Introduction
Sepsis is a systemic and deleterious inflammatory
response to noxious infection (1,2).
Sepsis causes ~18 million new cases and millions of deaths
worldwide annually; therefore, it is a major cause of morbidity and
mortality globally in critically ill patients (3,4). The
excessive activation of inflammation, complement and coagulation
systems may damage the host's own tissues and organs, leading to
multiple organ failure and death (5). In a group of patients diagnosed with
sepsis, the most common causative agents are Gram-positive and
Gram-negative bacteria (6,7).
Tang et al (8) used the microarray expression profile
of GSE6535 to identify the differentially expressed genes (DEGs)
between patients with Gram-positive and Gram-negative sepsis with
univariate F test according to the cut-off criteria of false
discovery rate (FDR) <0.05 and |log fold-change (FC)|>1.5 and
determined that Gram-positive sepsis and Gram-negative sepsis had a
common host response at the transcriptome level in critically ill
patients (8). However, a previous
study illustrated the different mechanisms of sepsis caused by
Gram-positive bacteria and Gram-negative bacteria.
Hypoxia-inducible factor 1α and Kruppel-like factor 2 have been
identified to be involved in Gram-positive endotoxin-mediated
sepsis by regulating cellular motility and proinflammatory gene
expression in myeloid cells (9).
In Gram-negative bacteria-induced sepsis, it has been determined
that the inhibition of caspase-1 and defective interleukin 1β
production are important immunological features (10). Additionally, α2-antiplasmin has
been identified to be a protective mediator during Gram-negative
sepsis by inhibiting bacterial growth, inflammation, tissue injury
and coagulation (11).
Furthermore, thrombomodulin-mediated protein C activation may
contribute to protective immunity in severe Gram-negative sepsis by
regulating inflammatory and procoagulant response (12). Despite the clinical importance of
the disease and extensive research, no specific treatment is
available for sepsis caused by Gram-positive and Gram-negative
bacteria. Therefore, it is necessary to screen the biomarkers for
sepsis.
The present study aimed to use the microarray data
of Tang et al (8) to screen
the DEGs in Gram-positive and Gram-negative samples compared with
control samples using the limma package based on a wide range of
thresholds (P<0.05 and |log2FC|>0.8). In addition,
specific genes were collected as biomarkers for sepsis caused by
Gram-positive and Gram-negative bacteria. A previous study has
proposed that analyses based on differential statistical tests may
lead to different outcomes (13).
Therefore, the findings of the present study may differ to those of
Tang et al (8).
Materials and methods
Microarray data
The microarray dataset of GSE6535 (8) was downloaded from the database of
gene expression omnibus (www.ncbi.nlm.nih.gov/geo), which was sequenced on the
platform GPL4274 NHICU Human 19K version 1.0. Probe annotation
information for mapping the probes into gene symbols was also
downloaded. From GSE6535 dataset, 17 neutrophil samples from
patients without sepsis, 18 neutrophil samples from patients with
Gram-positive sepsis, and 25 neutrophil samples from patients with
Gram-negative sepsis were selected. Tang et al (8) obtained whole blood samples from
critically ill patients on admission to the intensive care unit of
Nepean Hospital (Sydney, Australia). Using Ficoll-Paque density
gradient separation, neutrophils were isolated from the whole
blood. The patients with sepsis were diagnosed retrospectively
according to their medical record. According to the criteria
established by Calandra and Cohen (14), the patients with sepsis were
divided into Gram-positive and Gram-negative sepsis groups through
assessing various clinical features, including physical examination
and history and microbiological cultures, such as bronchoalveolar
washings, urine, blood and cerebrospinal fluid. GSE6535 was
deposited by Tang et al (8). The study of Tang et al was
approved by the Ethics Committee of Nepean Hospital and written
informed consent was provided by the patients or their families
(8).
Data preprocessing and differential
expression analysis
Based on the probe annotation information, probe IDs
were converted into their corresponding gene symbols. The average
value of multiple probes (that were corresponding to the same gene)
was used as the gene expression value. To eliminate inherent
expression differences between genes, the gene expression values
were performed with Z-score normalization as previously described
(15). Subsequently, the limma
package version 3.32.2 in R (16)
was used to screen the DEGs in the Gram-positive and Gram-negative
samples compared with the control samples. The P<0.05 and
|log2FC| >0.8 were used as the cut-off criteria for
screening DEGs. Using the VennDiagram in R (17), the common DEGs between
Gram-positive and Gram-negative samples, as well as the specific
DEGs in Gram-positive samples or Gram-negative samples were
identified. Gene Ontology (GO; www.geneontology.org) is a bioinformatics resource
that may be used to classify gene product function using
controlled, structured vocabularies (18). Using the Database for Annotation,
Visualization and Integrated Discovery (DAVID) (19), GO functional enrichment analysis
was performed on the common DEGs. The hierarchical cluster analysis
of the specific DEGs in Gram-positive or Gram-negative samples was
conducted using cluster version 3.0 software (20) and then visualized using TreeView
tool version 3 (21).
Similarity network construction
Pearson's correlation coefficient (PCC) (22), which determines the correlation
between two variables, was used to identify the positive or
negative correlations among different samples, with the threshold
of |PCC|>0.5. Using Cytoscape version 2.8 software (23), a similarity network was constructed
for the Gram-positive, Gram-negative and control samples.
Functional and pathway enrichment
analyses
Kyoto Encyclopedia of Genes and Genomes (KEGG;
www.genome.jp/kegg/), which integrates
genomic, chemical and systemic functional information, is a useful
resource for pathway mapping (24). Using the online tool DAVID
(19), GO functional and KEGG
pathway enrichment analyses were conducted for the DEGs. P<0.05
was used as the threshold.
Identification of significantly
differential functions using stochastic perturbations
The average expression value in Gram-positive or
Gram-negative samples was calculated for each gene enriched in the
same term (GO functions or KEGG pathways). Euclidean distance
(25) was used to calculate the
difference between the levels of all the terms between
Gram-positive and Gram-negative samples, according to the following
equation:
distance=∑i=1T(X¯Pi–X¯Ni)2
Where distance represents the Euclidean distance
between Gram-positive samples and Gram-negative samples;
X¯Pi stands for the average
expression value of gene i in Gram-positive samples; X¯Pi represents the average expression
value of gene i in Gram-negative samples; and T indicates the gene
number in each term.
Subsequently, stochastic perturbations were used
(26) to determine the
significance findings. The 18 Gram-positive and 25 Gram-negative
samples were randomly sorted. Subsequently, 18 samples were
randomly selected and defined as Gram-positive samples and the
remaining 25 samples were defined as Gram-negative samples. The
Euclidean distance between the newly defined Gram-positive samples
and Gram-negative samples was recalculated. This was repeated for
10,000 times and the Euclidean distance for 10,000 perturbations
were sorted from small to large and used as the background
distribution. The ranking order of the initial Euclidean distance
in the background distribution was calculated and converted to a
P-value. The terms with P<0.05 were considered significantly
differential functions between Gram-positive and Gram-negative
samples.
