (Whatman; GE Healthcare Life
Sciences, Chalfont, UK) and blocked with LI-COR blocking buffer for
1–2 h. The following primary antibodies were used: Anti-FXR
(1:1,000; cat. no. 12295; Cell Signaling Technology, Inc., Danvers,
MA, USA), anti-caspase 3 (1:800; cat. no. 9664; Cell Signaling
Technology, Inc.) and anti-GAPDH (1:1,000; cat. no. 2118; Bioworld
Technology, Inc., St. Louis Park, MN, USA). The secondary
antibodies of HRP-conjugated goat anti mouse IgG (1:10,000; cat.
no. 22855) were obtained from LI-COR Biosciences (Lincoln, NE,
USA). The protein bands were visualized using an Odyssey infrared
imaging system (LI-COR Biosciences). Protein levels were
semi-quantified on duplicate blots with standard densitometry using
Odyssey software version 3.0 (LI-COR Biosciences). The intensities
of protein bands were normalized to those of GAPDH and expressed as
fold increase relative to control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
As previously described (20,21),
total RNA from tissues and cardiomyocytes was extracted using
TRIzol® Reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) according to the manufacturer's protocol.
RNA was reverse transcribed into cDNA using M-MLV reverse
transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.). The
following primers were used for qPCR: GAPDH, forward
5′-TCACTGCCACCCAGAAGA-3′, reverse 5′-GACGGACACATTGGGGGTAG-3′;
collagen III, forward 5′-GACCAAAAGGTGATGCTGGACAG-3′, reverse
5′-CAAGACCTCGTGCTCCAGTTAG-3′; and matrix metalloproteinase (MMP)-9,
forward 5′-GCTGACTACGATAAGGACGGCA-3′ and reverse
5′-TAGTGGTGCAGGCAGAGTAGGA-3′. qPCR was performed on cDNA with a
Real-Time PCR system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA), using SYBR Premix Ex Taq™ (Takara Bio, Inc., Otsu, Japan).
The reactions of 10 µl volume contained 2X SYBR Premix Ex Taq™
(Takara Bio, Inc.) at 5 µl, each primer at 0.2 µl, and 0.2 µl of
cDNA template (1:5 diluted) at 1 µl, RNase-free dH2O at
3.6 µl. Amplification was performed under the following conditions:
Initial denaturation step at 95°C for 3 min, followed by 40 cycles
of denaturation at 95°C for 30 sec, annealing and extension at 60°C
for 30 sec. The relative expression levels for each gene were
normalized to GAPDH and analyzed using the 2−∆∆Cq method
(22).
TUNEL assay
Cellular apoptosis of left ventricular myocardium
was assessed using the ApopTag® Fluorescein In
Situ Apoptosis Detection kit (S7110; EMD Millipore, Billerica,
MA, USA), according to the manufacturer's protocol. Cell nuclei
were stained with 4′-6-diamidino-2-phenylindole (DAPI), and
incubated at room temperature for 10 min. Sections were mounted and
observed under a FluoView™ FV1000 confocal microscope (Olympus
Corporation, Tokyo, Japan). The number of TUNEL-positive
cardiomyocyte nuclei was manually determined. At least 3 fields per
section were analyzed, as previously described (23).
Immunofluorescence
Paraffin-embedded tissue sections were stained with
an anti-cluster of differentiation (CD) 31 primary antibody (1:50;
cat. no. GB13063; Wuhan Goodbio Technology Co., Ltd., Wuhan, China)
overnight at 4°C, washed three times with PBS and incubated with
fluorescein isothiocyanate-conjugated secondary donkey anti-rabbit
immunoglobulin G (1:200; cat. no. 705-165-003; Wuhan Goodbio
Technology Co., Ltd.) for 1 h at room temperature in a darkened
humidified chamber. Samples were counterstained with DAPI, and
incubated at room temperature for 10 min. Stained sections were
observed under a confocal laser scanning microscope using ×200 and
×400 magnifications.
Statistical analysis
The statistical significance of the difference
between two groups was performed by unpaired Student's t test. The
comparison of parameters among more than three groups was performed
by one-way analysis of variance with Tukey's HSD post hoc test.
Data are expressed as the mean ± standard error of the mean. The
experiments were repeated 5 times. P<0.05 was considered to
indicate a statistically significant difference. All analyses were
performed using GraphPad Prism software version 5.0 (GraphPad
Software, Inc., La Jolla, CA, USA).
