Introduction
Candida albicans (C. albicans) is a
common commensal fungus which is normally accumulated in the oral
cavity, respiratory tract and gastrointestinal systems in low
numbers in healthy individuals (1). C. albicans is one of the most
frequent opportunistic fungal pathogens of candidiasis. According
to data provided by the US National Health Care Safety Network,
C. albicans is the fourth most common microorganism
infection in US hospitals (2). It
has been estimated that ~250,000–400,000 individuals succumb to
C. albicans-associated mortality annually worldwide
(3). Although there are several
antifungal drugs available, including azoles, polyenes, allylamines
and echinocandins (4), the
efficiency of anti-infection therapy remains low and drug
resistance is a predominant problem in clinical therapy (5). Therefore, it is essential to
elucidatethe mechanism of C. albicans infection in order to
provide novel insights into the clinical treatment of
candidiasis.
Following the identification of C. albicans,
studies have investigated the processes involved in its infection
and methods against infection. Generally, candidiasis is not
detected in healthy individuals but is frequently detected in
individuals who are immunocompromised to a certain extent,
including babies, children, the elderly and patients suffering from
immune system disease (6). Raska
et al reported that the virulence factors secreted by C.
albicans are one of the main factors enabling them to damage
tissues and evade the immune system of the host (7). It has also been demonstrated that the
transition of a yeast-like phenotype to a hyphal phenotype is an
essential procedure for infection (8). pH, temperature and the ratio of
oxygen/carbon dioxide in the microenvironment are also crucial in
the process of C. albicans infection (9). Exposure to antifungal drugs is the
main therapeutic approach for the candidiasis, however, due to thee
overuse of antifungal drugs and the adaptive alteration of fungal
genomes, the efficiency of clinical treatment remains low and the
development of novel methods has been slow (10). Although, several studies have
focused on the relevant factors, including cytokines, chemokines
and effector cells, involved to obtain further insight into this
process, information is lacking due to the restricted analysis
(9). Therefore, it is important to
gain a wider understanding of C. albicans.
To further analyze the alterations to the C.
albicans transcriptome following treatment with macrocyclic
compound FR59, Sanglard et al (10) generated a microarray of C.
albicans (GSE65396) treated with 3 µM FR59 at the two time
points of 15 and 45 min (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65396).
In this microarray analysis, Sanglard et al showed that FR59
may induce a reduction in the expression of genes involved in cell
wall biosynthesis, and that ATP binding cassette transporters may
be involved in this process. This indicates that FR59 may be a
promising drug for the treatment of candidiasis. However, the
detailed reasons for this alteration to the cell wall remain to be
elucidated. In order to obtain amore detailed understanding of
C. albicans treated with FR59, the present study performed
further analysis of the GSE65396 microarray. By performing this
analysis, potential target genes or biofunctions may be identified,
which may provide novel insights into the treatment of
candidiasis.
Materials and methods
Gene expression profile
The gene expression profile of GSE65396 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65396)
for the macrocyclic compound RF59-treated C. albicans was
downloaded from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), which was sequenced
on the Agilent-037331 Copy of C. albicans assembly 21_017942
platform. This time profile contained three groups: 0 min control
group, 15 min RF59-treated group and 45 min-RF59 treated group,
with three repetitions in each.
Data processing
The Affy package (11) in R (Version 3.3.0, http://www.bioconductor.org/packages/release/bioc/html/affy.html)
was used to read the raw data, the and RMA (12,13)
method was used for data processing, including background
correction, normalization and expression calculation. Annotation of
the gene expression profile was performed to transform the probe
IDs into gene symbols. In cases of different probes mapping to one
gene, the mean value was considered to be the final expressed level
of this gene.
Identification of differentially
expressed genes (DEGs)
The Bayes test in the Limma (14) package was utilized to screen the
DEGs of the 15 min group, vs. control; and the 45 min group, vs.
control, P<0.05 and |log2 fold change >0.585 were
considered to be significant.
Pathway enrichment analysis for
DEGs
The Database for Annotation, Visualization and
Integrated Discovery (DAVID) is a free database which uses an
agglomeration algorithm to organize functionally related genes and
terms into a manageable number of biological modules (15). Kyoto Encyclopedia of Genes and
Genomes (KEGG) is a another free database for the systematic
analysis of gene functions by linking current knowledge to cellular
processes (16). In the present
study, the DAVID online tool (Version 6.8, https://david-d.ncifcrf.gov/) was used to perform KEGG
pathway enrichment analysis of DEGs. P<0.05 was set as the
significance cut-off.
Protein-protein interaction (PPI)
network
The Search Tool for Retrieval of Interacting Genes
(STRING) is a precompute global database for the analysis and
exploration of protein interaction (17). To further reveal the biofunctions
of the identified DEGs, STRING (version 10.0, http://www.string-db.org/) was used to examine the
interactions between differentially expressed proteins. The
threshold was set as a required confidence >0.4.
Based on the results of the STRING analysis,
Cytoscape (Version 3.2.0, http://www.cytoscape.org/) (18) was utilized to visualize the PPI
network of differentially expressed proteins and the CytoNCA
plug-in of Cytoscape (19) was
used to perform topological analysis, including degree centrality
(DC), betweenness centrality (BC) and closeness centrality (CC).
