Introduction
The chloride ion (Cl−) is a primary
intracellular anion, and is involved in a wide variety of cell and
intracellular organelle functions, including regulation of
electrical activity, intracellular pH, cell volume, apoptosis and
Ca2+ homeostasis (1,2).
Previous studies have demonstrated that an increase of
intracellular chloride ion concentration
([Cl−]i) serves an important role in the
pathogenesis of myocardial ischemia/reperfusion (I/R) injury
(3–8). However, increased
[Cl−]i can promote the release of
intracellular Ca2+ to elicit Ca2+ overload
through Cl−-increase-induced Ca2+ release
from intracellular stores (3,9,10);
on the other hand, the increased [Cl−]i can
also activate the Cl−-OH− exchanger to
increase intracellular reactive oxygen species (ROS) (3,10).
Furthermore, the increased [Cl−]i can induce
the mitochondrial permeability transition pore opening, which
results in ROS burst and subsequent oxidative stress (11). Therefore, inhibition of increased
[Cl−]i has been considered to be a reasonable
therapeutic strategy to alleviate I/R injury.
Anion exchange protein 3 (AE3), an embedded membrane
protein, belongs to the solute carrier 4 (SLC4) protein family and
is differentially expressed in excitable tissues (e.g. the brain,
heart and retina) (12). The main
function of AE3 is to mediate the reversible electroneutral
exchange of Cl− for HCO3− across
the plasma membrane (13–15). It means that AE3 protein has two
operating modes-the forward exchange mode and the reverse exchange
mode. The former is mainly an acidifying mechanism that promotes
the electroneutral efflux of bicarbonate in exchange for chloride
influx. However, the later contributes to sustaining intracellular
chloride homeostasis by promoting excess chloride efflux, which is
subject to regulation by intracellular pH, transmembrane
Cl−/HCO3− gradient and
phosphorylation state near the AE3 N-terminal region (13,16,17).
Notably, Alvarez et al (16,18)
demonstrated that AE3 is the protein kinase C (PKC)-sensitive anion
exchange protein of the heart, and that PKCε-dependent
phosphorylation of serine 67 on AE3 can promote
Cl−/HCO3− reverse exchange
activity.
Sasanquasaponin (SQS; 22-O-angeloyl camelliagenin C
3-O-[b-D-glucopyranosyl (1,2)]
[b-D-glucopyranosyl (1,2)-a-L-arabinopyranosyl (1,3)]-b-D-glucopyranosiduronic acid,
C58H92O26,) is a biologically
active ingredient extracted from the Chinese medicinal herb
Camellia oleifera Abel and has gained considerable attention
due to its wide range of biological and pharmacological properties;
in particular, its cardioprotective effect. Previous studies have
demonstrated that SQS effectively protects cardiomyocytes against
I/R injury by suppressing I/R-induced elevation of
[Cl−]i (19). Our
previous study further demonstrated that AE3 is required for SQS to
elicit cardioprotective effects against I/R injury, and the
inhibitory effect of SQS on I/R-induced elevation of
[Cl−]i is involved in the increase of
Cl−/HCO3− reverse exchange
activity of AE3 (20). However,
the molecular basis for SQS-induced increase of
Cl−/HCO3− reverse exchange of AE3
remains to be fully elucidated.
Therefore, the aim of the present study was to
identify the molecular basis for SQS activation of AE3 in H9c2
cells. It was revealed that SQS could promote phosphorylation of
the Ser67 of AE3 through activating PKCε in H9c2 cells undergoing
hypoxia/reoxygenation (H/R). Importantly, the PKCε-dependent
phosphorylation of serine 67 on AE3 was responsible for the
increase of Cl−/HCO3− exchange of
AE3 and intracellular chloride efflux by SQS.
Materials and methods
Chemicals and reagents
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS) and
3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2-tetrazolium bromide
(MTT) were purchased from Gibco (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). 5(6)-carboxy-20, 70-dichlorofluorescein
diacetate (cDCFH-DA) and
N-ethoxycarbonyl-methyl-6-methoxyquinolinium bromide (MQAE) were
from Invitrogen (Thermo Fisher Scientific, Inc.) εV1-2 (PKCε
inhibitor) and
8-[2-(2-pentyl-cyclopropyl-methyl)-cyclopropyl]-octanoic acid
(DCP-LA; PKCε activator) were purchased from Merck KGaA (Darmstadt,
Germany). 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein
(BCECF) and
1-[2-Amino-5-(2,7-dichloro-6-acetoxy-methoxy-3-oxo-9-xanthenyl)
phenoxy]-2-(2-amino-5-methylphenoxy) ethane-N,N,N', N'-tetraacetic
acid, tetra (acetoxymethyl) ester (fluo-3/AM) were purchased from
Beyotime Institute of Biotechnology (Shanghai, China). The primary
antibodies against β-Actin (rabbit, pAb; sc-130656; 1:1,000), PKCε
(C-15; rabbit, pAb; sc-214; 1:500), p-PKCε Ser 729 (goat, pAb;
sc-12355; 1:1,000) and a horseradish peroxidase (HRP)-conjugated
secondary antibody (mouse anti-rabbit IgG-HRP; sc-2357; 1:5,000)
were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). Antibodies against AE3 (rabbit, pAb; LS-C20639; 1:1,000),
were from LifeSpan BioScience, Inc. (Seattle, WA, USA), and
anti-phosphoserine (anti-p-ser; rabbit, pAb; AB1603; 1:500) were
from EMD Millipore (Billerica, MA, USA). SQS was kindly provided by
Professor Yongming Luo from Jiangxi Chinese Medical University
(Nanchang, China). Fluo-3/AM and all other chemicals were from
Sigma-Aldrich (Merck KGaA), unless otherwise stated.
Cell culture
H9c2 cells, a clonal line derived from embryonic rat
hearts, and HEK293 cells were obtained from the American Type
Culture Collection (Manassas, VA, USA) and maintained in DMEM
supplemented with 10% (v/v) FBS, 10 mM L-glutamine and 5 mg/ml
penicillin/streptomycin, in a humidified atmosphere of 95% air and
5% CO2 at 37°C.
Adenovirus and cell infections
The adenoviral vector expressing rat wild-type AE3
(Ad-AE3) and AE3 S67A mutant (Ad-AE3-S67A) were produced by
Shanghai GeneChem, Co. Ltd. (Shanghai, China). For cell infections,
80% confluent dishes of H9c2 or HEK293 cells were used. Cells were
infected at a multiplicity of infection of 25 for 24 h with
recombinant adenoviruses Ad-AE3 or Ad-AE3-S67A. The adenoviruses
were removed and cells were left to recover for 24 h in complete
medium. These conditions resulted in uniform expression of the
transgenes in close to 95% of the cells, as estimated by control
vector expressing only green fluorescent protein.
Cellular model of H/R injury
H/R was achieved as described previously (21–23).
Briefly, hypoxia was achieved by incubating the cells for 2 h in an
airtight chamber in which O2 was replaced by N2 with
glucose-free Tyrode's solution containing 139 mM NaCl, 4.7 mM KCl,
0.5 mM MgCl2, 1.0 mM CaCl2 and 5 mM HEPES, pH
7.4, at 37°C. Following incubation in hypoxic conditions, the cells
were provided with fresh medium and then moved to normoxic
conditions (5% CO2, 37°C) for 60 min for
reoxygenation.
Quantification of cell damage
Cell damage was determined by measuring cell
viability and the release of lactate dehydrogenase (LDH) and
creatine phosphokinase (CPK) into the cell culture medium. The
release of LDH and CPK were quantified using the CytoTox-ONE™
Homogenous Membrane Integrity assay (Promega Corporation, Madison,
WI, USA) according to the manufacturer's protocol. Cell viability
was determined by MTT assay. Briefly, cells were seeded into
96-well plates at a density of 1×105 cells/well.
Following treatment, cells were washed with warm phosphate buffered
saline (PBS) and incubated with 0.5 mg/ml MTT in PBS for 4 h at
37°C. The reaction was stopped by the addition of 150 µl
diphenylamine solution and the absorbance of the blue formazan
derivative was read at a wavelength of 570 nm using a
spectrophotometer.
Determination of anion exchange
activity of AE3
Anion exchange activity was determined by
2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF)
according to previously described protocols (18,24).
Briefly, H9c2 cells were cultured on coverslips in 60 mm dishes.