Results
FXR protein expression levels in
post-MI myocardial tissue
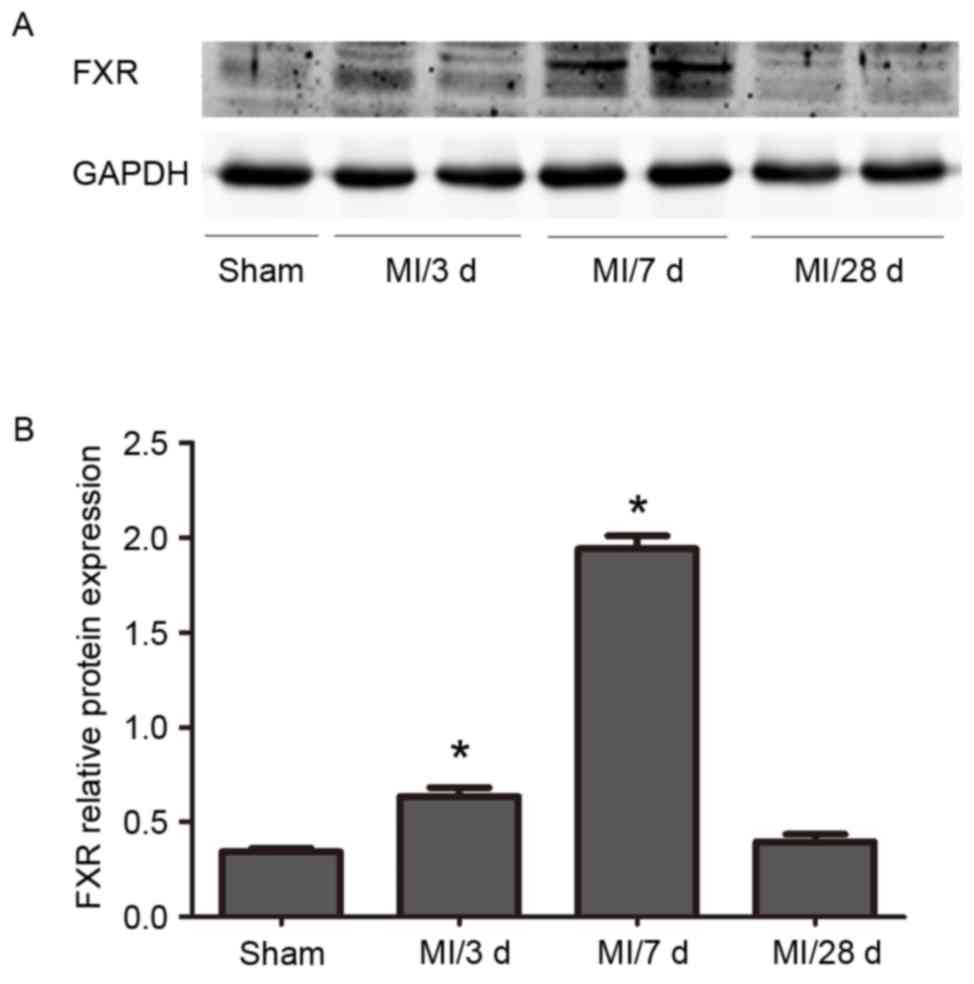
Western blot analysis revealed a progressive
increase in FXR protein expression levels in WT mice between 3
days, 7 days and 28 days following LAD ligation. FXR levels
declined at 28 days. (Fig. 1).
