Introduction
Cerebral infarction is a severe neurological disease
that is characterized by obstacles to the brain blood supply or
limitations in brain tissue and frequently leads to cerebral
ischemic necrosis anoxic lesions (1,2).
Previous studies have suggested that cerebral infarction may result
innerve dysfunction and is often associated with high blood
pressure, diabetes, hyperlipidemia and other risk factors (3,4).
Clinical investigations indicate that pathological changes of blood
vessel walls, blood composition changes and drug-induced or
injury-caused cerebral artery dissection contribute to the
initiation and development of cerebral infarction (5,6).
Thrombolytic treatments are the most commonly used therapy for
patients suffering from cerebral infarction and it is necessary to
understand the underlying therapeutic effects and potential
molecular mechanisms in the progression of cerebral infarction.
The imbalance between coagulation and the
fibrinolytic system serves an important role in the progression and
pathogenesis of arterial thrombosis (7). Aspirin is effective and safe in the
secondary prevention of cerebral infarction, as previously reported
in a number of randomized, double-blind trials (8,9).
Tissue plasminogen activator (tPA) is an effective drug approved by
US FDA for the treatment of ischemic stroke, and it exhibits
pleiotropic effects in addition to thrombolysis (10). Thrombin-activatable fibrinolysis
inhibitor (TAFI) is also an anti-fibrinolytic factor with an
increased risk of cerebral infarction as the levels of plasma TAFI
increase (11,12). Previous studies have suggested that
TAFI and plasminogen activator inhibitor (PAI)-1serve crucial roles
in the initiation and development of deep cerebral infarction
(13,14). Inflammation and apoptosis of
cerebrovascular endothelial cells increase the aggravation of
cerebral infarction and other syndromes inpatients (14).
The present study investigated the therapeutic
effects of aspirin on cerebral infarction in a mouse model.
Potential molecular mechanisms mediated by aspirin were analyzed in
cerebrovascular endothelial cells in a mouse model of cerebral
infarction. The anti-inflammatory and anti-apoptotic effects of
aspirin were also researched. The results indicated that aspirin
can inhibit inflammation and apoptosis in addition to its
profibrinolytic properties, as determined by in vitro and
in vivo analysis. These findings suggested that aspirin may
be a potential drug for cerebral infarction therapy, which may act
by downregulating toll-like receptor (TLR)4/nuclear factor
(NF)-κB-mediated endoplasmic reticulum (ER) stress.
Materials and methods
Ethical approval and participant
consent
The present preclinical study was approved by the
Ethics Committee of Dezhou People's Hospital (Shandong, China). All
surgeries and euthanasia of experimental animals were performed
under sodium pentobarbital anesthesia to minimize pain.
Cells and reagents
Cerebrovascular endothelial cells were isolated from
experimental mice according to a previous study (15) and cultured in minimum essential
medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) supplemented
with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Cells were cultured in a 37°C humidified
atmosphere of 5% CO2.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cerebrovascular
endothelial cells and serum using an RNA plus kit (Invitrogen;
Thermo Fisher Scientific, Inc.) following the manufacturer's
protocol, and treated with 2 U/µg DNase I (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C for 30 min. A total of 5 µg RNA
per sample was primed with Oligo dT primer (Takara Biotechnology
Co., Ltd., Dalian, China) and reverse transcribed using the AMV
Reverse Transcription System (Invitrogen; Thermo Fisher Scientific,
Inc.). For the PCR experiments, the following forward and reverse
primers were used: Forward, 5′-TGGCAGCAGTGACAGCAGCA-3′ and reverse,
5′-TACGGAGGTGGAGTGGGTGT-3′ for ADP; forward,
5′-AGCCGAGGAAGAACTATGAAC-3′ and reverse, 5′-ATTTGAGGGTGAGGAATGGG-3′
for PAI-1; forward, 5′-GAGGGCAGAATCATCACGAAGT-3′ and reverse,
5′-TGAGAGATCTGGTTCCCGAAAC-3′ for vWF; forward,
5′-CAAGGCAGAGGTGGGTTTGG-3′ and reverse, 5′-GGCACCTTTTCAGTTGCTCAC-3′
for thromboxane; forward, 5′-CAAAGGTGGATCAGATTCAAG-3′ and reverse,
5′-GGTGAGCATTATCACCCAGAA-3′ for the reference gene GAPDH. The
RT-qPCR mixture system comprised cDNA templates (1 µl; Invitrogen;
Thermo Fisher Scientific, Inc.), primers (2 µl eachof the forward
and reverse primers) and SYBR Green qPCR Master mix (5 µl;
Invitrogen; Thermo Fisher Scientific, Inc.). The PCR conditions
included an initial denaturation step of 94°C for 2 min, followed
by 30 cycles of 94°C for 30 sec, 59°C for 30 sec, 72°C for 2 min
and a final elongation step at 72°C for 10 min. Taq DNA polymerase
was purchased from Sigma-Aldrich; Merck KGaA. GAPDH was used as an
internal control to normalize gene expression. The relative gene
expression levels were calculated using the 2−∆∆Cq
method (16). All experiments were
repeated 3 times.
Western blot analysis
Cerebrovascular endothelial cells (1×104
cells) were lysed using radioimmunoprecipitation assay lysis buffer
(Invitrogen; Thermo Fisher Scientific, Inc.) and centrifuged (1,000
× g) at 4°C for 10 min. The protein concentrations of the cell
extracts were then measured using Bradford protein dye reagent
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). A total of 30
µg/lane protein was loaded and separated by 12% SDS-PAGE and
transferred to nitrocellulose membranes. The membranes were blocked
with 5% skimmed milk for 1 h at room temperature, washed in
Tris-buffered saline containing 0.1% Tween-20 (TBST) and incubated
with the following primary antibodies: Anti-mouse IL-6 (1:2,000;
cat no. ab7737; Abcam, Cambridge, UK); anti-mouse TNF-α (1:2,000;
cat no. ab6671; Abcam); anti-mouse IL-1β (1:2,000; cat no. ab 9722;
Abcam); anti-mouse caspase-3 (1:1,000; cat no. ab13847; Abcam);
anti-mouse caspase-9 (1:1,000; cat. no. ab18571; Abcam); anti-mouse
p53 (1:1,000; cat no. ab1431; Abcam); anti-mouse Bcl-2 (1:1,000;
cat no. ab194583; Abcam); anti-mousecaspase-12 (1:1,000; cat. no.
ab18766; Abcam); anti-mouse TLR-4 (1:1,000; cat no. ab13867;
Abcam); anti-mouse p65 (1:1,000; cat. no. ab16502; Abcam);
anti-mouse IKKβ (1:1,000; cat. no. ab53694; Abcam); anti-mouse IkBα
(1:1,000; cat. no. ab72429; Abcam); anti- protein kinase R-like
endoplasmic reticulum kinase (PERK; 1:1,000; cat. no. ab65142;
Abcam); anti-mouse eukaryotic translation initiation factor 2
subunit 1 (eIF2α; 1:1,000; cat. no. ab26197; Abcam); anti-mouse
CHOP (1:1,000; cat. no. ab10444; Abcam); anti-mouseGRP78 (1:1,000;
cat. no. ab32618; Abcam) and anti-β-actin (1:2,000; cat. no.
ab8226; Abcam) overnight at 4°C. The labeled membranes were then
washed three times with TBST, incubated for 2 h at room temperature
with secondary anti-primary IgG conjugated with horseradish
peroxidase (1:1,500; cat. no. ab6717; Abcam). The protein bands
labeled with the antibodies were visualized using the SuperSignal
West Pico Chemiluminescent Substrate Trial kit (Pierce Protein
Biology; Thermo Fisher Scientific, Inc.). Images were obtained
using the ChemiDoc XRS system with Quantity One software (Bio-Rad
Laboratories, Inc.). Protein expression was analyzed using BandScan
5.0 software (Glyko, Inc., Novato, CA, USA). All experiments were
repeated 3 times.
