Introduction
Diabetes is one of the greatest global health
emergencies of the 21st century. In 2015, 415 million adults had
diabetes, and this number is estimated to increase to 642 million
in 2040 (1). Among the
microvascular complications of the diabetes mellitus, diabetic
kidney disease (DKD) is a leading cause of end-stage renal disease
(2). However, the pathogenesis of
DKD remains unclear, and no effective treatments for this disease
are available. Thus, there is an urgent need to elucidate the
pathogenic mechanisms underlying DKD and to develop more effective
therapies for this disease.
Endothelial cells (ECs) serve critical roles in many
physiological functions, including cell proliferation and survival,
vascular tone modulation, blood cell trafficking, haemostatic
balance, permeability status and innate and adaptive immunity. EC
injury is a major event in diabetes and results in multiple macro-
and microvascular complications (3). Microvascular complications involve
small vessels, such as capillaries, whereas macrovascular
complications primarily involve large vessels, such as arteries and
veins. Nephropathy has been recognized as a common microvascular
complication of diabetes, and the underlying EC dysfunction is
often underestimated in cases of DKD (4).
Given the broad function of renal ECs and the
complicated endothelial pathophysiology of DKD, inflammation
biomarkers [interleukin (IL)-1β, IL-6 and tumor necrosis factor
(TNF)-α] are important tools in EC research (4). The microinflammatory state plays an
important role in DKD development and progression (5,6).
Adhesion molecules, such as intercellular adhesion molecule-1
(ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), have also
been reported to be upregulated in DKD (7,8), and
these factors promote inflammatory cell adhesion to the endothelium
and recruit circulating immune cells to diabetic kidneys.
Calcium dobesilate (CaD, calcium
2,5-dihydroxybenzenesulfonate) is considered an angioprotective
drug that can reduce blood viscosity, platelet activity and
capillary permeability, as well as alleviate microcirculatory and
hemorheologic abnormalities (9).
CaD is widely used to treat diabetic retinopathy (10), chronic venous insufficiency
(11) and various
microangiopathies (12). Moreover,
recent studies have demonstrated that CaD exerts protective effects
against diabetic nephropathy (13)
and gentamicin-induced acute kidney injury (14). Despite its broad use, very little
attention has been devoted to the molecular and cellular mechanisms
underlying the vasculoprotective effects of CaD.
Thus, the aim of the present study was to elucidate
the molecular and cellular mechanisms underlying the protective
effects of CaD against diabetes-induced endothelial dysfunction and
inflammation.
Materials and methods
Human umbilical vein endothelial cell
(HUVEC) culture
Primary HUVECs and EC growth medium were purchased
from ScienCell Research Laboratories (Carlsbad, CA, USA). Culture
flasks were coated with poly-l-lysine before use, and the cells
were cultured in EC supplemented with EC growth supplement, 5% FBS
and penicillin/streptomycin solution at 37°C in 5% CO2.
HUVECS in the 2nd to 5th passages were used in all experiments.
Cell migration
A Transwell migration apparatus (24 wells, 6.5 mm
internal diameter, 8 µm pore size; Corning Incorporated, Corning,
NY, USA) was used to measure HUVEC migration. HUVECs
(1×105 cells/well) were suspended in 100 µl complete
medium and loaded into the upper chamber of the Transwell
apparatus. The lower side of filter was coated with 0.1 mg/ml
gelatin. Control or experimental medium was added to the lower
chamber. Then, the chambers were incubated for 24 h at 37°C in 5%
CO2. Afterwards, the non-migrated cells were removed
from the top of the filter using cotton buds, whereas the migrated
cells adhering to the bottom of the filter were fixed and stained
with haematoxylin. The stained cells were counted in 10 randomly
chosen fields at ×200 magnification. Each experiment was repeated
three times (15).
Permeability assay
HUVECs were cultured on Transwell-COL membrane
inserts (12 wells, 1 cm2 filter area, 0.4 µm pore size;
Corning Incorporated). A total of 1×105 cells were
suspended in 0.5 ml complete medium and added to the upper
compartment, and 1.5 ml complete medium was added to the lower
compartment. After reaching 90% confluence, the HUVECs were starved
for 12 h in serum-free medium, which was subsequently replaced with
control or experimental medium for 24 h. After the cells were
washed three times with serum-free medium, a tracer solution of
fluorescein isothiocyanate (FITC)-labelled bovine serum albumin
(BSA, 250 µg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in
serum-free medium was added to the upper compartment of the
Transwell-COL insert, and 1.5 ml medium without FITC-BSA was added
to the lower compartment of the insert. Following 2 h of
equilibration of the tracer solution, 100 µl medium was removed
from the lower compartment. Sample fluorescence was measured using
a fluorescence spectrofluorophotometer (Tecan Safire 2™;
Tecan, Männedorf, Switzerland) at an emission/excitation wavelength
of 495/520 nm, and the rate of albumin transfer across the
monolayer was assessed by measuring the increase in FITC-BSA signal
in the lower well after 2 h. Albumin flux across the monolayer was
expressed as
P=([A]2/txV)/(Sx([A]1-[A]2)),
where V was the volume in the lower compartment, [A]2
was the albumin concentration in the lower compartment during the
time interval t, [A]1 was the albumin concentration in
the upper compartment, and S was the monolayer surface area. The
data were expressed as the permeability ratio
[P(experimental)/P(control)] (16). Each experiment was repeated in
triplicate.
Cell counting kit-8 (CCK-8) assay
HUVECs were seeded in 96-well culture plates
(1×104 cells/well). Following 24 h, the control or
experimental medium was added. At 12, 24, 48 and 72 h, 10 µl CCK-8
reagent (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was
added to each well for 1 h. Absorbance values were recorded at a
wavelength of 450 nm using a microplate reader (SpectraMax 250; GE
Healthcare Life Sciences, Chalfont, UK).