FXR−/− mice demonstrate
improved myocardial morphology and function post-MI
UCG was used to assess cardiac function 1 and 4
weeks post-MI (Tables I and
II). %FS was used as a systolic
function index and results indicated a significant improvement in
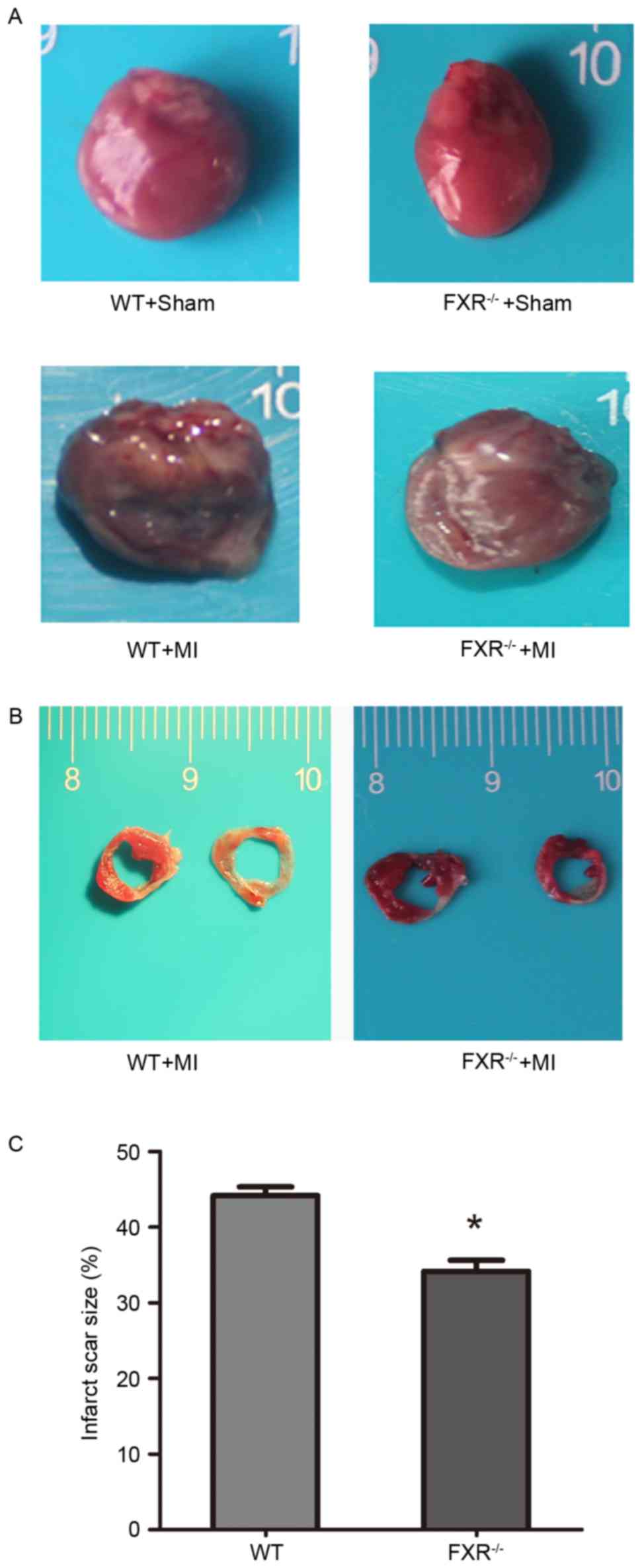
FXR−/− mice compared with in WT mice (Table II). Furthermore, FXR−/−
mice exhibited lower LVEDD and LVESD compared with WT mice
(Fig. 2A). In addition, no
significant difference in cardiac function was revealed between
FXR−/− and WT sham-operated mice (Tables I and II), suggesting that FXR knockout does
not affect cardiac function under physiological conditions.
 | Table I.Ultrasonic echocardiography
performance 1 week following MI surgery. |
Table I.
Ultrasonic echocardiography
performance 1 week following MI surgery.
| Variable | WT sham | FXR−/−
sham | WT MI | FXR−/−
MI |
|---|
| n | 6 | 7 | 8 | 8 |
| EF% |
68.32±1.05a | 65.28±2.52 |
44.41±1.54c |
47.63±1.42b |
| FS% |
38.29±0.46a | 39.18±0.53 |
21.57±1.23c |
26.08±1.46b |
| LVEDD (mm) |
3.47±0.10a | 3.38±0.09 |
4.05±0.13c |
3.85±0.12b |
| LVESD (mm) |
2.17±0.11a | 2.28±0.13 |
3.13±0.16c |
2.85±0.15b |
| Table II.Ultrasonic echocardiography
performance 4 weeks following MI surgery. |
Table II.