Animals
A total of 30 maleC57BL/6 mice (mean age of 6 months
and a body weight of 20–25 g) were purchased from Vital River
Laboratory Animal Technology Co. Ltd. (Shanghai, China). Mice were
maintained in a room with constant temperature (22±1°C) and a 12-h
light/dark cycle, and cages in groups <5 per cage with ad
libitum access to food and water. Mice were used to establish
the model of cerebral infarction by local atherosclerosis cerebral
ischemic necrosis according to previous study (17). Experimental mice were randomly
divided into two groups after cerebral infarction surgery (n=15 in
each group) and received aspirin or PBS treatment for 30 days.
Aspirin (20 mg/kg body weight) or PBS was administered in drinking
water once/day. The experimental mice were sacrificed under 1.5%
pentobarbital sodium (1 ml/kg; Lianshuoinc, Shanghai, China) on day
30 for further analysis.
Immunohistochemical analysis
Cerebrovascular tissues were isolated from
experimental mice following 30-day treatment with aspirin or PBS.
The tissues in cerebral infarction were soaked in mixed stationary
liquid (RongboBio, Shanghai, China) to fully fix for 24 h at room
temperature. It was then washed in flowing water for 24 h, and
underwent conventional gradient alcohol dehydration, and xylene
paraffin embedding. The wax blocks were cut into sections, with
each section cut at 4 µm thickness for immunohistochemical
staining. The sections were dewaxed by conventional methods and
underwent microwave antigen retrieval at 95°C for 10 min. After
cooling, they were washed with distilled water and blocked in
normal fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.)
at room temperature for 30 min. The primary antibody TLR4 (1:500;
cat no. ab13867; Abcam) and NF-κB (1:500; cat no. ab32360; Abcam)
was added and placed at 4°C for 20 h. The slides were allowed to
equilibrate for 1 h at room temperature, and soaked in PBS four
times for 5 min. Then, secondary antibody (1:1,000; cat. no.
ab6717; Abcam) was added and allowed to incubate at 37°C for 10
min, after which the slides underwent PBS cleaning three times for
5 min. Slides were observed with a BZ-9000 fluorescent video
microscope (Keyence Corporation, Osaka, Japan). All experiments
were repeated 3 times.
Assessment of thrombus formation in
vivo
Thrombus formation in experimental mice was measured
in using an Alexa Flour 488 (Invitrogen; Thermo Fisher Scientific,
Inc.) ex vivo labeled anti-fibrin antibody (1:1,000; cat.
no. ab34269; Abcam). Fluorescence intensity was quantified by
intravital video microscopy (BX51WI; Olympus Corporation, Tokyo,
Japan) using Image-Pro Plus (version 6.0, Media Cybernetics, Inc.,
Rockville, MD, USA).
ELISA
The expression of PERK (cat. no DEIA-XYA1959;
Creative Diagnostics, USA), NF-ΚB (cat no. JK-(a)-6261;
JingkangBioscience, Shanghai, China), and e eIF2α (cat no. 7952S;
Cell Signaling Technology, Inc., Danvers, MA, USA) in
cerebrovascular endothelial cells were analyzed by ELISA kits and
performed according to the manufacturer's protocols.
Knockdown of TLR4
TLR4 small interfering RNA (siRNA) and scramble
siRNA (as a negative control) were purchased from Shanghai
GenePharma Co., Ltd. (Shanghai, China). The sequences were as
follows: si-TLR4, forward 5′-GCAUCUCUACAUUCAAGAA-3′ and reverse
5′-UUCUUGAAUGUAGAGAUGC-3′; scrambled sequence (si-NC), forward
5′-UUCUCCGAACGUGUCACGU-3′ and reverse 5′-ACGUGACACGUUCGGAGAA-3′.
For transfection, cerebrovascular endothelial cells at a density of
1×105 cells/well were seeded in each well of a 24-well
microplate, grown for 24 h to reach 30–50% confluence and then
incubated with TLR4 siRNA or scrambled siRNA and Lipofectamine™
2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) in 100 µl
serum-free Dulbecco's Modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. The
CCN4 knockdown was confirmed by western blot analysis and
RT-qPCR.
Apoptosis analysis
Cerebrovascular endothelial cells (1×104
cells) were isolated from experimental mice, trypsinized and
collected by centrifugation (1,500 × g) for 10 min in order to
apoptosis analysis. Cell density was adjusted to 5×106
cells/ml with PBS, labeled with annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI) using the Annexin
V-FITC kit from BD Biosciences (Franklin Lakes, NJ, USA) and
analyzed with a FACScan flow cytometer (BD Biosciences). Apoptotic
rate of cerebrovascular endothelial cells was analyzed by flow
cytometry (BD, Biosciences) using WinMDI software (version 2.9; BD
Biosciences). All experiments were repeated 3 times.