Results
DEGs analysis
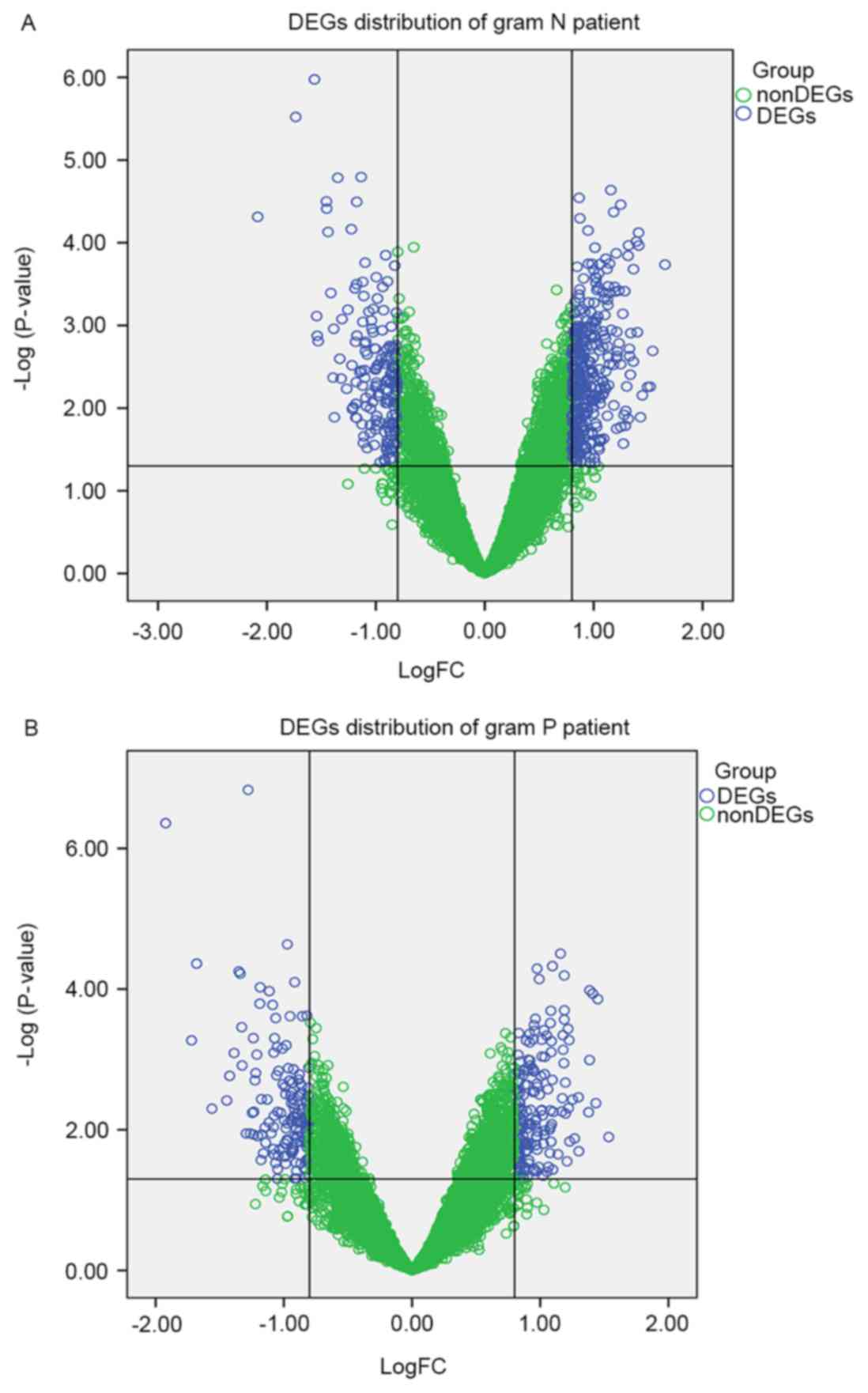
The gene distribution of Gram-negative (Fig. 1A) and Gram-positive samples
(Fig. 1B) are presented using a
volcano plot. Using the P<0.05 and |log2FC|>0.8 as
thresholds, a total of 340 DEGs, including 181 upregulated genes,
including large tumor suppressor kinase 2 (LATS2), NADH:ubiquinone
oxidoreductase subunit S4 (NDUFS4) and 159 downregulated genes,
including myeloid cell leukemia 1 (MCL1) and chitinase-like 1, were
obtained in Gram-positive samples compared with control samples. A
total of 485 DEGs were identified, 324 upregulated genes, including
NDUFS4 and NADH:ubiquinone oxidoreductase subunit B2 (NDUFB2) and
161 downregulated genes, including MCL1 and ecotropic viral
integration site 2B, were identified in Gram-negative samples
compared with the control samples. The top 10 significantly
upregulated genes and downregulated genes in the Gram-negative and
Gram-positive samples are presented in Table I.
 | Table I.Top 10 upregulated and downregulated
genes in patients with Gram-negative and Gram-positive sepsis. |
Table I.
Top 10 upregulated and downregulated
genes in patients with Gram-negative and Gram-positive sepsis.
| A,
Gram-negative |
|---|
|
|---|
| DEGs | -log2
(P-value) | logFC |
|---|
| Upregulated
genes |
|
RPL27 | 3.735442 |
1.654937 |
|
TM4SF1 | 2.691156 |
1.541493 |
|
SEC11A | 2.258718 |
1.518767 |
|
PLOD2 | 2.255042 |
1.494549 |
|
UQCRH | 2.154078 |
1.446315 |
|
AFP | 1.889918 |
1.429409 |
|
CDK5RAP2 | 3.965291 |
1.413672 |
|
EPB41L4A-AS1 | 4.122053 |
1.411759 |
|
SOD1 | 2.91784 |
1.40033 |
|
CANX | 4.013587 |
1.392226 |
| Downregulated
genes |
|
EVI2B | 4.312471 | −2.08489 |
|
MME | 5.521434 | −1.73605 |
|
ZBP1 | 5.974694 | −1.56361 |
|
LITAF | 3.113137 | −1.54349 |
|
CYTH4 | 2.874971 | −1.53818 |
|
FBXL5 | 2.808339 | −1.53 |
|
CHI3L1 | 4.498941 | −1.45407 |
|
QPCT | 4.411504 | −1.45154 |
|
TREM1 | 4.12983 | −1.43918 |
|
MXD1 | 3.392031 | −1.41323 |