Following calculation of the score of each node, the hub proteins
in the PPI network were screened out (20).
Functional subnetwork analysis of
DEGs
The ReactomeFI plug-in of Cytoscape is an
application with a loaded gene expression profile and can be
utilized to calculate the weights of edges in a PPI network. In the
present study, ReactomeFI was used to compute the correlations as
weights for the edges of the PPI network and was combined with the
Markov Cluster Algorithm method to map significant functional
subnetworks in the PPI network (21). Subsequently the DAVID online tool
was used to perform KEGG pathway enrichment analyses for the
selected subnetwork. P<0.05 was considered significant.
Results
Screening of DEGs
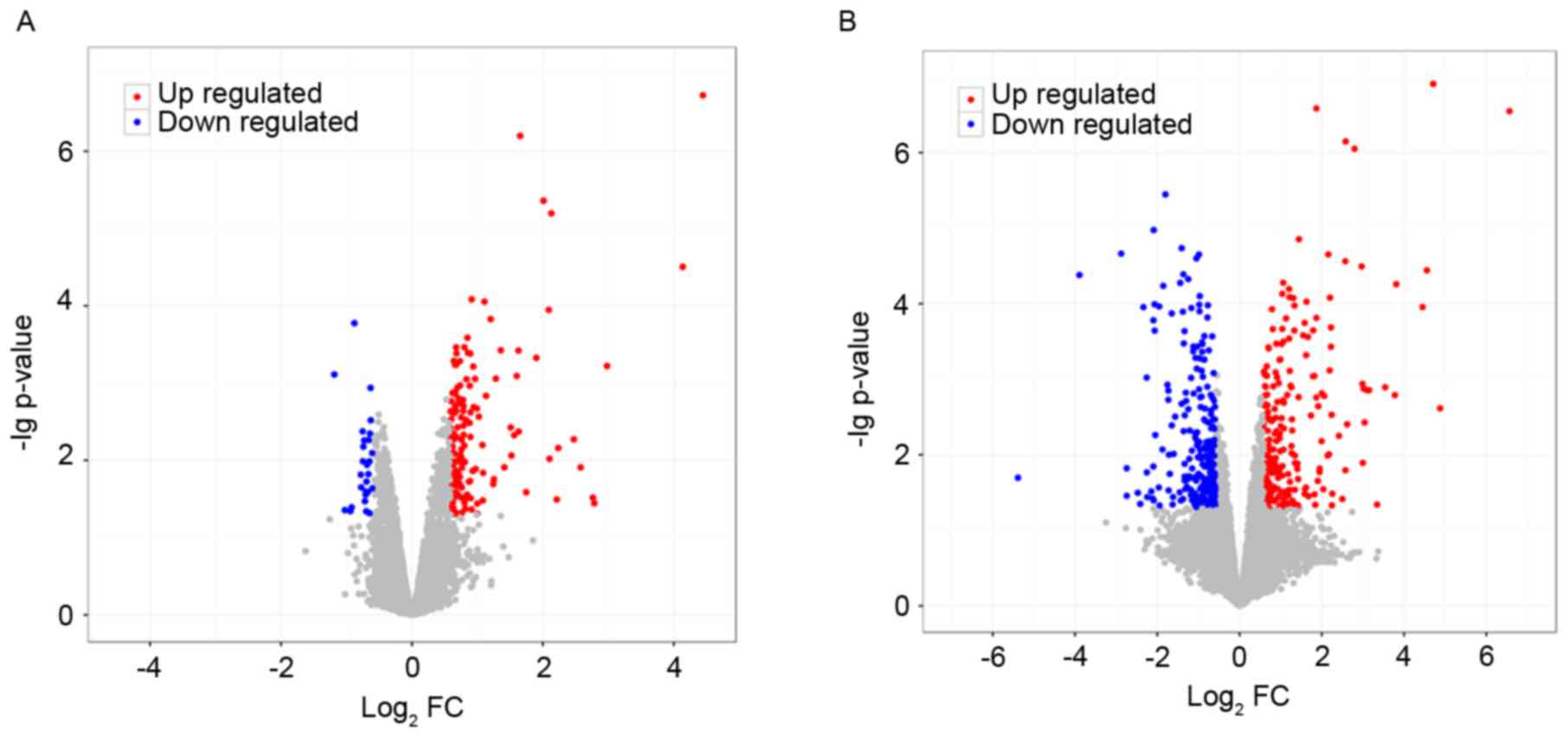
Following preprocessing and analysis, a total of 181
DEGs were identified in the 15-min treatment group, compared with
the control group, which included 154 upregulated and 27
downregulated genes (Fig. 1A). A
total of 468 DEGs were screened out in the 45-min treatment group,
compared with the control group, which included 235 upregulated and
233 downregulated genes (Fig.
1B).