VEGF, VEGFR, endocan, ICAM-1, MCP-1
and PTX3 mRNA expression
HUVECs were incubated in control or experimental
medium for 12, 24, 48 and 72 h. Total RNA was extracted after
medium removal using TRIzol reagent (Takara Bio Inc., Otsu, Japan)
according to the manufacturer's instructions. Vascular endothelial
growth factor (VEGF), vascular endothelial growth factor receptor 2
(VEGFR), endocan, ICAM-1, monocyte chemotactic protein 1 (MCP-1)
and pentraxin (PTX)3 mRNA expression was analysed via reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
using SYBR Green Master Mix (Takara Bio Inc.). RNA (1 µg) was
reverse-transcribed to cDNA using random primers (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and Moloney murine leukaemia
virus (MMLV) reverse transcriptase (Takara Bio Inc.). RT-qPCR was
performed using an Applied Biosystems TaqMan 7000 (Applied
Biosystems; Thermo Fisher Scientific, Inc.) in 384-well plates
containing 5 µl reaction mixtures consisting of 2.5 µl SYBR Green
Master Mix, 0.1 µl each of forward and reverse primers, 0.1 µl Rox,
1.7 µl H2O and 0.5 µl cDNA. The amplification program
consisted of 1 cycle of 95°C for 10 min, followed by 40 cycles of
95°C for 15 sec, 55°C for 30 sec and 72°C for 30 sec. The primer
sequences are presented in Table
I. The relative mRNA levels in the samples were normalized to
the mRNA level of GAPDH, and the data are expressed as fold
increases in mRNA expression.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Primer sequence
(5′-3′) | Product Size
(bp) |
|---|
| VEGF | Forward
5′-GGCCTTCGCTTACTCTCACC-3′ | 104 |
|
| Reverse
5′-CTGTCATGGGCTGCTTCTTC-3′ |
|
| VEGFR | Forward
5′-CCGTCAAGGGAAAGACTACG-3′ | 106 |
|
| Reverse
5′-CTCCTCCACAAATCCAGAGC-3′ |
|
| Endocan | Forward
5′-TGCTACCGCACAGTCTCA-3′ | 105 |
|
| Reverse
5′-GCAGATACCAAACTCTTCACCA-3′ |
|
| ICAM | Forward
5′-AAGTTGTTGGGCATAGAGACC-3′ | 126 |
|
| Reverse
5′-AGGGCAGTTTGAATAGCACATT-3′ |
|
| MCP-1 | Forward
5′-AAGTCTCTGCCGCCCTTCT-3′ | 170 |
|
| Reverse
5′-CTTGCTGCTGGTGATTCTTCTA-3′ |
|
| PTX3 | Forward
5′-CATCTCCTTGCGATTCTGTTT-3′ | 107 |
|
| Reverse
5′-CCATTGTCTATTTCGTTGTCCA-3′ |
|
Western blot analysis
The HUVECs were lysed with radioimmunoprecipitation
assay buffer (Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The
total protein levels in the cell lysates was detected using the
Pierce™ bicinchoninic acid protein assay kit according to the
manufacturer's protocol (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). Samples containing 50 µg protein were separated on 10%
SDS-PAGE gels. Separated proteins were transferred onto
nitrocellulose filter membranes (EMD Millipore, Billerica, MA, USA)
blocked for 2 h at room temperature in 5% non-fat dried milk in
buffer containing 10 mM Tris-HCl (pH 8.0), 150 mM NaCl and 0.05%
Tween-20, and then incubated with the primary antibody overnight at
4°C. The primary antibodies targeted to one of the following
proteins (with their respective dilutions): VEGF (1:200; Abcam,
Cambridge, UK; cat. no. ab1316), VEGFR (1:1,000; Abcam; cat. no.
ab39256), endocan (1:1,000; Abcam; cat. no. ab103590), ICAM-1
(1:200; Abcam; cat. no. ab20), MCP-1 (1:5,000; Abcam; cat. no.
ab151538) and PTX3 (1:1,000; Abcam; cat. no. ab125007). After
washing the membranes three times in TBST buffer (Tris-buffered
saline with 0.1% Tween-20), the membranes were incubated with a
horseradish peroxidase (HRP)-conjugated secondary antibody. The
signal on the nitrocellulose filter membranes were detected by the
Enhanced Chemiluminescence system using chemiluminescent HRP
substrates (EMD Millipore). The expression of proteins was
semi-quantitatively analyzed by Image-J image analysis system
(version 1.44p, National Institutes of Health, Bethesda, MD,
USA).
Statistical analysis
Continuous variables were expressed as mean values
(mean ± standard error of the mean). T-test was used to the
comparison between two groups. One-way analysis of variance was
used for the comparative analysis in multiple groups followed by
the least significant difference post hoc test. P<0.05 was
considered to indicate statistically significant differences. All
data were analyzed by SPSS 20.0 statistical software (version,
20.0; IBM SPSS, Armonk, NY, USA).
Results
HG induced VEGF, VEGFR-2, endocan,
ICAM-1, MCP-1 and PTX3 upregulation in cultured HUVECs
HUVECs were cultured with different D-glucose
concentrations (15, 25 and 35 mmol/l) for 48 h to determine the
effect of HG on HUVEC gene expression. Control cells were
maintained in medium containing 5.5 mmol/l D-glucose to provide the
cells with the basal needs for cell growth.
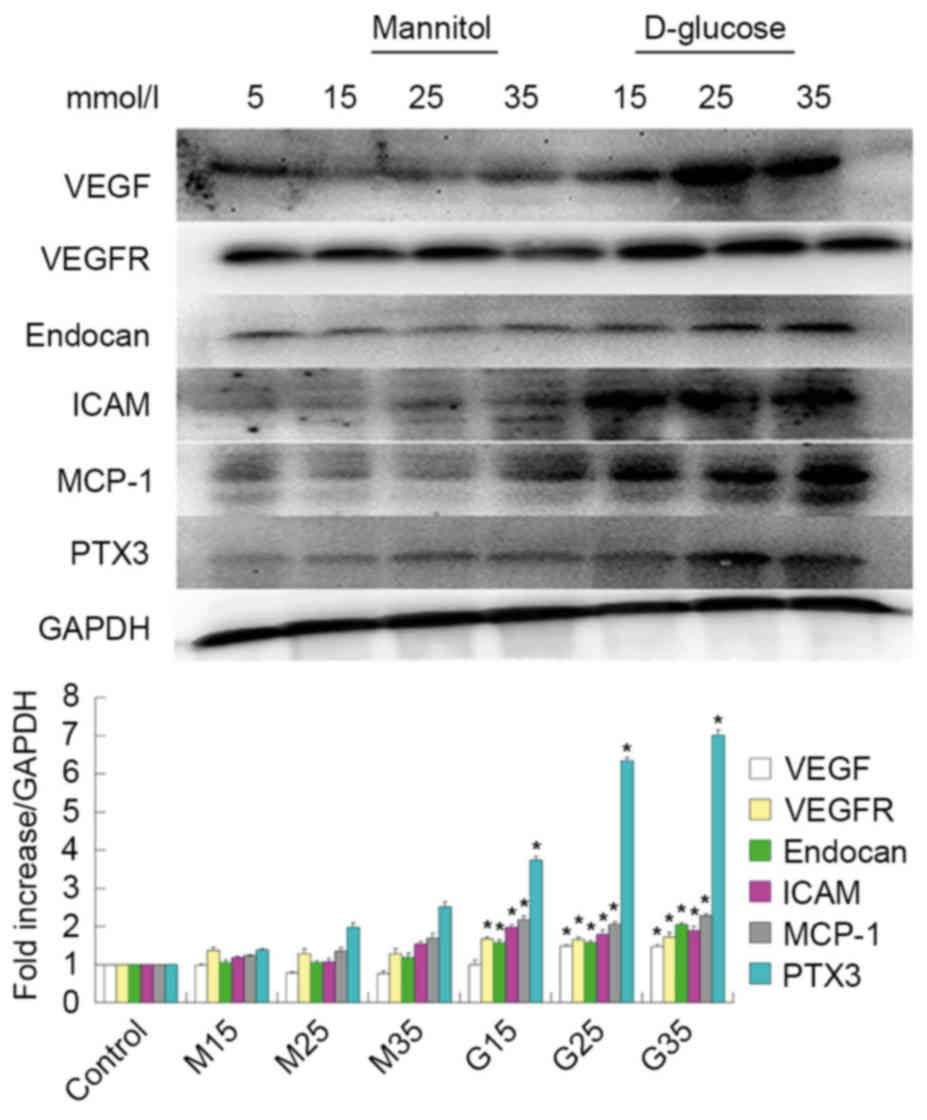
As illustrated in Fig.
