Introduction
Glioma is one of the most common malignant tumors
threatening human health and has increased in incidence (1,2).
Even with aggressive treatments, the average 5-year survival rate
remains poor (3,4). Detailed investigations have been
performed to understand the pathogenesis of glioma. However, the
underlying molecular network involved in the initiation and
progression of glioma remains to be fully elucidated.
CDGSH iron sulfur domain 2 (CISD2) belongs to the
CDGSH iron sulfur domain protein family (5). CISD2 contains a transmembrane domain,
a CDGSH domain and a conserved amino acid sequence for iron
binding. Located in the outer membrane of mitochondria, CISD2 is
important for mitochondrial integrity and lifespan (6–8).
CISD2 deficiency leads to mitochondrial damage, following which
autophagy is induced to eliminate the impaired mitochondria
(9). Compared with young mice, the
levels of CISD2 are significantly lower in older mice. In addition,
CISD2-knockout mice exhibit a significant premature aging
phenotype, characterized by opaque eyes, blindness, lordokyphosis,
osteopenia and skin atrophy (10).
CISD2 has been demonstrated to be important in tumor cells. CISD2
is reported to be elevated in human epithelial breast cancer cells,
and significantly promotes cell proliferation and tumor growth
(11). CISD2 has also been
identified as a novel marker correlating with metastasis and
prognosis in patients with early-stage cervical cancer (12). However, the role of CISD2 in glioma
remains to be elucidated.
Autophagy is a conserved process, which is
responsible for the turnover of long-life proteins or for the
removal of damaged organelles in eukaryotic cells (13). Autophagy is typically activated
under conditions of starvation, and autophagy marker proteins
include beclin-1, light chain 3 (LC3) and p62 (14–16).
p62 is a selective substrate of autophagy, and its accumulation is
observed when autophagy is inhibited (17). The role of autophagy in cancer
differs depending on the situation (18). Studies have shown that the
inhibition of autophagy promotes cancer initiation, however, others
have shown that it can also suppress the growth of certain
malignancies, including breast cancer and hepatocellular carcinoma
(19–22). Therefore, it is necessary to
investigate the involvement of autophagy in the function of CISD2
in glioma.
In the present study, the levels of CISD2 in glioma
tissues were evaluated, and the association between levels of CISD2
and the prognosis of patients with glioma was examined. In
addition, the role of CISD2 in the proliferation and carcinogenesis
of glioma cells was investigated in vitro and in
vivo. The downstream signaling pathway underlying the oncogenic
role of CISD2 in glioma cells was identified and the results
provided evidence that CISD2 was a direct target of miR-449a. Taken
together, the data suggested that CISD2 is important in the
proliferation of glioma and indicated that CISD2 may be a novel
therapeutic target for the treatment of glioma.
Materials and methods
Clinical sample collection
The present study was reviewed and approved by the
ethical review board of Xinxiang Medical University (Xinxiang,
China). All applicable international, national, and/or
institutional guidelines for the care and use of animals were
followed. All procedures performed involving human participants
were in accordance with the ethical standards of the institutional
and national research committee, and with the 1964 Helsinki
Declaration and its later amendments or comparable ethical
standards. The study was performed following the provision of
written informed consent from patients. A total of 72 fresh glioma
tissues and their adjacent non-glioma tissues were collected during
surgery between December 2014 and December, 2015. The levels of
CISD2 were evaluated in these paired tissues. In order to determine
the association between CISD2 and the prognosis of patients with
glioma, 120 paraffin-fixed glioma specimens were collected from
patients between January 2008 and January 2010, and these patients
were followed up 40 months later.
Cell culture
The U87 glioma cell line was purchased from the
Chinese Academy of Sciences (Shanghai, China) and was cultured in
DMEM (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% FBS (Thermo Fisher Scientific, Inc.). Please
note that the U87 cell line is known to be cross-contaminated with
another cell line, which is most likely to be a glioblastoma cell
line (23). The cells were
maintained in a humidified 37°C incubator containing 5%
CO2. For autophagy inhibition, the cells were treated
with 1 mM 3-MA (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for
24 h at 37°C.
Tumor graft
BALB/c nude male mice (6 weeks old) were purchased
from the Laboratory Animal Center of Xinxiang Medical University.
All mice were housed in a strictly pathogen-free conditions at room
temperature with free access to food and water and 12 h/12 h
light/dark cycle. The protocols for the experiments involving mice
complied with the Guide for the Care and Use of Laboratory Animals
(National Institutes of Health, Bethesda, MD, USA) and approved by
the Animal Ethics Committee of the Xinxiang Medical University.
Following being transfected with the small
interfering RNA to knock down CISD (si-CISD) or a scramble
construct, the U87 cells (5×107) were harvested and
subcutaneously inoculated into the right groin of the nude mice
(n=3 per group). Following growth for 40 days, the formed tumors
were carefully excised. The weight of the formed tumor was measured
and the volume was calculated using the following formula: Length ×
width2 × π/6.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
TRIzol reagent (Thermo Fisher Scientific, Inc.) was
used to extract RNA from samples. Equal quantities of RNA were then
reverse transcribed into cDNA with SuperScript® IV
Reverse Transcriptase (Thermo Fisher Scientific, Inc.). A total of
2 µg CISD2 cDNA was amplified under the following thermal
conditions: 95°C for 10 min; followed by 40 cycles of 95°C for 15
sec and 60°a for 1 min; and 4°C holding; the total duration was 1 h
48 min. GAPDH was used as an internal control. The final results
were calculated using the 2−ΔΔCq (24) method and presented as fold changes.
The following primers were used: CISD2, forward
5′-GCAAGGTAGCCAAGAAGTGC-3′ and reverse 5′-CCCAGTCCCTGAAAGCATTA-3′;
GAPDH, forward 5′-GCGAGATCGCACTCATCATCT-3′ and reverse
5′-TCAGTGGTGGACCTGACC-3.
Western blot analysis
Briefly, the tissues were lysed with
radioimmunoprecipitation assay lysis buffer containing inhibitor
cocktail for protease and phosphatase (Thermo Fisher Scientific,
Inc.). The concentration of protein sample was determined using the
bicinchoninic acid method. Following boiling for 15 min, 15-µg
protein samples were separated on a 10% SDS gel, blocked with 5%
nonfat milk and transferred onto a PVDF membrane (EMD Millipore,
Billerica, MA, USA). The membrane was incubated with primary
antibody overnight at 4°C. Following washing with TBST five times,
the membrane was incubated with secondary antibody for 2 h at 27°C.
The membrane was washed with TBST five times, following which the
membrane was visualized with Pierce™ Enhanced
Chemiluminescence Plus Western Blotting Substrate (Thermo Fisher
Scientific, Inc.). GAPDH was used as an internal control. The
protein band intensity was measured using Image J software (version
1.49; National Institutes of Health, Bethesda, MD, USA).
The primary antibodies used in the present study
were all purchased from Sigma-Aldrich (Merck KGaA) and were as
follows: Anti-CISD2 (1:1,000; cat no. AV44552), anti-LC3-II (1:800;
cat. no. ABC432), anti-beclin-1 (1:1,000; cat. no. SAB1306484),
anti-autophagy related 7 (Atg7) (1:1,000; cat. no. MABN1124),
anti-p62 (1:1,000; cat. no. MABC32) and anti-GAPDH (1:1,000; cat.
no. G9545). The secondary antibodies used were as follows:
Horseradish peroxidase (HRP)-labeled goat anti-rabbit
immunoglobulin (Ig)G (1:3,000; cat. no. A0208; Beyotime Institute
of Biotechnology, Haimen, China) and HRP-labeled goat anti-mouse
IgG (1:3,000; cat. no. A0216; Beyotime Institute of
Biotechnology).
Immunohistochemistry
Immunohistochemical staining was performed on the
collected clinical glioma tissues sections to detect the levels of
CISD2. Briefly, formalin and paraffin were used to fix and embed
the tissues, respectively. The samples were heated to retrieve the
antigen. The sections were incubated with anti-CISD2 primary
antibody (1:500; cat. no. AV44552; Sigma-Aldrich; Merck KGaA) at
4°C overnight. Following three washes with PBS, the sections with
incubated with HRP-labeled goat anti-rabbit IgG (1:1,000; cat. no.