Functional enrichment analysis of
DEGs
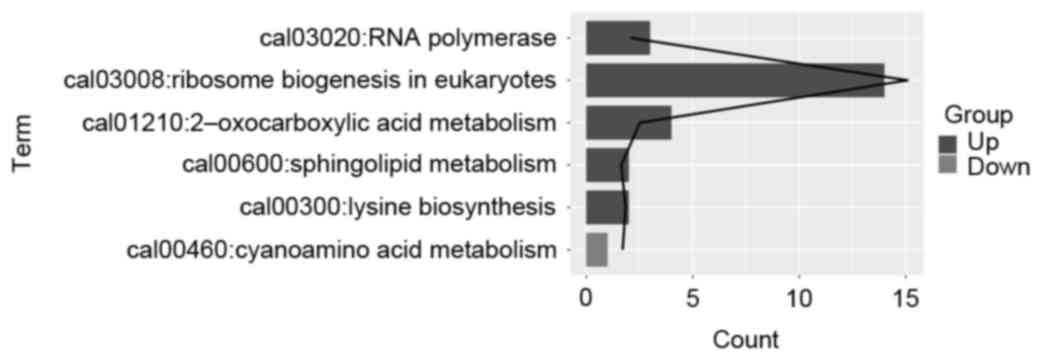
To further investigate the biofunctions of the DEGs,
KEGG pathway enrichment analysis was performed. The results showed
that the upregulated DEGs in the 15-min treatment group were
significantly enriched in functions, which included ribosome
biogenesis in eukaryotes (P=8.28E-16), oxocarboxylix acid
metabolism (P=0.003), and RNA polymerase (P=0.009). The
downregulated DEGs were significantly enriched in cyanoamino acid
metabolism (P=0.020), as show in Fig.
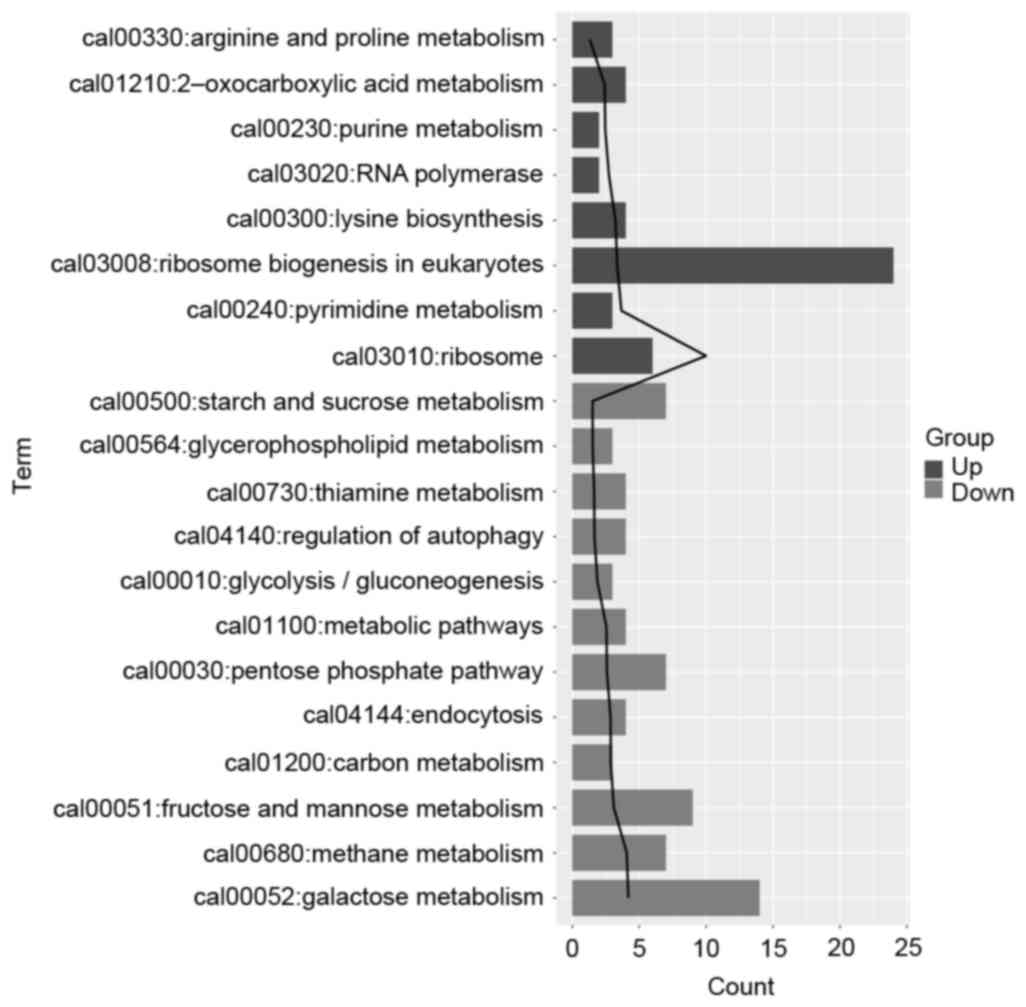
2. The upregulated DEGs in the 45-min treatment group were
significantly enriched in functions, which included ribosome
(P=1.03E-10), pyrimidine metabolism (P=2.14E-04) and ribosome
biogenesis in eukaryotes (P=4.39E-04). The downregulated DEGs were
significantly enriched in functions, which included galactose
metabolism (P=6.49E-05), methane metabolism (P=8.45E-05), and
fructose and mannose metabolism (P=7.88E-04), as shown in Fig. 3.
PPI network analysis
Due to the deficiency of C. albicans data in STRING,
the profile of Saccharomyces cerevisiae (S. cerevisiae) was used.