Following treatment, coverslips were incubated in 4 ml serum-free
medium containing 2 µM BCECF-AM (37°C, 30 min) and mounted in a
fluorescence cuvette. The cuvette was perfused at 3.5 ml/min
alternately with Ringer's buffer (5 mM glucose, 5 mM potassium
gluconate, 1 mM calcium gluconate, 1 mM MgSO4, 2.5 mM
NaH2PO4, 25 Mm NaHCO3, 10 mM
HEPES, pH 7.4) containing 140 mM sodium chloride (Cl−
buffer) or 140 mM sodium gluconate (Cl−-free buffer).
The two buffers were bubbled continuously with air containing 5%
CO2. Intracellular pH was monitored by measuring
fluorescence at excitation wavelengths of 440 and 502 nm and an
emission wavelength of 529 nm, using a spectrofluorometer.
Fluorescence data were converted to pHi by calibration using the
nigericin/high potassium method (25), with pH values of 6.4, 6.8 and 7.2.
Transport rates were determined by linear regression of the initial
linear rate of the change of pH.
Determination of
[Cl−]i
The levels of [Cl−]i were measured using
the Cl−-specific fluorescence probe MQAE, as previous
described (11,19,20).
Briefly, following treatment, cells were washed twice with
Cl−-free Tyrode solution (NaCl was replaced by equimolar
amounts of D-glucuronic acid; MgCl2 by MgSO4;
KCl by potassium gluconate), and loaded with 10 mM of MQAE in the
dark for 60 min at 37°C. Then the excess dye was washed off and the
cells were resuspended in Cl−-free Tyrode solution. The
fluorescence intensity of each group was determined by flow
cytometry (BD Biosciences, Franklin Lakes, NJ, USA) at excitation
and emission wavelengths of 355 and 460 nm and analyzed with FlowJo
software version 7.6 (FlowJo LLC, Ashland, OR, USA). Finally,
[Cl−]i was calculated in the light of the calibration
curve for the mean fluorescence intensity of MQAE which changed
with [Cl−]i.
Measurement of ROS generation
ROS generation was determined using the
cell-permeable probe cDCFH-DA, which is cleaved by cellular
esterases to nonfluorescent 2′,7′-dichlorofluorescin (DCFH) and
oxidized by intracellular ROS to a fluorescent product
dichlorofluorescein (DCF). Following the indicated treatments, the
cells were harvested and washed with cold PBS. Washed cells were
further incubated with 10 µM cDCFH-DA at 37°C for 20 min. The
excess dye was washed off and the cells were resuspended in PBS.
The fluorescent intensity was measured using a fluorescence
microscope (Olympus BX51; Olympus Corporation, Tokyo, Japan) at an
excitation wavelength of 488 nm and an emission wavelength of 525
nm (26).
Determination of
[Ca2+]i
The change in [Ca2+]i of cells was
determined by the fluorescence of the calcium-sensitive dye
fluo-3/AM as previously described (27). Briefly, cells in 60-mm plastic
petri dishes were incubated for 30 min at 37°C in the absence of
light in loading buffer [20 mM HEPES, pH 7.4, 130 mM NaCl, 5 mM
KCl, 2 mM CaCl2, 1 mM MgSO4, 0.8 mM
Na2HPO4, 0.2 mM
NaH2PO4, 25 mM mannose and 1 mg/ml bovine
serum albumin (BSA)] containing 2 mM Fluo-3/AM and 0.008% Pluronic
F-127 dissolved in DMSO. Following incubation, the monolayers were
washed in detaching buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 5 mM
KCl, 0.55 mM MgCl2 and 3 mM EDTA) and incubated in the
same buffer at 37°C for 10 min. Detached cells were harvested by
low-speed centrifugation (1,000 × g) at room temperature for 5 min,
re-suspended in assay buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 5
mM KCl, 0.55 mM MgCl2 and 1 mM CaCl2) and
analyzed on a flow cytometer (BD Biosciences) with FlowJo software
version 7.6 (FlowJo LLC).
Western blot analysis
Total proteins were extracted from cells using a
Protein Extraction kit (Pierce; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Protein content was
determined by the Lowry method using a DC protein assay kit
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Immunoblotting
analysis was performed as described previously (21,22).
In brief, 30 µg proteins were separated by 9% SDS-PAGE and
transferred to polyvinylidene difluoride membranes. Following
blocking with 5% non-fat milk or BSA at room temperature for 2 h,
the blots were then incubated with primary antibodies against AE3
(1:1,000), PKCε (1:1,000), p-PKCε (1:1,000), p-ser (1:500) and
β-actin (1:1,000) at 4°C overnight. The bound antibodies were
detected by an appropriate HRP-conjugated secondary antibody at
room temperature for 1 h and visualized using an enhanced
chemiluminescence system (EMD Millipore). The levels of each
protein were standardized to the loading control (β-actin) and were
quantified using ImageJ software version 1.41 (National Institutes
of Health, Bethesda, MD, USA).
Measurement of AE3
phosphorylation
AE3 phosphorylation was detected in AE3
immunoprecipitates with an anti-phosphoserine antibody. Briefly,
whole-cell lysates containing 0.5 mg proteins were pre-cleared with
protein A/G plus-agarose (Santa Cruz Biotechnology, Inc.) at 4°C
for 2 h. Aliquots (40 µl) of pre-clarified lysates were then
incubated with anti-AE3 antibodies at 4°C overnight followed by
incubation with protein A/G plus-agarose for 4 h. The agarose beads
were collected by centrifugation (500 × g at 4°C for 5 min), washed
4 times with cold PBS (4°C) and heated to 95°C for 5 min after
adding Laemmli buffer. The resulting immunoprecipitates were
separated by SDS-PAGE and probed with anti-AE3 or
anti-phosphoserine antibodies in western blotting as described
above. In brief, 40 µl proteins were separated by 10% SDS-PAGE and
transferred to polyvinylidene difluoride membranes. Following
blocking with 5% non-fat milk or BSA in room temperature for 2 h,
the blots were then incubated with primary antibodies against AE3
(rabbit, pAb; LS-C20639; 1:1,000; LifeSpan BioScience, Inc.), p-Ser
(rabbit, pAb; AB1603; 1:500; EMD Millipore) and β-actin (rabbit,
pAb; sc-130656; 1:1,000; Santa Cruz Biotechnology, Inc.) at 4°C
overnight. The bound antibodies were detected by an appropriate
HRP-conjugated secondary antibody (mouse anti-rabbit IgG-HRP;
sc-2357; 1:5,000; Santa Cruz Biotechnology, Inc.) at room
temperature for 1 h. The blots were analyzed densitometrically and
the ratio of phosphorylated AE3 to total AE3 protein was determined
to normalize for differences in loading by using ImageJ software
version 1.41 (National Institutes of Health).
Statistical analysis
All data are expressed as the mean ± standard error.