|
| B,
Gram-positive |
|
| DEGs | −log2
(P-value) | logFC |
|
| Upregulated
genes |
|
SSBP1 | 1.896196279 |
1.5341469 |
|
LAIR1 | 3.8569852 |
1.4491332 |
|
MRPS18A | 2.377785977 |
1.4338711 |
|
NDUFC2 | 3.935542011 |
1.4093212 |
|
CTSC | 3.982966661 |
1.3851966 |
|
MT1L | 2.991399828 |
1.3843636 |
|
TM4SF1 | 2.249491605 |
1.3772427 |
|
FCHSD2 | 1.694648631 |
1.301164 |
|
CYP1B1 | 2.460923901 |
1.297874 |
|
NDUFA4 | 1.876148359 |
1.267824 |
| Downregulated
genes |
|
CHI3L1 | 6.359519 | −1.92246 |
|
EVI2B | 3.271646 | −1.71993 |
|
MME | 4.36251 | −1.68036 |
|
KCNB1 | 2.300162 | −1.56111 |
|
LITAF | 2.415669 | −1.44533 |
|
FUS | 2.767004 | −1.42285 |
|
QPCT | 3.090444 | −1.38819 |
|
CKAP4 | 4.251812 | −1.35146 |
|
MCL1 | 4.221126 | −1.34034 |
|
EFHC2 | 3.458421 | −1.32892 |
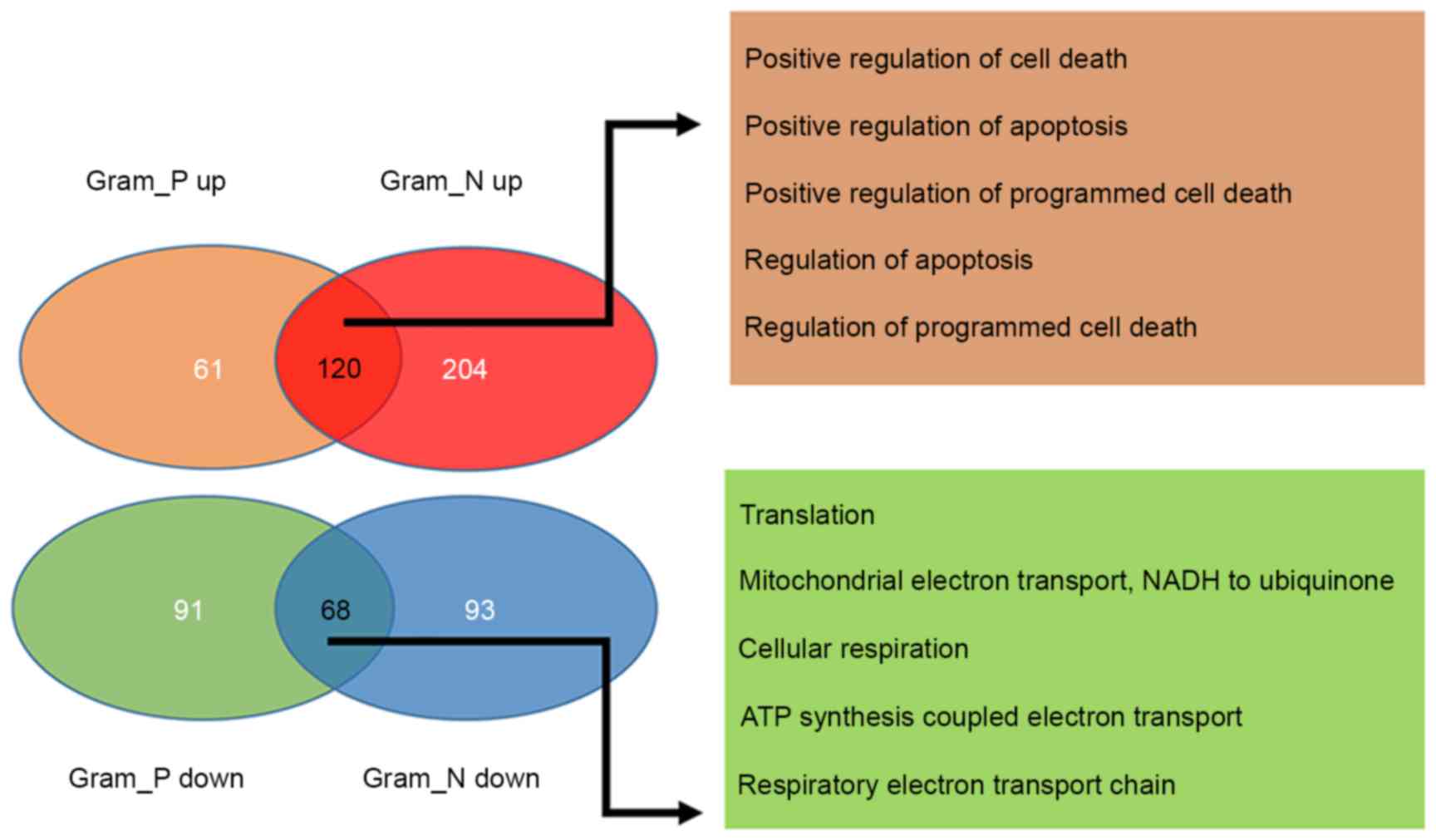
A total of 188 common DEGs, including 120
upregulated and 68 downregulated were identified between
Gram-positive and Gram-negative samples. Additionally, 152 specific
DEGs, including 61 upregulated and 91 downregulated genes in the
Gram-positive samples and 297 specific DEGs, including 204
upregulated and 93 downregulated genes in Gram-negative samples
were also screened (Fig. 2). GO
functional enrichment analysis was performed on the common DEGs,
consisted of 120 upregulated and 68 downregulated genes in the
Gram-positive and Gram-negative samples and the top 5 terms for
each sample type were presented in Fig. 2. The findings revealed that the
common upregulated genes were primarily associated with the
regulation of apoptosis and cell death, whereas the common
downregulated genes were primarily associated with cellular
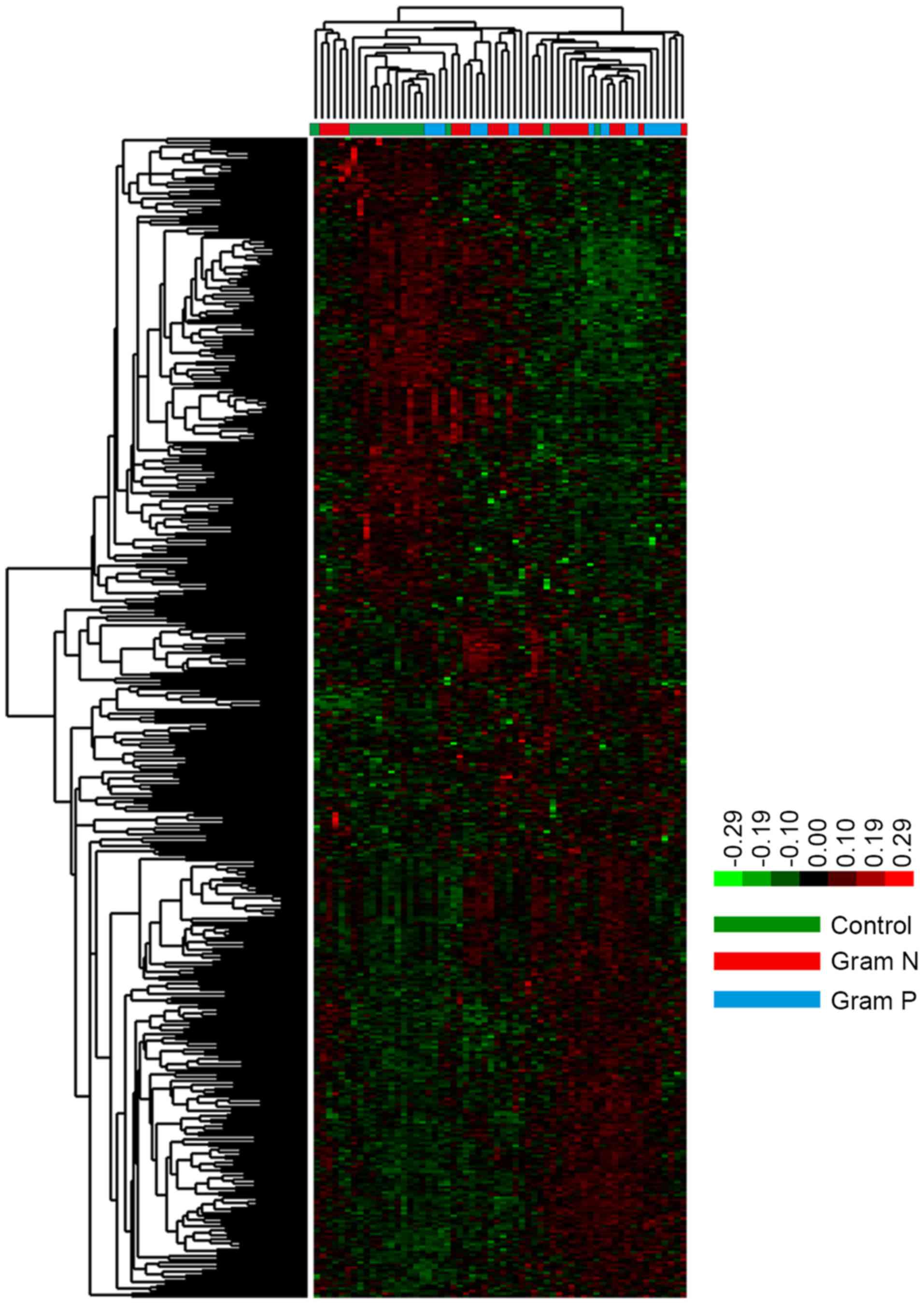
respiration (Fig. 2). Hierarchical
cluster analysis of the specific DEGs revealed that there were
significant differences between control and sepsis samples.
However, no significant difference was identified between the
Gram-positive and Gram-negative samples (Fig. 3).