Ultrasonic echocardiography
performance 4 weeks following MI surgery.
| Variable | WT sham | FXR−/−
sham | WT MI | FXR−/−
MI |
|---|
| n | 9 | 9 | 10 | 10 |
| EF% |
72.13±0.86a | 70.35±1.13 | 37.64±0.77 |
47.31±1.08b |
| FS% |
37.25±0.63a | 38.23±0.76 | 20.63±0.71 |
25.96±0.62b |
| LVEDD (mm) |
3.45±0.12a | 3.67±0.11 | 4.78±0.22 |
3.84±0.13b |
| LVESD (mm) |
2.09±0.12a | 2.21±0.10 | 3.99±0.12 |
3.10±0.10b |
Infarct scar size appeared significantly reduced in
the FXR−/− compared with in the WT group (Fig. 2B and C). A 10% relative reduction
in infarct size was reported in FXR−/− mice compared
with in WT mice. LV cavity size was preserved in the
FXR−/− group compared with in the WT group, as
demonstrated by the significantly smaller LV diameters in
FXR−/− mice (Table
II).
Microvesicular density and
fibrosis
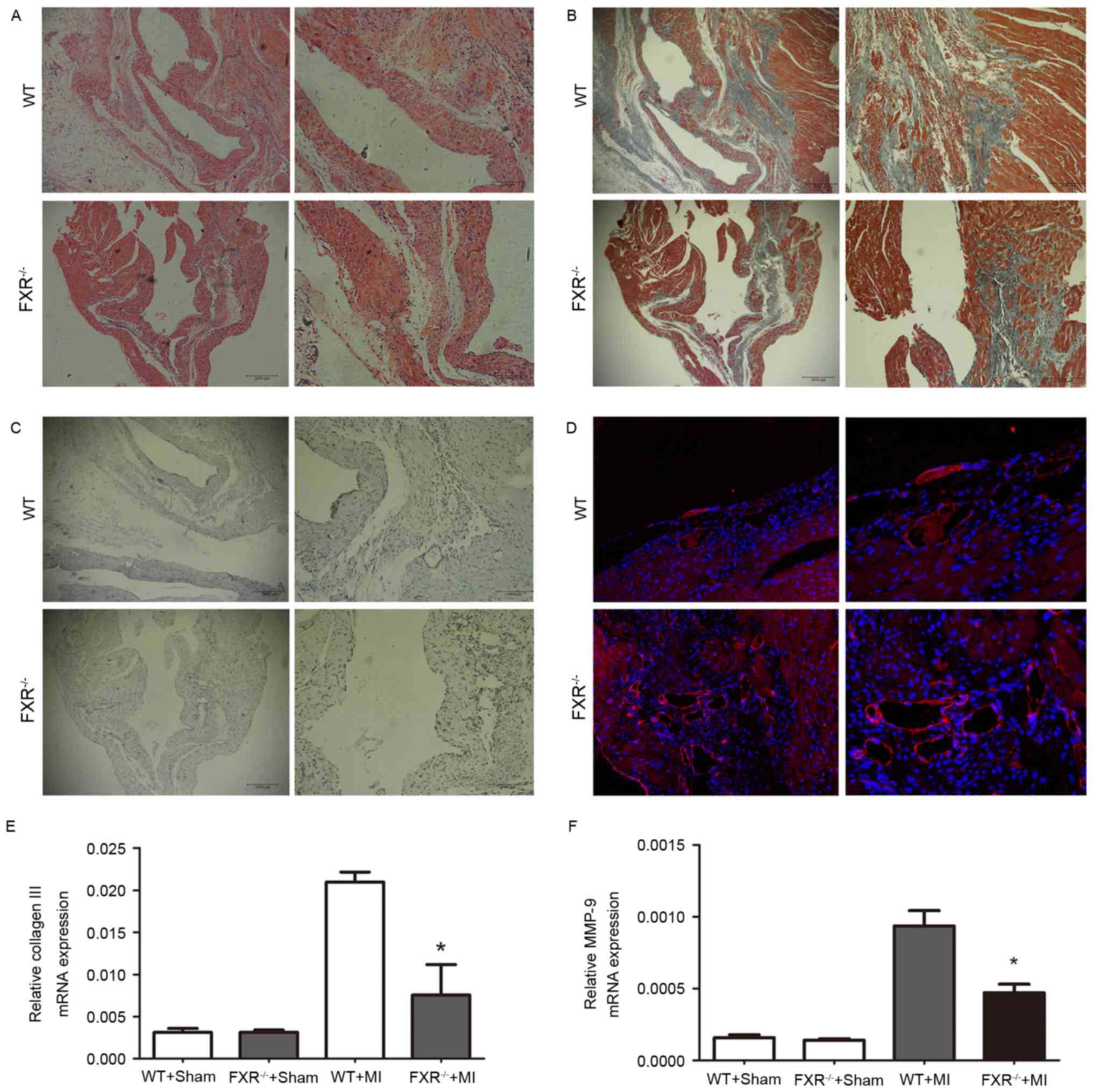
Masson's trichrome staining for interstitial
fibrosis in the border zone is demonstrated in Fig. 3B in representative samples from the
WT and FXR−/− groups. The border zone was revealed by
H&E staining in serial sections (Fig. 3A). Expression of α-smooth muscle
actin (α-SMA) is considered a marker for the conversion of
fibroblasts into myofibroblasts in myocardial tissue.