Statistical analysis
All data were presented as mean ± standard error of
the mean. Statistical significance was established using two-tailed
Student's t-test using SPSS software version 19.0 (IBM Corp.,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
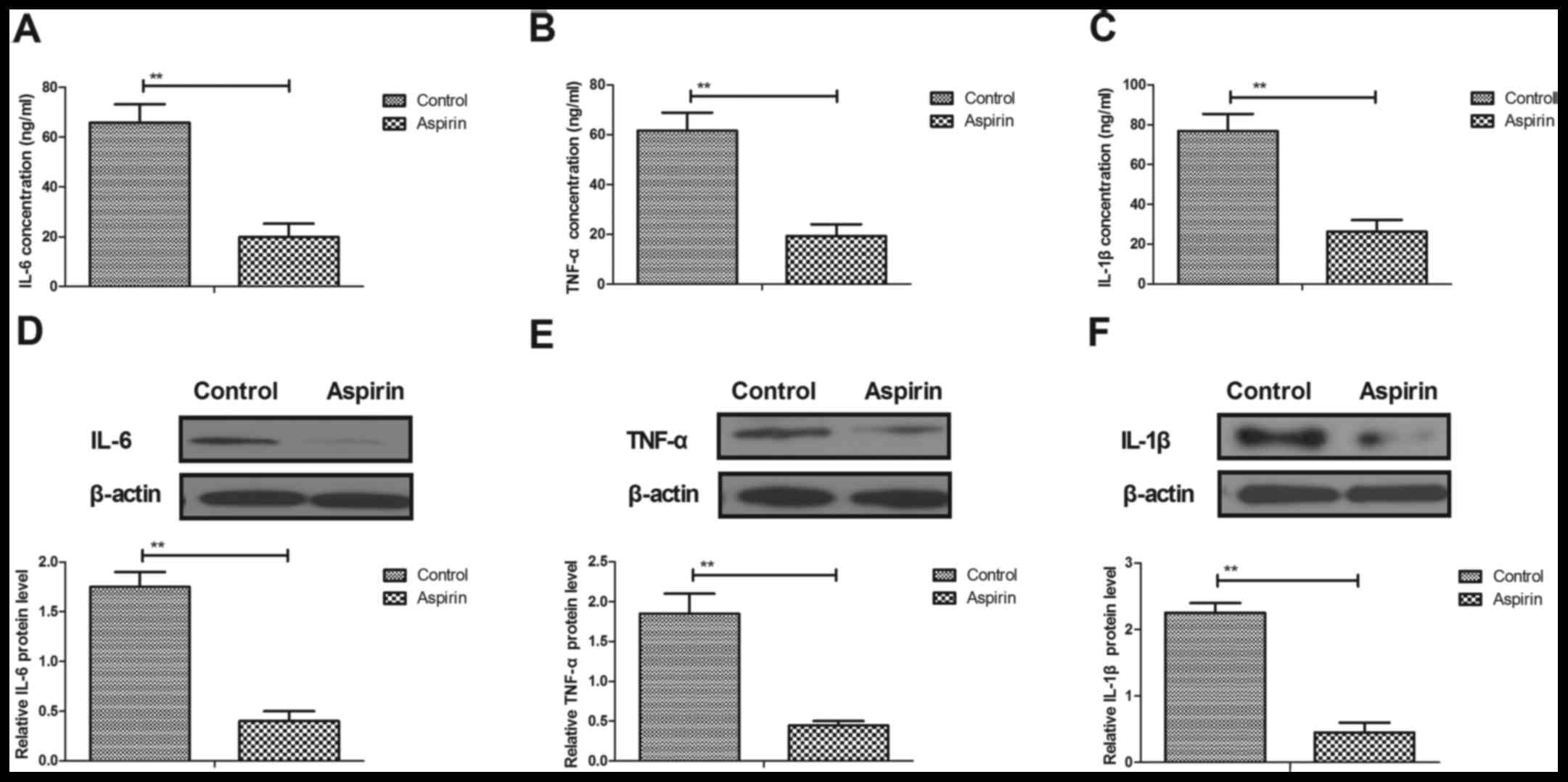
Aspirin inhibits inflammatory
cytokines in cerebrovascular endothelial cells and serum in a mouse
model of cerebral infarction
Inflammation has been regarded as a therapeutic
target in cerebral infarction (17). The present study analyzed the
cellular inflammatory response and the levels of inflammatory
mediators in cerebrovascular endothelial cells and serum in mice
with cerebral infarction. Serum concentrations of interleukin
(IL)-6, tumor necrosis factor (TNF)-α and IL-1β were significantly
decreased in aspirin-treated cerebral infarction mice compared with
expression levels in control mice (Fig. 1A-C, respectively). Western blot
analysis demonstrated that protein expression levels of IL-6, TNF-α
and IL-1β were downregulated in cerebrovascular endothelial cells
following treatment with aspirin (Fig.
1D-F, respectively). These results suggested that aspirin may
inhibit inflammatory cytokine expression in cerebrovascular
endothelial cells and serum in a mouse model of cerebral
infarction.
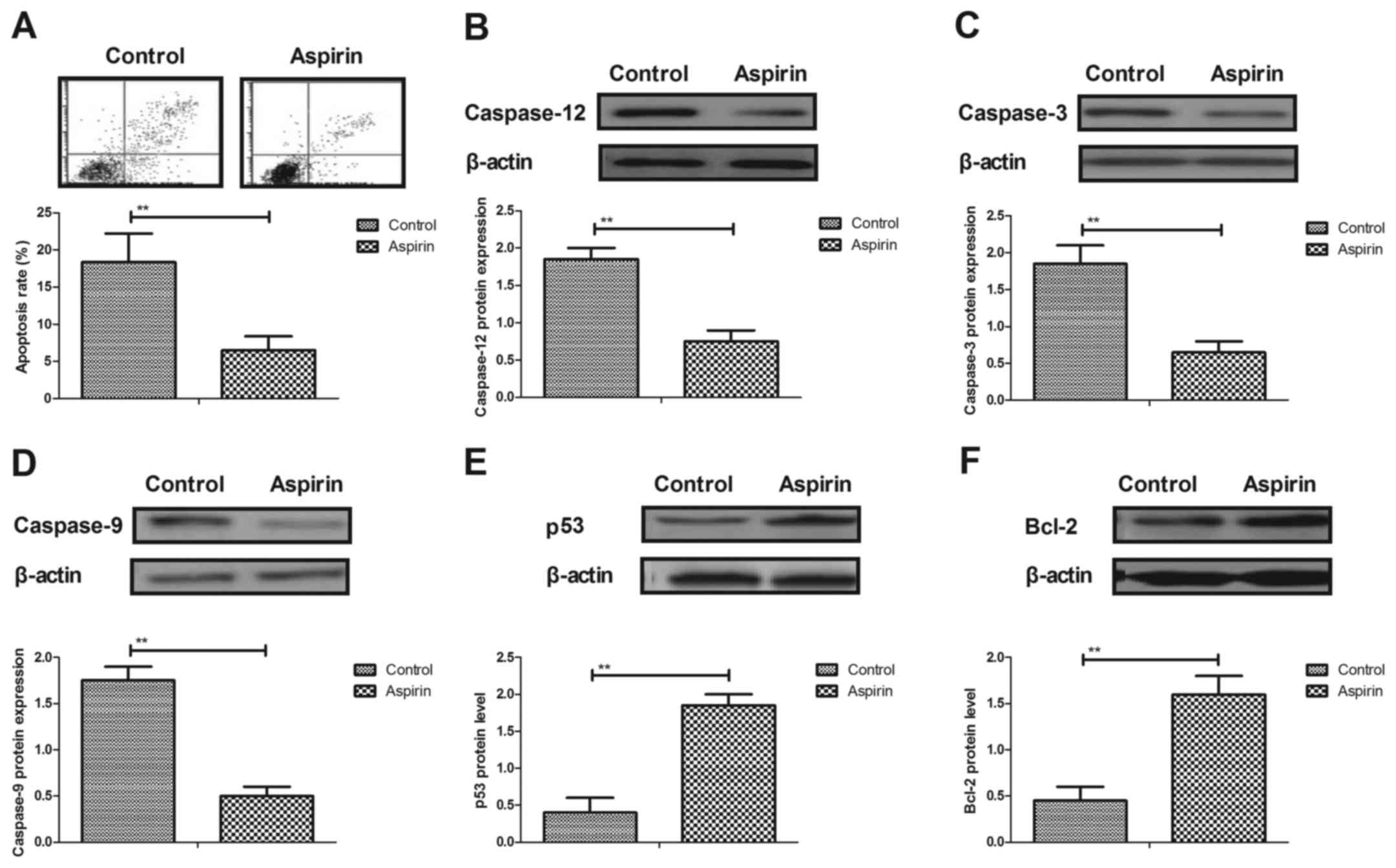
Aspirin inhibits apoptosis of
cerebrovascular endothelial cells in a mouse model of cerebral
infarction
The anti-apoptotic effects of aspirin in a cerebral
infarction mouse model were evaluated. The rate of apoptosis of
cerebrovascular endothelial cells was significantly decreased by
aspirin treatment, compared with control cells (Fig. 2A). Results from western blot
analysis demonstrated that caspase-12, caspase-3 and caspase-9
expression levels were downregulated in aspirin-treated mice
compared with control mice (Fig.