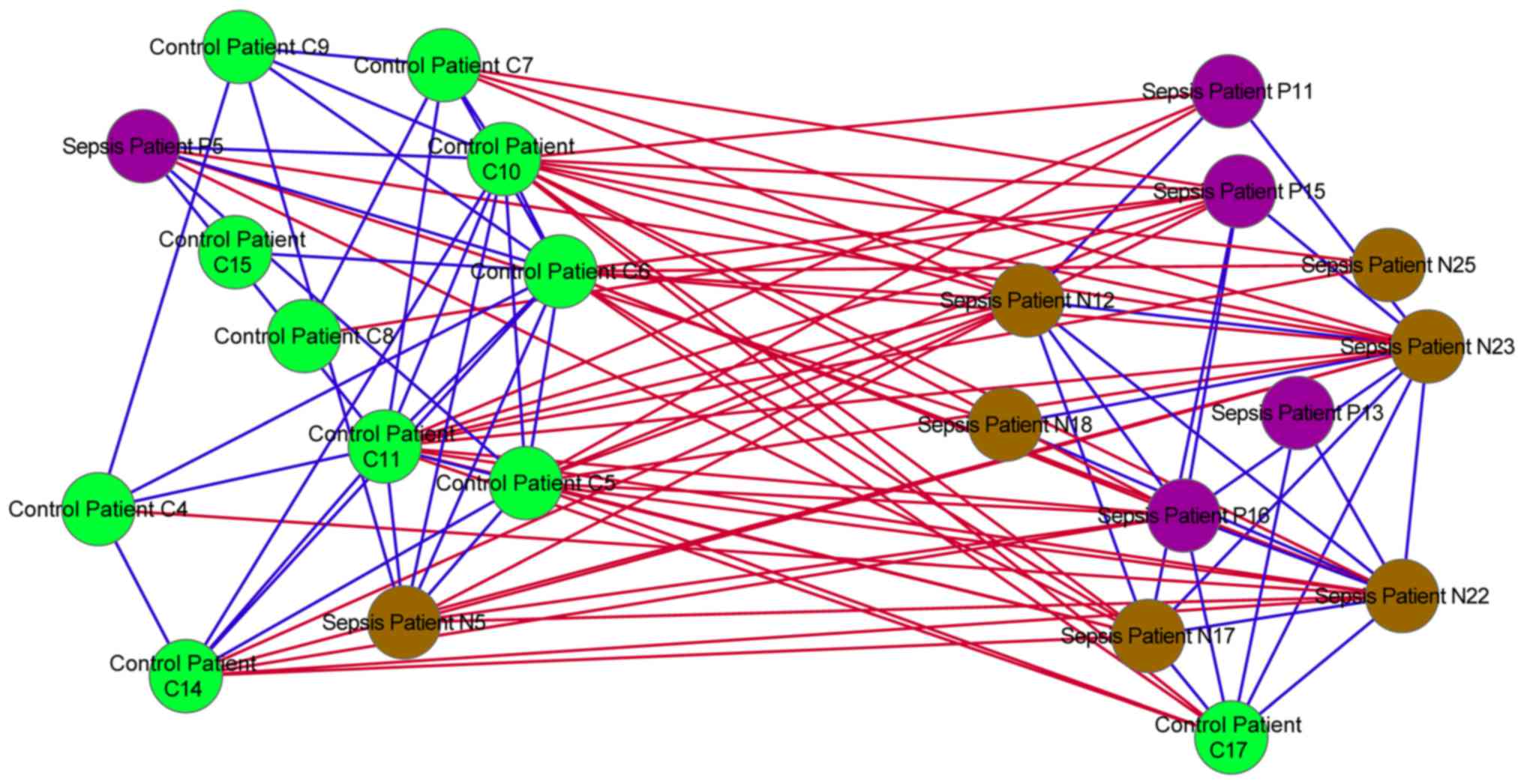
Similarity network analysis
In the similarity network, positive associations
were identified between the majority of the control and sepsis
samples. However, negative associations were also identified
between the control and sepsis samples (Fig. 4).
Functional and pathway enrichment
analyses
Functional enrichment analysis was performed on the
upregulated and downregulated genes in the Gram-positive or
Gram-negative samples separately. For the downregulated genes in
the Gram-positive samples and Gram-negative samples, MCL1 was
significantly associated with the functions of apoptosis and
programmed cell death regulation. For the upregulated genes in the
Gram-positive and Gram-negative samples, NDUFS4 was significantly
associated with mitochondrial respiratory chain complex I assembly.
Additionally, NDUFB2, NDUFB8 and ubiquinol-cytochrome c reductase
hinge protein (UQCRH) were significantly enriched in the functions
of cellular respiration, ATP synthesis coupled electron transport
and respiratory electron transport chain in Gram-negative samples.
LATS2 was significantly associated with the G1/S transition of the
mitotic cell cycle in Gram-positive samples (Tables II and III). KEGG pathway enrichment analysis
was also conducted for up and downregulated genes in Gram-positive
and Gram-negative samples (Tables
IV and V). NDUFS4 was
significantly enriched in the pathway of oxidative
phosphorylation.
| Table II.Top 10 enriched GO terms for the
upregulated and the downregulated genes in Gram-positive
samples. |
Table II.
Top 10 enriched GO terms for the
upregulated and the downregulated genes in Gram-positive
samples.
| A, Upregulated
genes |
|---|
|
|---|
| Term | Function | Count | P-value | Gene symbol |
|---|
| GO:0006412 | Translation | 22 |
3.52×10−11 | AIMP1, EEF1A2,
GARS, MRPS10, RPS27L, MRPS18C, MRPL15, MRPS18A, RPL22, MRPL27,
EEF1E1, NARS2, EIF2S2, HARS, MRPL19, MRPL36, RPL10, RPL11, RSL24D1,
MRPL32, EEFSEC, MRPL33 |
| GO:0006120 | Mitochondrial
electron transport, NADH to ubiquinone | 7 |
4.78×10−6 | NDUFA4, NDUFS6,
NDUFS5, NDUFS4, NDUFA8, NDUFA9, NDUFC2 |
| GO:0042773 | ATP synthesis
coupled electron transport | 7 |
2.62×10−5 | NDUFA4, NDUFS6,
NDUFS5, NDUFS4, NDUFA8, NDUFA9, NDUFC2 |
| GO:0022904 | Respiratory
electron transport chain | 7 |
5.65×10−5 | NDUFA4, NDUFS6,
NDUFS5, NDUFS4, NDUFA8, NDUFA9, NDUFC2 |
| GO:0045333 | Cellular
respiration | 8 |
7.34×10−5 | NDUFA4, NDUFS6,
NDUFS5, NDUFS4, NDUFA8, NDUFA9, NDUFC2, MDH1 |
| GO:0044267 | Cellular protein
metabolic process | 44 |
1.09×10−4 | FASTK, FKBP3,
PRDX4, MRPS10, RPS27L, CANX, LATS2, VRK1, PSMB7, PSMB6, MRPL15,
PLOD2, NARS2, MRPL36, B3GNT1, MRPL19, RPL10, RPL11, RSL24D1,
MRPL32, LOXL1, FGF2, MRPL33, HSP90AA1, AIMP1, EEF1A2, GARS, SOD1,
LRPAP1, IKBKE, MAST4, HSP90B1, PPIH, MRPS18C, PSMA5, MRPL27, RPL22,
MRPS18A, EEF1E1, EIF2S2, HARS, DSP, EEFSEC, FKBP2 |
| GO:0016310 |
Phosphorylation | 19 |
1.87×10−3 | NDUFA4, NDUFA8,
MVD, NDUFA9, FASTK, HK2, NDUFC2, PRDX4, SOD1, LATS2, NDUFS6, IKBKE,
MAST4, VRK1, NDUFS5, NDUFS4, ATP5C1, ATP5A1, FGF2 |
| GO:0000079 | Regulation of
cyclin-dependent protein kinase activity | 5 |
2.51×10−3 | GTPBP4, CDKN2C,
CKS2, CDKN3, LATS2 |
| GO:0033365 | Protein
localization in organelle | 7 |
4.53×10−3 | PPIH, MTX2,
NDUFA13, FGF2, TIMM44, SEC61G, NR5A1 |
| GO:0010257 | NADH dehydrogenase
complex assembly | 3 |
4.73×10−3 | NDUFAF4, NDUFS5,
NDUFS4 |
|
| B, Downregulated
genes |
|
| Term | Function | Count | P-value | Gene symbol |
|
| GO:0010942 | Positive regulation
of cell death | 17 |
1.51×10−6 | PTGS2, PREX1,
STK17B, RRAGA, PRKDC, NLRP3, NLRP1, SERINC3, NOTCH1, EI24, SSTR3,
CASP4, DUSP1, CASP8, BNIP3L, FGD3, KALRN |
| GO:0043065 | Positive regulation
of apoptosis | 16 |
6.31×10−6 | PTGS2, PREX1,
STK17B, PRKDC, NLRP3, NLRP1, SERINC3, NOTCH1, EI24, SSTR3, CASP4,
DUSP1, CASP8, BNIP3L, FGD3, KALRN |
| GO:0043068 | Positive regulation
of programmed cell death | 16 |
6.86×10−6 | PTGS2, PREX1,
STK17B, PRKDC, NLRP3, NLRP1, SERINC3, NOTCH1, EI24, SSTR3, CASP4,
DUSP1, CASP8, BNIP3L, FGD3, KALRN |
| GO:0042981 | Regulation of
apoptosis | 20 |
8.53×10−5 | PTGS2, MCL1,
PREX1, STK17B, PRKDC, PIM2, NLRP3, NLRP1, SERINC3, NOTCH1, EI24,
SSTR3, CASP4, DUSP1, IGF2R, BNIP3L, CASP8, DLG5, FGD3,
KALRN |
| GO:0043067 | Regulation of
programmed cell death | 20 |
9.73×10−5 | PTGS2, MCL1,
PREX1, STK17B, PRKDC, PIM2, NLRP3, NLRP1, SERINC3, NOTCH1, EI24,
SSTR3, CASP4, DUSP1, IGF2R, BNIP3L, CASP8, DLG5, FGD3,
KALRN |
| GO:0012502 | Induction of
programmed cell death | 12 |
1.38×10−4 | SERINC3, EI24,
CASP4, SSTR3, PREX1, BNIP3L, CASP8, STK17B, NLRP3, NLRP1, FGD3,
KALRN |
| GO:0009966 | Regulation of
signal transduction | 20 |
2.68×10−4 | LITAF, PREX1,
KLK5, CYTH4, RABGAP1L, PIM2, TBC1D22A, OSM, ECE1, CXCR4, SOSTDC1,
CASP8, GPSM1, RAMP1, RAPGEF1, RUNX2, ARAP1, FGD3, GNG7,
KALRN |
| GO:0008277 | Regulation of
G-protein coupled receptor protein signaling pathway | 5 |
1.10×10−3 | ECE1, KLK5,
GPSM1, RAMP1, GNG7 |
| GO:0051056 | Regulation of small
GTPase mediated signal transduction | 8 |
7.45×10−3 | PREX1, CYTH4,
RABGAP1L, RAPGEF1, FGD3, ARAP1, TBC1D22A, KALRN |
| GO:0010647 | Positive regulation
of cell communication | 8 |
2.82×10−2 | OSM, LAMA2,
ECE1, PTGS2, LITAF, KLK5, CASP8, PIM2 |
| Table III.Top 10 enriched GO terms for the
upregulated and the downregulated genes in Gram-negative
samples. |
Table III.