FXR−/− mice exhibited a significant reduction in the
number of α-SMA positive myofibroblasts in peri-infarct areas
compared with WT mice, as assessed on week 4 following MI (Fig. 3C). Microvesicular density was
assessed in the border zone using CD31 staining and was revealed to
be increased in the FXR−/− compared with in the WT group
(Fig. 3D).
The mRNA expression levels of collagen III and MMP-9
were evaluated using RT-qPCR on day 28 following MI. As presented
in Fig. 3E and F, collagen III and
MMP-9 mRNA levels were upregulated in mice that underwent MI
compared with in the sham-operated groups. Furthermore,
FXR−/− mice exhibited a marked reduction in collagen III
and MMP-9 mRNA levels compared with WT mice 28 days following
MI.
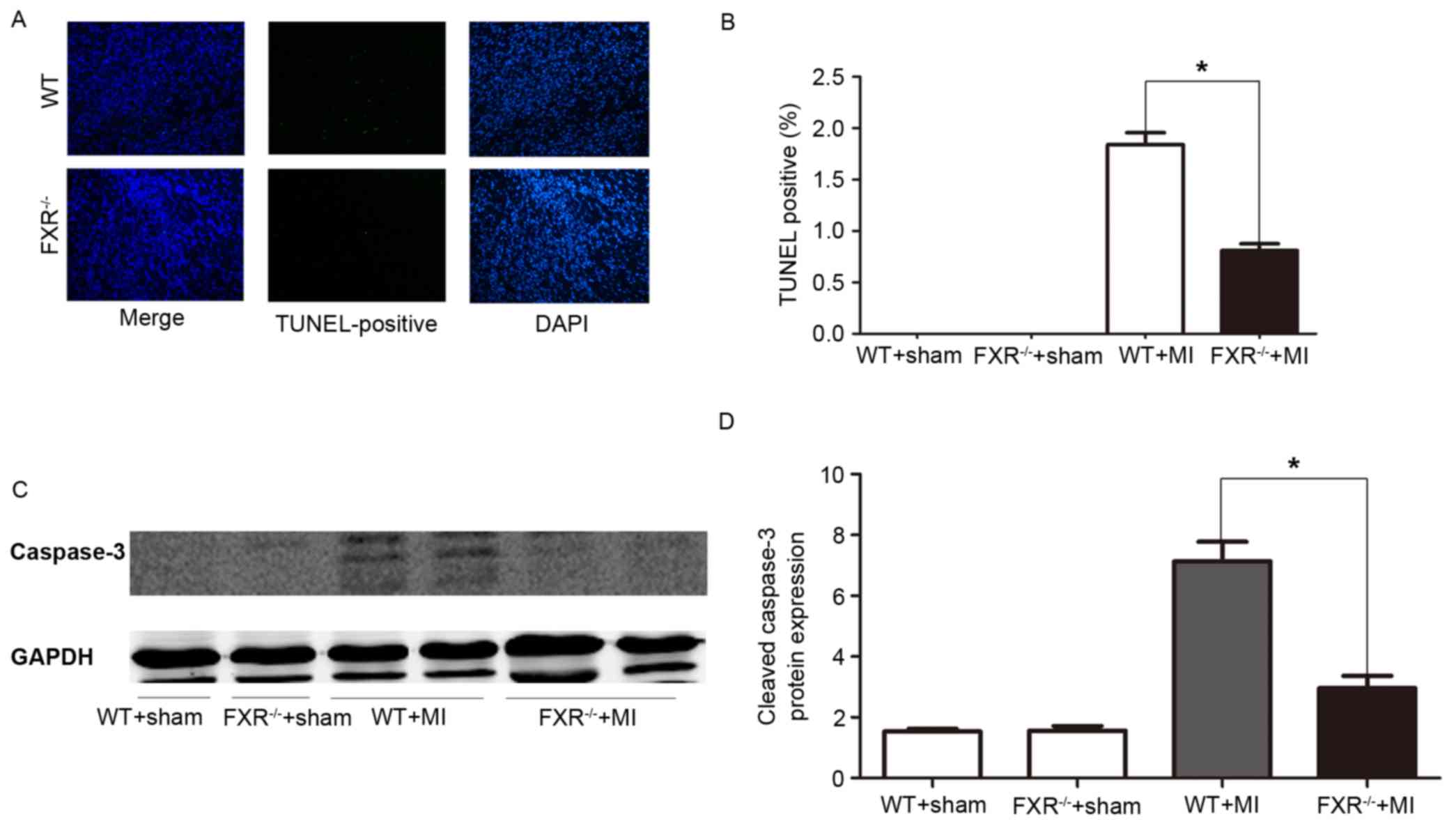
Cardiomyocyte apoptosis
Cardiomyocyte apoptosis was assessed using a TUNEL
assay. In the border zone, the percentage of TUNEL-positive, i.e.,
apoptotic, cardiomyocytes was significantly reduced in
FXR−/− mice compared with in WT mice (Fig. 4A and B). Western blot analysis
revealed a significant reduction in cleaved caspase-3 protein
levels in FXR−/− mice compared with in WT mice, as
assessed on day 7 post-MI (Fig. 4C and
D).
Discussion
Patients who recover from MI often develop cardiac
fibrosis, which can lead to a decline in cardiac function and
ultimately in heart failure (1,24).
Cardiac remodeling is a critical process in the development of
heart failure following MI (25,26).
The mechanisms underlying cardiac remodeling are complex; however,
matrix turnover has been reported to serve a central role during
the early days and weeks following MI (27,28).
It has previously been suggested that the pharmacological
inhibition or genetic ablation of FXR may reduce cardiomyocyte
apoptosis, decrease infarct size and improve cardiac function in
myocardium that has undergone ischemia/reperfusion (13). Therefore, it may be hypothesized
that FXR-deficient mice are characterized by deficits in cardiac
remodeling and FXR may serve an important role in cardiomyocyte
apoptosis following MI. The present results demonstrated an
increase in FXR expression in the infarct region that peaked on day