Statistical comparisons among groups were performed by one-way
analysis of variance followed by the least significant difference
post hoc test (two-tailed). Statistical calculations were performed
using SPSS software version 11.0 (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
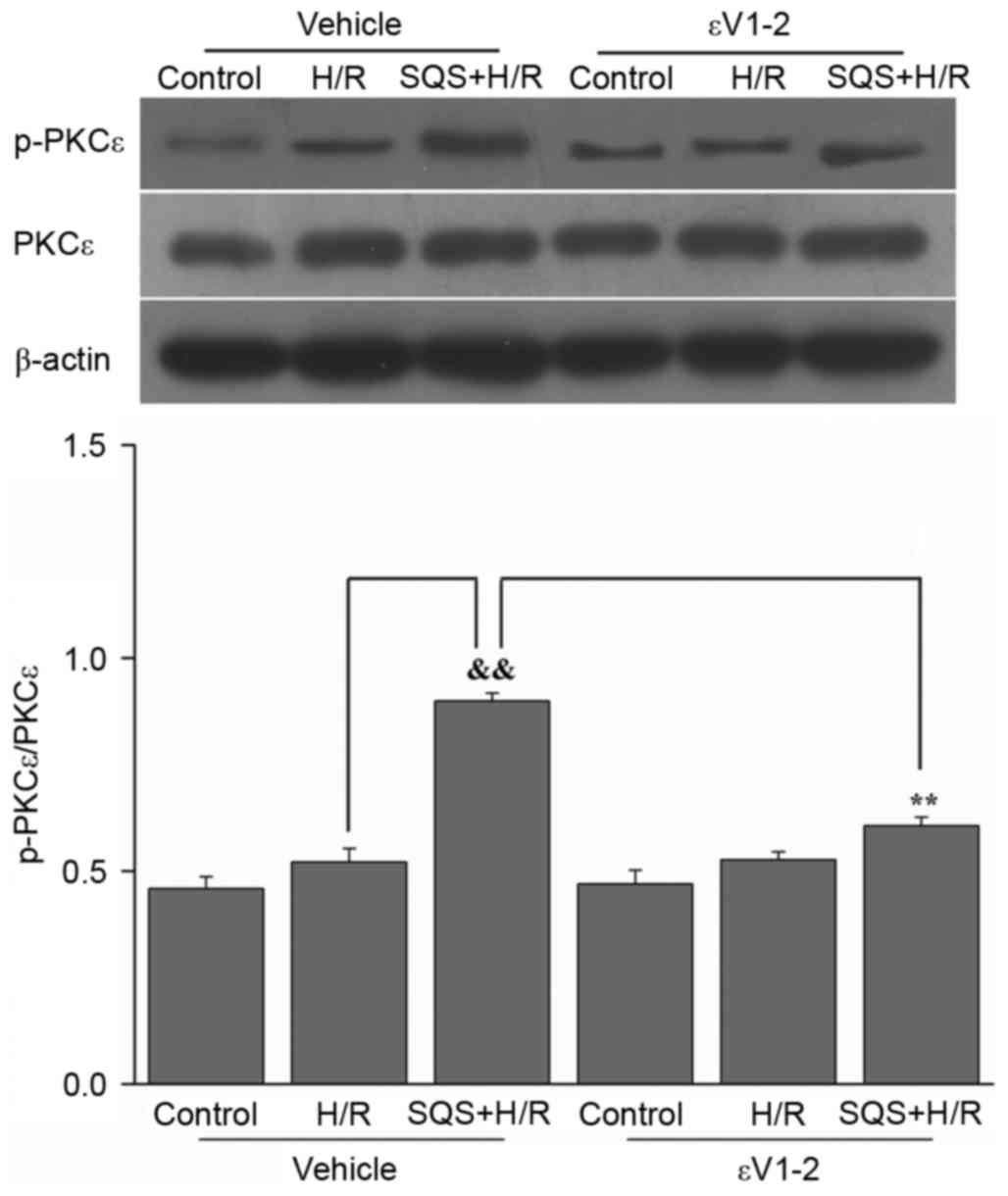
SQS induces PKCε activation in H9c2
cells undergoing H/R
Initial experiments were conducted to determine the
effect of SQS on PKCε activation. H9c2 cells were pretreated with
10 µM SQS for 24 h, followed by H/R. Subsequently, PKCε activation
was determined by western blot analysis. The results demonstrated
that SQS increased PKCε phosphorylation, whereas the expression of
total PKCε proteins remained unchanged (Fig. 1). These results suggested that SQS
pretreatment can activate PKCε in H9c2 cells subjected to H/R. In
addition, when administered 60 min before and during SQS
pretreatment, 1.0 µM εV1-2 (PKCε inhibitor) eradicated the
activation of PKCε induced by SQS (Fig. 1).
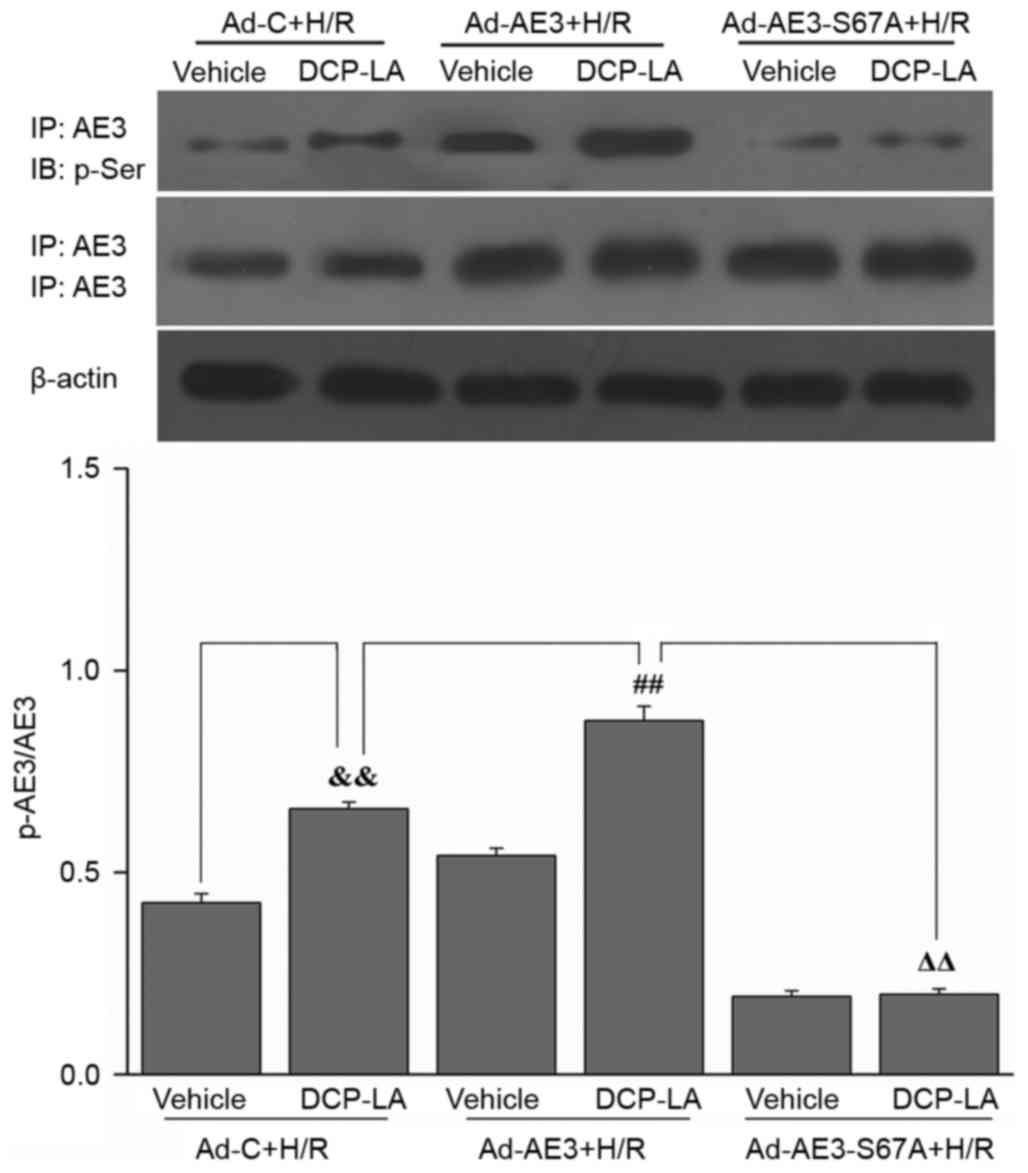
SQS promotes phosphorylation of Ser67
of AE3 via the PKCε-dependent signaling pathway in H9c2 cells
undergoing H/R
The effect SQS on AE3 phosphorylation in H9c2 cells
undergoing H/R was examined. SQS upregulated AE3 expression and
increased AE3 phosphorylation in H9c2 cells undergoing H/R.
Interestingly, pretreatment with 1.0 µM εV1-2 (PKCε inhibitor)
blocked SQS-induced AE3 phosphorylation but did not affect AE3
expression (Fig. 2A), suggesting
that SQS can promote AE3 phosphorylation via activation of PKCε.
Notably, in Ad-AE3-S67A-transfected H9c2 cells, the S67A mutation
completely annulled the SQS-induced phosphorylation of AE3
(Fig. 2B). By contrast, the
negative control virus Ad-C did not alter phosphorylation of AE3 by
SQS. These data indicated that SQS-induced AE3 phosphorylation
appears to be dependent on Ser67. To further support the fact that
activation of PKCε and Ser67 of AE3 are required for SQS-induced
AE3 phosphorylation, the effect of the PKCε activator, DCP-LA, on
AE3 phosphorylation was examined in Ad-AE3 and
Ad-AE3-S67A-transfected H9c2 cells. As expected, it was observed
that DCP-LA treatment resulted in phosphorylation of AE3 in
Ad-AE3-transfected H9c2 cells but not in Ad-AE3-S67A-transfected
H9c2 cells (Fig. 3). Taken
together, these results demonstrated that SQS increases the
phosphorylation of AE3 in a manner that is dependent on Ser67 and
PKCε activation.