A0516; Beyotime Institute of Biotechnology) for 2 h. Following
three washes with PBS, the sections were visualized with DBA
solution, followed by counterstaining of nuclei with hematoxylin.
Images were captured using a microscope (Eclipse Ci-E; Nikon,
Tokyo, Japan).
Co-immunoprecipitation assay
A Pierce Co-IP kit (Thermo Fisher Scientific, Inc.)
was used to examine the binding activity between CISD2 and beclin-1
in the indicated groups. The general procedure was performed
according to the manufacturer's protocol, as previously described
(25). The protein extracts were
precipitated using anti-CISD2 (Sigma-Aldrich; Merck KGaA), and the
precipitated protein was evaluated using western blot analysis with
anti-beclin-1 (Sigma-Aldrich; Merck KGaA).
Immunofluorescence
Immunofluorescence was also used to determine the
levels of CISD2 in the glioma tissue sections. Briefly, following
fixation with 4% paraformaldehyde, the sections were incubated with
anti-CISD2 primary antibody overnight at 4°C. Following three
washes with PBS, the sections were incubated with Cy3-labeled goat
anti-rabbit IgG (1:1,000; cat. no. A0516; Beyotime Institute of
Biotechnology) for 2 h at 27°C. Images were captured using a laser
confocal microscope (A1; Nikon Corporation, Tokyo, Japan).
Plasmid transfection
Briefly, when the cells reached a confluence of 70%,
the cells were transfected with negative scramble siRNA, a
CISD2-overexpression plasmid or an si-CISD2 and/or si-beclin-1
plasmid using Lipofectamine® 2000 transfection reagent
(Thermo Fisher Scientific, Inc.). After 6 h, the DMEM was replaced
with normal medium. All plasmids were purchased from GenePharma
Co., Ltd. (Shanghai, China).
Detection of proliferation rates
The U87 glioma cells were transfected with the
scramble or si-CISD2 plasmid and seeded into a 96-well plate at a
confluence of 30% (3×104 cells/well). At the indicated
time points, the number of cells in each well was determined using
a Scepter Handheld Automated Cell Counter (EMD Millipore).
TUNEL assay
A TUNEL assay was used to measure apoptosis.
Briefly, cells grown on a cover slip were fixed with 4%
paraformaldehyde. Following washing with PBS, the cells were
incubated with 0.3% H2O2 to block endogenous
peroxidase activity. The cells were then incubated with TUNEL
reaction solution (Sigma-Aldrich; Merck KGaA) for 1 h at 37°C.
Images were captured under a laser confocal microscope (A1;
Nikon).
Wound-healing assay
The U87 glioma cells were transfected with scramble
or si-CISD2 plasmid and seeded into a 6-well plate at a confluence
of 30% (3×104 cells/well). When cell confluence reached
95%, the cells were starved for 12 h. A 100-ml pipette tip was then
used to scratch a straight line in the cell layer. Following
incubation for another 24 h, the cells were fixed and images were
captured under a microscope (Eclipse Ci-E; Nikon Corporation). The
length of the wound was measured using Image J software.
Luciferase reporter assay
CISD2 wild-type 3-untranslated region (UTR) and
mutated 3′-UTR constructs were sub-cloned into the pGL3 Luciferase
Promote Vector (Sangon Biotech Co., Ltd., Shanghai, China) with
XbaI and NotI restriction sites. Using
Lipofectamine® 2000 transfection reagent (Thermo Fisher
Scientific, Inc.), the pGL3 vector containing the CISD2 wild-type
3′-UTR or mutated form was co-transfected with or without miR-449a
mimic (GenePharma Co., Ltd.) into the U87 cells. At 48 h
post-transfection, the luciferase activity was measured using a
Luciferase Reporter Assay kit (Sangon Biotech Co., Ltd.).
Statistical analysis
Data are expressed as the mean ± standard deviation
of at least three independent experiments. Comparisons between two
groups were analyzed using Student's t-test (two-tailed).
Comparisons among groups were analyzed using one-way analysis of
variance followed by Student-Newman-Keuls test. All analyses were
performed with SPSS 19.0 software (IBM SPSS, Armonk, NY, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
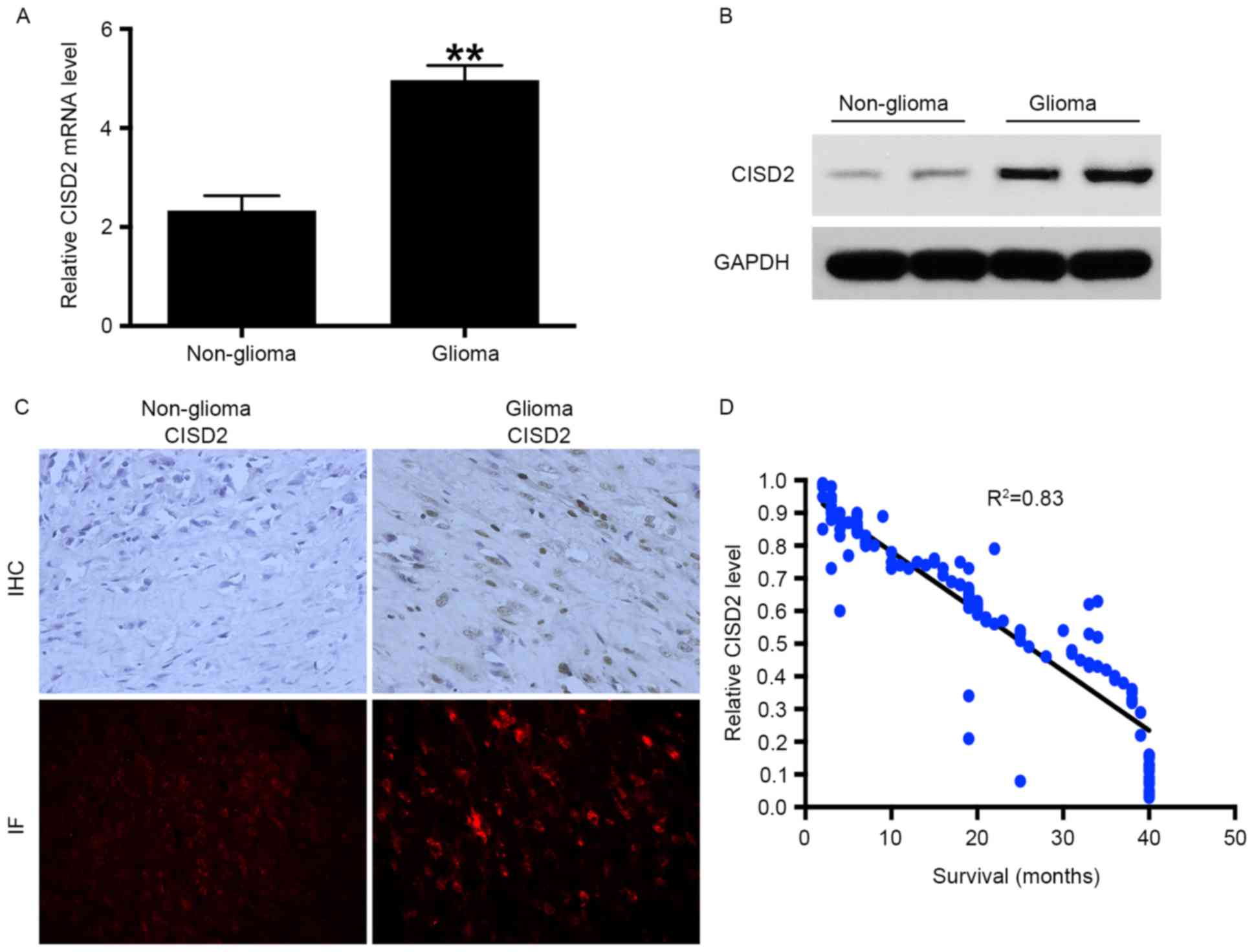
CISD2 is increased in glioma samples
and negatively correlated with survival rates of patients
RT-qPCR analysis was performed to measure the mRNA
levels of CISD2 in 72 fresh glioma tissue samples and corresponding
non-glioma tissue samples. The results showed that the mRNA level
of CISD2 was significantly increased in the glioma tissues,
compared with that in the non-glioma tissues (Fig. 1A). In addition, elevated expression
of CISD2 in glioma tissues at the protein level was confirmed using
western blot analysis (Fig. 1B),
immunohistochemistry and immunofluorescence (Fig. 1C). Taken together, the above data
demonstrated that the mRNA and protein levels of CISD2 were
markedly elevated in glioma tissues, compared with non-glioma
tissues. As CISD2 was significantly increased in the glioma
tissues, it was hypothesized that CISD2 may be a predictor of the
survival rates of patients with glioma. A total of 120
paraffin-fixed glioma specimens were collected between January 2008
and January 2010 and the corresponding survival rates of the
patients were recorded. The results showed that the level of CISD2
was negatively correlated with the survival rates of the patients
(Fig. 1D). The associations
between the level of CISD2 and clinicopathological characteristics
were also analyzed in these patients (Table I). A high level of CISD2 was
associated with advanced clinical stage (P<0.05), relapse
(P<0.05), vascular invasion (P<0.05) and increased tumor size
(P<0.05), but not differentiation (P>0.05).
 | Table I.Association between the expression of
CISD2 and clinicopathological parameters in 120 patients with
glioma. |
Table I.