According to the biofunctions of the DEGs, the PPI network of the
15-min treatment group was constructed, which contained 76 nodes
and 975 edges (Fig. 4). Based on
the scores from the topological analysis, PRP5 (DC=48, BC=273.754,
CC=0.185), RCL1 (DC=44, BC=456.142, CC=0.184), NOP13 (DC=46,
BC=116.109, CC=0.184), MRT4 (DC=47, BC=87.329, CC=0.184) and NOP4
(DC=46, BC=40.931, CC=0.184) were the top five nodes in the PPI
network (Table I). The PPI network
of the 45-min treatment group was also constructed, containing 177
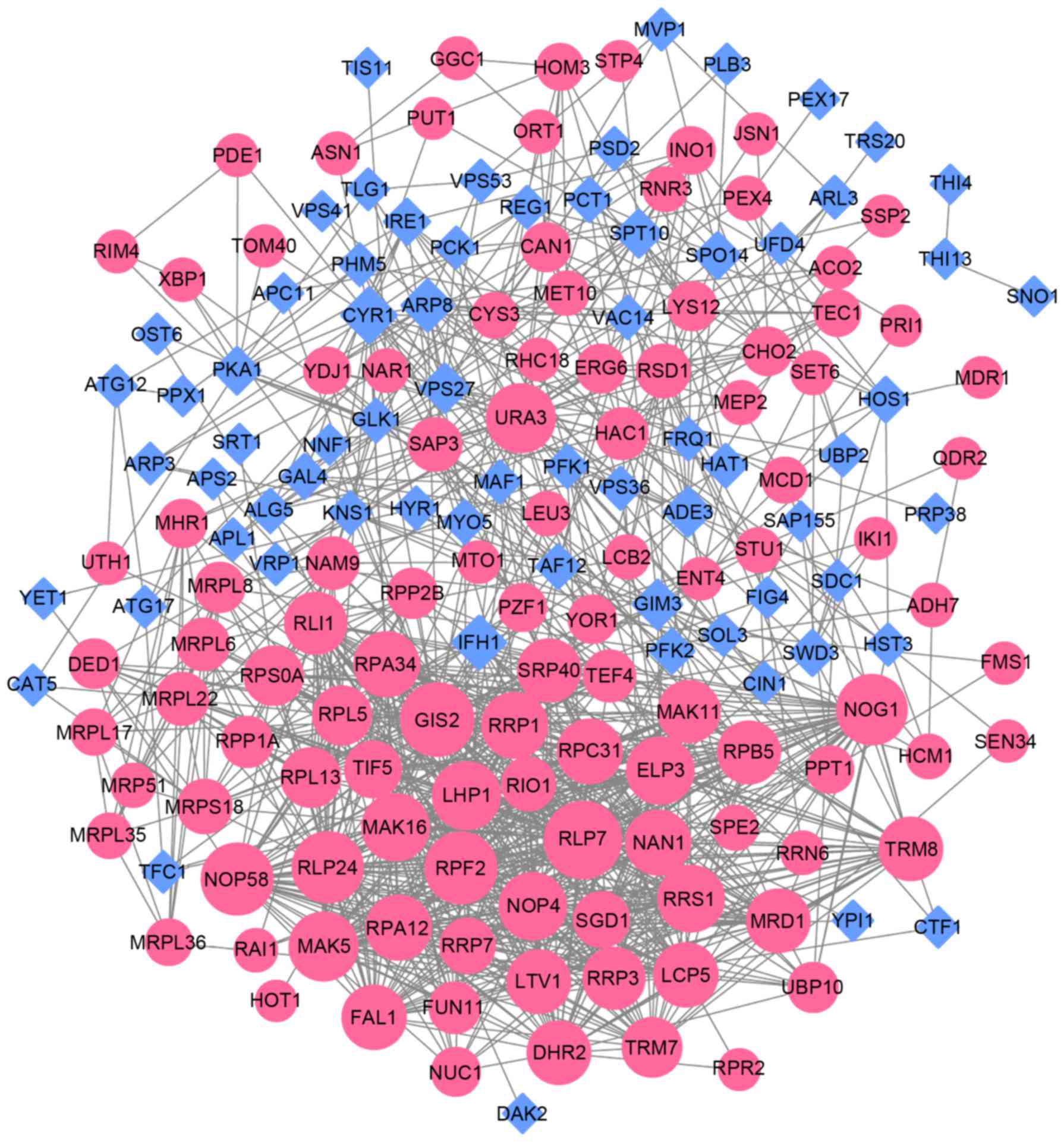
nodes and 1,051 edges (Fig. 5).
According to the scores from the topological analysis, GIS2 (DC=44,
BC=4461.883, CC=0.202), URA3 (DC=38, BC=4507.660, CC=0.203), NOP58
(DC=42, BC=806.258, CC=0.200), ELP3 (DC=36, BC=1725.969, CC=0.198)
and PLP7 (DC=49, BC=1129.925, CC=0.197) were the top five nodes in
the PPI network (Table II). NOP4
and ELP3 were two shared genes within the top 20 of the two
comparisons.
 | Table I.Top 15 nodes identified in the
topological analysis of the 15-min treatment protein-protein
interaction network. |
Table I.