2B-D, respectively). However, expression levels of
antiapoptoticp53 and B-cell lymphoma 2(Bcl-2) proteins were
significantly increased in cerebrovascular endothelial cells in the
aspirin-treated mice compared with the controls (Fig. 2E and F). These results indicated
that aspirin may inhibit apoptosis in cerebrovascular endothelial
cells in a mouse model of cerebral infarction.
Aspirin treatment suppresses TLR4,
NF-κB, p65, IκBα, IKKβ, ADP, PAI-1, VWF and thromboxane expression
in cerebrovascular endothelial cells
To explore the therapeutic effects of aspirin, TLR4
and NF-κB expression in cerebrovascular endothelial cells was
analyzed. The protein expression level of TLR4 was downregulated in
cerebrovascular endothelial cells in aspirin-treated mice, compared
with control mice (Fig. 3A). The
results also demonstrated that aspirin treatment reduced the
protein expression levels of NF-κB p65, inhibitor of NF-κB kinase
(IKK)β and NF-κB inhibitor (IκB)α in cerebrovascular endothelial
cells compared with control-treated cells (Fig. 3B-D, respectively). RT-qPCR analysis
revealed that the mRNA expression levels of activator inhibitor
type-1 (ADP), (PAI-1), von Willebrand factor (VWF) and thromboxane
were significantly reduced in cerebrovascular endothelial cells
(Fig. 3E) and in serum (Fig. 3F) in experimental mice. These
results demonstrated that aspirin treatment may suppress TLR4NF-κB,
p65, ADP, PAI, VWF and thromboxane expression in cerebrovascular
endothelial cells.
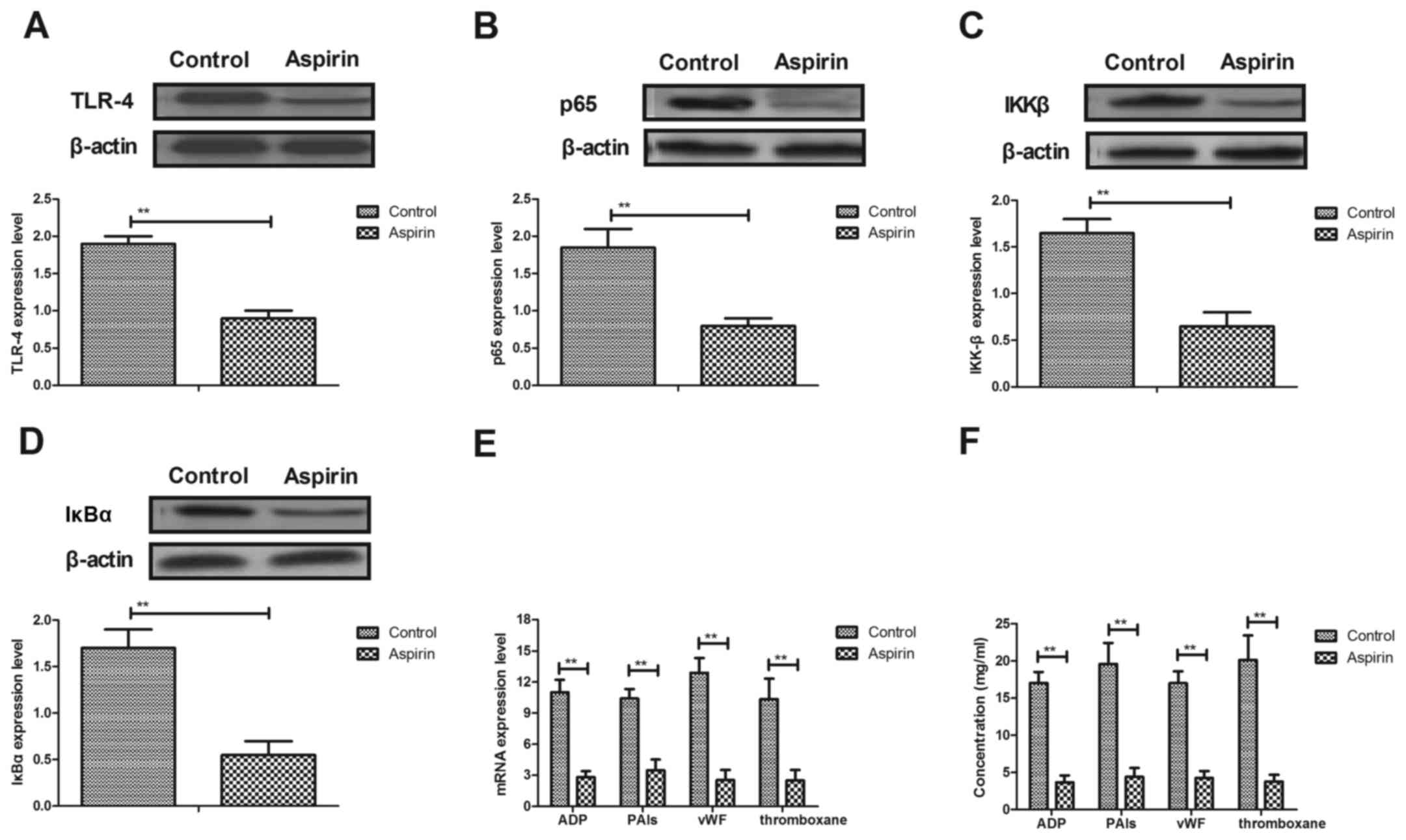 | Figure 3.Aspirin inhibits TLR4 and NF-κB
expression in cerebrovascular endothelial cells. (A) Aspirin
inhibited TLR4 expression levels in cerebrovascular endothelial
cells. (B-D) Effects of aspirin on protein expression levels of (B)
NF-κBp65, (C) IKKβ and (D) IκBα in cerebrovascular endothelial
cells in a mouse model of cerebral infarction. (E) mRNA expression
and (F) serum concentration levels of ADP, PAIs, vWF and
thromboxanein experimental mice. ADP, adenosine diphosphate; IKBα,
NF-κB inhibitor α; IKKβ, inhibitor of NF-κB kinase β; NF-κB,
nuclear factor κB; PAI, plasminogen activator inhibitor; TLR,
toll-like receptor; vWF, von Willebrand factor. **P<0.01 vs.