7 and declined at day 28 post-MI. These results suggested that FXR
may be implicated in the primary stages of cardiac healing
processes that follow MI. To the best of our knowledge, the present
study is the first to demonstrate that FXR knockdown attenuated
cardiac remodeling following MI. FXR knockdown appeared to reduce
infarct size and improve cardiac function following MI, thus
suggesting that FXR is implicated in the mechanisms underlying
post-MI cardiac remodeling. The observed improvement in post-MI
remodeling in FXR-deficient hearts may be attributed to the
reduction of cardiomyocyte apoptosis and interstitial fibrosis, and
increasing angiogenesis.
It has previously been reported that cardiomyocyte
apoptosis is a major mechanism implicated in unfavorable LV
remodeling (29). Myocardial cell
apoptosis mediated by mitochondria have an important role in the
repair of myocardial infarction. FXR activation may lead to
significant cardiomyocyte apoptosis and mitochondrial death
signaling characterized by caspase-3 activation (13). The present study demonstrated that
myocyte apoptosis was suppressed in FXR-deficient mice 7 days
post-MI. Additionally, a significant reduction in cleaved caspase-3
protein levels in FXR−/− mice on day 7 post-MI was
determined, thus suggesting that the absence of FXR may be
associated with the improvement of LV remodeling and cardiac
function following MI.
Neoangiogenesis is an essential component of cardiac
remodeling processes. Following MI, the existing vasculature is not
able to meet the enhanced oxygen demands of the viable myocardium,
which may lead to adjacent viable myocardial tissue necrosis,
progressive infarct extension and fibrous replacement (25). In the present study, angiogenesis
via detection of CD31 appeared to be enhanced in FXR−/−
compared with in WT mice, which may contribute to the improvement
of cardiac function and LV remodeling following MI.
Previous studies have reported that cardiac
extracellular matrix (ECM) dysregulation may participate in
progressive LV remodeling, and may lead to excessive collagen
deposition, fibrosis and collagen degradation, followed by LV
dilatation (27,30,31).