Top 15 nodes identified in the
topological analysis of the 15-min treatment protein-protein
interaction network.
| Node | Value |
|---|
| Degree
centrality |
|
|
RRP5 | 48 |
|
MRT4 | 47 |
|
NOP13 | 46 |
|
ENP1 | 46 |
|
RLP24 | 46 |
|
NOP4 | 46 |
|
TSR1 | 46 |
|
NOP15 | 46 |
|
PWP2 | 45 |
|
RPA43 | 45 |
|
SPB1 | 45 |
|
DBP3 | 45 |
|
LSG1 | 45 |
|
RCL1 | 44 |
|
RRP8 | 44 |
| Betweenness
centrality |
|
|
LEU1 | 499.743 |
|
RCL1 | 456.152 |
|
AAH1 | 315.751 |
|
RRP8 | 311.275 |
|
RRP5 | 273.754 |
|
PUS1 | 260.075 |
|
PAB1 | 186.937 |
|
PWP2 | 182.521 |
|
LYS4 | 149.977 |
|
NEP1 | 147.078 |
|
UTP6 | 146.198 |
|
TEC1 | 141.955 |
|
RSM22 | 140.000 |
|
ELP3 | 136.432 |
|
NOP13 | 116.109 |
| Closeness
centrality |
|
|
RRP5 | 0.185185 |
|
RCL1 | 0.184275 |
|
NOP13 | 0.183824 |
|
MRT4 | 0.183824 |
|
NOP4 | 0.183824 |
|
TSR1 | 0.183824 |
|
PWP2 | 0.183374 |
|
ENP1 | 0.183374 |
|
RLP24 | 0.183374 |
|
SPB1 | 0.183374 |
|
DBP3 | 0.183374 |
|
NOP15 | 0.183374 |
|
RRP8 | 0.182927 |
|
RPA43 | 0.182927 |
|
LSG1 | 0.182927 |
| Table II.Top 15 nodes identified in
topological analysis of the 45-min treated protein-protein
interaction network. |
Table II.
Top 15 nodes identified in
topological analysis of the 45-min treated protein-protein
interaction network.
| Node | Value |
|---|
| Degree
centrality |
|
|
RLP7 | 49 |
|
GIS2 | 44 |
|
NOP58 | 42 |
|
RPF2 | 42 |
|
RLP24 | 41 |
|
NOG1 | 40 |
|
URA3 | 38 |
|
MAK5 | 38 |
|
LHP1 | 37 |
|
MAK16 | 36 |
|
NOP4 | 36 |
|
ELP3 | 36 |
|
RRS1 | 35 |
|
RRP1 | 34 |
|
RPA12 | 33 |
| Betweenness
centrality |
|
|
URA3 | 4507.660 |
|
GIS2 | 4461.883 |
|
CYR1 | 2211.606 |
|
ELP3 | 1725.969 |
|
ARP8 | 1309.269 |
|
VPS27 | 1287.934 |
|
SAP3 | 1233.091 |
|
RLP7 | 1129.925 |
|
RSD1 | 1121.732 |
|
ADE3 | 1080.301 |
|
HAC1 | 937.495 |
|
GIM3 | 906.974 |
|
RPS0A | 886.321 |
|
SPT10 | 881.783 |
|
NOP58 | 806.258 |
| Closeness
centrality |
|
|
URA3 | 0.202532 |
|
GIS2 | 0.201835 |
|
NOP58 | 0.199773 |
|
ELP3 | 0.198422 |
|
LHP1 | 0.197753 |
|
RLP7 | 0.196648 |
|
CYR1 | 0.194906 |
|
RPF2 | 0.194690 |
|
RPB5 | 0.194261 |
|
MAK5 | 0.193833 |
|
RPC31 | 0.193407 |
|
RRP1 | 0.193407 |
|
RLP24 | 0.193194 |
|
RPA34 | 0.193194 |
|
RPS0A | 0.192982 |
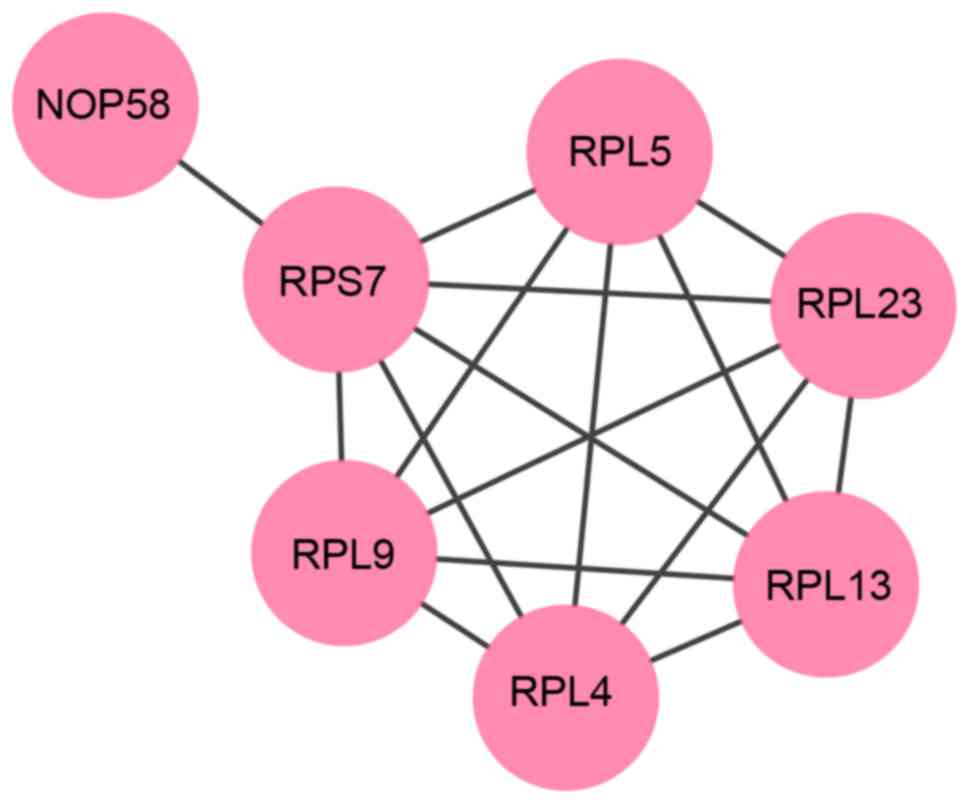
Functional subnetwork analysis
Following analysis using the ReactomeFI plugin, a
significantly functional subnetwork of the 45-min treatment group
was identified with an average correlation of 0.969. In this
subnetwork, seven nodes and 16 edges were included (Fig. 6). All of these nodes were
upregulated and significantly enriched in the ribosome pathway
(P=2.08E-11).