Top 10 enriched GO terms for the
upregulated and the downregulated genes in Gram-negative
samples.
| A, Upregulated
genes |
|---|
|
|---|
| Term | Function | Count | P-value | Gene symbol |
|---|
| GO:0045333 | Cellular
respiration | 15 |
8.97×10−9 | UQCRC2, NDUFA8,
NDUFB8, NDUFA6, NDUFA7, CYCS, NDUFC2, NDUFB2, NDUFS6, UQCR10,
NDUFS5, NDUFS4, UQCRH, UQCRB, MDH1 |
| GO:0042773 | ATP synthesis
coupled electron transport | 12 |
1.31×10−8 | NDUFS6, NDUFS5,
UQCR10, NDUFS4, NDUFA8, UQCRH, NDUFB8, NDUFA6, NDUFA7, NDUFC2,
UQCRB, NDUFB2 |
| GO:0022904 | Respiratory
electron transport chain | 12 |
5.68×10−8 | NDUFS6, NDUFS5,
UQCR10, NDUFS4, NDUFA8, UQCRH, NDUFB8, NDUFA6, NDUFA7, NDUFC2,
UQCRB, NDUFB2 |
| GO:0006412 | Translation | 24 |
2.78×10−7 | MRPL3, RPL19,
EEF1B2, EEF1A2, HBS1L, RPL15, MRPS10, RPL27, RPS27L, RPL22L1,
IARS2, RPS7, MRPS18C, MRPL15, MRPS18A, RPL22, MRPL27, EEF1E1,
MRPL19, RPL11, RSL24D1, MRPL32, MRPL33, RPS23 |
| GO:0006457 | Protein
folding | 17 |
6.33×10−7 | HSP90AA1, FKBP5,
FKBP4, FKBP3, TTC9, PDIA5, CCT3, LMAN1, CANX, LRPAP1, CCT7, PPIH,
HSP90B1, SIL1, RUVBL2, HSPD1, FKBP2 |
| GO:0006120 | Mitochondrial
electron transport, NADH to ubiquinone | 9 |
1.70×10−6 | NDUFS6, NDUFS5,
NDUFS4, NDUFA8, NDUFB8, NDUFA6, NDUFA7, NDUFC2, NDUFB2 |
| GO:0051437 | Positive regulation
of ubiquitin-protein ligase activity during mitotic cell cycle | 9 |
6.75×10−5 | CDK1, PSMB7,
PSMD14, PSMB6, PSMA6, PSMA5, PSMC2, PSMA4, PSMD1 |
| GO:0051351 | Positive regulation
of ligase activity | 9 |
1.12×10−4 | CDK1, PSMB7,
PSMD14, PSMB6, PSMA6, PSMA5, PSMC2, PSMA4, PSMD1 |
| GO:0051438 | Regulation of
ubiquitin-protein ligase activity | 9 |
1.80×10−4 | CDK1, PSMB7,
PSMD14, PSMB6, PSMA6, PSMA5, PSMC2, PSMA4, PSMD1 |
| GO:0051436 | Negative regulation
of ubiquitin-protein ligase activity during mitotic cell cycle | 8 |
3.37×10−4 | PSMB7, PSMD14,
PSMB6, PSMA6, PSMA5, PSMC2, PSMA4, PSMD1 |
|
| B, Downregulated
genes |
|
| Term | Function | Count | P-value | Gene symbol |
|
| GO:0043067 | Regulation of
programmed cell death | 22 |
3.47×10−6 | IFIH1, PTGS2,
MCL1, PREX1, TGFBR1, BCL2A1, STK17B, IFI16, NLRP3, NLRP1, TNFRSF9,
CASP4, DUSP1, BTG2, IGF2R, BNIP3L, CHST11, CASP8, LRRK2, MX1, IFI6,
KALRN |
| GO:0010942 | Positive regulation
of cell death | 16 |
3.57×10−6 | PTGS2, PREX1,
TGFBR1, STK17B, RRAGA, IFI16, NLRP3, NLRP1, TNFRSF9, CASP4, DUSP1,
CASP8, BNIP3L, MX1, LRRK2, KALRN |
| GO:0042981 | Regulation of
apoptosis | 21 |
1.10×10−5 | IFIH1, PTGS2,
MCL1, PREX1, TGFBR1, BCL2A1, STK17B, IFI16, NLRP3, NLRP1, TNFRSF9,
CASP4, DUSP1, BTG2, IGF2R, BNIP3L, CHST11, CASP8, MX1, IFI6,
KALRN |
| GO:0043068 | Positive regulation
of programmed cell death | 15 |
1.61×10−5 | PTGS2, PREX1,
TGFBR1, STK17B, IFI16, NLRP3, NLRP1, TNFRSF9, CASP4, DUSP1, CASP8,
BNIP3L, MX1, LRRK2, KALRN |
| GO:0043065 | Positive regulation
of apoptosis | 14 |
6.62×10−5 | PTGS2, TGFBR1,
PREX1, STK17B, IFI16, NLRP3, NLRP1, TNFRSF9, CASP4, DUSP1, CASP8,
BNIP3L, MX1, KALRN |
| GO:0012502 | Induction of
programmed cell death | 12 |
8.32×10−5 | TNFRSF9, CASP4,
PREX1, TGFBR1, BNIP3L, CASP8, STK17B, IFI16, MX1, NLRP3, NLRP1,
KALRN |
| GO:0031401 | Positive regulation
of protein modification process | 7 |
5.07×10−3 | OSM, CCND3,
TGFBR1, CD4, RICTOR, UBE2D1, LRRK2 |
| GO:0010562 | Positive regulation
of phosphorus metabolic process | 5 |
1.02×10−2 | OSM, CCND3,
TGFBR1, CD4, RICTOR |
| GO:0045937 | Positive regulation
of phosphate metabolic process | 5 |
1.02×10−2 | OSM, CCND3,
TGFBR1, CD4, RICTOR |
| GO:0019048 | Virus-host
interaction | 3 |
1.22×10−2 | IRF7, RRAGA,
CD4 |
| Table IV.Enriched pathways for the upregulated
and the downregulated genes in Gram-negative samples. |
Table IV.