Association between the expression of
CISD2 and clinicopathological parameters in 120 patients with
glioma.
|
|
| Expression of
CISD2 |
|
|---|
|
|
|
|
|
|---|
| Parameter | Total (n) | High (n) | Low (n) | P-value |
|---|
| Total | 120 | 83 | 37 |
|
| Age (years) |
|
|
| NS |
|
<50 | 53 | 36 | 17 |
|
|
<50 | 67 | 47 | 20 |
|
| Gender |
|
|
| NS |
|
Male | 56 | 38 | 18 |
|
|
Female | 64 | 45 | 19 |
|
| Clinical stage |
|
|
| <0.05 |
|
I–II | 33 | 11 | 22 |
|
|
III–IV | 87 | 72 | 15 |
|
| Relapse |
|
|
| <0.05 |
| No | 41 | 20 | 21 |
|
|
Yes | 79 | 63 | 16 |
|
| Vascular
invasion |
|
|
| <0.05 |
| No | 72 | 40 | 32 |
|
|
Yes | 48 | 43 | 5 |
|
|
Differentiation |
|
|
| >0.05 |
|
Well | 77 | 53 | 24 |
|
|
Moderate | 43 | 29 | 14 |
|
| Tumor size |
|
|
| <0.05 |
| <3
cm | 62 | 36 | 26 |
|
| >3
cm | 58 | 47 | 11 |
|
CISD2 promotes the proliferation and
survival of glioma cells
To examine the role of CISD2 in glioma cells, the
level of CISD2 was upregulated using a CISD2 expression construct
or knocked down using siRNA in U87 glioma cells. These effects were
validated using western blot analysis (Fig. 2A). Compared with the vector control
cells, the upregulation of CISD2 significantly increased the
proliferation rate of the U87 cells (Fig. 2B). The overexpression of CISD2 also
significantly increased the mean number of colonies in the colony
formation assay (Fig. 2C).
However, the knock down of CISD2 significantly reduced the
proliferation rate and number of colonies of glioma cells, as
indicated in the proliferation and colony formation assays,
compared with the control or CISD2 upregulation groups (Fig. 2B and C). In addition, the TUNEL
assay revealed that the inhibition of CISD2 markedly increased the
apoptosis of U87 cells, compared with the control or CISD2
overexpression groups (Fig. 2D).
The wound-healing assay showed that, compared with the control
group, the upregulation of CISD2 enhanced the invasive ability of
the U87 cells, whereas si-CISD2 significantly inhibited the
invasion of U87 cells (Fig. 2E).
Taken together, these results suggested that CISD2 promoted the
proliferation and survival of glioma cells.
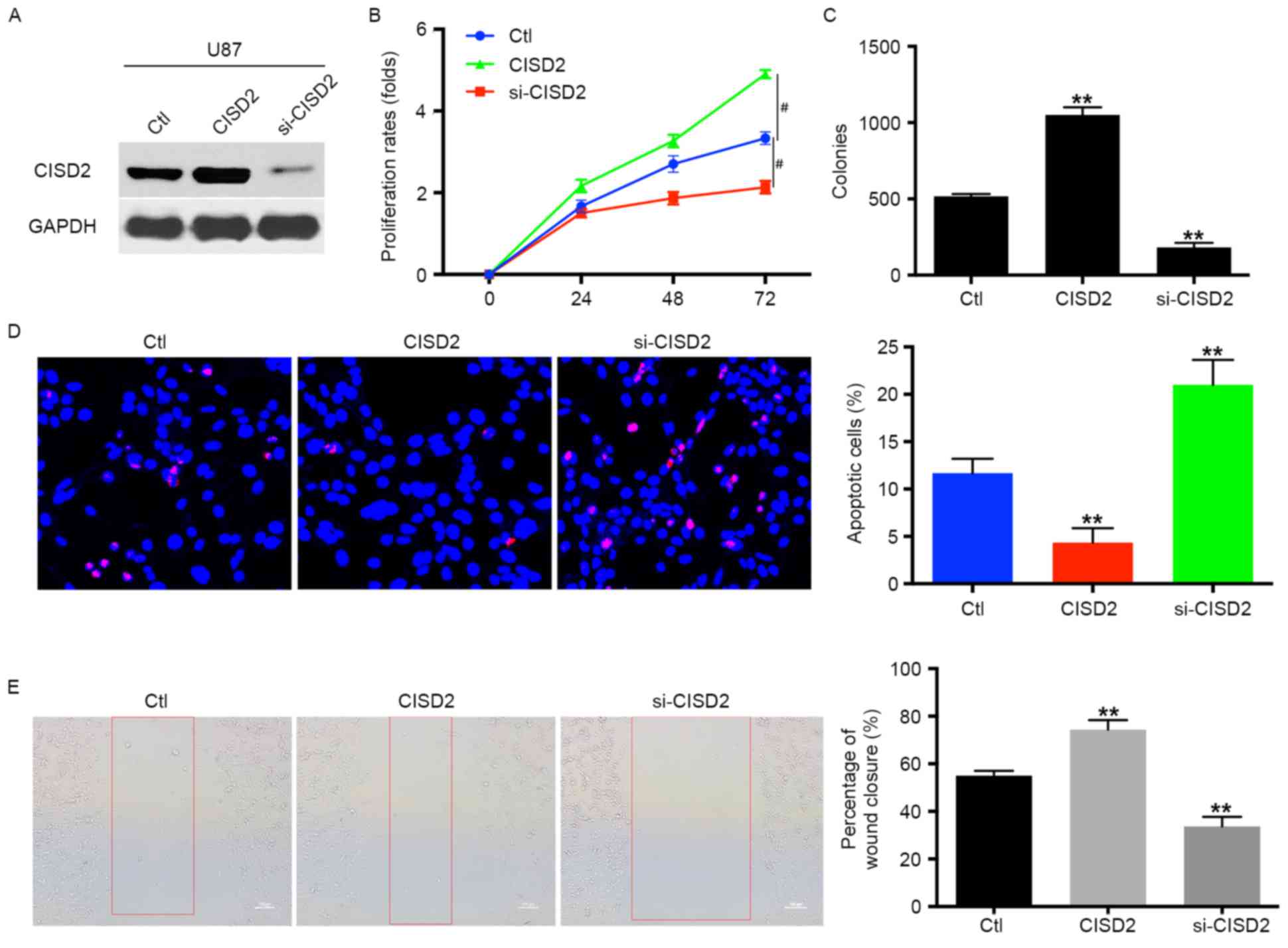 | Figure 2.CISD2 promotes proliferation and
survival of glioma cells. (A) In U87 cells, a CISD2 expression
construct was applied to upregulate CISD2, whereas siRNA was used
to knock down CISD2 (si-CISD2). The efficiency of modulating CISD2
was confirmed using western blot analysis. The effects of
modulating CISD2 on (B) proliferation rate and (C) colony formation
were evaluated. (D) A TUNEL assay was used to detect the apoptosis