control group. |
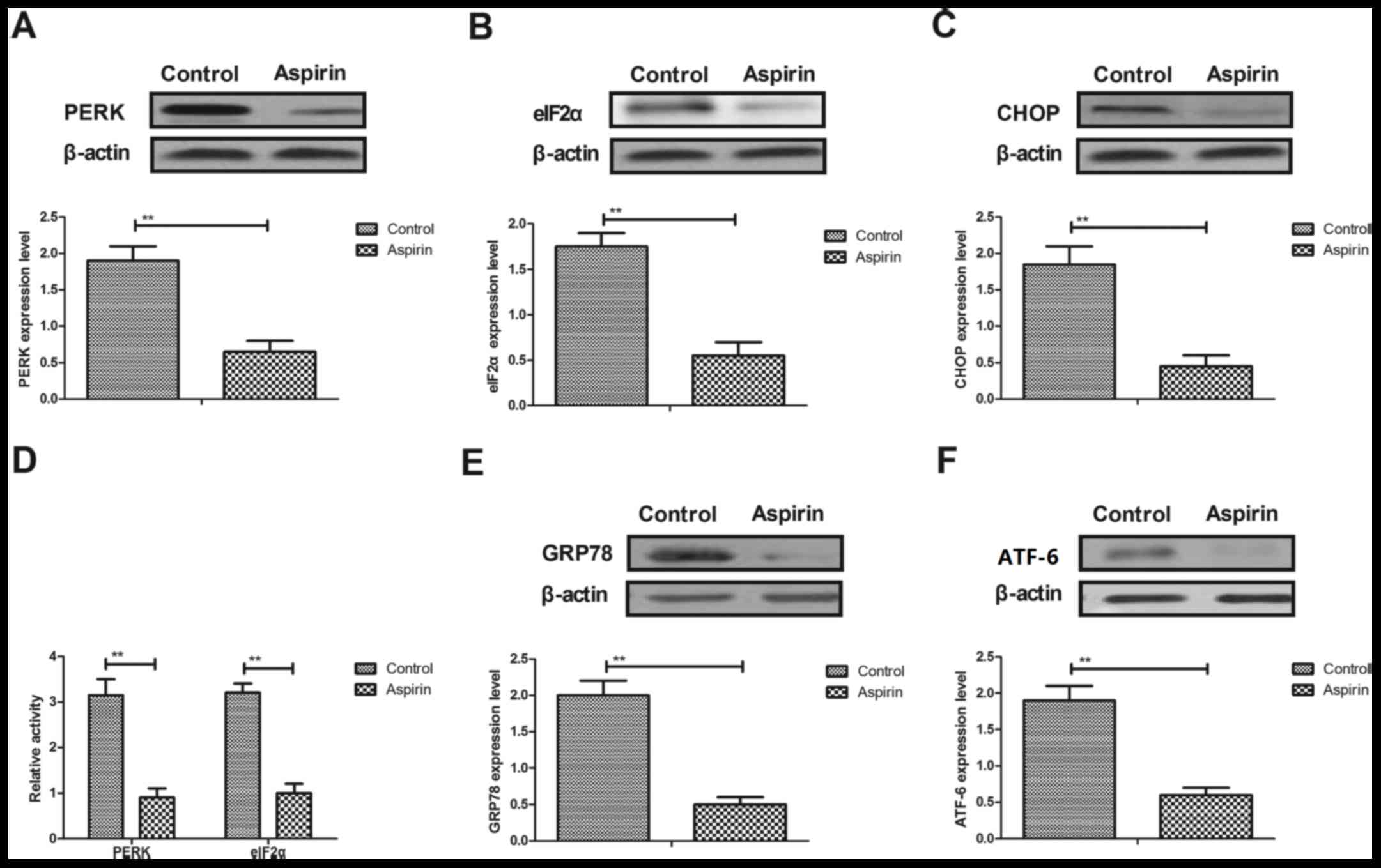
Aspirin treatment decreases ER stress
in cerebrovascular endothelial cells in a mouse model of cerebral
infarction
Changes in ER stress in cerebrovascular endothelial
cells in a mouse model of cerebral infarction were investigated. As
demonstrated in Fig. 4A-C, the
protein expression levels of PERK, eIF2α and C/EBP homologous
protein (CHOP) were significantly downregulated by aspirin
treatment in cerebrovascular endothelial cells, compared with
expression levels in control cells. The data also indicated that
PERK and eIF2α activity were downregulated by aspirin in
cerebrovascular endothelial cells (Fig. 4D). In addition, the protein
expression levels of glucose-regulated protein (GRP)78 and
activating transcription factor (ATF)-6 were significantly
downregulated by aspirin treatment in cerebrovascular endothelial
cells (Fig. 4E and F,
respectively). These results suggest that aspirin treatment
decreases ER stress in cerebrovascular endothelial cells in a mouse
model of cerebral infarction.
Aspirin treatment regulates ER stress
through TLR4-NF-κB signaling pathway in cerebrovascular endothelial
cells
The potential mechanism of aspirin-mediated
improvements of ER stress was investigated. The results
demonstrated that knockdown of TLR4by si-TLR4 transfection
inhibited the protein expression levels of p65, IKKβ and IκBα
(Fig. 5A-C, respectively), and
increased NF-κB activity in cerebrovascular endothelial cells
(Fig. 5D). Aspirin-inhibited
expression levels of GRP78, ATF-6 and CHOP were canceled by TLR4
knockdown in cerebrovascular endothelial cells (Fig. 5E-G). TLR4 knockdown also inhibited
aspirin-downregulated PERK and eIF2α activity in cerebrovascular
endothelial cells (Fig. 5H). These
results demonstrated that aspirin treatment downregulated ER stress
through the TLR4-NF-κB signaling pathway in cerebrovascular
endothelial cells.