Enriched pathways for the upregulated
and the downregulated genes in Gram-negative samples.
| A, Upregulated
genes |
|---|
|
|---|
| Term | Function | Count | P-value | Gene symbol |
|---|
| hsa05012 | Parkinson's
disease | 19 |
9.00×10−9 | UQCRC2, NDUFA8,
SLC25A5, NDUFA4L2, NDUFB8, SLC25A6, NDUFA6, COX7B, NDUFA7, CYCS,
NDUFC2, NDUFB2, NDUFS6, UQCR10, NDUFS5, NDUFS4, UQCRH, ATP5A1,
UQCRB |
| hsa05016 | Huntington's
disease | 21 |
7.73×10−8 | UQCRC2, NDUFA8,
SLC25A5, NDUFA4L2, NDUFB8, POLR2K, SLC25A6, NDUFA6, CYCS, COX7B,
NDUFA7, NDUFC2, SOD1, NDUFB2, NDUFS6, UQCR10, NDUFS5, NDUFS4,
UQCRH, ATP5A1, UQCRB |
| hsa00190 | Oxidative
phosphorylation | 17 |
4.25×10−7 | UQCRC2, NDUFA8,
NDUFA4L2, NDUFB8, NDUFA6, COX7B, NDUFA7, NDUFC2, NDUFB2, NDUFS6,
UQCR10, NDUFS5, NDUFS4, UQCRH, ATP5A1, ATP5I, UQCRB |
| hsa05010 | Alzheimer's
disease | 18 |
1.96×10−6 | UQCRC2, NDUFA8,
NDUFA4L2, NDUFB8, NDUFA6, COX7B, NDUFA7, CYCS, NDUFC2, NAE1,
NDUFB2, NDUFS6, UQCR10, NDUFS5, NDUFS4, UQCRH, ATP5A1,
UQCRB |
| hsa03050 | Proteasome | 9 |
3.61×10−5 | PSMB7, PSMD14,
PSMB6, PSMA6, PSMA5, PSMC2, PSMA4, SHFM1, PSMD1 |
| hsa03010 | Ribosome | 10 |
6.12×10−4 | RPL19, RPL22,
RPL15, RPL27, RPS27L, RPL11, RSL24D1, RPL22L1, RPS23, RPS7 |
| hsa00620 | Pyruvate
metabolism | 6 |
4.60×10−3 | LDHA, ACYP1,
GLO1, ACAT2, PCK1, MDH1 |
| hsa04260 | Cardiac muscle
contraction | 7 |
2.05×10−2 | UQCRC2, UQCR10,
UQCRH, COX7B, ATP1A2, TNNI3, UQCRB |
| hsa04110 | Cell cycle | 9 |
2.21×10−2 | CDK1, YWHAG,
CDKN2C, YWHAQ, TFDP2, PCNA, CDK6, GADD45A, SMC3 |
| hsa04115 | p53 signaling
pathway | 6 |
3.94×10−2 | CDK1, CYCS,
CDK6, PERP, IGFBP3, GADD45A |
|
| B, Downregulated
genes |
|
| Term | Function | Count | P-value | Gene symbol |
|
| hsa04622 | RIG-I-like receptor
signaling pathway | 5 |
5.32×10−3 | IFIH1, ISG15,
IRF7, CASP8, IFNA8 |
| hsa04612 | Antigen processing
and presentation | 5 | 9.21×10–3 | HSPA6, CD4,
IFNA8, CTSS, HLA-F |
| hsa04620 | Toll-like receptor
signaling pathway | 4 |
1.99×10−2 | IRF7, CASP8,
IFNA8, CD14 |
| hsa04660 | T cell receptor
signaling pathway | 4 |
2.33×10−2 | PTPN6, RAF1,
CD4, MAP3K14 |
| Table V.Enriched pathways for the upregulated
and the downregulated genes in Gram-positive samples. |
Table V.
Enriched pathways for the upregulated
and the downregulated genes in Gram-positive samples.
| A, Upregulated
genes |
|---|
|
|---|
| Term | Function | Count | P-value | Gene symbol |
|---|
| hsa05012 | Parkinson's
disease | 10 |
3.37×10−5 | NDUFA4, NDUFS6,
NDUFS5, NDUFS4, NDUFA8, SLC25A5, NDUFA9, NDUFC2, ATP5C1,
ATP5A1 |
| hsa05016 | Huntington's
disease | 11 |
9.07×10−5 | NDUFA4, NDUFS6,
NDUFS5, NDUFS4, NDUFA8, SLC25A5, NDUFA9, NDUFC2, ATP5C1, ATP5A1,
SOD1 |
| hsa00190 | Oxidative
phosphorylation | 9 |
2.43×10−4 | NDUFA4, NDUFS6,
NDUFS5, NDUFS4, NDUFA8, NDUFA9, NDUFC2, ATP5C1, ATP5A1 |
| hsa05010 | Alzheimer's
disease | 9 |
1.11×10−3 | NDUFA4, NDUFS6,
NDUFS5, NDUFS4, NDUFA8, NDUFA9, NDUFC2, ATP5C1, ATP5A1 |
| hsa03010 | Ribosome | 5 |
2.58×10−2 | RPL22, RPL10,
RPS27L, RPL11, RSL24D1 |
| hsa04612 | Antigen processing
and presentation | 4 |
9.23×10−2 | HSP90AA1, IFI30,
HSPA4, CANX |
| hsa00970: | Aminoacyl-tRNA
biosynthesis | 3 | 9.85×10–2 | NARS2, HARS,
GARS |
|
| B, Downregulated
genes |
|
| Term | Function | Count | P-value | Gene symbol |
|
| hsa04650 | Natural killer cell
mediated cytotoxicity | 6 |
9.37×10−3 | PTPN6, ICAM2,
RAF1, IFNA8, NFATC2, KLRD1 |
| hsa04621 | NOD-like receptor
signaling pathway | 4 |
2.25×10−2 | IL8, CASP8,
NLRP3, NLRP1 |
| hsa04660 | T cell receptor
signaling pathway | 4 |
2.91×10−2 | PTPN6, RAF1,
NFATC2, MAP3K14 |
Significantly differential functions
screening
Based on the Euclidean distance of the biological
functions, as well as the P-values of the 10,000 stochastic
perturbations between Gram-positive samples and Gram-negative
samples, a total of 10 significantly differential functions were
obtained, including cellular respiration
(P<1.00×10−8, Euclidean distance=1.156277), ATP
synthesis coupled electron transport (P<1.00×10−8,
Euclidean distance=1.156277) and G1/S transition of mitotic cell
cycle (P=0.015, Euclidean distance = 0.554799; Table VI).
| Table VI.Top 10 significant differential
functions between Gram-negative samples and Gram-positive
samples. |
Table VI.
Top 10 significant differential
functions between Gram-negative samples and Gram-positive
samples.