in U87 cells. Compared with the control group, CISD2-overexpression
decreased apoptosis, whereas si-CISD2 increased apoptosis. (the
magnification is 200 times). (E) A wound healing assay showed that
the overexpression of CISD2 enhanced the invasive ability of U87
cells, whereas si-CISD2 weakened invasive ability, compared with
the control group (magnification, ×100). Data are presented as the
mean ± standard deviation from at least three independent
experiments. **P<0.01, compared with the Ctl group;
#P<0.05, compared with the indicated groups. CISD2,
CDGSH iron sulfur domain 2; si-CISD2, small interfering RNA
targeting CISD2; Ctl, control. |
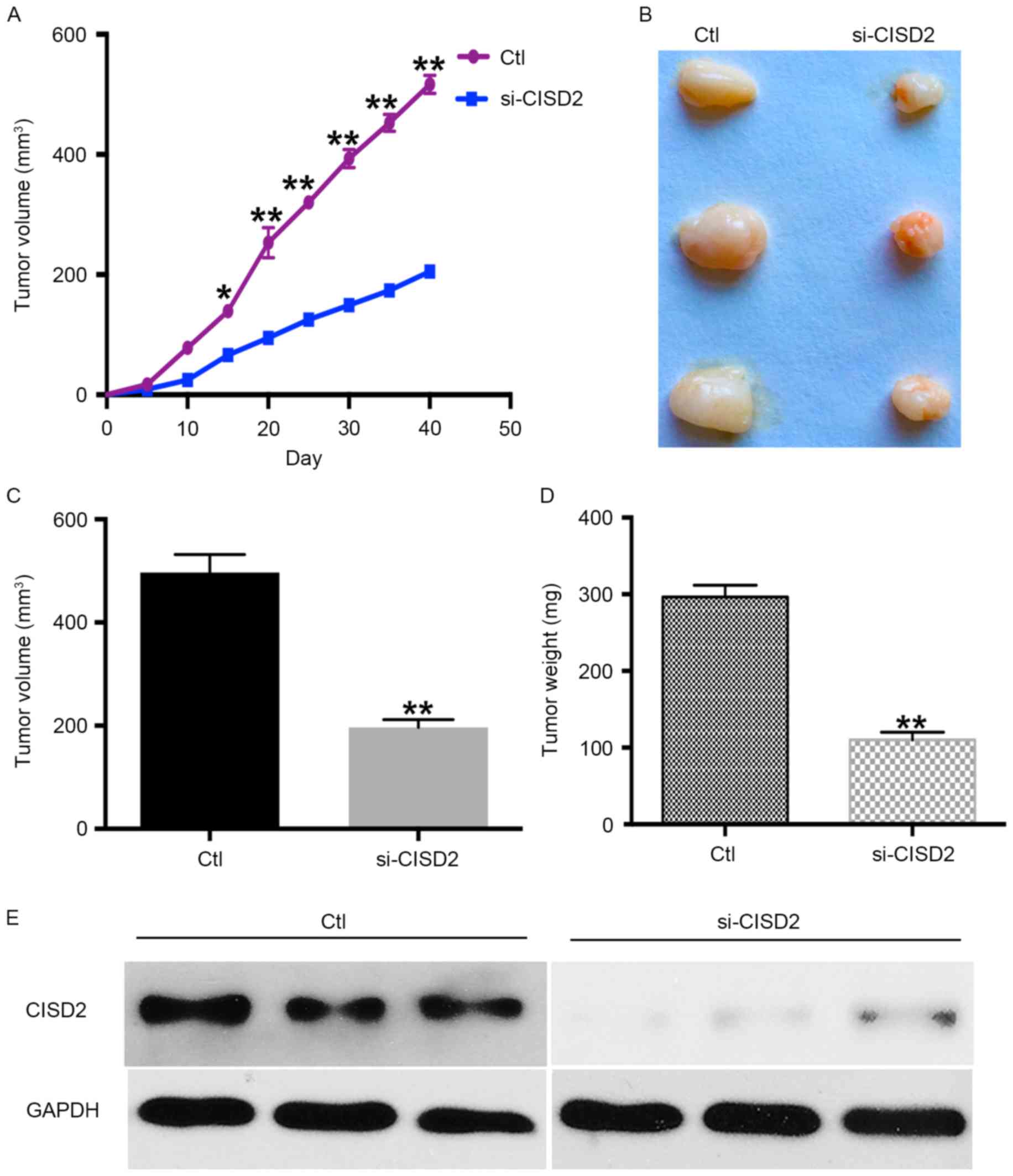
CISD2 silencing inhibits
carcinogenesis of glioma cells in a xenograft model
The present study also established a xenograft model
in BALB/C nude mice using U87 cells to determine the in vivo
effects of silencing CISD2. The results showed the silencing of
CISD2 led to a significantly slower growth rate, compared with that
in the control group (Fig. 3A). In
addition, the CISD2-deficit U87 cells formed smaller tumors,
compared with those in the control vector-transfected cells
(Fig. 3B). The average volume and
weight of tumors were significantly lower in the CISD2-deficit
group, compared with those in the control group (Fig. 3C and D). Western blot analysis
confirmed that CISD2 was effectively knocked down in the formed
tumors of the si-CISD2 group, compared with that in the control
group (Fig. 3E). Taken together,
these data demonstrated that silencing of CISD2 significantly
reduced the ability of glioma cells to form tumors in
vivo.
CISD2 silencing activates autophagy in
glioma cells
In order to examine the involvement of autophagy in
the tumor-inhibitory effects of si-CISD2 in glioma cells, CISD2 was
manipulated and the levels of autophagy activity markers were
evaluated using western blot analysis. The resulting data showed
that, compared with the control group, silencing of CISD2 led to
significant increases in the levels of LC3-II, beclin-1 and Atg7,
and a decrease in selective autophagy target p62; this was reversed
by the overexpression of CISD2 (Fig.
4A-E). The above data demonstrated that the silencing of CISD2
activated autophagy whereas the overexpression of CISD2 inhibited
autophagy in glioma cells.
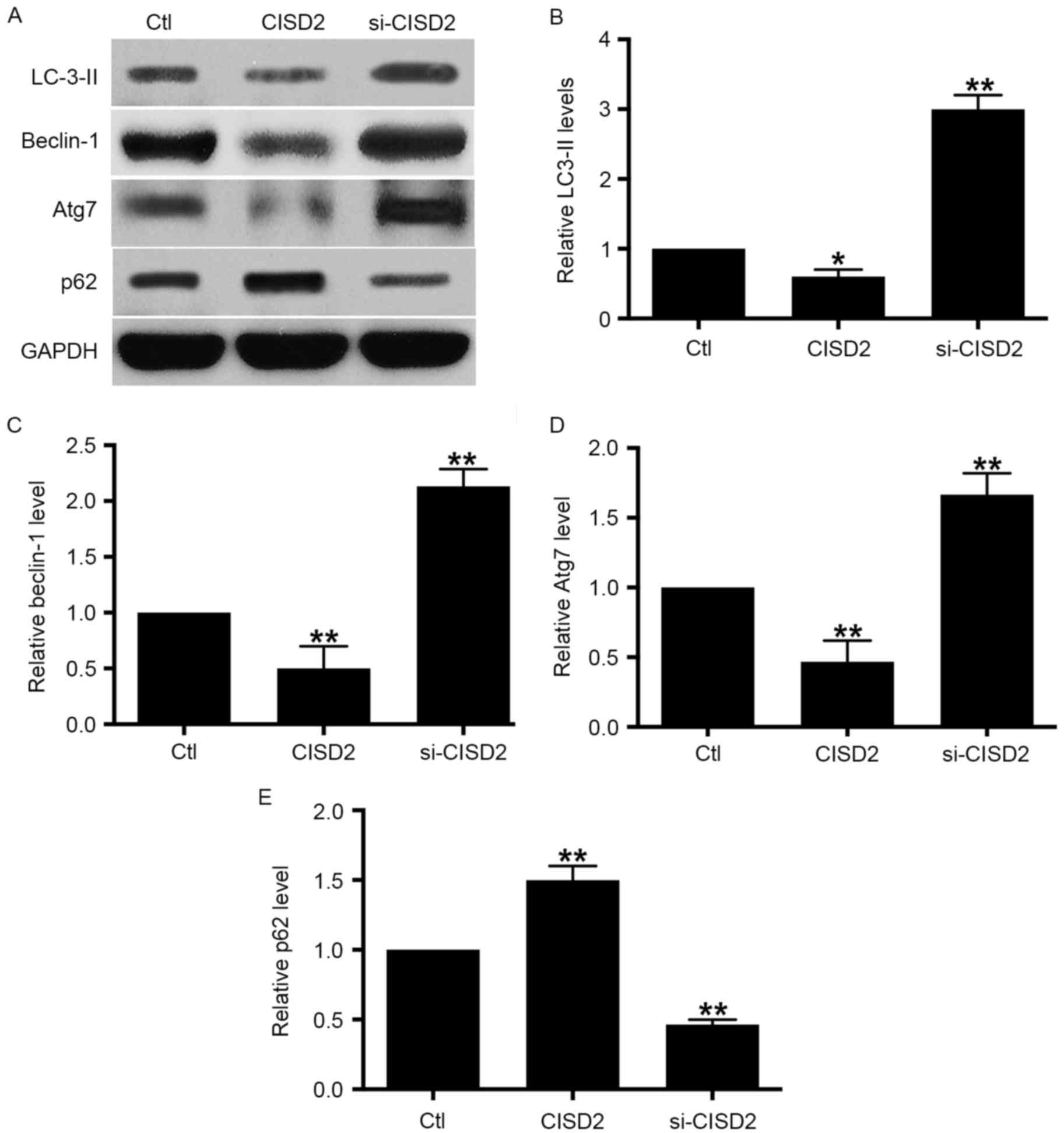 | Figure 4.Silencing CISD2 activates autophagy in
glioma cells. (A) In the U87 cells, the CISD2 expression construct
was used to upregulate CISD2 and siRNA was used to knock down
CISD2. Effects of manipulating CISD2 on autophagic markers LC3-II,
beclin-1, Atg7 and p62 were determined using western blot analysis.