1, VEGF, VEGFR and endocan mRNA expression levels gradually
increased under HG conditions in HUVECs, but were not significantly
altered in mannitol-treated cells compared with normally treated
control cells, indicating that HG modulates VEGF, VEGFR and endocan
mRNA expression in cultured HUVECs. Western blot analyses also
revealed that HG modulates VEGF, VEGFR and endocan expression, as
demonstrated in Fig. 2.
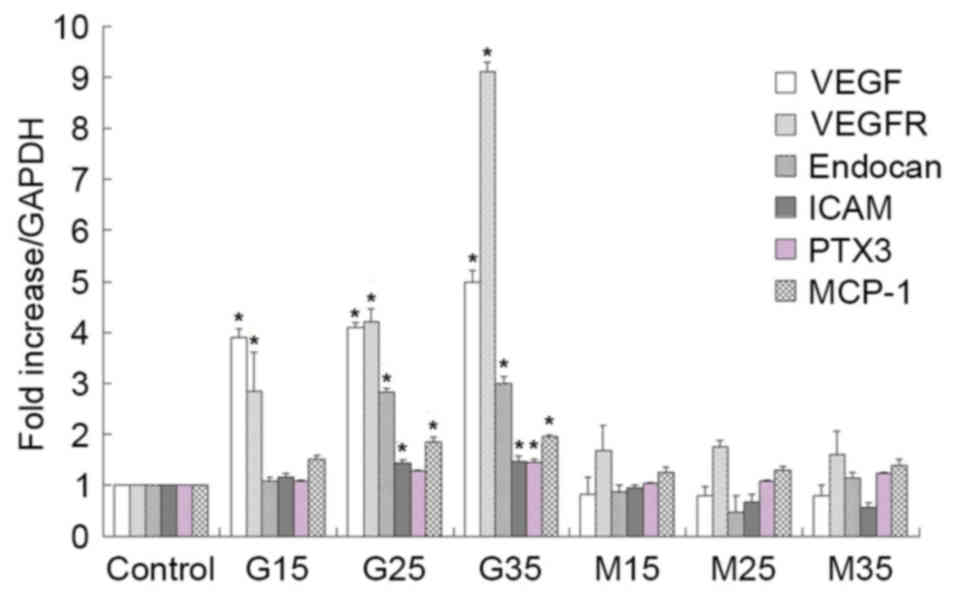 | Figure 1.mRNA expression of VEGF, VEGFR,
endocan, ICAM, MCP-1 and PTX3 mRNA in high glucose conditions in
human umbilical vein endothelial cells. mRNA expression gradually
increased. *P<0.05 vs. respective control. VEGF, vascular
endothelial growth factor; VEGFR, vascular endothelial growth
factor receptor; ICAM, intercellular adhesion molecule; MCP-1,
monocyte chemotactic protein 1; PTX3, pentraxin 3; G15, G25, G35,
D-glucose concentrations (15, 25 and 35 mmol/l, respectively); M15,
M25, M35, mannitol concentrations (15, 25 and 35 mmol/l,
respectively). |
In addition to examining the effects of HG treatment
on endothelial dysfunction marker expression, the authors examined
the effects of HG treatment on inflammatory marker expression.
Similar to the above results, they observed that ICAM-1, MCP-1 and
PTX3 mRNA and protein expression levels increased under HG
conditions in HUVECs, as demonstrated in Figs. 1 and 2.
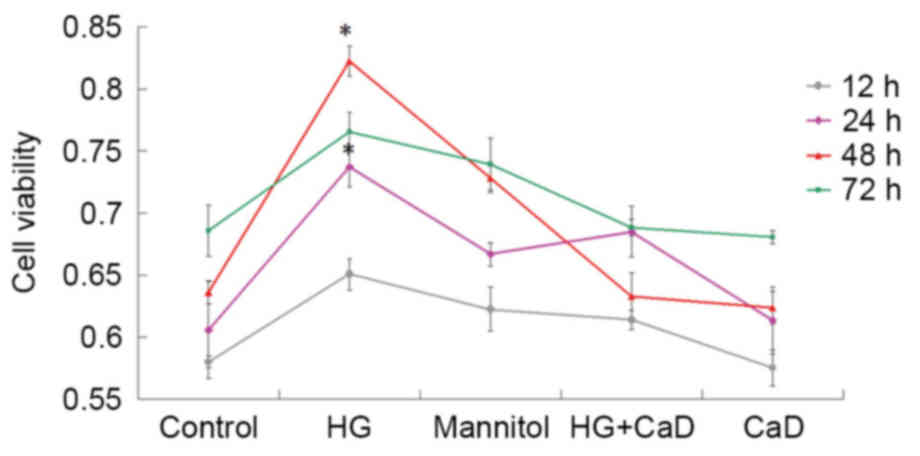
HUVEC proliferation
Cells were incubated with HG or mannitol, which
served as an osmotic control, for 3 days. HUVEC viability at 12,
24, 48 and 72 h was tested via CCK-8 assays. The results indicated
that HG treatment induces increases in cell proliferation in a
time-dependent manner at 24 and 48 h (P<0.05), followed by a
decrease in cell proliferation at 72 h (P>0.05).
Endothelial cells were also incubated with 100
µmol/l CaD for 3 days to determine the effects of CaD on HUVEC
viability. HUVEC viability was nearly identical across all time
points between 12 and 72 h following 100 µmol/l CaD treatment. As
demonstrated in Fig. 3, compared
with HG treatment (35 mmol/l), CaD (100 µmol/l) application
significantly attenuated abnormal HUVEC proliferation.