| GO ID | Term | Euclidean
distance | P-value | Gene symbols |
|---|
| GO:0006120 | Mitochondrial
electron transport, NADH to ubiquinone | 1.156277 |
<1.00×10−8 | NDUFB8, NDUFB2,
NDUFA6, NDUFA7, NDUFA9, NDUFA4 |
| GO:0042773 | ATP synthesis
coupled electron transport | 1.156277 |
<1.00×10−8 | UQCRH, NDUFB8,
NDUFB2, UQCRB, NDUFA6, NDUFA7, UQCR10, NDUFA9, NDUFA4 |
| GO:0022904 | Respiratory
electron transport chain | 1.156277 |
<1.00×10−8 | UQCRH, NDUFB8,
NDUFB2, UQCRB, NDUFA6, NDUFA7, UQCR10, NDUFA9, NDUFA4 |
| GO:0045333 | Cellular
respiration | 1.156277 |
<1.00×10−8 | UQCRH, NDUFB8,
NDUFB2, UQCRC2, UQCRB, NDUFA6, NDUFA7, UQCR10, CYCS, NDUFA9,
NDUFA4 |
| GO:0016310 |
Phosphorylation | 1.413364 |
1.00×10−3 | UQCRH, PAK4,
FGFR1, NDUFB8, NDUFB2, CDK1, CDK6, UQCRC2, GHR, IGFBP3, UQCRB,
NDUFA6, NDUFA7, UQCR10, MET, ATP5I, MVD, NDUFA9, LATS2, MAST4,
NDUFA4, IKBKE, ATP5C1 |
| GO:0042981 | Regulation of
apoptosis | 1.508092 |
2.70×10−3 | IFI16, TNFRSF9,
IFIH1, MX1, BTG2, CHST11, TGFBR1, BCL2A1, IFI6, LGALS1, PRDX1,
PHLDA1, HSPD1, SORT1, MAL, DHCR24, GLO1, ITGB3BP, CDK1, GHR,
SERPINB2, NQO1, ANXA1, IGFBP3, SMO, CADM1, CD44, KRT18, CYCS, NAE1,
PERP, DLG5, EI24, PRKDC, NOTCH1, SERINC3, FGD3, PIM2,
SSTR3 |
| GO:0043067 | Regulation of
programmed cell death | 1.517301 |
3.60×10−3 | IFI16, TNFRSF9,
IFIH1, MX1, BTG2, CHST11, LRRK2, TGFBR1, BCL2A1, IFI6, LGALS1,
PRDX1, PHLDA1, HSPD1, SORT1, MAL, DHCR24, GLO1, ITGB3BP, CDK1, GHR,
SERPINB2, NQO1, ANXA1, IGFBP3, SMO, CADM1, CD44, KRT18, CYCS, NAE1,
PERP, DLG5, EI24, PRKDC, NOTCH1, SERINC3, FGD3, PIM2,
SSTR3 |
| GO:0000082 | G1/S transition of
mitotic cell cycle | 0.554799 |
1.50×10−2 | LATS2 |
| GO:0044267 | Cellular protein
metabolic process | 2.267426 |
2.37×10−2 | RPS23, IARS2,
HSPD1, PAK4, FGFR1, SEC11A, HEXB, FKBP5, PSMA4, MRPL3, RPL27,
PSMC2, SIL1, SUPT3H, RPL27, RPS7, RPL19, FKBP4, LMAN1, PTPN1, CDK1,
CDK6, GHR, RPL22L1, PDIA5, CCT7, FBXO7, ANXA1, IGFBP3, PSMA6,
PSMD1, CCT3, HERC3, TTC9, MET, NAE1, PSMD14, HBS1L, RABGGTB,
EEF1B2, RPL15, RUVBL2, AIMP1, LATS2, MAST4, EEFSEC, LOXL1, NARS2,
HARS, B3GNT1, MRPL36, IKBKE, EIF2S2, DSP, RPL10, GARS |
| GO:0007517 | Muscle organ
development | 1.050407 |
4.15×10−2 | FAM65B, LAMA2,
ANKRD2, FOXP1, CACNB4 |
Discussion
In line with the results of Tang et al
(8), the present study determined
that there was no significant difference in the expression profile
between Gram-positive and gram-negative samples from hierarchical
clustering analysis. In the Gram-positive and Gram-negative
samples, the GO functional enrichment analysis revealed that MCL1
was significantly associated with the regulation of apoptosis and
programmed cell death. A previous study has determined that the
apoptosis of T-cells may induce the breakdown of defense mechanisms
resulting in sepsis (27).
Additionally, the inhibition of programmed cell death may reverse
T-cell exhaustion and thus eradicate the invading pathogens which
cause sepsis (28). Additionally,
MCL1 may also be associated with the reduction of apoptosis of
neutrophils in patients with sepsis (29). Therefore, it is possible for MCL1
to be involved in sepsis via the regulation of T-cell apoptosis and
programmed T-cell death in both Gram-positive and Gram-negative
sepsis.
Additionally, the present study also determined that
NDUFS4 was significantly associated with mitochondrial respiratory
chain complex I assembly. Mitochondrial dysfunction may lead to
oxidative stress and failure of energy production, which may result
in organ dysfunction in sepsis (30). The KEGG pathway enrichment analysis
revealed that NDUFS4 was significantly enriched in oxidative
phosphorylation. Lee and Hüttemann (31) have determined that the inhibition
of oxidative phosphorylation may lead to a reduction of the
mitochondrial membrane potential, resulting in a lack of energy,
which may cause organ failure and death in septic patients
(31). NDUFS4 has been previously
reported to be an important subunit of complex I which has a key
role in oxidative phosphorylation (32). Additionally, NDUFS4 may participate
in the regulation of sepsis induced by Gram-negative and
Gram-positive bacteria through regulation of oxidative
phosphorylation.
However, the present study identified specific DEGs
in Gram-positive and Gram-negative samples compared with normal
samples. According to the Euclidean distance and the stochastic
perturbations performed between Gram-positive and Gram-negative
samples, NDUFB2, NDUFB8 and UQCRH were significantly upregulated in
the Gram-negative samples, whereas they were not upregulated in the
Gram-positive samples. In addition, functional annotation revealed
that they were significantly associated with cellular respiration,
ATP synthesis coupled electron transport and mitochondrial electron
transport, ubiquinol to cytochrome c. NDUFB2 and NDUFB8 are parts
of the multisubunit mitochondrial NADH ubiquinone oxidoreductase
(complex I) which has an important role in mitochondrial
functioning (33,34). A previous study determined that a
dysfunction of respiratory chain complex I may be associated with
reactive oxygen species (ROS) production (35). Additionally, previous studies
reported that ROS are toxic oxygen-containing molecules that may
damage the cells and the antioxidant defense system, which is the
pathogenesis of sepsis (36,37).
UQCRH, which encodes the cytochrome b-c1 complex subunit 6 of
complexes III (cytochrome c-oxidoreductase), is involved in the
mitochondrial oxidative phosphorylation and the dysfunction of
UQCRH may lead to breast and ovarian cancer by altering the
function of the mitochondria (38,39).
To the best of our knowledge, this is the first study investigating
the functions of NDUFB2, NDUFB8 and UQCRH in Gram-negative
bacteria-induced sepsis. The present study concluded that NDUFB2,
NDUFB8 and UQCRH may be involved in the Gram-negative
bacteria-induced sepsis by altering mitochondrial oxidative
phosphorylation and may also be potential targets for the treatment
of Gram-negative bacterial sepsis.
In addition, the function of the G1/S transition of
the mitotic cell cycle was also determined to be significantly
different between the Gram-positive and Gram-negative samples.
LATS2 was enriched in this function and was significantly
upregulated in patients with Gram-positive sepsis, whereas it was
not significantly expressed in Gram-negative patients. LATS2,
encoding serine/threonine-protein kinase, has been identified to
inhibit the G1/S transition in the cell cycle of tumor cells
(40). Additionally, G1 cell cycle
arrest may be important for the initiation of kidney injury in
sepsis (41). Therefore, LATS2 may
be associated with Gram-negative bacterial sepsis by the modulation
of G1/S transition in cell cycle.
In conclusion, MCL1, NDUFS5 and NDUFS4 may be
potential target genes for the treatment of Gram-positive and
Gram-negative bacterial sepsis. Additionally, NDUFB2, NDUFB8 and
UQCRH may also be associated with Gram-negative bacterial sepsis.
LATS2 may contribute to the progression of Gram-negative bacterial
sepsis. However, further studies are still required in order to
elucidate their action mechanisms in sepsis.
Glossary
Abbreviations
Abbreviations:
|
DEGs
|
differentially expressed genes
|
|
FDR
|
false discovery rate
|
|
FC
|
fold-change
|
|
ICU
|
intensive care unit
|
|
PCC
|
Pearson's correlation coefficient
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
References
|
1
|
Taylor FB Jr, Kinasewitz GT and Lupu F:
Pathophysiology, staging and therapy of severe sepsis in baboon
models. J Cell Mol Med. 16:672–682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brodská H, Malíčková K, Adámková V,
Benáková H, Šťastná MM and Zima T: Significantly higher
procalcitonin levels could differentiate Gram-negative sepsis from
Gram-positive and fungal sepsis. Clin Exp Med. 13:165–170. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deutschman C and Tracey K: Sepsis: Current
dogma and new perspectives. Immunity. 40:463–475. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lyle NH, Pena OM, Boyd JH and Hancock REW:
Barriers to the effective treatment of sepsis: Antimicrobial
agents, sepsis definitions, and host-directed therapies. Ann N Y
Acad Sci. 1323:101–114. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hotchkiss RS and Karl IE: The
pathophysiology and treatment of sepsis. N Engl J Med. 348:138–150.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Koh EM, Lee SG, Kim CK, Kim M, Yong D, Lee
K, Kim JM, Kim DS and Chong Y: Microorganisms isolated from blood
cultures and their antimicrobial susceptibility patterns at a
university hospital during 1994–2003. Korean J Lab Med. 27:265–275.