Type I and III collagen are among the main components of the ECM,
which provides the structure and mechanical support for the heart,
and participates in signaling among cardiomyocytes (27). The physiological integrity of the
ECM lies under the control of the MMP family of endopeptidases and
their specific tissue inhibitors (15). Enhanced expression and activity of
MMP-9 has been associated with collagen formation, whereas MMP-9
deletion has been reported to attenuate myocardial fibrosis
(28). The results of the present
study revealed that mRNA expression levels of type III collagen and
MMP-9 were significantly upregulated in WT compared with
FXR−/− mice 4 weeks following MI. It may suggest FXR
deletion may reduce the collagen deposition following MI and
improve the LV remodeling.
In conclusion, the present results suggested a
central role for FXR in myocardial injury and remodeling. To the
best of our knowledge, this is the first study to demonstrate the
effects of FXR deletion on the preservation of cardiac function
following MI and in the suppression of abnormal remodeling.
Therefore, FXR may represent a potential novel therapeutic target
for the treatment of patients following MI.
References
|
1
|
Ma Y, Halade GV, Zhang J, Ramirez TA,
Levin D, Voorhees A, Jin YF, Han HC, Manicone AM and Lindsey ML:
Matrix metalloproteinase-28 deletion exacerbates cardiac
dysfunction and rupture after myocardial infarction in mice by
inhibiting M2 macrophage activation. Circ Res. 112:675–688. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al:
Heart disease and stroke statistics-2014 update: A report from the
American Heart Association. Circulation. 129:e28–e292. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Krishnamurthy P, Thal M, Verma S, Hoxha E,
Lambers E, Ramirez V, Qin G, Losordo D and Kishore R:
Interleukin-10 deficiency impairs bone marrow-derived endothelial
progenitor cell survival and function in ischemic myocardium. Circ
Res. 109:1280–1289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu S, Yin R, Ernest R, Li Y, Zhelyabovska
O, Luo J, Yang Y and Yang Q: Liver X receptors are negative
regulators of cardiac hypertrophy via suppressing NF-kappaB
signalling. Cardiovasc Res. 84:119–126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kuipers I, Li J, Vreeswijk-Baudoin I,
Koster J, van der Harst P, Silljé HH, Kuipers F, van Veldhuisen DJ,
van Gilst WH and de Boer RA: Activation of liver X receptors with
T0901317 attenuates cardiac hypertrophy in vivo. Eur J Heart Fail.
12:1042–1050. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He Q, Pu J, Yuan A, Lau WB, Gao E, Koch
WJ, Ma XL and He B: Activation of liver-X-receptor α but not
liver-X-receptor β protects against myocardial ischemia/reperfusion
injury. Circ Heart Fail. 7:1032–1041. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lei P, Baysa A, Nebb HI, Valen G, Skomedal
T, Osnes JB, Yang Z and Haugen F: Activation of Liver X receptors
in the heart leads to accumulation of intracellular lipids and
attenuation of ischemia-reperfusion injury. Basic Res Cardiol.
108:3232013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Son NH, Yu S, Tuinei J, Arai K, Hamai H,
Homma S, Shulman GI, Abel ED and Goldberg IJ: PPARγ-induced
cardiolipotoxicity in mice is ameliorated by PPARα deficiency
despite increases in fatty acid oxidation. J Clin Invest.
120:3443–3454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Makishima M, Okamoto AY, Repa JJ, Tu H,
Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ and Shan B:
Identification of a nuclear receptor for bile acids. Science.
284:1362–1365. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Swales KE, Korbonits M, Carpenter R, Walsh
DT, Warner TD and Bishop-Bailey D: The farnesoid X receptor is
expressed in breast cancer and regulates apoptosis and aromatase
expression. Cancer Res. 66:10120–10126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Parks DJ, Blanchard SG, Bledsoe RK,
Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki
AM, Moore DD and Lehmann JM: Bile Acids: Natural ligands for an
orphan nuclear receptor. Science. 284:1365–1368. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Forman BM, Goode E, Chen J, Oro AE,
Bradley DJ, Perlmann T, Noonan DJ, Burka LT, McMorris T, Lamph WW,
et al: Identification of a nuclear receptor that is activated by
farnesol metabolites. Cell. 81:687–693. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pu J, Yuan A, Shan P, Gao E, Wang X, Wang
Y, Lau WB, Koch W, Ma XL and He B: Cardiomyocyte-expressed
farnesoid-X-receptor is a novel apoptosis mediator and contributes
to myocardial ischaemia/reperfusion injury. Eur Heart J.