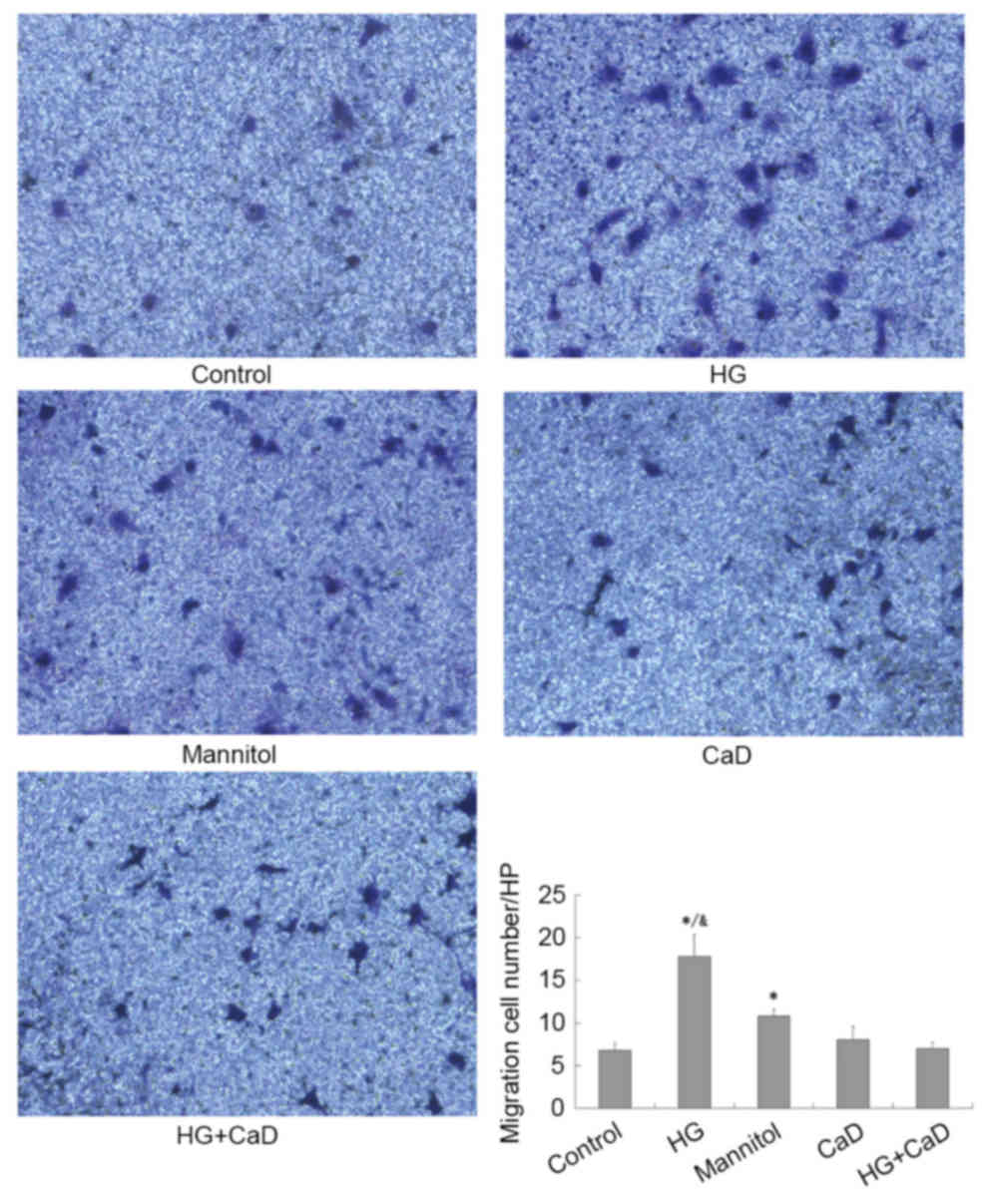
CaD prevented migration of HUVECs
partially induced by diabetes or high glucose
As presented in Fig.
4, HG significantly enhanced HUVEC migration compared to the
control treatment (18±3 cells/HPF vs. 7±2 cells/HPF, P<0.05).
CaD partially inhibited HUVEC migration compared to HG (8±2
cells/HPF vs. 18±3 cells/HPF, P<0.05) but did not completely
inhibit HVUEC migration compared to the control treatment (7±1
cells/HPF vs. 7±2 cells/HPF, P>0.05).
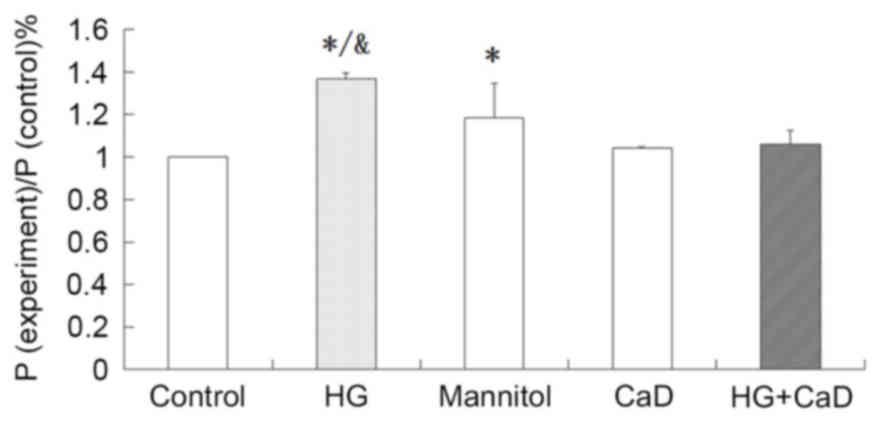
HG increased the permeability of
HUVECs, and CaD blocked this increase
HG treatment increased FITC-BSA permeability in
HUVECs compared to the control treatment (137±3% vs. 100%,
P<0.05). Addition of CaD prevented the increase in permeability
induced by HG (137±3% vs. 105±7%, P<0.05), although CaD alone
did not alter the permeability of HUVECs to FITC-BSA compared to
the control treatment (104±1% vs. 100%, P>0.05). Osmotic control
treatment increased FITC-BSA permeability, although to a
significantly lesser extent than HG (118±16% vs. 137±3%, P<0.05;
Fig. 5).
CaD suppressed HG-induced VEGF,
VEGFR-2, endocan and inflammatory marker overexpression in
HUVECs
Endothelial function is mediated by interactions
among VEGF, VEGFR-2 and endocan, which are expressed in HUVECs. HG
treatment (35 mmol/l), but not mannitol treatment (osmotic
control), increased VEGF, VEGFR-2 and endocan protein and mRNA
expression levels in a time-dependent manner over 48 h in HUVECs.
However, these increases diminished at 72 h of HG treatment. In
addition, 100 µmol/l CaD prevented the increases in the VEGF,
VEGFR-2 and endocan levels induced by HG (Figs. 6 and 7).