2007.(In Korean). View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Glik J, Kawecki M, Gaździk T and Nowak M:
The impact of the types of microorganisms isolated from blood and
wounds on the results of treatment in burn patients with sepsis.
Pol Przegl Chir. 84:6–16. 2012.PubMed/NCBI
|
|
8
|
Tang BM, McLean AS, Dawes IW, Huang SJ,
Cowley MJ and Lin RC: Gene-expression profiling of gram-positive
and gram-negative sepsis in critically ill patients. Crit Care Med.
36:1125–1128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mahabeleshwar GH, Qureshi MA, Takami Y,
Sharma N, Lingrel JB and Jain MK: A myeloid hypoxia-inducible
factor 1α-Krüppel-like factor 2 pathway regulates gram-positive
endotoxin-mediated sepsis. J Biol Chem. 287:1448–1457. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Giamarellos-Bourboulis EJ, van de Veerdonk
FL, Mouktaroudi M, Raftogiannis M, Antonopoulou A, Joosten LA,
Pickkers P, Savva A, Georgitsi M, van der Meer JW and Netea MG:
Inhibition of caspase-1 activation in Gram-negative sepsis and
experimental endotoxemia. Crit Care. 15:R272011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kager LM, Weehuizen TA, Wiersinga WJ,
Roelofs JJ, Meijers JC, Dondorp AM, van't Veer C and van der Poll
T: Endogenous α2-antiplasmin is protective during severe
gram-negative sepsis (melioidosis). Am J Respir Crit Care Med.
188:967–975. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kager LM, Wiersinga WJ, Roelofs JJ, de
Boer OJ, Weiler H, van't Veer C and van der Poll T: A
thrombomodulin mutation that impairs active protein C generation is
detrimental in severe pneumonia-derived gram-negative sepsis
(melioidosis). PLoS Negl Trop Dis. 8:e28192014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Afsari B, Geman D and Fertig EJ: Learning
dysregulated pathways in cancers from differential variability
analysis. Cancer Inform. 13:(Suppl 5). S61–S67. 2014.
|
|
14
|
Calandra T, Cohen J, et al: International
Sepsis Forum Definition of Infection in the ICU Consensus
Conference: The international sepsis forum consensus conference on
definitions of infection in the intensive care unit. Crit Care Med.
33:1538–1548. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Murie C, Barette C, Lafanechère L and
Nadon R: Control-plate regression (CPR) normalization for
high-throughput screens with many active features. J Biomol Screen.
19:661–671. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen H and Boutros PC: VennDiagram: A
package for the generation of highly-customizable Venn and Euler
diagrams in R. BMC Bioinformatics. 12:352011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gene Ontology Consortium, . Blake JA,
Dolan M, Drabkin H, Hill DP, Li N, Sitnikov D, Bridges S, Burgess
S, Buza T, et al: Gene ontology annotations and resources. Nucleic
Acids Res. 41:(Database issue). D530–D535. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2008. View Article : Google Scholar
|
|
20
|
Zhou J, Dong X, Zhou Q, Wang H, Qian Y,
Tian W, Ma D and Li X: microRNA expression profiling of heart
tissue during fetal development. Int J Mol Med. 33:1250–1260.
2014.PubMed/NCBI
|
|
21
|
Zhai Y, Tchieu J and Saier MH Jr: A
web-based tree view (TV) program for the visualization of
phylogenetic trees. J Mol Microbiol Biotechnol. 4:69–70.
2002.PubMed/NCBI
|
|
22
|
Weaver B and Wuensch KL: SPSS and SAS
programs for comparing Pearson correlations and OLS regression
coefficients. Behav Res Methods. 45:880–895. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Demchak B, Hull T, Reich M, Liefeld T,
Smoot M, Ideker T and Mesirov JP: Cytoscape: The network
visualization tool for GenomeSpace workflows. F1000Res.
3:1512014.PubMed/NCBI
|
|
24
|
Kotera M, Moriya Y, Tokimatsu T, Kanehisa
M and Goto S: KEGG and GenomeNet, New Developments. Metagenomic
Analysis. 329–339. 2015.
|
|
25
|
Liberti L, Lavor C, Maculan N and
Mucherino A: Euclidean distance geometry and applications.
Quantitative Biol. 56:3–69. 2012.
|
|
26
|
Bódai T, Altmann EG and Endler A:
Stochastic perturbations in open chaotic systems: Random versus
noisy maps. Phys Rev E Stat Nonlin Soft Matter Phys. 87:0429022013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schmidt MV, Paulus P, Kuhn AM, Weigert A,
Morbitzer V, Zacharowski K, Kempf VA, Brüne B and von Knethen A:
Peroxisome proliferator-activated receptor γ-induced T cell
apoptosis reduces survival during polymicrobial sepsis. Am J Respir
Crit Care Med. 184:64–74. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chang K, Svabek C, Vazquez-Guillamet C,
Sato B, Rasche D, Wilson S, Robbins P, Ulbrandt N, Suzich J, Green
J, et al: Targeting the programmed cell death 1: Programmed cell
death ligand 1 pathway reverses T cell exhaustion in patients with
sepsis. Crit Care. 18:R32014. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Härter L, Mica L, Stocker R, Trentz O and
Keel M: Mcl-1 correlates with reduced apoptosis in neutrophils from
patients with sepsis. J Am Coll Surg. 197:964–973. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Galley H: Oxidative stress and
mitochondrial dysfunction in sepsis. Br J Anaesth. 107:57–64. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee I and Hüttemann M: Energy crisis: The
role of oxidative phosphorylation in acute inflammation and sepsis.
Biochim Biophys Acta. 1842:1579–1586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Petruzzella V, Vergari R, Puzziferri I,
Boffoli D, Lamantea E, Zeviani M and Papa S: A nonsense mutation in
the NDUFS4 gene encoding the 18 kDa (AQDQ) subunit of complex I
abolishes assembly and activity of the complex in a patient with
Leigh-like syndrome. Hum Mol Genet. 10:529–535. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ugalde C, Hinttala R, Timal S, Smeets R,
Rodenburg RJ, Uusimaa J, van Heuvel LP, Nijtmans LG, Majamaa K and
Smeitink JA: Mutated ND2 impairs mitochondrial complex I assembly
and leads to Leigh syndrome. Mol Genet Metab. 90:10–14. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bottley A, Phillips NM, Webb TE, Willis AE
and Spriggs KA: eIF4A inhibition allows translational regulation of
mRNAs encoding proteins involved in Alzheimer's disease. PLoS One.
5:pii: e13030. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dröse S and Brandt U: Molecular mechanisms
of superoxide production by the mitochondrial respiratory
chainMitochondrial Oxidative Phosphorylation. Springer; pp.
145–169. 2012, View Article : Google Scholar
|
|
36
|
Van Raamsdonk JM and Hekimi S: Superoxide
dismutase is dispensable for normal animal lifespan. Proc Natl Acad
Sci USA. 109:5785–5790. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schulte J, Struck J, Köhrle J and Müller
B: Circulating levels of peroxiredoxin 4 as a novel biomarker of
oxidative stress in patients with sepsis. Shock. 35:460–465. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Owens KM, Kulawiec M, Desouki MM,
Vanniarajan A and Singh KK: Impaired OXPHOS complex III in breast
cancer. PLoS One. 6:e238462011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Modena P, Testi MA, Facchinetti F,
Mezzanzanica D, Radice MT, Pilotti S and Sozzi G: UQCRH gene
encoding mitochondrial Hinge protein is interrupted by a
translocation in a soft-tissue sarcoma and epigenetically
inactivated in some cancer cell lines. Oncogene. 22:4586–4593.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Y, Pei J, Xia H, Ke H, Wang H and Tao
W: Lats2, a putative tumor suppressor, inhibits G1/S transition.
Oncogene. 22:4398–4405. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang QH, Liu DW, Long Y, Liu HZ, Chai WZ
and Wang XT: Acute renal failure during sepsis: Potential role of
cell cycle regulation. J Infect. 58:459–464. 2009. View Article : Google Scholar : PubMed/NCBI
|