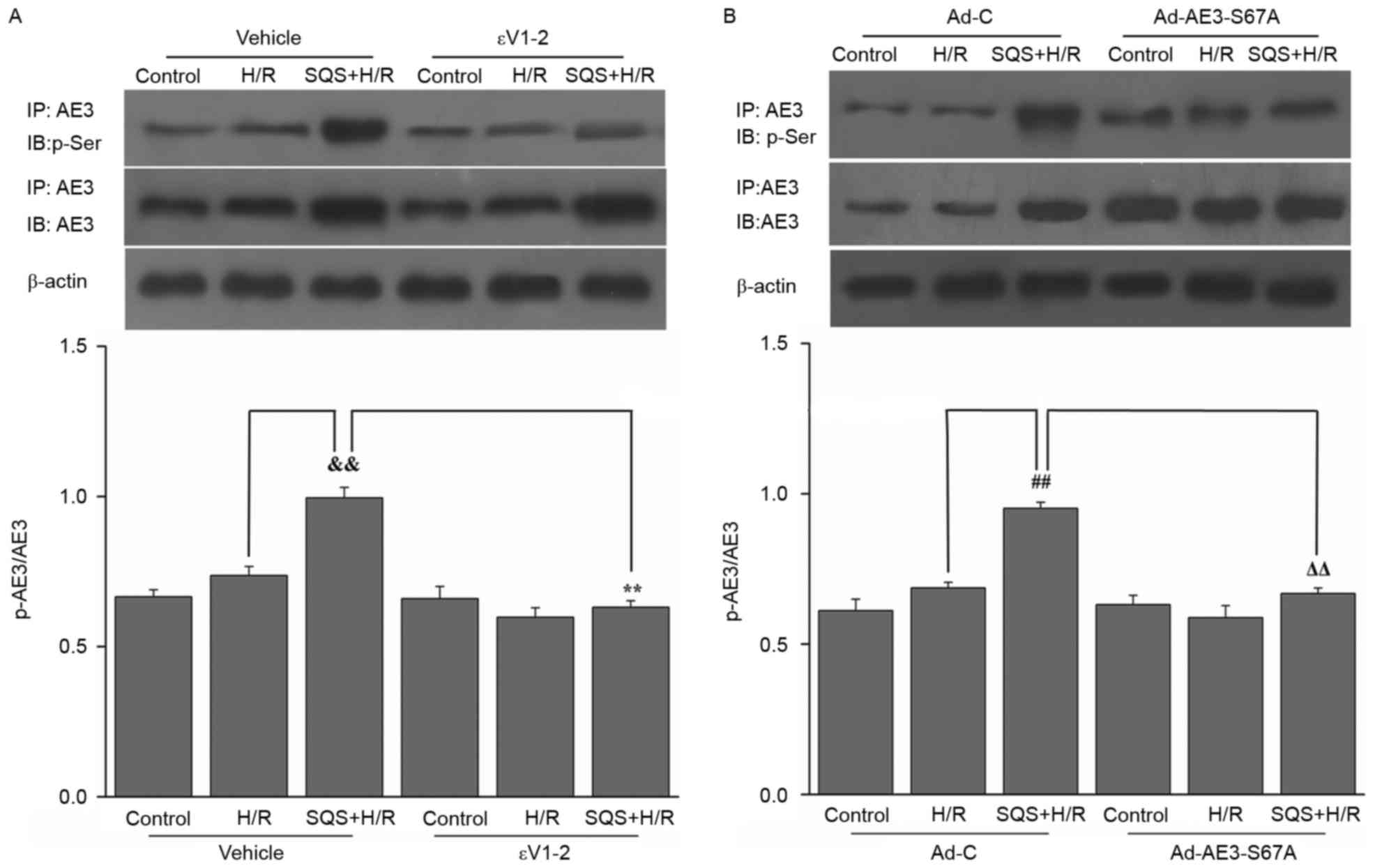 | Figure 2.Effects of SQS on phosphorylation of
AE3 in H9c2 cells undergoing H/R were assessed by
immunoprecipitation. Cell lysates were immunoprecipitated (IP) with
anti-AE3 antibody, and immunoblot (IB) analysis was performed with
anti-p-Ser or anti-AE3. Representative western blot images and
quantification of protein expression levels of AE3 and p-AE3 in
H9c2 cells (A) incubated for 24 h with SQS (10 µM) alone or
combination with the protein kinase Cε inhibitor εV1-2 (1.0 µM),
followed by H/R and (B) transfected with Ad-AE3-S67A 24 h before
SQS (10 µM, 24 h) pretreatments, followed by H/R. β-actin served as
an internal control. Data are presented as the mean ± standard
error of least four independent experiments.
&&P<0.01, **P<0.01,
##P<0.01, ΔΔP<0.01. SQS,
sasanquasaponin; AE3, anion exchanger 3; H/R,
hypoxia/reoxygenation; p, phosphorylated; Ad-C, negative control
adenovirus. |
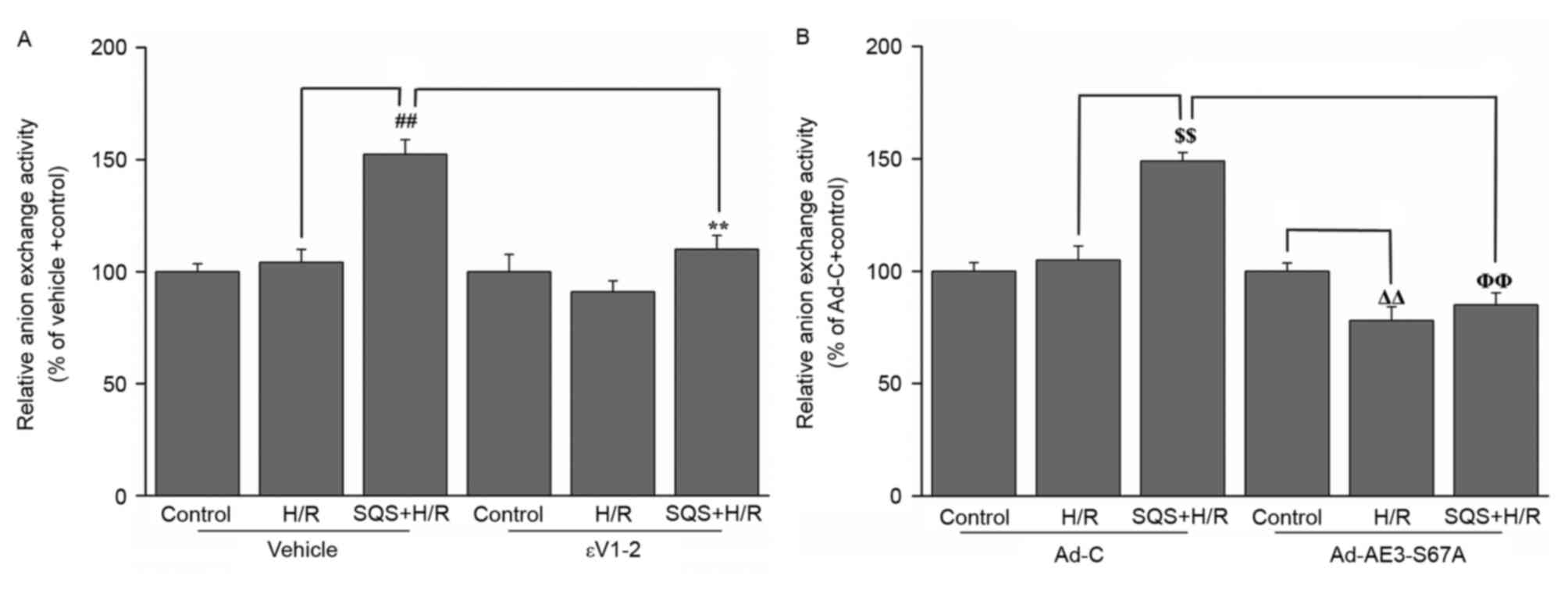
PKCε-dependent phosphorylation of
serine 67 on AE3 is responsible for SQS increasing the
Cl−/HCO3− exchange activity of
AE3
To investigate whether PKCε-dependent
phosphorylation of AE3 Ser67 is associated with SQS-induced
increase of Cl−/HCO3− exchange
activity of AE3, the effect of SQS on AE3 exchange activity in H9c2
cells with or without 1.0 µM εV1-2 pretreatment or Ad-AE3-S67A
transfection was detected. SQS elicited increased anion exchange
activity of AE3 in H9c2 cells undergoing H/R, which was blocked by
pretreatment with εV1-2 (Fig. 4A).