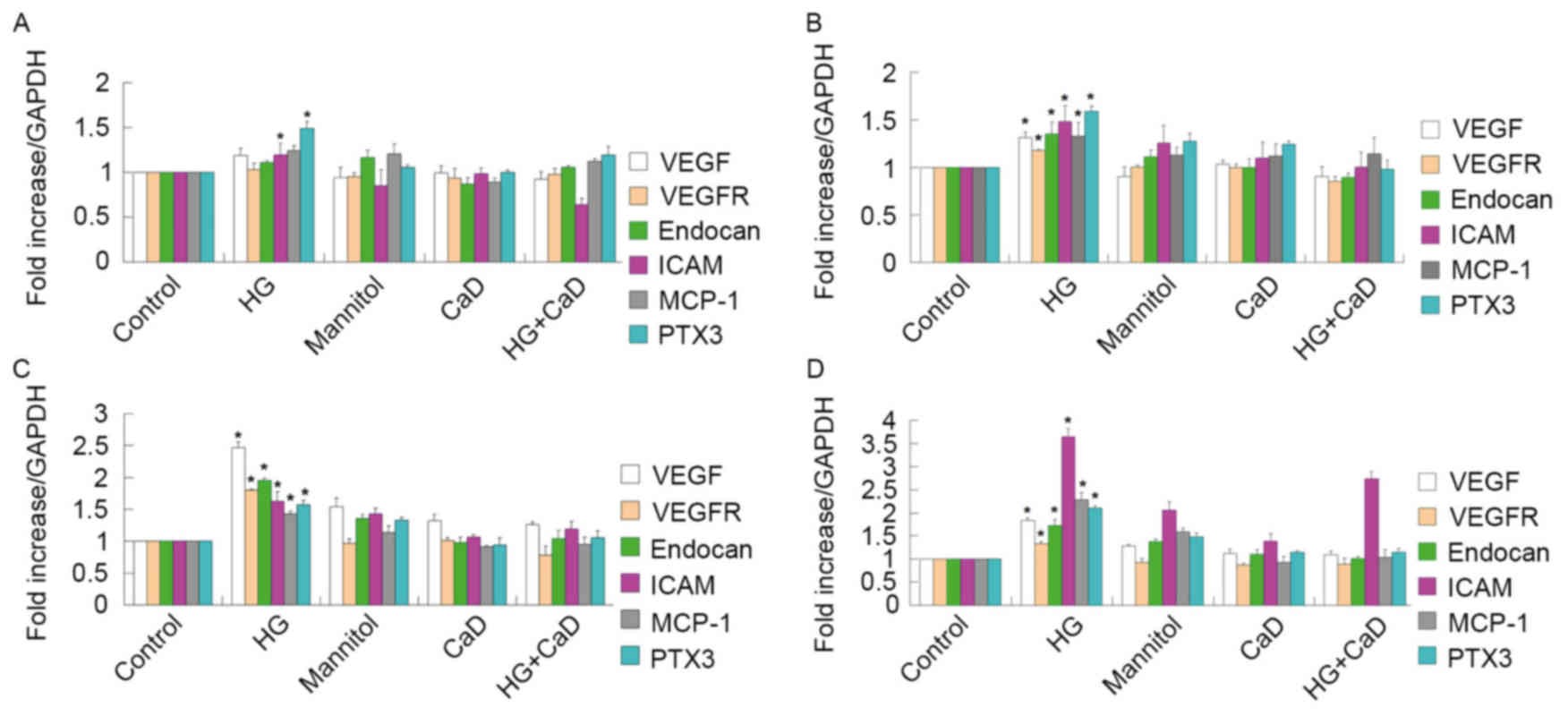 | Figure 6.HG (35 mmol/l) increased the mRNA
levels of VEGF, VEGFR-2, endocan in a time-dependent manner from 12
to 48 h. However, the growth trend began to slow down in 72 h. 100
µmol/l CaD prevented the increase of VEGF, VEGFR-2 and endocan
levels induced by HG. HG also increased the mRNA levels of ICAM,
MCP-1 and PTX3 in a time-dependent manner from 12 to 72 h. 100
µmol/l CaD prevented the increase the expression of these
inflammatory factors induced by HG. (A) HUVECs were incubated with
control or experimental medium for 12 h. (B) HUVECs were incubated
with control or experimental medium for 24 h. (C) HUVECs were
incubated with control or experimental medium for 48 h. (D) HUVECs
were incubated with control or experimental medium for 72 h.
*P<0.05 vs. respective control or respective mannitol, HG+CaD,
CaD group. HG, high glucose; VEGF, vascular endothelial growth
factor; VEGFR, vascular endothelial growth factor receptor; ICAM,
intercellular adhesion molecule; MCP-1, monocyte chemotactic
protein 1; PTX3, pentraxin 3; CaD, calcium dobesilate; HUVECs,
human umbilical vein endothelial cells. |
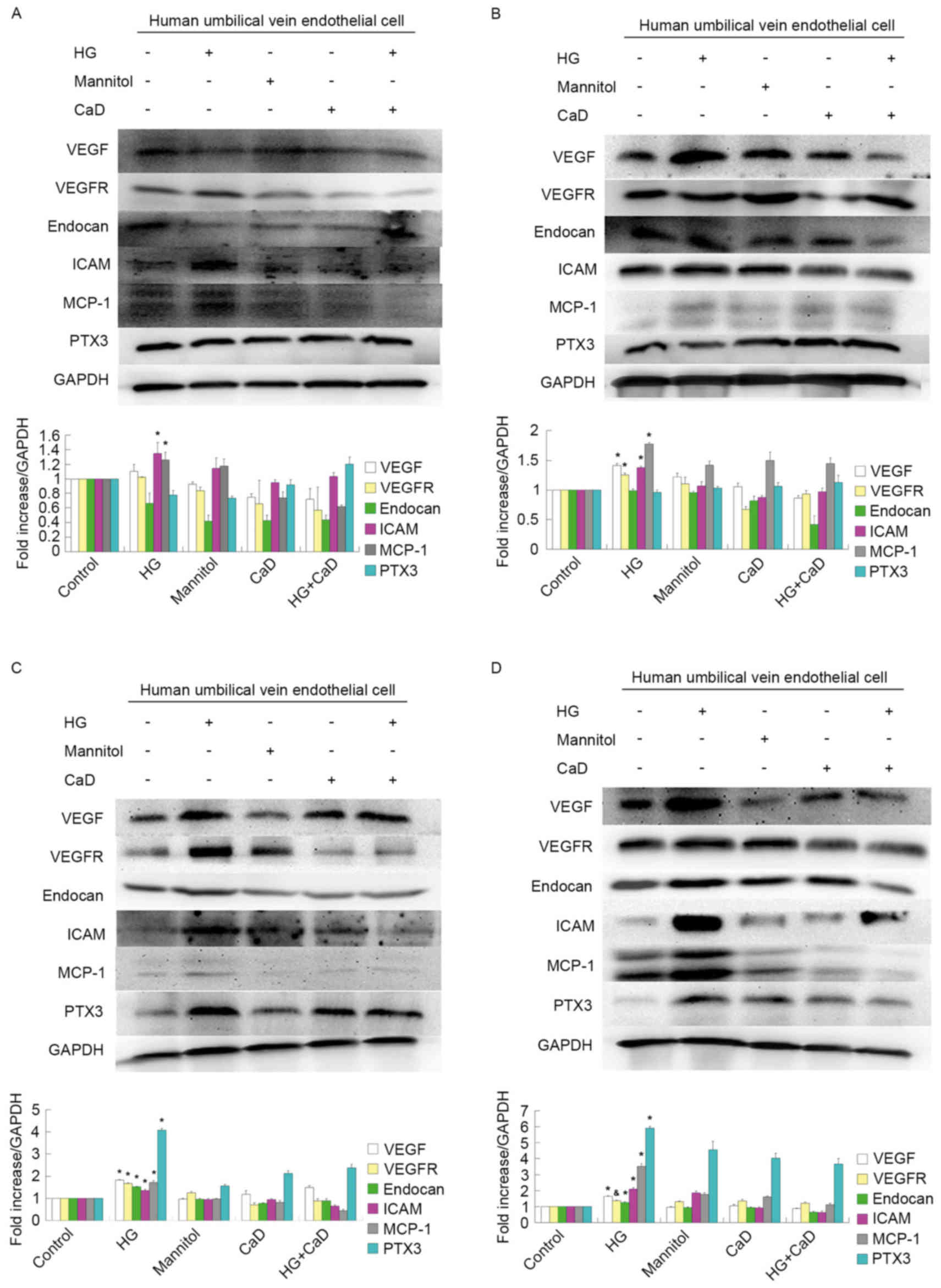 | Figure 7.HG (35 mmol/l) increased the protein
levels of VEGF, VEGFR-2, endocan in a time-dependent manner from 12
to 48 h. However, the growth trend began to slow down in 72 h. 100
µmol/l CaD prevented the increase of VEGF, VEGFR-2 and endocan
levels induced by HG. HG also increased the protein levels of ICAM,
MCP-1 and PTX3 in a time-dependent manner from 12 to 72 h. 100
µmol/l CaD prevented the increase the expression of these
inflammatory factors induced by HG. (A) HUVECs were incubated with
control or experimental medium for 12 h. (B) HUVECs were incubated
with control or experimental medium for 24 h. (C) HUVECs were
incubated with control or experimental medium for 48 h. (D) HUVECs
were incubated with control or experimental medium for 72 h.
*P<0.05 vs. control, mannitol, HG+CaD and CaD;
&P<0.05 vs. control group. HG, high glucose;
VEGF, vascular endothelial growth factor; VEGFR, vascular
endothelial growth factor receptor; ICAM, intercellular adhesion
molecule; MCP-1, monocyte chemotactic protein 1; PTX3, pentraxin 3;
CaD, calcium dobesilate; HUVECs, human umbilical vein endothelial
cells. |
Furthermore, HG treatment increased ICAM-1, MCP-1
and PTX3 mRNA and protein expression in a time-dependent manner
from 12 to 72 h. However, 100 µmol/l CaD treatment prevented these
HG-induced increases in inflammatory marker expression (Figs. 6 and 7).
Discussion
To the best of the authors' knowledge, the current
study is the first to demonstrate that CaD inhibits changes in
endothelial and inflammatory protein levels. Specifically, CaD
significantly inhibited the expression of VEGF and VEGFR, which are
known to mediate abnormal HUVEC proliferation (17). CaD also prevented the HG-induced
increases in HUVEC migration and permeability, most likely by
suppressing ICAM-1, PTX3 and MCP-1 expression.
CaD, an antioxidant and angioprotective agent,
blocks the increase in capillary permeability induced by reactive
oxygen species, enhances the activity of nitric oxide synthase in
endothelial cells, and inhibits apoptosis and inflammatory cytokine
expression in blood vessels and other tissues (18). CaD effectively treats diabetic