Relative levels of (B) LC3-II, (C) beclin-1, (D) Atg7 and (E) p62
were measured using Image J software and normalized to GAPDH. Data
are presented as the mean ± standard deviation from at least three
independent experiments. *P<0.05 and **P<0.01, compared with
the Ctl group. CISD2, CDGSH iron sulfur domain 2; si-CISD2, small
interfering RNA targeting CISD2; Ctl, control; LC3-II, light chain
3 II; Atg7, autophagy related 7. |
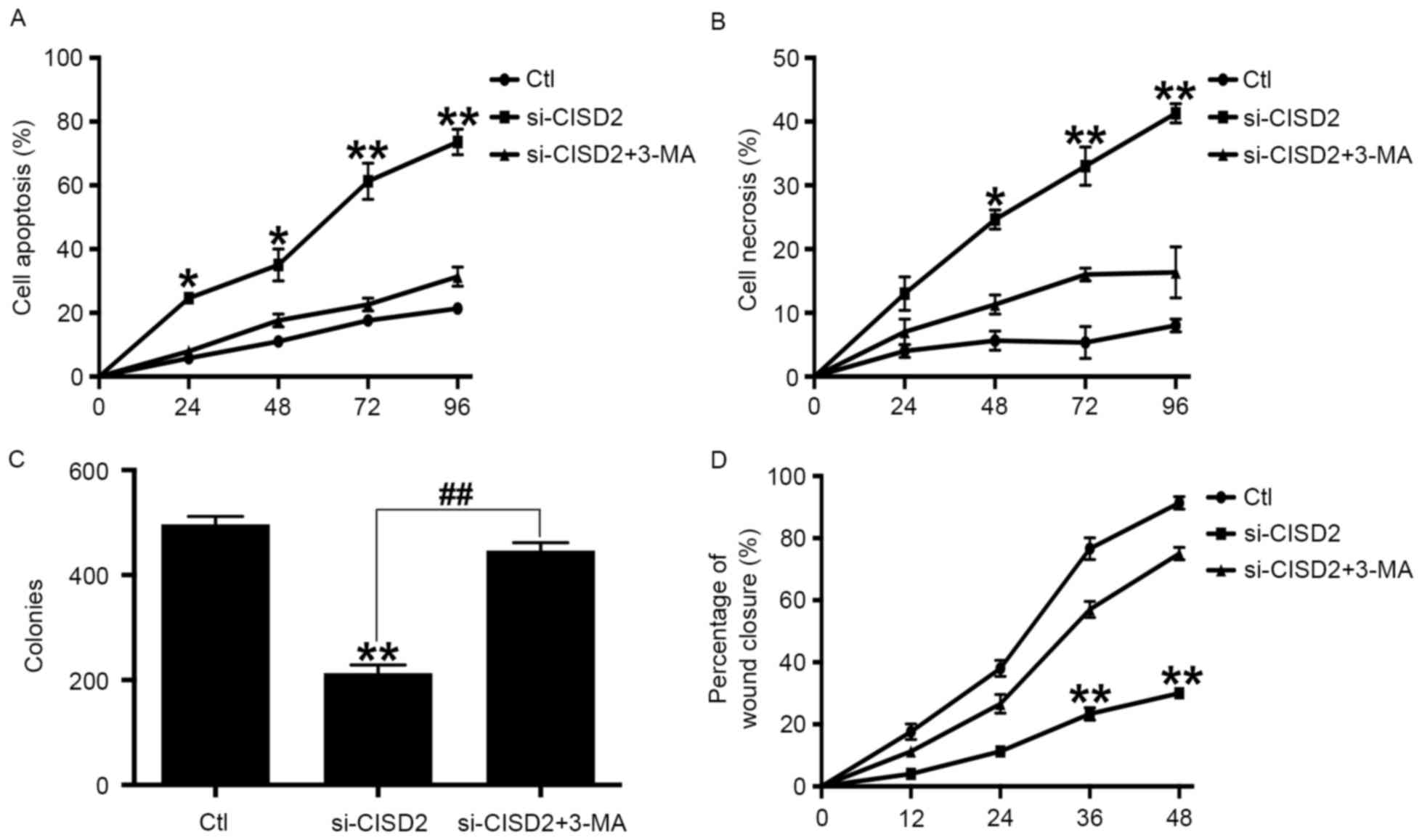
Inhibiting autophagy alleviates CISD2
silencing-induced glioma cell death
In order to confirm whether CISD2 silencing-induced
glioma cell death was dependent on autophagy, the specific
autophagy inhibitor, 3-MA, was used. Compared with the control
group, si-CISD2 led to significant increases in cell apoptosis
(Fig. 5A) and necrosis (Fig. 5B), however, these effects were
eliminated by 3-MA. Consistently, the colony formation assay showed
that si-CISD2 markedly reduced the mean number of colonies,
compared with that in the control group, which was reversed by 3-MA
(Fig. 5C). Finally, the
wound-healing assay revealed that si-CISD significantly attenuated
the invasive ability of U87 cells, which was also abrogated by 3-MA
(Fig. 5D). Taken together, these
data suggested that inhibiting autophagy significantly alleviated
the CISD2 silencing-induced suppression of glioma cell
proliferation and survival.
Activation of CISD2 silencing-induced
autophagy is mediated by beclin-1
Previous studies have shown that beclin-1 is vital
in the activation of autophagy (26). Therefore, the present study
hypothesized that the enhanced autophagy by CISD2 silencing is
dependent on beclin-1. The Co-IP assay showed that endogenous CISD2
was able to bind with beclin-1 (Fig.
6A). In addition, U87 cells with si-CISD2 and si-beclin-1
vectors were co-transfected, and the activity of autophagy activity
was evaluated using western blot analysis. The results of the
western blot analysis confirmed that CISD2 and beclin-1 were
effectively knocked down (Fig.
6B-D). The data revealed that the downregulation of beclin-1
significantly eliminated the si-CISD2-induced increase in LC3-II
and Atg7, and decrease in p62 in the U87 cells (Fig. 6E-G). Taken together, these data
demonstrated that the CISD2 silencing-induced activation of
autophagy was mediated by beclin-1.