In addition, the increased transport activity elicited by SQS was
also attenuated in Ad-AE3-S67A-transfected H9c2 cells (Fig. 4B). This indicated that the increase
of AE3 activity on treatment with SQS is mediated via
PKCε-dependent phosphorylation of AE3 Ser67.
PKCε-dependent phosphorylation of AE3
serine 67 is responsible for the inhibitory effect of SQS on
H/R-induced elevation of [Cl−]i
Subsequently, the effect of SQS on H/R-induced
elevation of [Cl−]i in H9c2 cells with or without 1.0 µM
εV1-2 pretreatment or Ad-AE3-S67A transfection was detected. As
presented in Fig. 5, when cells
were subjected to H/R, the [Cl−]i was significantly
increased and the peak of [Cl−]i values was 49.7±5.1 mM,
compared with 26.8±3.8 mM in the control (P<0.01). Treatment
with 10 µM SQS produced a significant reduction in intracellular
Cl− concentration in H9c2 cells undergoing H/R, whereas
pretreatment with 1.0 µM εV1-2 abrogated the inhibitory effect of
SQS on H/R-induced elevation of [Cl−]i (Fig. 5A). These results suggested that SQS
can attenuate H/R-induced elevation of [Cl−]i in a
PKCε-dependent manner. Additionally, in Ad-AE3-S67A-transfected
H9c2 cells, SQS did not delay the H/R-induced increase in
[Cl−]i (Fig. 5B). These
results indicated that PKCε-dependent phosphorylation of AE3 Ser67
is required for SQS preconditioning to inhibit H/R-induced
elevation of [Cl−]i.
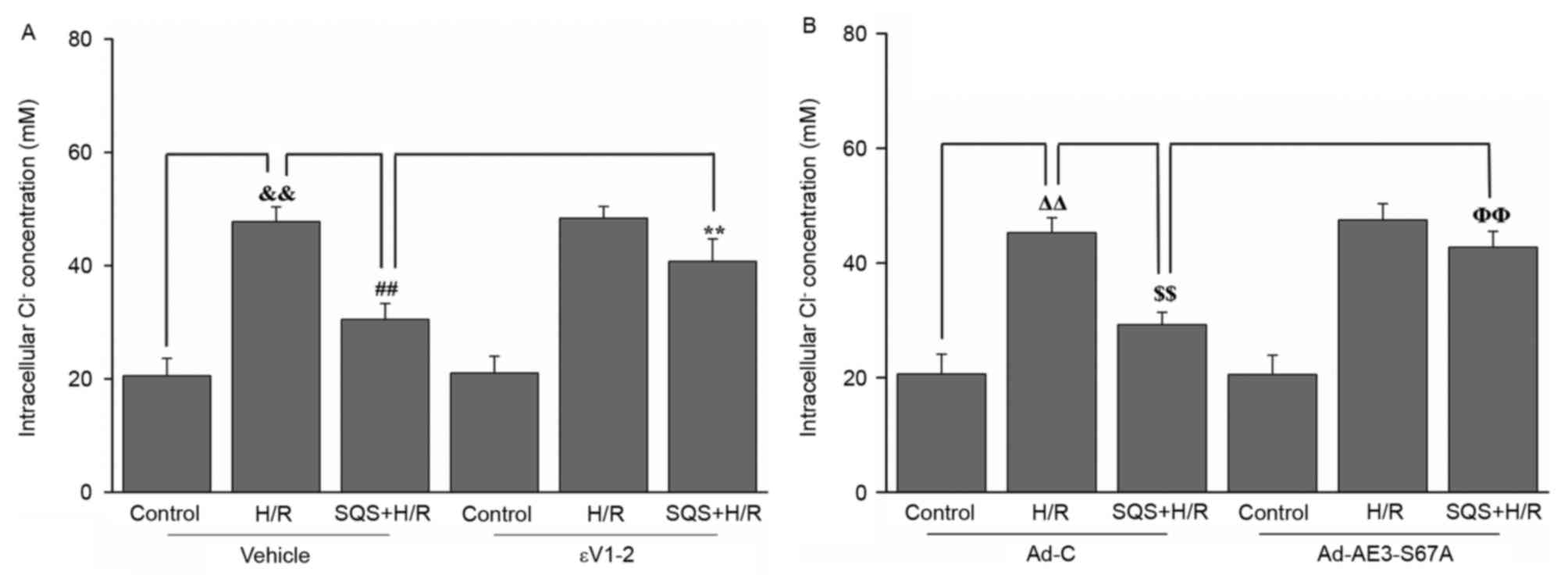 | Figure 5.Effects of SQS on [Cl−]i
in εV1-2-pretreated or Ad-AE3-S67A-transfected H9c2 cells
undergoing H/R. [Cl−]i in H9c2 cells (A) incubated with
εV1-2 and (B) transfected with Ad-AE3-S67A. Data are presented as
the mean ± standard error of four independent experiments.
##P<0.01, **P<0.01, ΔΔP<0.01,
$$P<0.01, ΦΦP<0.01. SQS,
sasanquasaponin; H/R, hypoxia/reoxygenation; p, phosphorylated;
AE3, anion exchanger 3; Ad-C, negative control adenovirus;
[Cl−]i, intracellular Cl− concentration. |
PKCε-dependent phosphorylation of
serine 67 on AE3 is responsible for SQS inhibiting H/R-induced
Ca2+ overload
Subsequently, the effect of SQS on H/R-induced
Ca2+ overload in H9c2 cells with or without 1.0 µM εV1-2
pretreatment or Ad-AE3-S67A transfection was further detected. As
presented in Fig. 6, when the H9c2
cells were subjected to H/R, intracellular Ca2+ was
significantly increased. Following preincubation with 10 µM SQS for
24 h, the H/R-induced increase in [Ca2+]i was
suppressed, whereas pretreatment with 1.0 µM εV1-2 eliminated the
inhibitory effect of SQS on H/R-induced Ca2+ overload
(Fig. 6A). These results suggested
that SQS can attenuate H/R-induced Ca2+ overload in a
PKCε-dependent manner. However, in Ad-AE3-S67A-transfected H9c2
cells, SQS did not delay the H/R-induced increase in
[Ca2+]i (Fig. 6B).
These results indicated that SQS can attenuate H/R-induced the
elevation of [Ca2+]i through PKCε signaling pathway and
phosphorylation Ser67 of AE3.