retinopathy at the systemic and local ocular levels and is
therefore widely used to treat this disorder (18). DKD and retinopathy often coexist,
develop in parallel, and have similar pathogeneses (19). Zhang et al (13) observed that CaD exerted therapeutic
effects on type 2 diabetes patients with microalbuminuria; however,
the mechanism underlying the effects of CaD on diabetic nephropathy
is unclear.
It is generally known that diabetic nephropathy is
characterized by an early increase followed by a gradual decrease
in the glomerular filtration rate (GFR). The increase in GFR may be
caused by capillary angiogenesis. A streptozotocin-induced diabetes
model demonstrated increases in glomerular capillary number,
capillary length and capillary cross-sectional area (20–22).
Correspondingly, neoangiogenesis was observed in a human study
(23). Immature neovessels exhibit
thin basement membranes and endothelial swelling, which may limit
permselectivity and lead to microalbuminuria (23–27).
New blood vessel formation involves EC proliferation and migration
(28). The present study
demonstrated that HG treatment induces concentration- and
time-dependent increases in cell proliferation at 12 and 48 h of
treatment followed by decreases in cell proliferation at 72 h of
treatment. In addition, the authors indicated that CaD ameliorates
these abnormal increases in cell proliferation. CaD also prevents
the migration of HUVECs partially induced by HG. The abovementioned
early increased and subsequent gradual decrease in the number of
glomerular capillaries may be attributed to corresponding changes
in VEGF expression (26). The
present study also demonstrated that HG induced early increases in
VEGF, VEGFR-2 and endocan, although the rate of these expression
increases was reduced at 72 h, which conformed to the pattern of
cell proliferation. The important role of VEGF in diabetic
microangiopathy has been studied extensively in the setting of
proliferative diabetic retinopathy, which is characterized by the
formation of new vessels inside the retina along with abnormal
vascular architecture and permeability. In the kidney, VEGF is
produced primarily by podocytes and is upregulated in early
diabetic nephropathy, promoting new blood vessel formation
(26,29). In contrast, VEGF expression and
activity are downregulated in advanced diabetic nephropathy,
leading to glomerular capillary rarefaction and GFR decreases,
possibly due to podocyte loss (30–33).
CaD suppressed HG-induced VEGF, VEGFR-2 and endocan overexpression
in the current study. These results demonstrated that CaD has
angioprotective properties.
Diabetic nephropathy is caused by not only
uncontrolled hemodynamics and hyperglycaemia but also low-grade
inflammation and chronically activated innate immunity (34,35).
Studies indicate that the expression levels of inflammatory
cytokines, such as TNF-α, IL-1, IL-6 and IL-18, are elevated in the
kidneys of diabetes models (36,37).
In diabetic rat models, the elevated expression levels of TNF-α and
IL-6 are associated with increases in kidney weight and urinary
albumin excretion (35). Serum
IL-18 and TNF-α levels were higher in patients with DKD than in
controls and positively correlated with the degree of albuminuria
in patients with diabetes (38,39).