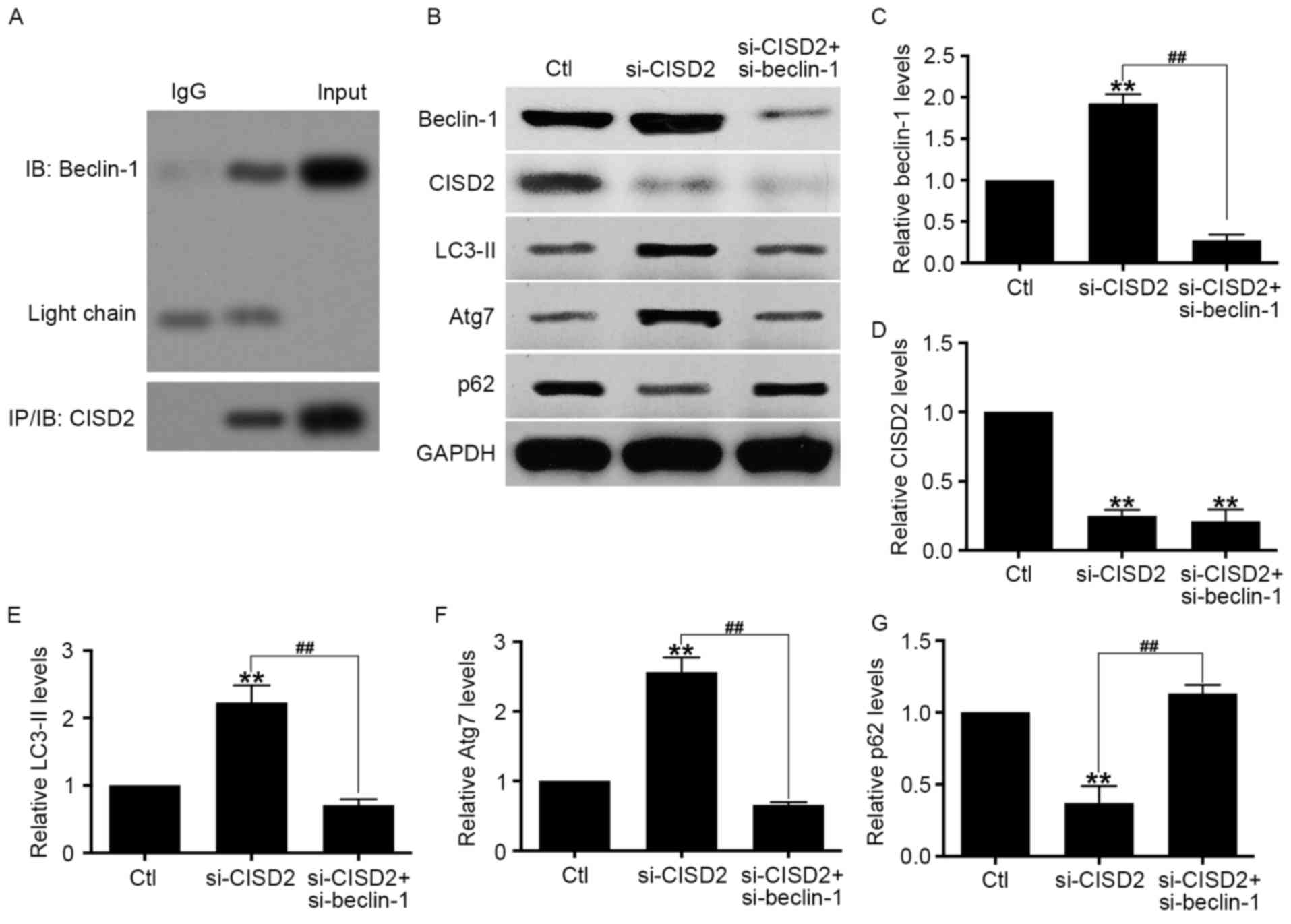 | Figure 6.CISD2 silencing-induced activation of
autophagy is mediated by beclin-1. (A) Co-immunoprecipitation assay
showed the interaction between endogenous CISD2 and beclin-1. (B)
U87 cells were co-transfected with si-CISD2 and si-beclin-1
plasmids, and the activity of autophagy was determine using western
blot analysis. Relative levels of (C) beclin-1, (D) CISD2, (E)
LC3-II, (F) Atg7 and (G) p62 were measured using Image J software
and normalized to GAPDH. Data are presented as the mean ± standard
deviation from at least three independent experiments. **P<0.01,
compared with Ctl group; ##P<0.01, compared with the
indicated groups. CISD2, CDGSH iron sulfur domain 2; si-CISD2,
small interfering RNA targeting CISD2; LC3-II, light chain 3 II;
Atg7, autophagy related 7; Ctl, control. |
CISD2 is a direct target of
miRNA-449a
Using TargetScan, the present study found that CISD2
was one of the potential targets of miR-449a. The predicted binding
site of miR-449a with the CISD2 3′-UTR is shown in Fig. 7A. To confirm the interaction
between miR-449a and CISD2, the CISD2 complementary sites, with or
without mutations, were cloned into the 3′-UTR of the firefly
luciferase gene and co-transfected with miR-449a mimics into U87
cells. The results revealed that the presence of miR-449a led to a
significant reduction in the relative luciferase activity of the
wild-type construct of the CISD2 3′-UTR in U87 cells. However, the
mutant construct of the CISD2 3′-UTR eliminated the suppressive
effect of miR-449a in the U87 cells (Fig. 7B). The above results demonstrated
that CISD2 is a direct target of miR-449a.
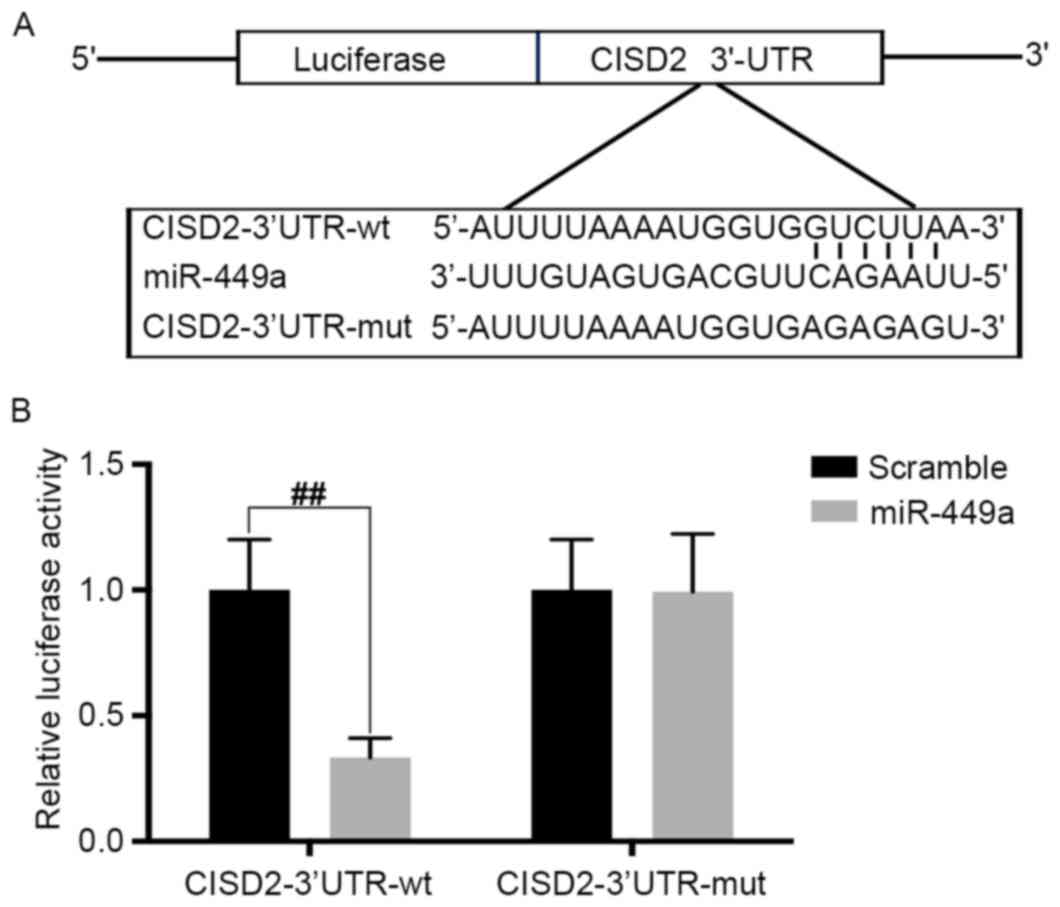 | Figure 7.CISD2 is a direct target of miR-449a.