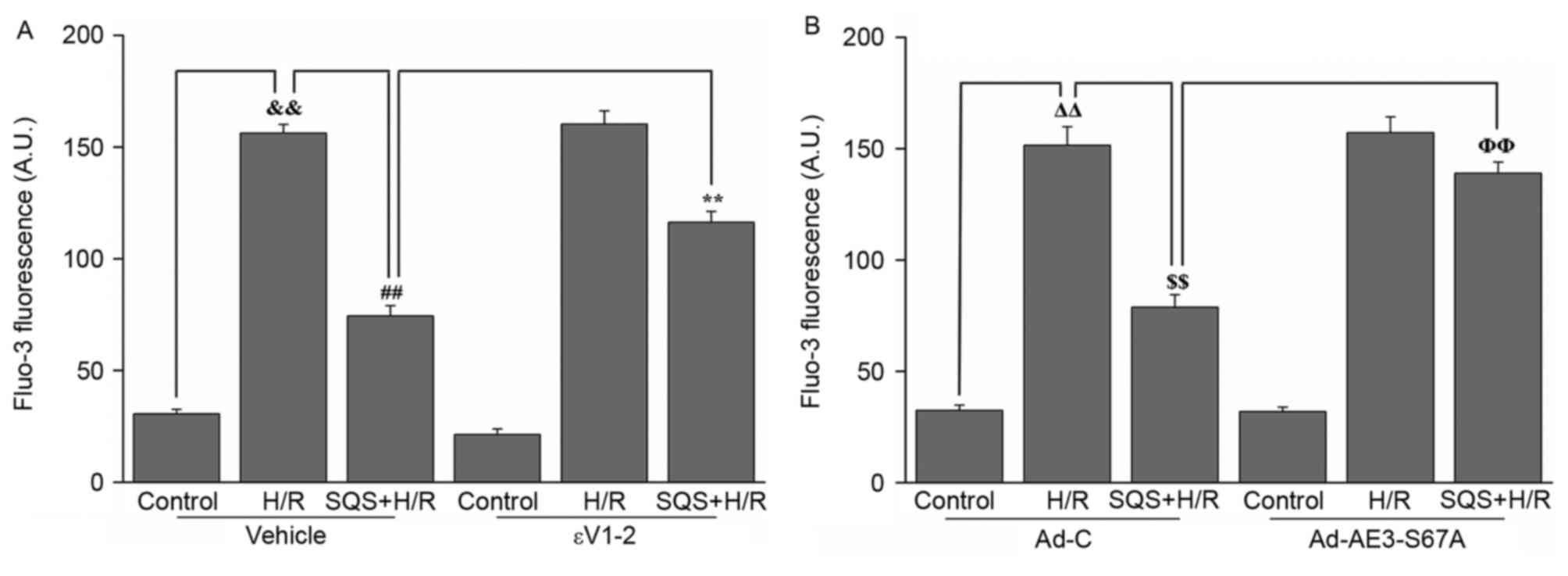 | Figure 6.Effects of SQS on [Ca2+]i
in εV1-2-pretreated or Ad-AE3-S67A-transfected H9c2 cells
undergoing H/R. [Ca2+]i in H9c2 cells (A) incubated with
εV1-2 and (B) transfected with Ad-AE3-S67A. Data are presented as
the mean ± standard error of four independent experiments.
##P<0.01, **P<0.01, ΔΔP<0.01,
$$P<0.01, ΦΦP<0.01. SQS,
sasanquasaponin; H/R, hypoxia/reoxygenation; p, phosphorylated;
AE3, anion exchanger 3; Ad-C, negative control adenovirus;
[Ca2+]i, intracellular Ca2+
concentration. |
PKCε-dependent phosphorylation of AE3
serine 67 is necessary for SQS to reduce H/R-induced ROS
production
In addition, the effect of SQS on H/R-induced ROS
production in H9c2 cells with or without 1.0 µM εV1-2 pretreatment
or Ad-AE3-S67A transfection was assessed. As presented in Fig. 7, H/R induced marked intracellular
ROS production, whereas simultaneous pretreatment with 10 µM SQS
inhibited ROS production. In the presence of εV1-2 (Fig. 7A) or Ad-AE3-S67A (Fig. 7B), SQS lost its inhibitory effect
on ROS production induced by H/R, indicating that PKCε-dependent
phosphorylation of AE3 Ser67 is required for SQS preconditioning to
inhibit H/R-induced ROS production.
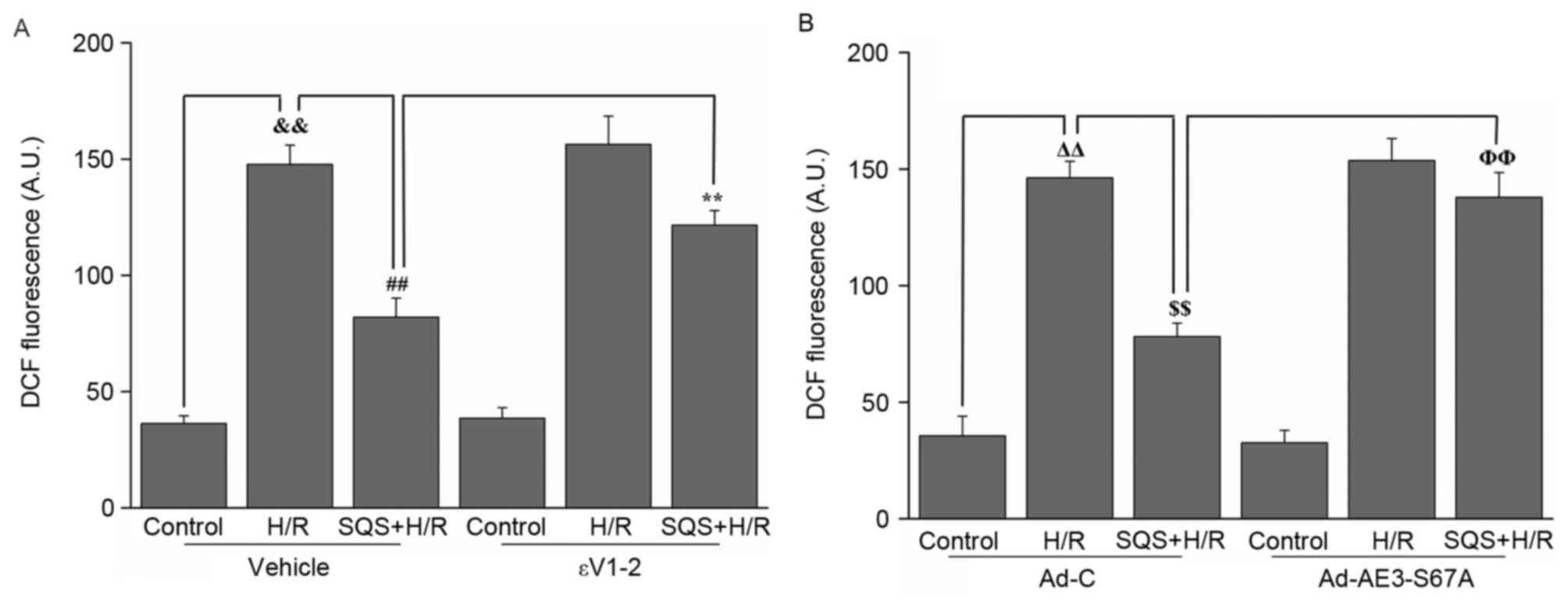 | Figure 7.Effects of SQS on ROS generation in
εV1-2-pretreated or Ad-AE3-S67A-transfected H9c2 cells undergoing
H/R. DCF fluorescence in H9c2 cells (A) incubated with εV1-2 and
(B) transfected with Ad-AE3-S67A. Data are presented as the mean ±
standard error of four independent experiments.
##P<0.01, **P<0.01, ΔΔP<0.01,
$$P<0.01, ΦΦP<0.01. SQS,
sasanquasaponin; H/R, hypoxia/reoxygenation; p, phosphorylated;
AE3, anion exchanger 3; Ad-C, negative control adenovirus; ROS,
reactive oxygen species; DCF, 2′,7′-dichlorofluorescein. |
PKCε-dependent phosphorylation of AE3
serine 67 is essential for SQS to elicit its cardioprotective
effect
Finally, to determine whether PKCε-dependent
phosphorylation of AE3 Ser67 is required for SQS-induced
cardioprotection against H/R injury, H9c2 cells were pretreated
with 10 µM SQS for 24 h in the absence or presence of εV1-2 or
Ad-AE3-S67A, followed by H/R. CPK and LDH release and cell
viability were used as indexes of cellular injury. As presented in
Fig. 8, SQS pretreatment
attenuated H/R-induced viability loss and CPK and LDH leakage.
However, in the presence of εV1-2 (Fig. 8A) or Ad-AE3-S67A (Fig. 8B), SQS did not induce
cardioprotection against H/R injury, indicating that PKCε-dependent
phosphorylation of AE3 Ser67 is required for the cardioprotective
effect elicited by SQS.