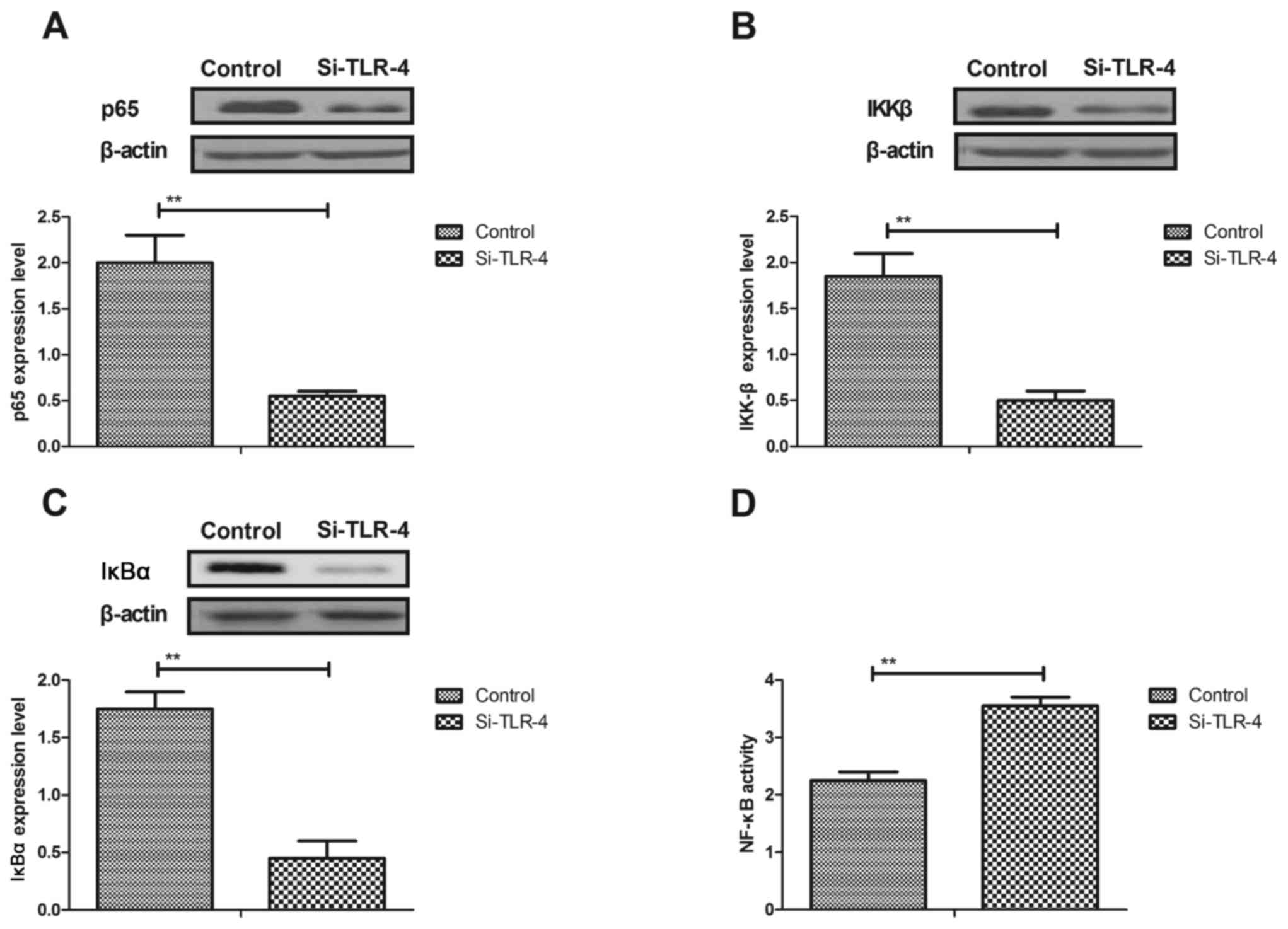 | Figure 5.Aspirin regulates ER stress through
the TLR4/NF-κB signaling pathway in cerebrovascular endothelial
cells. (A and B) Knockdown of TLR4 (by si-TLR4 transfection)
inhibited the protein expression levels of (A) NF-κBp65, (B) IKKβ
and (C) IκBα in cerebrovascular endothelial cells. (D) si-TLR4
transfected cells also exhibited downregulated NF-κB activity in
cerebrovascular endothelial cells. (E and G) si-TLR4 treatment
increased the aspirin-inhibited expression levels of (E) GRP78, (F)
ATF-6 and (G) CHOP in cerebrovascular endothelial cells. (H)
si-TLR4 reversed the aspirin-induced inhibition of PERK and eIF2α
activity in cerebrovascular endothelial cells. ATF, activating
transcription factor; CHOP, C/EBP homologous protein; eIF2α,
eukaryotic translation initiation factor 2 subunit 1; GRP,
glucose-regulated protein; IKBα, NF-κB inhibitor α; IKKβ, inhibitor
of NF-κB kinase β; NF-κB, nuclear factor-κB; PERK, protein kinase
R-like endoplasmic reticulum kinase; si, small interfering RNA;
TLR, toll-like receptor. **P<0.01 vs. control group. |
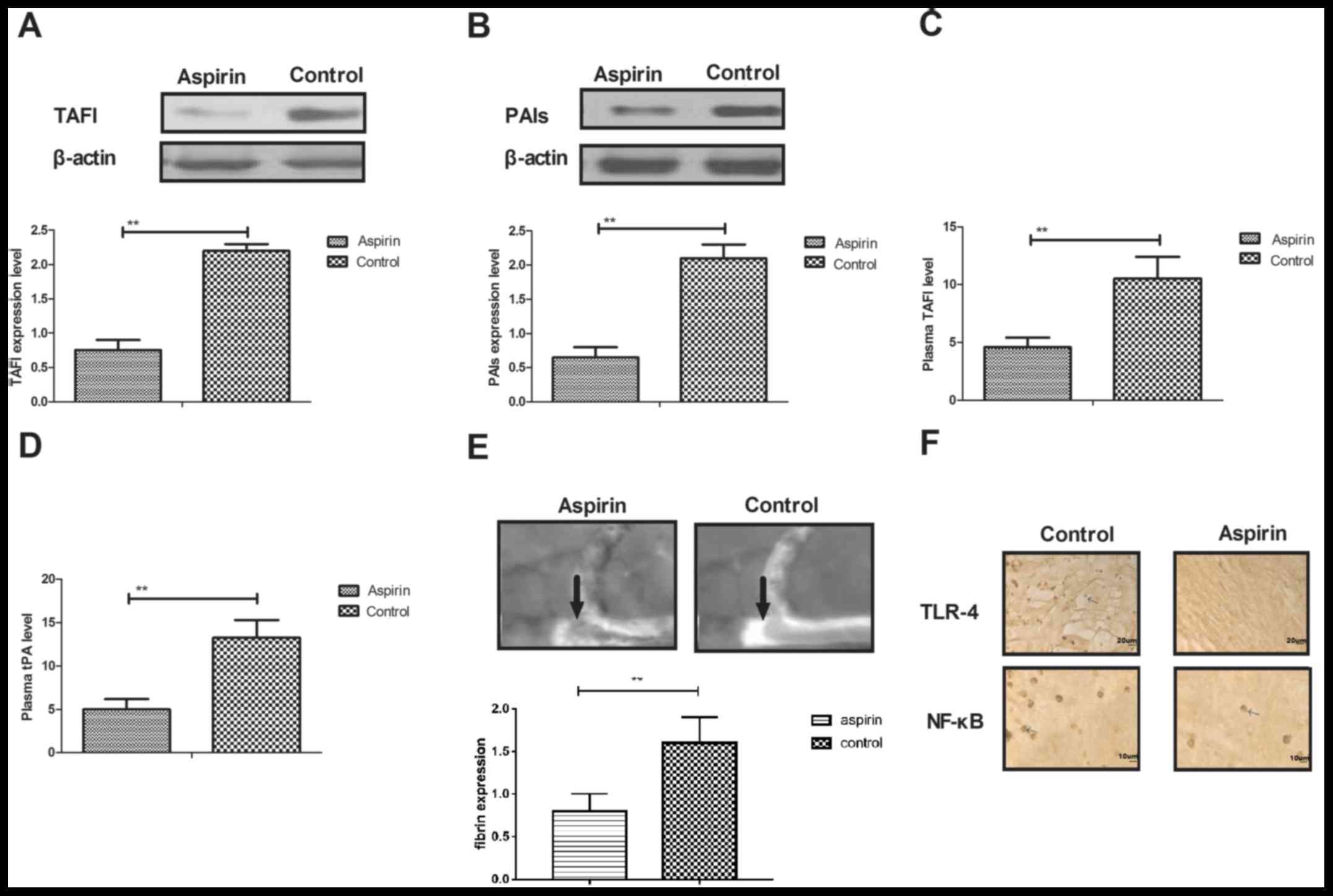
In vivo efficacy of aspirin for a
mouse model of cerebral infarction
The present study further analyzed the in
vivo efficacy of aspirin for cerebral infarction in a mouse
model. The results demonstrated that aspirin treatment may suppress
thrombolysis through increasing expression levels of TAFI and PAI-1
in cerebrovascular endothelial cells (Fig. 6A and B, respectively). TAFI and tPA
plasma concentration levels were significantly decreased in
cerebral infarction model mice following treatment with aspirin,
compared with control-treated mice (Fig. 6C and D, respectively). The result
revealed that aspirin could suppress thrombolysis in a mouse model
of cerebral infarction (Fig. 6E).