These cytokines promote vascular endothelial cell permeability
in vitro, leading to glomerular basement membrane
thickening, contributing to EC apoptosis, and even exerting a
direct toxic effect on renal cells (40–48).
In addition, VCAM-1 was identified to be upregulated in DKD
patients (8) and may promote
inflammatory cell adhesion to the endothelium and recruit
circulating immune cells to diabetic kidneys. The results of the
present study are consistent with the above research findings. HG
induced increases in HUVEC permeability, as well as concentration-
and time-dependent increases in ICAM-1, MCP-1 and PTX3 protein and
mRNA expression from 12 to 72 h of treatment. Furthermore, CaD
prevented the HG-induced increases in the expression of these
inflammatory factors and ameliorated the HG-mediated increase in
HUVEC permeability. These results indicated that CaD protects ECs
partly by ameliorating HG-induced inflammation.
In conclusion, CaD suppressed HG-induced VEGF,
VEGFR-2 and endocan overexpression, indicating that CaD has
angioprotective properties. CaD also protected ECs partly by
ameliorating HG-induced inflammation. The current findings
demonstrated the potential application of CaD to the treatment of
diabetic nephropathy, particularly during the early stages of this
disease.
Acknowledgements
The present study was supported in part by the
National Natural Science Foundation of China (grant nos. 81370794
and 81570604) as well as by a grant (grant no. ZHYY-ZXYJHZX-1-02)
from the Shanghai Municipal Commission of Health and Family
Planning. The authors would like to thank Shanghai Zhaohui
pharmaceutical Co., Ltd, who provided the original powder of
CaD.
Glossary
Abbreviations
Abbreviations:
|
DKD
|
diabetic kidney disease
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
|
HG
|
high glucose
|
|
CaD
|
calcium dobesilate (calcium
2,5-dihydroxybenzenesulfonate)
|
|
CCK-8
|
Cell Counting Kit-8
|
|
FITC
|
fluorescein isothiocyanate
|
|
BSA
|
bovine serum albumin
|
|
ECs
|
endothelial cells
|
|
ICAM-1
|
intercellular cell adhesion
molecule-1
|
|
VCAM-1
|
vascular cell adhesion molecule-1
|
|
ECM
|
endothelial cells growth medium
|
|
GFR
|
glomerular filtration rate
|
|
VEGF
|
vascular endothelial growth factor
|
|
VEGFR
|
vascular endothelial growth factor
receptor 2
|
|
MCP-1
|
monocyte chemotactic protein 1
|
|
PTX3
|
pentraxin 3
|
References
|
1
|
International Diabetes Federation, .
Diabetes Atlas (7th edition). 2015.https://www.diabetesatlas.org
|
|
2
|
Tuttle KR, Bakris GL, Bilous RW, Chiang
JL, de Boer IH, Goldstein-Fuchs J, Hirsch IB, Kalantar-Zadeh K,
Narva AS, Navaneethan SD, et al: Diabetic kidney disease: A report
from an ADA Consensus Conference. Am J Kidney Dis. 64:510–533.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fu J, Lee K, Chuang PY, Liu Z and He JC:
Glomerular endothelial cell injury and cross talk in diabetic
kidney disease. Am J Physiol Renal Physiol. 308:F287–F297. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheng H and Harris RC: Renal endothelial
dysfunction in diabetic nephropathy. Cardiovasc Haematol Disord
Drug Targets. 14:22–33. 2014. View Article : Google Scholar
|
|
5
|
Navarro-González JF, Mora-Fernández C, de
Fuentes M Muros and García-Pérez J: Inflammatory molecules and
pathways in the pathogenesis of diabetic nephropathy. Nat Rev
Nephrol. 7:327–340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Galkina E and Ley K: Leukocyte recruitment
and vascular injury in diabetic nephropathy. J Am Soc Nephrol.
17:368–377. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sugimoto H, Shikata K, Hirata K, Akiyama
K, Matsuda M, Kushiro M, Shikata Y, Miyatake N, Miyasaka M and
Makino H: Increased expression of intercellular adhesion molecule-1
(ICAM-1) in diabetic rat glomeruli: Glomerular hyperfiltration is a
potential mechanism of ICAM-1 upregulation. Diabetes. 46:2075–2081.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seron D, Cameron JS and Haskard DO:
Expression of VCAM-1 in the normal and diseased kidney. Nephrol
Dial Transplant. 6:917–922. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tejerina T and Ruiz E: Calcium dobesilate:
Pharmacology and future approaches. Gen Pharmacol. 31:357–360.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Javadzadeh A, Ghorbanihaghjo A, Adl FH,
Andalib D, Khojasteh-Jafari H and Ghabili K: Calcium dobesilate
reduces endothelin-1 and high-sensitivity C-reactive protein serum
levels in patients with diabetic retinopathy. Mol Vis. 19:62–68.
2013.PubMed/NCBI
|
|
11
|
Rabe E, Jaeger KA, Bulitta M and Pannier
F: Calcium dobesilate in patients suffering from chronic venous
insufficiency: A double-blind, placebo-controlled, clinical trial.
Phlebology. 26:162–168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hall JF: Modern management of hemorrhoidal
disease. Gastroenterol Clin North Am. 42:759–772. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang X: Therapeutic effects of calcium
dobesilate on diabetic nephropathy mediated through reduction of
expression of PAI-1. Exp Ther Med. 5:295–299. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jafarey M, Ashtiyani S Changizi and Najafi
H: Calcium dobesilate for prevention of gentamicin-induced
nephrotoxicity in rats. Iran J Kidney Dis. 8:46–52. 2014.PubMed/NCBI
|
|
15
|
Pan SL, Guh JH, Huang YW, Chern JW, Chou
JY and Teng CM: Identification of apoptotic and antiangiogenic
activities of terazosin in human prostate cancer and endothelial
cells. J Urol. 169:724–729. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Irwin DC, van Patot MC Tissot, Tucker A
and Bowen R: Direct ANP inhibition of hypoxia-induced inflammatory
pathways in pulmonary microvascular and macrovascular endothelial
monolayers. Am J Physiol Lung Cell Mol Physiol. 288:L849–L859.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Demirtas S, Caliskan A, Guclu O, Yazici S,
Karahan O, Yavuz C and Mavitas B: Can calcium dobesilate be used
safely for peripheral microvasculopathies that require
neoangiogenesis? Med Sci Monit Basic Res. 19:253–257. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang X, Liu W, Wu S, Jin J, Li W and Wang
N: Calcium dobesilate for diabetic retinopathy: A systematic review
and meta-analysis. Sci China Life Sci. 58:101–107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Krakoff J, Lindsay RS, Looker HC, Nelson
RG, Hanson RL and Knowler WC: Incidence of retinopathy and
nephropathy in youth-onset compared with adult-onset type 2
diabetes. Diabetes Care. 26:76–81. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seyer-Hansen K: Renal hypertrophy in
experimental diabetes mellitus. Kidney Int. 23:643–646. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Seyer-Hansen K: Renal hypertrophy in
streptozotocin-diabetic rats. Clin Sci Mol Med. 51:551–555.