Discussion
According to the analytical criteria, a total of 181
DEGs were identified in the 15-min treatment group, compared with
the control group, which included 154 upregulated and 27
downregulated genes. A total of 468 DEGs were identified in the
45-min treatment group, compared with the control group, which
included 235 upregulated and 233 downregulated genes. Between these
two comparisons, the upregulated DEGs were significantly enriched
in ribosome related pathways. Based on the PPI network analysis,
PRP5, RCL1, NOP13, MRT4 and NOP4 were the top five nodes in the PPI
network of the 15-min treatment group. GIS2, URA3, NOP58, ELP3 and
PLP7 were the top five nodes in the PPI network of the 45-min
treatment group. NOP58 showed significant involvement in the
subnetwork, which was significantly enriched in the ribosome
pathway.
Based on the PPI network analysis, RCL1 and MRT4
were two hub nodes in the 15-min treatment group. RCL1 is an RNA
3′-terminal phosphate cyclase, which is similar to S.
cerevisiae RCL1 involved in rRNA processing (22). It has been demonstrated that RCL1
can interact with an important GTPase, Bms1, having an essential
role in the process of 18S rRNA biogenesis (23). In addition, Delprato et al
reported that the RCL1-Bms1complex is crucial in the pre-ribosomal
RNA processing of yeast (24). In
the present study, a significant upregulation was identified in
C. albicans following treatment with RF59 for 15 min.
Enjalbert et al demonstrated that RCL1 is a core stress
genes under oxidative, osmotic and heavy metal stress in C.
albicans (25). These results
indicate that the upregulation of RCL1 may be key in rRNA
biosynthetic processing. With this alteration, C. albicans
may be able to adjust to a physiological status enabling improved
response to stress from RF59. MRT4 was also identified as an
upregulated node involved in the processing of rRNA in the PPI
network of the 15-min treatment group. MRT4 is a non-essential
nucleolar protein, which is involved in the biogenesis of the 60S
r-subunit (26). Being homologous
to the N-terminal domain of P0, Rodríguez-Mateos et al
reported that MRT4 may have the same rRNA binding site as P0 in the
process of ribosome synthesis (27). Rodriguez-Mateos et al also
showed that MRT4 is a notable nucleolar component of the ribosome
assembly process and shares a binding domain of 25S rRNA with P0 in
S. cerevisiae (28). A
study by Kressler et al indicated that MRT4 may also
function in the assembly of the 60S stalk (29). Based on the above findings, MRT4
may be crucial in rRNA assembly under the restriction of RF59. With
the combined biofunctions of RCL1 and MRT4, the increased
biosynthesis of rRNA may be the initial response of C.
albicans to RF59, and this transformation may assist in
adapting to RF59 in the short-term exposure environment. The
results of the KEGG analysis also showed that the upregulated genes
were significantly enriched in the ribosomal and related
pathways.
According to the PPI and subnetwork analyses, GIS2
and NOP58 were identified as two hub nodes in the 45-min treatment
group. GIS, the DBA-binding zinc-finger protein (30), showed significant upregulation
following treatment with RF59 for 45 min. Sammons et al
reported that GIS2 can be upregulated by limiting glucose, and acts
as a translational activator for mRNA within the internal ribosome
entry site in S. cerevisiae (31). GIS2 has also been reported to be
associated with the initiation and degradation of protein
translation (32), and is reported
to be key in ribosome biogenesis and cell size control in yeast
(33). Taken together, it is
hypothesized that GIS2 maybe crucial in ribosome related pathways.