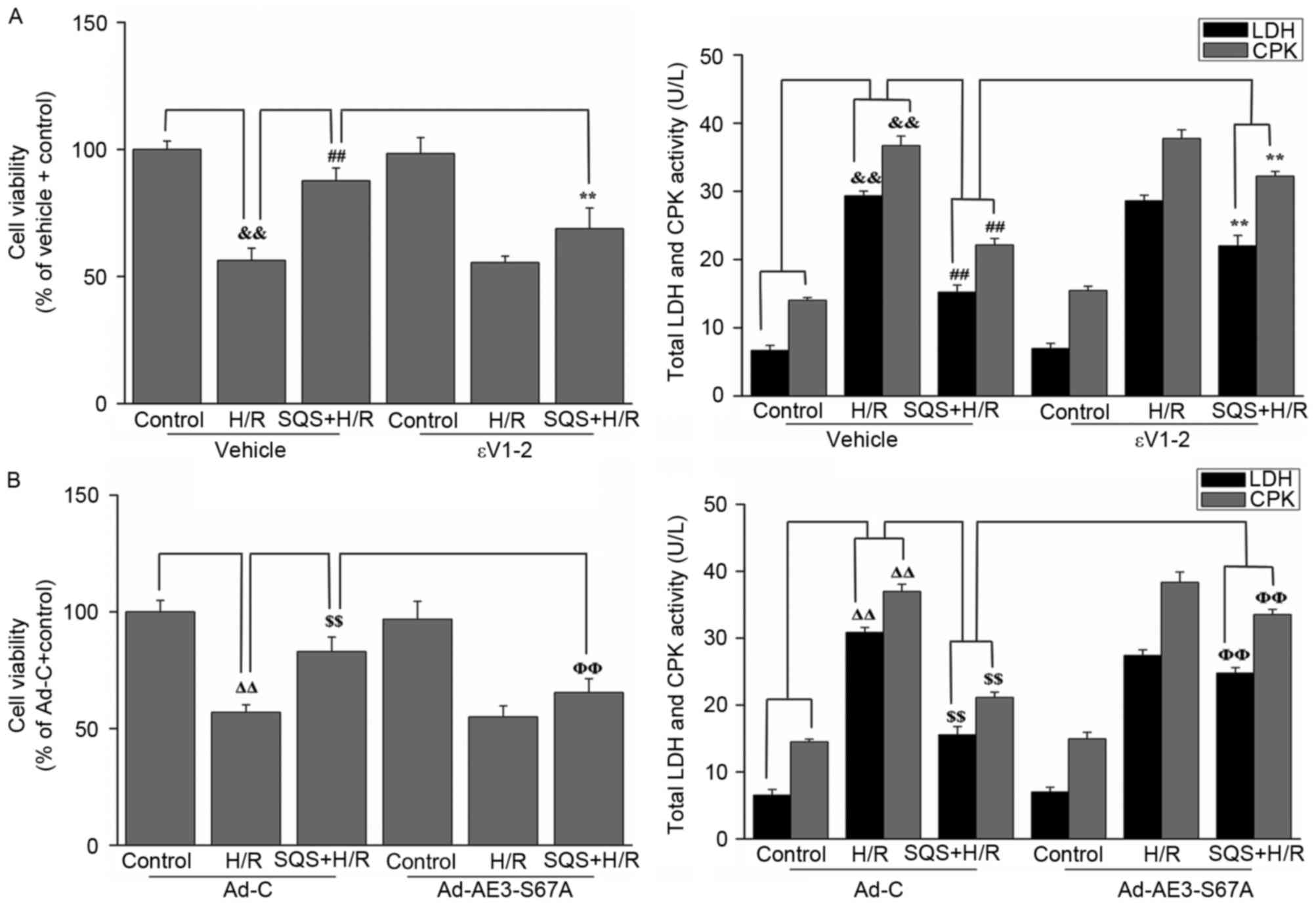 | Figure 8.Effects of SQS on cell viability and
release of LDH and CPK in εV1-2-preteated or
Ad-AE3-S67A-transfected H9c2 cells undergoing hypoxia/reoxygenation
(H/R). Cell viability and LDH and CPK release in H9c2 cells (A)
incubated with εV1-2 and (B) transfected with Ad-AE3-S67A. Data are
presented as the mean ± standard error of four independent
experiments. ##P<0.01, **P<0.01,
ΔΔP<0.01, $$P<0.01,
ΦΦP<0.01. SQS, sasanquasaponin; H/R,
hypoxia/reoxygenation; p, phosphorylated; AE3, anion exchanger 3;
Ad-C, negative control adenovirus; LDH, lactate dehydrogenase; CPK,
creatine phosphokinase. |
Discussion
The present study demonstrated that PKCε-dependent
phosphorylation of serine 67 on AE3 is a critical molecular basis
by which SQS increases activity of
Cl−/HCO3− exchange of AE3,
promotes the intracellular chloride efflux and elicits
cardioprotection in H9c2 cells undergoing H/R. This was indicated
by results demonstrating that SQS promotes PKCε activation and
enhances the level of phosphorylation of AE3 in H9c2 cells
subjected to H/R. Additionally, SQS-induced AE3 phosphorylation was
blocked by the PKCε selective inhibitor, εV1-2, and S67A mutation
of AE3. Furthermore, inhibition of PKCε by εV1-2 and the S67A
mutation of AE3 abrogated SQS-induced increase of AE3 activity,
reversed the inhibitory effect of SQS on H/R-induced elevation of
[Cl−]i, Ca2+ overload and ROS production, and
eliminated the cardioprotection induced by SQS in H9c2 cells. Taken
together, the present study expanded the current understanding of
the cardioprotective effects of SQS by identifying a novel
regulatory process induced by SQS, which stimulates phosphorylation
of Ser67 of AE3 via PKCε activation and, as a result, increases AE3
activity and attenuates H/R-induced elevation of [Cl−]i
in H9c2 cells.
SQS has profound cardioprotective effects on
ischemia/reperfusion injury (19).
Our previous study reported that upregulation and activation of AE3
may contribute to SQS cardioprotection (20). The present study demonstrated that
SQS can promote intracellular chloride efflux by increasing
Cl−/HCO3− exchange of AE3 and can
elicit cardioprotection in H9c2 cells subjected to H/R. However,
the molecular basis for SQS-induced increase of
Cl−/HCO3− exchange of AE3 remains
unclear; therefore, the present study focused on elucidating the
underlying mechanisms.
AE3 is known to serve crucial roles in maintaining
intracellular chloride homeostasis by facilitating the reversible
electroneutral exchange of Cl− for
HCO3− across the plasma membrane. Notably,
AE3 has been reported to be a PKCε-sensitive anion exchange protein
of the heart, and the PKCε-dependent phosphorylation of serine 67
on AE3 can obviously promote the
Cl−/HCO3− exchange activity
(16,18). Notably, our previous study
indicated that SQS exerts its cardioprotective effects in
association with PKCε activation (28). On this basis, it was hypothesized
that PKCε-dependent phosphorylation of serine 67 on AE3 may be
responsible for the increase of
Cl−/HCO3− exchange activity of AE3
by SQS and contribute to the inhibitory effect of SQS on
H/R-induced elevation of [Cl−]i in H9c2 cells. To
investigate this, the present study first determined the effects of
SQS on PKCε activation and subsequent AE3 phosphorylation. The
results demonstrated that SQS promotes PKCε activation and
increases AE3 phosphorylation, whereas both inhibition of PKCε by
εV1-2 and the S67A mutation of AE3 exhibited adverse effects. These
findings suggested that SQS can promote phosphorylation of Ser67 on
AE3 via the PKCε-dependent signaling pathway.
To further verify the contribution of PKCε-dependent
phosphorylation of Ser67 on AE3 in the SQS-induced increase of
Cl−/HCO3− exchange activity of AE3
and intracellular chloride efflux in H9c2 cells subjected to H/R,
the present study investigated the effect of inhibition of PKCε by
εV1-2 and the S67A mutation of AE3 on AE3 exchange activity, and
H/R-induced elevation of [Cl−]i in H9c2 cells. As
expected, the results demonstrated that SQS pretreatment led to a
significant increase of AE3 activity and a significant reduction in
[Cl−]i in H9c2 cells undergoing H/R. However, these
effects were attenuated in both εV1-2-pretreated and
Ad-AE3-S67A-transfected H9c2 cells, suggesting that PKCε-dependent
phosphorylation of AE3 Ser67 is essential for SQS to increase the
Cl−/HCO3− exchange activity of AE3
and to inhibit H/R-induced elevation of [Cl−]i.