Immunohistochemical analysis demonstrated that TLR4 and NF-κB
expression levels were downregulated by aspirin treatment in
cerebrovascular lesions (Fig. 6F).
These results suggested that aspirin may be beneficial for the
treatment of cerebral infarction.
Discussion
Cerebral infarction is a combination of
pathophysiological processes that are induced by local
atherosclerosis cerebral ischemic necrosis (18,19).
Thrombolytic treatments for cerebral infarction are efficient in
clearing cerebrovascular congestion in patients (5,20).
The therapeutic effects of aspirin for cerebral infarction by
anti-platelet aggregation have been investigated in previous
studies (21,22). The present study investigated the
potential mechanism of aspirin-mediated treatments of cerebral
infarction in an animal model. The present study indicated that
aspirin inhibits inflammation and apoptosis of cerebrovascular
endothelial cells in experimental mice induced with autologous
arterial blood clot. The results also indicated that aspirin
treatment may inhibit ER stress through the suppression of
TLR4-mediated NF-κB signaling in cerebrovascular endothelial cells,
which may contribute to thrombolysis in a mouse model of cerebral
infarction
Inflammation is associated with the progression of
cerebral infarction. A previous study demonstrated that plasma
concentration levels of IL-6, TNF-α and IL-1β were upregulated in
patients with cerebral infarction (17,23,24).
Another study revealed that the inflammation-related NF-κB
signaling pathway was activated in patients with acute cerebral
infarction (25). It has also been
indicated that cytokine production and NF-κB activation were
upregulated in peripheral blood mononuclear cells of patients with
cerebral infarction (26). In the
present study, the data indicated that aspirin treatment
downregulated inflammation and NF-κB signaling in cerebrovascular
endothelial cells in a mouse model of cerebral infarction.
It has been reported that apoptosis and ER stress
are associated with aggravation of cerebrovascular diseases
(27,28). Increased ER stress in
cerebrovascular endothelial cells contributes to apoptosis of cells
and aggravation of cerebral infarction (29). Chen et al (30) demonstrated that the TLR4/NF-κB
pathway was involved in cognitive impairment, and
neuro-inflammatory and apoptotic responses. The results of the
present study demonstrated that aspirin treatment downregulated
TLR4 and NF-κB expression, which further decreased ER stress in
cerebrovascular endothelial cells in a mouse model of cerebral
infarction.
The present study investigated the efficacy of
aspirin treatment in vivo and demonstrated that aspirin
treatment improved thrombolysis by increasing the expression levels
of TAFI and PAI-1 in cerebrovascular endothelial cells. One
previous study provided further insight into the mechanism of
activated TAFI self-destruction, which indicated that TAFI deletion
mutant appears to be more stable than the activated TAFI control
(31). The tPA plasma
concentration levels were increased in the mouse model of cerebral
infarction following treatment with aspirin in the present study.
In addition, previous studies have also has indicated the
PAI-1regulates the balance of the plasma fibrinolytic and the blood
coagulation systems, and further initiates or promotes the
progression of cardiovascular disease (32,33).
The findings of the present study have suggested that aspirin
promoted thrombolysis in a mouse model of cerebral infarction
through decreasing TLR4 and NF-κB expression levels in
cerebrovascular lesions.
In conclusion, the present study examined the
therapeutic effects and potential mechanisms of aspirin in the
treatment of cerebral infarction. The results suggested that in a
mouse model of cerebral infarction aspirin treatment improved
cerebral infarction through decreasing inflammation and ER stress
in cerebrovascular endothelial cells and may have also promoted
thrombolysis through increasing the expression levels of ADP, PAIs,
VWF and thromboxane. Compared with previous studies, the present
study suggested that aspirin may modify cerebral infarction by
decreasing TLR4 and NF-κB expression levels via mediated ER stress
in a mouse model. This suggested that aspirin may contribute to
thrombolysis through the regulation of TLR4/NF-κB-mediated ER
stress in mice model.
References
|
1
|
García Serramito R, Santín Amo JM, Pena
Román P, Pita Buezas L, Gómez González L and Allut García A:
Cerebral infarction after pituitary apoplexy: Description of a case
and review of the literature. Neurocirugia (Astur). 27:310–314.
2016.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Romi F and Naess H: Spinal cord infarction
in clinical neurology: A review of characteristics and long-term
prognosis in comparison to cerebral infarction. Eur Neurol.
76:95–98. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arch AE and Sheth KN: Malignant cerebral
edema after large anterior circulation infarction: A review. Curr
Treat Options Cardiovasc Med. 16:2752014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Russek NS and Jensen MB: Histological
quantification of brain tissue inflammatory cell infiltration after
focal cerebral infarction: A systematic review. Int J Neurosci.
124:160–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hagiwara S, Yoshida A, Omata Y, Tsukada Y,
Takahashi H, Kamewada H, Koike S, Okuzumi K, Hishinuma A, Kobayashi
K and Nakano M: Desulfovibrio desulfuricans bacteremia in a patient
hospitalized with acute cerebral infarction: Case report and
review. J Infect Chemother. 20:274–277. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chiba F, Makino Y, Motomura A, Inokuchi G,
Ishii N, Torimitsu S, Sakuma A, Nagasawa S, Saito H, Yajima D, et
al: Bilateral middle cerebral artery infarction associated with
traumatic common carotid artery dissection: A case report and
review of literature. Forensic Sci Int. 236:e1–e4. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi J, Zhi P, Chen J, Wu P and Tan S:
Genetic variations in the thrombin-activatable fibrinolysis
inhibitor gene and risk of cardiovascular disease: A systematic
review and meta-analysis. Thromb Res. 134:610–616. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shinohara Y, Nishimaru K, Sawada T,
Terashi A, Handa S, Hirai S, Hayashi K, Tohgi H, Fukuuchi Y,
Uchiyama S, et al: Sarpogrelate-aspirin comparative clinical study
for efficacy and safety in secondary prevention of cerebral
infarction (S-ACCESS): A randomized, double-blind,
aspirin-controlled trial. Stroke. 39:1827–1833. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li YS: Aspirin in secondary prevention of
cerebral infarction. Zhonghua Nei Ke Za Zhi. 46:625–627. 2007.(In
Chinese). PubMed/NCBI
|
|
10
|
Dong MX, Hu QC, Shen P, Pan JX, Wei YD,
Liu YY, Ren YF, Liang ZH, Wang HY, Zhao LB and Xie P: Recombinant
tissue plasminogen activator induces neurological side effects
independent on thrombolysis in mechanical Animal models of focal
cerebral infarction: A systematic review and meta-analysis. PloS
One. 11:e01588482016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dubis J, Zuk N, Grendziak R, Zapotoczny N,
Pfanhauser M and Witkiewicz W: Activity of thrombin-activatable
fibrinolysis inhibitor in the plasma of patients with abdominal
aortic aneurysm. Blood Coagul Fibrinolysis. 25:226–231. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Naderi M, Dorgalaleh A, Alizadeh S, Khatib
Kashani Z, Tabibian S, Kazemi A, Dargahi H and Bamedi T:
Polymorphism of thrombin-activatable fibrinolysis inhibitor and
risk of intracranial haemorrhage in factor XIII deficiency.