KEGG enrichment analysis for the subnetwork of the 45-min treatment
group also showed that the involved genes were significantly
enriched in the ribosome pathway. NOP58 was significantly
upregulated in the 45-min treatment group. As a box C/D snoRNA
binding protein, NOP58 and its homologue, NOP56, are required for
pre-rRNA splicing and processing (34). NOP58 is also involved in the
methylation of 18S rRNA (35). It
has been reported that the overexpression of NOP58 in S.
cerevisiae induces an increased resistance to CANBEF-13, which
is a member of a novel family of selective antifungal compounds
(35). These results indicate that
NOP58 is crucial for rRNA processing and assembly. KEGG analysis
for the subnetwork also revealed that the involved genes were
significantly enriched in the ribosome pathway. Due to its
sensitivity to RF-59, NOP58 maybe an important gene for
RF-59-resistance.
NOP4 was identified as a shared upregulated protein
in the two PPI networks. Sun and Woolford reported that NOP4 is a
crucial NOP for the accumulation of 60S ribosomal subunits and
pre-RNA processing (36). It has
also been reported that NOP4 has four essential RNA recognition
motifs, which are necessary for the biogenesis of ribosomes
(37). With a deficiency of NOP4,
the production of 27S pre-RNAs is normalized but degraded rapidly,
and this result may lead to a deficiency of mature 25S rRNA
synthesis in yeast (36,38). However, the detailed biofunctions
of NOP4 in C. albicans remain to be fully elucidated, and
further investigation of NOP4 in C. albicans is
required.
From the in silico analysis, it was
identified that RF59 can act as an antifungal drug to inhibit the
infection of C. albicans via affecting ribosomal and related
pathways, however, there were limitations to the present study.
Although a series of DEGs were found to be important in the drug
resistance of C. albicans, there remains a lack of
experimental verification of these. In addition, the sample size in
each group was three, and this maybe not sufficient to draw a valid
conclusion. Finally, as drug resistance was found to be the result
of long-term drug use, a longer treated duration may be
considered.
In conclusion, RF59 maybe a promising therapeutic
drug for candidiasis via acting on the ribosome and its associated
pathways in C. albicans. RCL1, NOP4, MRT4, GIS2 and NOP58
may be important during this procedure. It is essential to further
investigate the detailed biofunctions of these genes in C.
albicans in order to provide novel insights into the
pathogenesis of candidiasis and its therapy.
Acknowledgements
The present study was supported by the Research and
Development Key Projects of Shanxi Province (grant no.
201603D321063), the Basic Research Project supported by ShanXi
Province, China (grant no. 201701D121171), the Institutions of
Higher Learning Innovation Project of Shanxi Province, China (grant
no. 20161118), and the Research Project Supported by the Health and
Family Planning Commission of Shanxi Province, China (grant no.
201601050).
Glossary
Abbreviations
Abbreviations:
|
DEGs
|
differentially expressed genes
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
DAVID
|
Database for Annotation, Visualization
and Integrated Discovery
|
|
DC
|
degree centrality
|
|
BC
|
betweenness centrality
|
|
CC
|
closeness centrality
|
References
|
1
|
Lara HH, Romero-Urbina DG, Pierce C,
Lopez-Ribot JL, Arellano-Jiménez MJ and Jose-Yacaman M: Effect of
silver nanoparticles on Candida albicans biofilms: An
ultrastructural study. J Nanobiotechnology. 13:912015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yapar N: Epidemiology and risk factors for
invasive candidiasis. Ther Clin Risk Manag. 10:95–105. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kullberg BJ and Arendrup MC: Invasive
candidiasis. N Engl J Med. 373:1445–1456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Odds FC, Brown AJ and Gow NA: Antifungal
agents: Mechanisms of action. Trends Microbiol. 11:272–279. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sharma M, Biswas D, Kotwal A, Thakuria B,
Kakati B, Chauhan BS and Patras A: Ibuprofen-mediated reversal of
fluconazole resistance in clinical isolates of Candida. J Clin
Diagn Res. 9:DC20–DC22. 2015.PubMed/NCBI
|
|
6
|
Magnuson JG: Potential role of RTA3 and
GNP3 transport genes in the quorum sensing response of Candida
albicans. 2015.
|
|
7
|
Raska M, Běláková J, Krupka M and Weigl E:
Candidiasis-Do we need to fight or to tolerate the Candida fungus?
Folia Microbiol (Praha). 52:297–312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Soll DR: The role of phenotypic switching
in the basic biology and pathogenesis of Candida albicans. J Oral
Microbiol. 6:2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vandeputte P, Ischer F, Sanglard D and
Coste AT: In vivo systematic analysis of Candida albicans Zn2-Cys6
transcription factors mutants for mice organ colonization. PLoS
One. 6:e269622011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sanglard D, Coste A and Ferrari S:
Antifungal drug resistance mechanisms in fungal pathogens from the
perspective of transcriptional gene regulation. FEMS Yeast Res.
9:1029–1050. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bolstad BM, Irizarry RA, Astrand M and
Speed TP: A comparison of normalization methods for high density
oligonucleotide array data based on variance and bias.
Bioinformatics. 19:185–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Smyth GK: LIMMA: Linear models for
microarray dataBioinformatics and Computational Biology Solutions
Using R and Bioconductor. Gentleman R, Carey V, Huber W, Irizarry R
and Dudoit S: Springer-Verlag New York; New York, NY: pp. 397–420.