1976.PubMed/NCBI
|
|
22
|
Nyengaard JR and Rasch R: The impact of
experimental diabetes mellitus in rats on glomerular capillary
number and sizes. Diabetologia. 36:189–194. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Osterby R and Nyberg G: New vessel
formation in the renal corpuscles in advanced diabetic
glomerulopathy. J Diabet Complications. 1:122–127. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wehner H and Nelischer G: Morphometric
investigations on intrarenal vessels of streptozotocin-diabetic
rats. Virchows Arch A Pathol Anat Histopathol. 419:231–235. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakagawa T, Kosugi T, Haneda M, Rivard CJ
and Long DA: Abnormal angiogenesis in diabetic nephropathy.
Diabetes. 58:1471–1478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanesaki Y, Suzuki D, Uehara G, Toyoda M,
Katoh T, Sakai H and Watanabe T: Vascular endothelial growth factor
gene expression is correlated with glomerular neovascularization in
human diabetic nephropathy. Am J Kidney Dis. 45:288–294. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Min W and Yamanaka N: Three-dimensional
analysis of increased vasculature around the glomerular vascular
pole in diabetic nephropathy. Virchows Arch A Pathol Anat
Histopathol. 423:201–207. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yuan J, Fang W, Lin A, Ni Z and Qian J:
Angiopoietin-2/Tie2 signaling involved in TNF-α induced peritoneal
angiogenesis. Int J Artif Organs. 35:655–662. 2012.PubMed/NCBI
|
|
29
|
Cooper ME, Vranes D, Youssef S, Stacker
SA, Cox AJ, Rizkalla B, Casley DJ, Bach LA, Kelly DJ and Gilbert
RE: Increased renal expression of vascular endothelial growth
factor (VEGF) and its receptor VEGFR-2 in experimental diabetes.
Diabetes. 48:2229–2239. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hohenstein B, Hausknecht B, Boehmer K,
Riess R, Brekken RA and Hugo CP: Local VEGF activity but not VEGF
expression is tightly regulated during diabetic nephropathy in man.
Kidney Int. 69:1654–1661. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Baelde HJ, Eikmans M, Doran PP, Lappin DW,
de Heer E and Bruijn JA: Gene expression profiling in glomeruli
from human kidneys with diabetic nephropathy. Am J Kidney Dis.
43:636–650. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Baelde HJ, Eikmans M, Lappin DW, Doran PP,
Hohenadel D, Brinkkoetter PT, Van der Woude FJ, Waldherr R,
Rabelink TJ, de Heer E and Bruijn JA: Reduction of VEGF-A and CTGF
expression in diabetic nephropathy is associated with podocyte
loss. Kidney Int. 71:637–645. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lindenmeyer MT, Kretzler M, Boucherot A,
Berra S, Yasuda Y, Henger A, Eichinger F, Gaiser S, Schmid H,
Rastaldi MP, et al: Interstitial vascular rarefaction and reduced
VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol.
18:1765–1776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
García-García PM, Getino-Melián MA,
Domínguez-Pimentel V and Navarro-González JF: Inflammation in
diabetic kidney disease. World J Diabetes. 5:431–443. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pickup JC: Inflammation and activated
innate immunity in the pathogenesis of type 2 diabetes. Diabetes
Care. 27:813–823. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Navarro JF, Milena FJ, Mora C, León C and
García J: Renal pro-inflammatory cytokine gene expression in
diabetic nephropathy: Effect of angiotensin-converting enzyme
inhibition and pentoxifylline administration. Am J Nephrol.
26:562–570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sekizuka K, Tomino Y, Sei C, Kurusu A,
Tashiro K, Yamaguchi Y, Kodera S, Hishiki T, Shirato I and Koide H:
Detection of serum IL-6 in patients with diabetic nephropathy.
Nephron. 68:284–285. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moriwaki Y, Yamamoto T, Shibutani Y, Aoki
E, Tsutsumi Z, Takahashi S, Okamura H, Koga M, Fukuchi M and Hada
T: Elevated levels of interleukin-18 and tumor necrosis
factor-alpha in serum of patients with type 2 diabetes mellitus:
Relationship with diabetic nephropathy. Metabolism. 52:605–608.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Navarro JF, Mora C, Muros M and García J:
Urinary tumour necrosis factor-alpha excretion independently
correlates with clinical markers of glomerular and
tubulointerstitial injury in type 2 diabetic patients. Nephrol Dial
Transplant. 21:3428–3434. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Royall JA, Berkow RL, Beckman JS,
Cunningham MK, Matalon S and Freeman BA: Tumor necrosis factor and
interleukin 1 alpha increase vascular endothelial permeability. Am
J Physiol. 257:L399–L410. 1989.PubMed/NCBI
|
|
41
|
Navarro-González JF and Mora-Fernández C:
The role of inflammatory cytokines in diabetic nephropathy. J Am
Soc Nephrol. 19:433–442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vestra M Dalla, Mussap M, Gallina P,
Bruseghin M, Cernigoi AM, Saller A, Plebani M and Fioretto P:
Acute-phase markers of inflammation and glomerular structure in
patients with type 2 diabetes. J Am Soc Nephrol. 16 Suppl
1:S78–S82. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ruef C, Budde K, Lacy J, Northemann W,
Baumann M, Sterzel RB and Coleman DL: Interleukin 6 is an autocrine
growth factor for mesangial cells. Kidney Int. 38:249–257. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mariño E and Cardier JE: Differential
effect of IL-18 on endothelial cell apoptosis mediated by TNF-alpha
and Fas (CD95). Cytokine. 22:142–148. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Stuyt RJ, Netea MG, Geijtenbeek TB,
Kullberg BJ, Dinarello CA and van der Meer JW: Selective regulation
of intercellular adhesion molecule-1 expression by interleukin-18
and interleukin-12 on human monocytes. Immunology. 110:329–334.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bertani T, Abbate M, Zoja C, Corna D,
Perico N, Ghezzi P and Remuzzi G: Tumor necrosis factor induces
glomerular damage in the rabbit. Am J Pathol. 134:419–430.
1989.PubMed/NCBI
|
|
47
|
DiPetrillo K, Coutermarsh B and Gesek FA:
Urinary tumor necrosis factor contributes to sodium retention and
renal hypertrophy during diabetes. Am J Physiol Renal Physiol.
284:F113–F121. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dipetrillo K and Gesek FA: Pentoxifylline
ameliorates renal tumor necrosis factor expression, sodium
retention, and renal hypertrophy in diabetic rats. Am J Nephrol.
24:352–359. 2004. View Article : Google Scholar : PubMed/NCBI
|