In addition, as elevation of [Cl−]i
induced by H/R contributes to Ca2+ overload and ROS
production, the present study further detected the effects of SQS
on Ca2+ overload and ROS production in H9c2 cells with
or without εV1-2 pretreatment or Ad-AE3-S67A transfection. As
expected, it was observed that SQS attenuated the Ca2+
overload and ROS production that normally follows H/R injury,
accompanied by reduction of LDH and CPK release and an increase in
cell viability. However, in the presence of εV1-2 or Ad-AE3-S67A,
the inhibitory effects of SQS onCa2+ overload and ROS
production were reversed, and the cardioprotective effects of SQS
were attenuated, suggesting that the PKCε-dependent phosphorylation
of AE3 Ser67 is a prerequisite for SQS to attenuate H/R-induced
Ca2+ overload and ROS production and to elicit
cardioprotection.
In conclusion, to the best of our knowledge, the
present study was the first to investigate the underlying mechanism
of regulation of AE3 activity by SQS. It was demonstrated that SQS
promotes phosphorylation of Ser67 of AE3 via a PKCε-dependent
regulatory signaling pathway. Notably, it was revealed that
PKCε-dependent phosphorylation of serine 67 on AE3 is responsible
for the increase of Cl−/HCO3−
exchange of AE3 and intracellular chloride efflux by SQS, and
contributes to the cardioprotection of SQS against H/R in H9c2
cells. These findings may be beneficial in further understanding
the molecular mechanism associated with SQS-induced
cardioprotection, and elucidating the cellular effect and
pharmacological profiles of SQS.
Acknowledgements
The present study was supported by the Natural
Scientific Foundation of China (grant nos. 30660209 and
81260491).
Glossary
Abbreviations
Abbreviations:
|
SQS
|
sasanquasaponin
|
|
AE3
|
anion exchanger 3
|
|
H/R
|
hypoxia/reoxygenation
|
|
I/R
|
ischemia/reperfusion
|
|
[Cl−]i
|
intracellular Cl−
concentration
|
|
PKCε
|
protein kinase Cε
|
References
|
1
|
Hume JR, Duan D, Collier ML, Yamazaki J
and Horowitz B: Anion transport in heart. Physiol Rev. 80:31–81.
2000.PubMed/NCBI
|
|
2
|
Okada Y, Shimizu T, Maeno E, Tanabe S,
Wang X and Takahashi N: Volume-sensitive chloride channels involved
in apoptotic volume decrease and cell death. J Membr Biol.
209:21–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lai ZF: The relationship between
intracellular chloride concentration and ischemia
reperfusion-induced arrhythmias in myocardial cells. Zhongguo Yi
Xue Ke Xue Yuan Xue Bao. 24:190–196. 2002.(In Chinese). PubMed/NCBI
|
|
4
|
Lai ZF and Nishi K: Intracellular chloride
activity increases in guinea pig ventricular muscle during
simulated ischemia. Am J Physiol. 275:H1613–H1619. 1998.PubMed/NCBI
|
|
5
|
Lai ZF, Liu J and Nishi K: Effects of
stilbene derivatives SITS and DIDS on development of intracellular
acidosis during ischemia in isolated guinea pig ventricular
papillary muscle in vitro. Jpn J Pharmacol. 72:161–174. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kawasaki H, Otani H, Mishima K, Imamura H
and Inagaki C: Involvement of anion exchange in the
hypoxia/reoxygenation-induced changes in pH(i) and. Eur J
Pharmacol. 411:35–43. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen J, Liu D, Chen HP, Liao ZP, Lai ZF
and He M: Mechanisms of chloride in anoxia-reoxygenation injury of
cultured rat ventricular myocytes. Chin Pharmacological Bulletin.
23:724–729. 2007.
|
|
8
|
Liu D, He H, Li GL, Chen J, Yin D, Liao
ZP, Tang L, Huang QR, Lai ZF and He M: Mechanisms of chloride in
cardiomyocyte anoxia-reoxygenation injury: The involvement of
oxidative stress and NF-kappaB activation. Mol Cell Biochem.
355:201–209. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Campbell KP and Shamoo AE:
Chloride-induced release of actively loaded calcium from light and
heavy sarcoplasmic reticulum vesicles. J Membr Biol. 54:73–80.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sukhareva M, Morrissette J and Coronado R:
Mechanism of chloride-dependent release of Ca2+ in the
sarcoplasmic reticulum of rabbit skeletal muscle. Biophys J.
67:751–765. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang QR, Li Q, Chen YH, Li L, Liu LL, Lei
SH, Chen HP, Peng WJ and He M: Involvement of anion exchanger-2 in
apoptosis of endothelial cells induced by high glucose through an
mPTP-ROS-Caspase-3 dependent pathway. Apoptosis. 15:693–704. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Romero MF, Fulton CM and Boron WF: The
SLC4 family of HCO 3-transporters. Pflugers Archiv. 447:495–509.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alper SL, Darman RB, Chernova MN and Dahl
NK: The AE gene family of Cl/HCO3-exchangers. J Nephrol.
15:(Suppl 5). S41–S53. 2002.PubMed/NCBI
|
|
14
|
Cordat E and Casey JR: Bicarbonate
transport in cell physiology and disease. Biochem J. 417:423–439.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alper SL: Molecular physiology of SLC4
anion exchangers. Exp Physiol. 91:153–161. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alper SL, Chernova MN and Stewart AK:
Regulation of Na+-independent Cl-/HCO3-exchangers by pH.
JOP. 2:(4 Suppl). S171–S175. 2001.
|
|
17
|
Hume JR and Harvey RD: Chloride
conductance pathways in heart. Am J Physiol. 261:C399–C412.
1991.PubMed/NCBI
|
|
18
|
Alvarez BV, Fujinaga J and Casey JR:
Molecular basis for angiotensin ii-induced increase of
chloride/bicarbonate exchange in the myocardium. Circ Res.
89:1246–1253. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lai ZF, Shao Z, Chen YZ, He M, Huang Q and
Nishi K: Effects of sasanquasaponin on ischemia and reperfusion
injury in mouse hearts. J Pharmacol Sci. 94:313–324. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen HP, He M, Mei ZJ, Huang QR, Peng W
and Huang M: Anion exchanger 3 is required for sasanquasaponin to
inhibit ischemia/reperfusion-induced elevation of intracellular
Cl-concentration and to elicit cardioprotection. J Cell Biochem.
112:2803–2812. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu HH, Xu Q, Chen HP, Wang S, Huang XS,
Huang QR and He M: Stable overexpression of DJ-1 protects H9c2
cells against oxidative stress under a hypoxia condition. Cell
Biochem Funct. 31:643–651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang XS, Chen HP, Yu HH, Yan YF, Liao ZP
and Huang QR: Nrf2-dependent upregulation of antioxidative enzymes:
A novel pathway for hypoxic preconditioning-mediated delayed
cardioprotection. Mol Cell Biochem. 385:33–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mizukami Y, Iwamatsu A, Aki T, Kimura M,
Nakamura K, Nao T, Okusa T, Matsuzaki M, Yoshida K and Kobayashi S:
ERK1/2 regulates intracellular ATP levels through alpha-enolase
expression in cardiomyocytes exposed to ischemic hypoxia and
reoxygenation. J Biol Chem. 279:50120–50131. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
de Camilión Hurtado MC, Alvarez BV, Pérez
NG, Ennis IL and Cingolani HE: Angiotensin II activates
Na+-independent Cl-HCO3-exchange in ventricular
myocardium. Circ Res. 82:473–481. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thomas JA, Buchsbaum RN, Zimniak A and
Racker E: Intracellular pH measurements in Ehrlich ascites tumor
cells utilizing spectroscopic probes generated in situ.
Biochemistry. 18:2210–2218. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Parinandi NL, Kleinberg MA, Usatyuk PV,
Cummings RJ, Pennathur A, Cardounel AJ, Zweier JL, Garcia JG and
Natarajan V: Hyperoxia-induced NAD(P)H oxidase activation and
regulation by MAP kinases in human lung endothelial cells. Am J
Physiol Lung Cell Mol Physiol. 284:L26–L38. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen HP, He M, Huang QR, Liu D and Huang
M: Sasanquasaponin protects rat cardiomyocytes against oxidative
stress induced by anoxia-reoxygenation injury. Eur J Pharmacol.
575:21–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qiu LY, Chen HP, Yan YF, Li YY, Wang H,
Liao ZP and Huang QR: Sasanquasaponin promotes cellular chloride
efflux and elicits cardioprotection via the PKCε pathway. Mol Med
Rep. 13:3597–3603. 2016.PubMed/NCBI
|