2005, View Article : Google Scholar
|
|
15
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Von Mering C, Huynen M, Jaeggi D, Schmidt
S, Bork P and Snel B: STRING: A database of predicted functional
associations between proteins. Nucleic Acids Res. 31:258–261. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang Y, Li M, Wang J, Pan Y and Wu FX:
CytoNCA: A cytoscape plugin for centrality analysis and evaluation
of protein interaction networks. Biosystems. 127:67–72. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He X and Zhang J: Why do hubs tend to be
essential in protein networks? PLoS Genet. 2:e882006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu G, Dawson E, Duong A, Haw R and Stein
L: ReactomeFIViz: A Cytoscape app for pathway and network-based
data analysis. Version 2. F1000Res. 3:1462014.PubMed/NCBI
|
|
22
|
Genschik P, Billy E, Swianiewicz M and
Filipowicz W: The human RNA 3′-terminal phosphate cyclase is a
member of a new family of proteins conserved in Eucarya, Bacteria
and Archaea. EMBO J. 16:2955–2967. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Horn DM, Mason SL and Karbstein K: Rcl1
protein, a novel nuclease for 18 S ribosomal RNA production. J Biol
Chem. 286:34082–34087. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Delprato A, Al Kadri Y, Pérébaskine N,
Monfoulet C, Henry Y, Henras AK and Fribourg S: Crucial role of the
Rcl1p-Bms1p interaction for yeast pre-ribosomal RNA processing.
Nucleic Acids Res. 42:10161–10172. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Enjalbert B, Smith DA, Cornell MJ, Alam I,
Nicholls S, Brown AJ and Quinn J: Role of the Hog1 stress-activated
protein kinase in the global transcriptional response to stress in
the fungal pathogen Candida albicans. Mol Biol Cell. 17:1018–1032.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huh WK, Falvo JV, Gerke LC, Carroll AS,
Howson RW, Weissman JS and O'Shea EK: Global analysis of protein
localization in budding yeast. Nature. 425:686–691. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rodríguez-Mateos M, Abia D, García-Gómez
JJ, Morreale A, de la Cruz J, Santos C, Remacha M and Ballesta JP:
The amino terminal domain from Mrt4 protein can functionally
replace the RNA binding domain of the ribosomal P0 protein. Nucleic
Acids Res. 37:3514–3521. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rodríguez-Mateos M, García-Gómez JJ,
Francisco-Velilla R, Remacha M, de la Cruz J and Ballesta JP: Role
and dynamics of the ribosomal protein P0 and its related
trans-acting factor Mrt4 during ribosome assembly in Saccharomyces
cerevisiae. Nucleic Acids Res. 37:7519–7532. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kressler D, Hurt E, Bergler H and Bassler
J: The power of AAA-ATPases on the road of pre-60S ribosome
maturation-molecular machines that strip pre-ribosomal particles.
Biochim Biophys Acta. 1823:92–100. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jang Y, Lee H, Lee SW, Choi YS, Ahn BJ,
Kim GH and Kim JJ: Cu(II)-induced molecular and physiological
responses in the brown-rot basidiomycete Polyporales sp. KUC9061. J
Appl Microbiol. 113:790–797. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sammons MA, Samir P and Link AJ:
Saccharomyces cerevisiae Gis2 interacts with the translation
machinery and is orthogonal to myotonic dystrophy type 2 protein
ZNF9. Biochem Biophys Res Commun. 406:13–19. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Matia-González AM and Gerber AP:
Approaches for dissecting RNA-binding protein networks. Fungal RNA
Biology. 347–370. 2014. View Article : Google Scholar
|
|
33
|
Scherrer T, Femmer C, Schiess R, Aebersold
R and Gerber AP: Defining potentially conserved RNA regulons of
homologous zinc-finger RNA-binding proteins. Genome Biol.
12:R32011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Anderson I, Göker M, Nolan M, Lucas S,
Hammon N, Deshpande S, Cheng JF, Tapia R, Han C, Goodwin L, et al:
Complete genome sequence of the hyperthermophilic
chemolithoautotroph Pyrolobus fumarii type strain (1A). Stand
Genomic Sci. 4:381–392. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mircus G, Albert N, Ben-Yaakov D,
Chikvashvili D, Shadkchan Y, Kontoyiannis DP and Osherov N:
Identification and characterization of a novel family of selective
antifungal compounds (CANBEFs) that interfere with fungal protein
synthesis. Antimicrob Agents Chemother. 59:5631–5640. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun C and Woolford JL Jr: The yeast NOP4
gene product is an essential nucleolar protein required for
pre-rRNA processing and accumulation of 60S ribosomal subunits.
EMBO J. 13:3127–3135. 1994.PubMed/NCBI
|
|
37
|
Sun C and Woolford JL Jr: The yeast
nucleolar protein Nop4p contains four RNA recognition motifs
necessary for ribosome biogenesis. J Biol Chem. 272:25345–25352.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Harnpicharnchai P, Jakovljevic J, Horsey
E, Miles T, Roman J, Rout M, Meagher D, Imai B, Guo Y, Brame CJ, et
al: Composition and functional characterization of yeast 66S
ribosome assembly intermediates. Mol Cell. 8:505–515. 2001.
View Article : Google Scholar : PubMed/NCBI
|