(A) Predicted binding site of miR-449a with CISD2 3′-UTR. (B) pGL3
vector containing CISD2 wild-type 3′-UTR or mutated form was
co-transfected with or without miR-449a mimic into U87 cells. At 48
h post-transfection, the luciferase activity was measured. Data are
presented as the mean ± standard deviation from at least three
independent experiments. ##P<0.01, compared with the
indicated groups. CISD2, CDGSH iron sulfur domain 2; si-CISD2,
small interfering RNA targeting CISD2; LC3-II, light chain 3 II;
Atg7, autophagy related 7; 3′-UTR, 3′-untranslated region; miR,
microRNA; wt, wild-type; mut, mutant. |
Discussion
The present study provided the first evidence, to
the best of our knowledge, that the mRNA and protein levels of
CISD2 were upregulated in glioma tissues, compared with matched
normal tissues. The inhibition of CISD2 via siRNA suppressed the
proliferation and survival of glioma cells. Furthermore, the
results demonstrated that the silencing of CISD2 inhibited the
carcinogenesis of glioma cells in a xenograft model.
Mechanistically, it was found that si-CISD2 significantly increased
the activity of autophagy, whereas upregulated CISD2 markedly
suppressed autophagy. Inhibiting autophagy with 3-MA significantly
alleviated the CISD2 silencing-induced suppression of proliferation
and survival of glioma cells. It was also found that endogenous
CISD2 was able to bind with beclin-1. The downregulation of
beclin-1 significantly eliminated the si-CISD2-induced increase of
LC3-II and Atg7, and decrease of p62 in the U87 cells. This
suggested that the activation of autophagy induced by silencing
CISD2 was mediated by beclin-1. The results also revealed that
CISD2 is a target of miR-449a.
Increasing evidence indicates that CISD2 is closely
associated with multiple tumors. CISD2 is increased in human
epithelial breast cancer cells, and suppressing CISD2 significantly
inhibits tumor growth (11). CISD2
is upregulated and associated with poor prognosis in early stage
cervical cancer (12), and CISD2
promotes the tumorigenesis and proliferation of gastric cancer cell
through activating the AKT pathway (27). In hepatocellular carcinoma, the
downregulation of CISD2 suppresses the proliferation of hepatoma
cells (28). However, the role of
CISD2 in glioma remains to be fully elucidated. To the best of our
knowledge, the present study provided the first evidence that the
mRNA and protein levels of CISD2 were upregulated in glioma
tissues, compared with paired normal tissues. It was also
demonstrated that si-CISD2 inhibited the proliferation, survival
and invasion of glioma cells. These results suggested that CISD2
acts as an oncogene in glioma and that CISD2 may be a promising
therapeutic target for glioma treatment.
The molecular mechanisms underlying the oncogenic
role of CISD2 in tumors has been investigated previously. In breast
cancer, CISD2 eliminates reactive oxygen and protects mitochondrial
function (11). In gastric cancer,
CISD2 activates the AKT signaling pathway to promote tumorigenesis
(27). However, there remains no
evidence demonstrating whether or not autophagy is involved in the
tumorigenic role of CISD2. In the present study, it was found that
si-CISD2 significantly increased the activity of autophagy, whereas
upregulated levels of CISD2 markedly suppressed autophagy.
Inhibiting autophagy with 3-MA significantly alleviated the CISD2
silencing-induced suppression of proliferation and survival of
glioma cells. Therefore, the present study confirmed that CISD2
promoted tumorigenesis through inhibiting autophagic activity.
However, whether or not the CISD2/autophagy pathway works in other
tumors requires further investigation.
Studies have shown that beclin-1 is a mediator for
the activation of autophagy in multiple conditions. In cerebral
ischemic injury, beclin-1-mediated autophagy can alleviate neuronal
damage (29). In addition,
beclin-1-mediated autophagy contributes to the radiosensitivity of
in pancreatic cancer cells (30).
Beclin-1/autophagy is closely associated with prognosis in patients
with non-metastatic renal cell carcinoma (31). Consistently, the present study
showed that the downregulation of beclin-1 significantly eliminated
the si-CISD2-induced increase of LC3-II and Atg7, and decrease of
p62 in the U87 cells, suggesting that the activation of autophagy
induced by CISD2 silencing was mediated by beclin-1. However,
studies have shown that cyclin-dependent kinase 5 and heme
oxygenase-1 are also mediators for the activation of autophagy
(32–34). Consequently, it is necessary to
investigate whether other mediators are involved in the
si-CISD2-induced activation of autophagy.
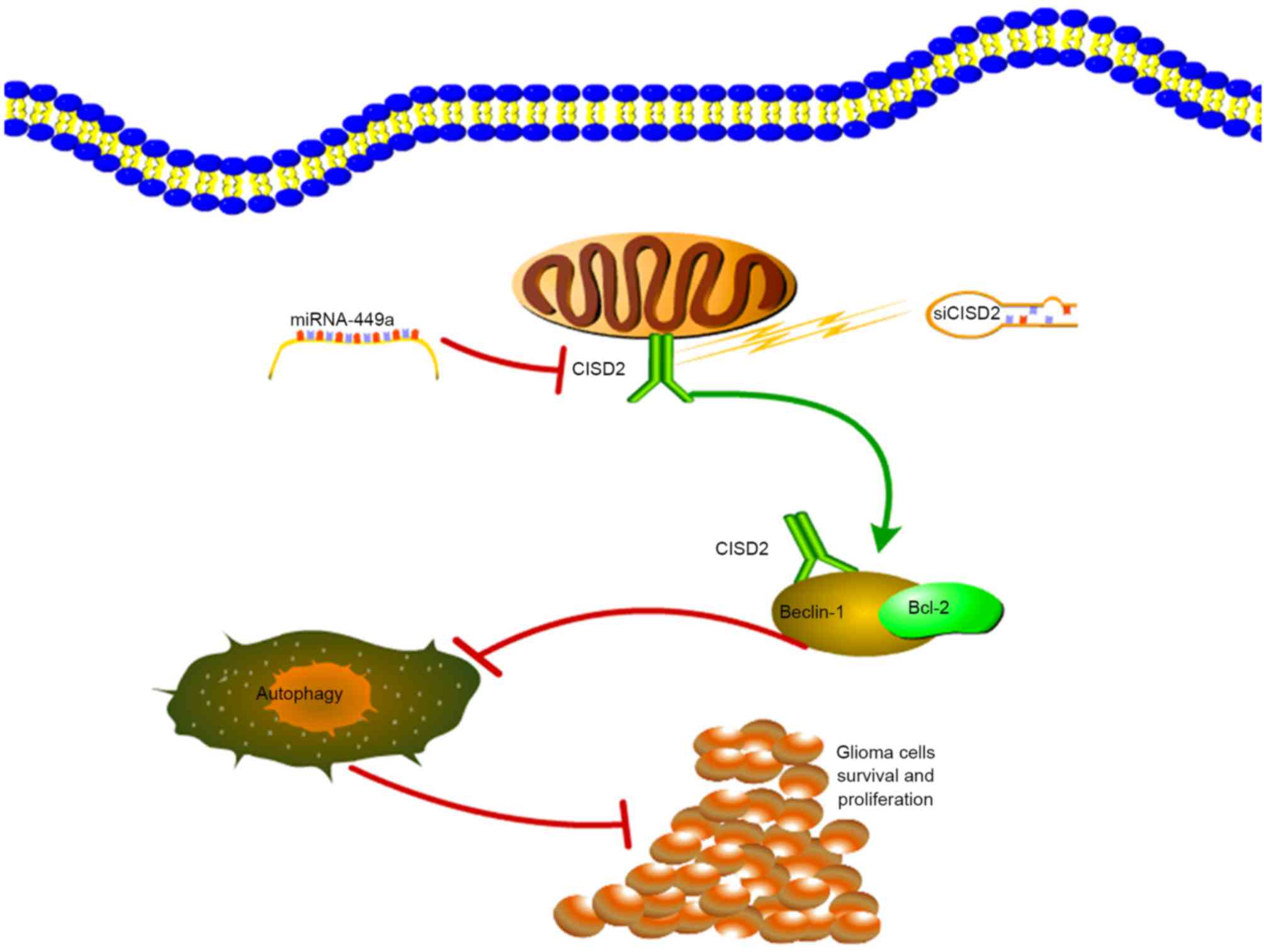
In conclusion, the present study demonstrated that
CISD2 was increased in glioma samples, and was associated with poor
prognosis and aggressive tumor behavior. The
miR-449a/CISD2/beclin-1-mediated autophagy regulatory network
contributed to the proliferation of glioma cells (Fig. 8). Therefore, targeting this pathway
may be a promising strategy for glioma therapy.
Acknowledgements
The authors would like to thank Dr Haipeng Xu from
Fudan University (Fudan, China) for proofreading of the
manuscript.