34:1834–1845. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hageman J, Herrema H, Groen AK and Kuipers
F: A role of the bile salt receptor FXR in atherosclerosis.
Arterioscler Thromb Vasc Biol. 30:1519–1528. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bishop-Bailey D, Walsh DT and Warner TD:
Expression and activation of the farnesoid X receptor in the
vasculature. Proc Natl Acad Sci USA. 101:3668–3673. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ye L, Jiang Y and Zuo X:
Farnesoid-X-receptor expression in monocrotaline-induced pulmonary
arterial hypertension and right heart failure. Biochem Biophys Res
Commun. 467:164–170. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gao E, Lei YH, Shang X, Huang ZM, Zuo L,
Boucher M, Fan Q, Chuprun JK, Ma XL and Koch WJ: A novel and
efficient model of coronary artery ligation and myocardial
infarction in the mouse. Circ Res. 107:1445–1453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu X, Gao J, Xia Q, Lu T and Wang F:
Increased mortality and aggravation of heart failure in liver X
receptor-α knockout mice after myocardial infarction. Heart
Vessels. 31:1370–1379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pfeffer MA, Pfeffer JM, Fishbein MC,
Fletcher PJ, Spadaro J, Kloner RA and Braunwald E: Myocardial
infarct size and ventricular function in rats. Circ Res.
44:503–512. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang M, Zhong M, Shang Y, Lin H, Deng J,
Jiang H, Lu H, Zhang Y and Zhang W: Differential regulation of
collagen types I and III expression in cardiac fibroblasts by AGEs
through TRB3/MAPK signaling pathway. Cell Mol Life Sci.
65:2924–2932. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Woo YJ, Panlilio CM, Cheng RK, Liao GP,
Atluri P, Hsu VM, Cohen JE and Chaudhry HW: Therapeutic delivery of
cyclin A2 induces myocardial regeneration and enhances cardiac
function in ischemic heart failure. Circulation. 114:(1 Suppl).
I206–I213. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He K, Chen X, Han C, Xu L, Zhang J, Zhang
M and Xia Q: Lipopolysaccharide-induced cross-tolerance against
renal ischemia-reperfusion injury is mediated by hypoxia-inducible
factor-2α-regulated nitric oxide production. Kidney Int.
85:276–288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cohn JN, Ferrari R and Sharpe N: Cardiac
remodeling-concepts and clinical implications: A consensus paper
from an international forum on cardiac remodeling. Behalf of an
International Forum on Cardiac Remodeling. J Am Coll Cardiol.
35:569–582. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ding L, Dong L, Chen X, Zhang L, Xu X,
Ferro A and Xu B: Increased expression of integrin-linked kinase
attenuates left ventricular remodeling and improves cardiac
function after myocardial infarction. Circulation. 120:764–773.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Q, Xie J, Li R, Shi J, Sun J, Gu R,
Ding L, Wang L and Xu B: Overexpression of microRNA-99a attenuates
heart remodelling and improves cardiac performance after myocardial
infarction. J Cell Mol Med. 18:919–928. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bowers SL, Banerjee I and Baudino TA: The
extracellular matrix: At the center of it all. J Mol Cell Cardiol.
48:474–482. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chiao YA, Ramirez TA, Zamilpa R, Okoronkwo
SM, Dai Q, Zhang J, Jin YF and Lindsey ML: Matrix
metalloproteinase-9 deletion attenuates myocardial fibrosis and
diastolic dysfunction in ageing mice. Cardiovasc Res. 96:444–455.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Abbate A, Biondi-Zoccai GG, Bussani R,
Dobrina A, Camilot D, Feroce F, Rossiello R, Baldi F, Silvestri F,
Biasucci LM and Baldi A: Increased myocardial apoptosis in patients
with unfavorable left ventricular remodeling and early symptomatic
post-infarction heart failure. J Am Coll Cardiol. 41:753–760. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Souders CA, Bowers SL and Baudino TA:
Cardiac fibroblast: The renaissance cell. Circ Res. 105:1164–1176.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jourdan-Lesaux C, Zhang J and Lindsey ML:
Extracellular matrix roles during cardiac repair. Life Sci.
87:391–400. 2010. View Article : Google Scholar : PubMed/NCBI
|