Haemophilia. 20:e89–e92. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kazanci Yaroglu S, Yesilbas O, Ersoy M,
Kihtir HS, Yildirim HM and Sevketoglu E: Cerebral infarction and
femoral venous thrombosis detected in a patient with diabetic
ketoacidosis and heterozygous factor V Leiden G1691A and PAI-1
4G/5G mutations. J Pediatr Endocrinol Metab. 28:1183–1186.
2015.PubMed/NCBI
|
|
14
|
Akatsu H, Yamagata H, Chen Y, Miki T,
Kamino K, Takeda M, Campbell W, Kondo I, Kosaka K, Yamamoto T and
Okada H: TAFI polymorphisms at amino acids 147 and 325 are not risk
factors for cerebral infarction. Br J Haematol. 127:440–447. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei YP, Kita M, Shinmura K, Yan XQ,
Fukuyama R, Fushiki S and Imanishi J: Expression of IFN-gamma in
cerebrovascular endothelial cells from aged mice. J Interferon
Cytokine Res. 20:403–409. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cuenca-López MD, Brea D, Segura T, Galindo
MF, Antón-Martínez D, Agulla J, Castillo J and Jordán J:
Inflammation as a therapeutic agent in cerebral infarction:
cellular inflammatory response and inflammatory mediators. Rev
Neurol. 50:349–359. 2010.(In Spanish). PubMed/NCBI
|
|
18
|
Yan Z, Yu T, Wang Y, Wang M and Liang H:
Literature review and case report of intravenous thrombolysis in
acute cerebral infarction attributed to cervical arterial
dissection. J Stroke Cerebrovasc Dis. 24:e265–e269. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Taylor B, Lopresti M, Appelboom G and
Sander Connolly E Jr: Hemicraniectomy for malignant middle cerebral
artery territory infarction: An updated review. J Neurosurg Sci.
59:73–78. 2015.PubMed/NCBI
|
|
20
|
Sha D, Fan G and Zhang J: Multiple
cerebral infarction as the initial manifestation of left atrial
myxoma: A case report and literature review. Acta Cardiol.
69:189–192. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cohen LK and Jensen MB: Scaffolds for
intracerebral grafting of neural progenitor cells after cerebral
infarction: A systematic review. Arch Neurosci. 2:e253642015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang C, Feng F, Zhu Y, Wang R and Xing B:
Cerebral infarction caused by pituitary apoplexy: Case report and
review of literature. Turk Neurosurg. 24:782–787. 2014.PubMed/NCBI
|
|
23
|
Shimamura N, Matsuda N, Kakuta K, Narita A
and Ohkuma H: A model of rat embolic cerebral infarction with a
quantifiable, autologous arterial blood clot. Transl Stroke Res.
4:564–570. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fang MF, Tan F and Zhang X: Effects of
Huatan Tongluo Granule on SOCS-3 and TNF-α expressions in patients
with acute cerebral infarction. Zhongguo Zhong Xi Yi Jie He Za Zhi.
30:1142–1145. 2010.(In Chinese). PubMed/NCBI
|
|
25
|
Jiang Y and Lian YJ: Effects of Danhong
injection on hemodynamics and the inflammation-related NF-κB
signaling pathway in patients with acute cerebral infarction. Genet
Mol Res. 14:16929–16937. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim SJ, Jeong HJ, Lee KM, Moon PD, Yun JM,
Cho KH, Moon BS, Lee HJ, Hong SH, Kim HM and Um JY: The effect of
SHJKS on cytokines production and NF-κB activation in the
peripheral blood mononuclear cells of patients with cerebral
infarction. Immunopharmacol Immunotoxicol. 28:557–570. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li M, Peng J, Wang MD, Song YL, Mei YW and
Fang Y: Passive movement improves the learning and memory function
of rats with cerebral infarction by inhibiting neuron cell
apoptosis. Mol Neurobiol. 49:216–221. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li B, Tian J, Sun Y, Xu TR, Chi RF, Zhang
XL, Hu XL, Zhang YA, Qin FZ and Zhang WF: Activation of NADPH
oxidase mediates increased endoplasmic reticulum stress and left
ventricular remodeling after myocardial infarction in rabbits.
Biochim Biophys Acta. 1852:805–815. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li M, Peng J, Song Y, Liang H, Mei Y and
Fang Y: Electro-acupuncture combined with transcranial magnetic
stimulation improves learning and memory function of rats with
cerebral infarction by inhibiting neuron cell apoptosis. J Huazhong
Univ Sci Technolog Med Sci. 32:746–749. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen L, Hu L, Zhao J, Hong H, Feng F, Qu W
and Liu W: Chotosan improves Aβ1-42-induced cognitive impairment
and neuroinflammatory and apoptotic responses through the
inhibition of TLR-4/NF-κB signaling in mice. J Ethnopharmacol.
191:398–407. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Plug T and Meijers JC: New clues regarding
the mysterious mechanism of activated thrombin-activatable
fibrinolysis inhibitor self-destruction. J Thromb Haemost.
13:1081–1083. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oztuzcu S, Ergun S, Ulaşlı M, Nacarkahya
G, Iğci YZ, Iğci M, Bayraktar R, Tamer A, Çakmak EA and Arslan A:
Evaluation of factor V G1691A, prothrombin G20210A, factor XIII
V34L, MTHFR A1298C, MTHFR C677T and PAI-1 4G/5G genotype
frequencies of patients subjected to cardiovascular disease (CVD)
panel in south-east region of Turkey. Mol Biol Rep. 41:3671–3676.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hilbers FS, Boekel NB, van den Broek AJ,
van Hien R, Cornelissen S, Aleman BM, van't Veer LJ, van Leeuwen FE
and Schmidt MK: Genetic variants in TGFβ-1 and PAI-1 as possible
risk factors for cardiovascular disease after radiotherapy for
breast cancer. Radiother Oncol. 102:115–121. 2012. View Article : Google Scholar : PubMed/NCBI
|