References
|
1
|
Chen Y, Wu Y, Huang X, Qu P, Li G, Jin T,
Xing J and He S: Leukocyte telomere length: A novel biomarker to
predict the prognosis of glioma patients. J Cancer Res Clin Oncol.
141:1739–1747. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wachsberger PR, Lawrence YR, Liu Y, Rice
B, Feo N, Leiby B and Dicker AP: Hsp90 inhibition enhances PI-3
kinase inhibition and radiosensitivity in glioblastoma. J Cancer
Res Clin Oncol. 140:573–582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gulati S, Jakola AS, Johannesen TB and
Solheim O: Survival and treatment patterns of glioblastoma in the
elderly: A population-based study. World Neurosurg. 78:518–526.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fukushima T, Kawaguchi M, Yorita K, Tanaka
H, Takeshima H, Umezawa K and Kataoka H: Antitumor effect of
dehydroxymethylepoxyquinomicin, a small molecule inhibitor of
nuclear factor-κB, on glioblastoma. Neuro Oncol. 14:19–28. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin J, Zhang L, Lai S and Ye K: Structure
and molecular evolution of CDGSH iron-sulfur domains. PLoS One.
6:e247902011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu CY, Chen YF, Wang CH, Kao CH, Zhuang
HW, Chen CC, Chen LK, Kirby R, Wei YH, Tsai SF and Tsai TF: A
persistent level of Cisd2 extends healthy lifespan and delays aging
in mice. Hum Mol Genet. 21:3956–3968. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang CH, Kao CH, Chen YF, Wei YH and Tsai
TF: Cisd2 mediates lifespan: Is there an interconnection among
Ca2+ homeostasis, autophagy, and lifespan? Free Radic
Res. 48:1–1114. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsai PH, Chien Y, Chuang JH, Chou SJ,
Chien CH, Lai YH, Li HY, Ko YL, Chang YL, Wang CY, et al:
Dysregulation of mitochondrial functions and osteogenic
differentiation in cisd2-deficient murine induced pluripotent stem
cells. Stem Cells Dev. 24:2561–2576. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chang NC, Nguyen M, Bourdon J, Risse PA,
Martin J, Danialou G, Rizzuto R, Petrof BJ and Shore GC:
Bcl-2-associated autophagy regulator Naf-1 required for maintenance
of skeletal muscle. Hum Mol Genet. 21:2277–2287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen YF, Kao CH, Chen YT, Wang CH, Wu CY,
Tsai CY, Liu FC, Yang CW, Wei YH, Hsu MT, et al: Cisd2 deficiency
drives premature aging and causes mitochondria-mediated defects in
mice. Genes Dev. 23:1183–1194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sohn YS, Tamir S, Song L, Michaeli D,
Matouk I, Conlan AR, Harir Y, Holt SH, Shulaev V, Paddock ML, et
al: NAF-1 and mitoNEET are central to human breast cancer
proliferation by maintaining mitochondrial homeostasis and
promoting tumor growth. Proc Natl Acad Sci USA. 110:pp.
14676–14681. 2013; View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu L, Xia M, Wang J, Zhang W, Zhang Y and
He M: CISD2 expression is a novel marker correlating with pelvic
lymph node metastasis and prognosis in patients with early-stage
cervical cancer. Med Oncol. 31:1832014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang B, Cai Z, Tao K, Zeng W, Lu F, Yang
R, Feng D, Gao G and Yang Q: Essential control of mitochondrial
morphology and function by chaperone-mediated autophagy through
degradation of PARK7. Autophagy. 12:1215–1228. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang JD, Cao YL, Li Q, Yang YP, Jin M,
Chen D, Wang F, Wang GH, Qin ZH, Hu LF and Liu CF: A pivotal role
of FOS-mediated BECN1/Beclin 1 upregulation in dopamine D2 and D3
receptor agonist-induced autophagy activation. Autophagy.
11:2057–2073. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choi J, Jung W and Koo JS: Expression of
autophagy-related markers beclin-1, light chain 3A, light chain 3B
and p62 according to the molecular subtype of breast cancer.
Histopathology. 62:275–286. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Komatsu M and Ichimura Y: Physiological
significance of selective degradation of p62 by autophagy. FEBS
Lett. 584:1374–1378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
He WS, Dai XF, Jin M, Liu CW and Rent JH:
Hypoxia-induced autophagy confers resistance of breast cancer cells
to ionizing radiation. Oncol Res. 20:251–258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mohapatra P, Preet R, Das D, Satapathy SR,
Choudhuri T, Wyatt MD and Kundu CN: Quinacrine-mediated autophagy
and apoptosis in colon cancer cells is through a p53- and
p21-dependent mechanism. Oncol Res. 20:81–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sasazawa Y, Sato N, Umezawa K and Simizu
S: Conophylline protects cells in cellular models of
neurodegenerative diseases by inducing mammalian target of
rapamycin (mTOR)-independent autophagy. J Biol Chem. 290:6168–6178.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brassesco MS, Pezuk JA, Morales AG, de
Oliveira JC, Valera ET, da Silva GN, de Oliveira HF, Scrideli CA,
Umezawa K and Tone LG: Cytostatic in vitro effects of
DTCM-glutarimide on bladder carcinoma cells. Asian Pac J Cancer
Prev. 13:1957–1962. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Allen M, Bjerke M, Edlund H, Nelander S
and Westermark B: Origin of the U87MG glioma cell line: Good news
and bad news. Sci Transl Med. 8:354re32016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yuan J, Zhang Y, Sheng Y, Fu X, Cheng H
and Zhou R: MYBL2 guides autophagy suppressor VDAC2 in the
developing ovary to inhibit autophagy through a complex of
VDAC2-BECN1-BCL2L1 in mammals. Autophagy. 11:1081–1098. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carchman EH, Rao J, Loughran PA, Rosengart
MR and Zuckerbraun BS: Heme oxygenase-1-mediated autophagy protects
against hepatocyte cell death and hepatic injury from
infection/sepsis in mice. Hepatology. 53:2053–2062. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang L, Ouyang F, Liu X, Wu S, Wu HM, Xu
Y, Wang B, Zhu J, Xu X and Zhang L: Overexpressed CISD2 has
prognostic value in human gastric cancer and promotes gastric
cancer cell proliferation and tumorigenesis via AKT signaling
pathway. Oncotarget. 7:3791–3805. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen B, Shen S, Wu J, Hua Y, Kuang M, Li S
and Peng B: CISD2 associated with proliferation indicates negative
prognosis in patients with hepatocellular carcinoma. Int J Clin Exp
Pathol. 8:13725–13738. 2015.PubMed/NCBI
|
|
29
|
Wang P, Liang J, Li Y and Li J, Yang X,
Zhang X, Han S, Li S and Li J: Down-regulation of miRNA-30a
alleviates cerebral ischemic injury through enhancing beclin
1-mediated autophagy. Neurochem Res. 39:1279–1291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang X, Shi H, Lin S, Ba M and Cui S:
MicroRNA-216a enhances the radiosensitivity of pancreatic cancer
cells by inhibiting beclin-1-mediated autophagy. Oncol Rep.
34:1557–1564. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nishikawa M, Miyake H, Liu B and Fujisawa
M: Expression pattern of autophagy-related markers in
non-metastatic clear cell renal cell carcinoma: Association with
disease recurrence following radical nephrectomy. J Cancer Res Clin
Oncol. 141:1585–1591. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wong AS, Lee RH, Cheung AY, Yeung PK,
Chung SK, Cheung ZH and Ip NY: Cdk5-mediated phosphorylation of
endophilin B1 is required for induced autophagy in models of
Parkinson's disease. Nat Cell Biol. 13:568–579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wen Z, Shu Y, Gao C, Wang X, Qi G, Zhang
P, Li M, Shi J and Tian B: CDK5-mediated phosphorylation and
autophagy of RKIP regulate neuronal death in Parkinson's disease.
Neurobiol Aging. 35:2870–2880. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Y, Shen J, Xiong X, Xu Y, Zhang H,
Huang C, Tian Y, Jiao C, Wang X and Li X: Remote ischemic
preconditioning protects against liver ischemia-reperfusion injury
via heme oxygenase-1-induced autophagy. PLoS One. 9:e988342014.
View Article : Google Scholar : PubMed/NCBI
|