Introduction
Alzheimer's disease (AD) is a late-onset
neurodegenerative disorder that is characterized by the deposition
of extracellular amyloid-β protein and construction of
intracellular neurofibrillary tangles, and results in a decline in
memory and cognition as well as alterations in behavior and
personality (1–3). Plaques and tangles present primarily
in the hippocampus, entorhinal cortex, amygdala and basal
forebrain, and induce neuronal cell death, which is associated with
AD progression. Thus, protecting neuronal cells against cell death
induced by various types of stress is essential in delaying the
progression of neurodegenerative diseases.
The HT22 mouse hippocampal cell line has been widely
used as an in vitro model for investigating oxidative
stress-induced cell death. It is established that the HT22 cells
lack an ionotropic glutamate receptor; thus, when the cells are
treated with a high concentration of glutamate, intracellular
glutathione is depleted and reactive oxygen species (ROS) levels
increase, and this process is independent of the ionotropic
glutamate receptor (4–6). In addition, it has been reported that
oxidative stress-induced neuronal cytotoxicity is mediated by the
mitogen-activated protein kinase (MAPK) signaling pathway and
apoptosis-inducing factor (AIF) (7–11),
and the activities of these pathways are widely associated with
cellular ROS levels. Conversely, antioxidant enzymes such as
superoxide dismutase 2 effectively protect against oxidative
stress-induced cell death in HT22 cells (12), indicating the involvement of
antioxidant enzymes in oxidative stress-induced neuronal cell
death.
Peroxiredoxins (Prxs) are a ubiquitous family of
antioxidant enzymes that catalyze the reduction of hydrogen
peroxide or alkyl peroxides. Previously, the function of Prxs has
interested researchers due to their ability to reduce ROS and
peroxynitrite, and their modulation of cytokines and growth factor
signaling cascades associated with cell apoptosis and proliferation
(13–15). Prxs have been reported to be
present in several cellular compartments, including the
mitochondria, nucleus, cytosol and peroxisomes. Prx V, also termed
atypical 2-Cys-Prx, is the smallest member of the family and has
only 10% sequence identity with typical 2-Cys-Prxs, and is widely
expressed in cellular compartments (16–19).
According a previous study, Prx V regulated the microglial
activation process by modulating the generation of ROS/nitrogen
oxide, which was regulated by the activation of thec-Jun N-terminal
kinase (JNK) signaling cascade activation (20). Reports have demonstrated that
recombinant Prx V attenuated ibotenate-mediated mouse brain
neurotoxicity and inhibited p53-induced HeLa cell apoptosis
(21,22). In addition, peroxisomal
overexpression of human Prx V protected against glial cell death
induced by ROS, while knockdown of Prx V inhibited osteoarthritic
chondrocyte growth via wnt/β-catenin signaling (23,24).
Furthermore, mitochondrial Prx V may regulate
the1-methyl-4-phenylpyridinium (MPP)+mitochondrial
pathway of apoptosis via crosstalk between the endoplasmic
reticulum and mitochondria (25).
It was also reported that Prx V is widely expressed in cellular and
subcellular structures in mammalian cells, which indicated the
important roles of Prx V in normal peripheral nerves (2,25).
These reports indicate that Prx V may be involved in
apoptosis-associated neuronal damage. However, the role of Prx V in
glutamate-induced HT22 cell apoptosis is not well understood at
present.
The present study investigated the role of Prx V in
the glutamate-mediated apoptosis of HT22 cells. The results
revealed that Prx V protein expression levels were upregulated
partially in a time- and dose-dependent manner by glutamate
stimulation. Furthermore, knockdown of Prx V increased apoptosis in
HT22 cells, potentially via extracellular signal-regulated kinase
(ERK) and AIF signaling pathways, which were mediated by glutamate.
The findings of the current study may provide a novel therapeutic
target for glutamate-induced hippocampal cell apoptosis.
Materials and methods
Chemicals
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS), L-glutamic acid and N-acetyl-L-cysteine (NAC)
were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
ERK, p38 MAPK and JNK inhibitors (PD98059, SB203580 and SP600125,
respectively) were all purchased from EMD Millipore (Billerica, MA,
USA).
Cell culture
HT22 cells, which were immortalized from murine
hippocampal neuroncells was kindly provide by Professor Dong-Seok
Lee, were maintained in DMEM supplemented with 10% (v/v) FBS and
penicillin and streptomycin (100 U/ml and 100 µg/ml respectively),
and incubated at 37°C and 5% CO2. Cells were
sub-cultured once every two days.
Construction of stable Prx V knockdown
HT22 cells
Short hairpin RNA (shRNA) specific to Prx V (shPrx V
LV3, H1/GFP&Puro) and control shRNA LV3 (H1/GFP&Puro)
lentivirus vectors were purchased from Shanghai GenePharma Co.,
Ltd. (Shanghai, China). The targeted sequence of shPrx V was
5′-GGAATCGACGTCTCAAGAGGT-3′ and the targeted sequence of the
negative control was 5′-GTTCTCCGAACGTGTCACGT-3′. To construct the
Prx V knockdown cells, 2×105/well of the HT22 cells were
seeded in a 6-well tissue culture plate for 24 h (37°C and 5%
CO2) prior to infection. The culture medium was replaced
by polybrene (5 µg/ml; Shanghai GenePharma Co., Ltd.) and the
packed lentivirus with a multiplicity of infection of 20 for 12 h,
and subsequently replaced with complete culture medium (DMEM with
10% FBS and antibiotics). Infected cells were selected by treatment
with puromycin and sub-cultured every 5–7 days (26). The expression of Prx V protein
levels were examined by western blotting 3 days after infection.
The proficiency of shRNA lentiviruses in selectively suppressing
Prx V expression, with no effects on Prx II expression, was
determined in virus-infected cells by western blotting.
Cell treatments
HT22 cells at a density of ~70% were treated with
various doses (0, 2, 4, 6, 8 and 10 mM) glutamate (Sigma-Aldrich,
Merck KGaA, Darmstadt, Germany) for 12 h at 37°C and 5%
CO2 to analyze cell viability and apoptosis. The
selection of doses for glutamate in the current study was based on
other reports that treated HT22 cells with glutamate to analyze the
toxicity of glutamate on neuronal cell death (9–12).
For NAC treatment, HT22 cells in were pretreated with NAC (1 or 5
mM) for 30 min at 37°C then flowed by glutamate (4 mM) treatment
for 12 h. For MAPK inhibitor treatment, approximately at 70%
density of the mock and short hairpin (sh)RNA-transfected HT22
cells were pretreated with SB203580 (5 µM, for p38 MAPK
inhibition), PD98059 (5 µM, for ERK MAPK inhibition) and SP600125
(5 µM, for JNK MAPK inhibition) for 30 min (37°C and 5%
CO2), followed by glutamate (4 mM) treatments (12 h,
37°C and 5% CO2).
Cell viability assay
Cell viability was examined via an MTT assay. HT22
cells were seeded at a density of 5,000 cells per well in 96-well
plates and treated with glutamate at 0–10 mM for 24 h (37°C and 5%
CO2) and the control cells were treated with media alone
without glutamate. The accumulation of formazan (the
dimethylsulfoxide was used as solvent) was determined following the
addition of MTT reagent (5 mg/ml) and the absorbance was measured
at a wavelength of 560 nm. Absorbance was detected by a UV max
Kineticmicroplate reader (Molecular Devices, LLC, Sunnyvale, CA,
USA).
Cell apoptosis detection
To determine cell apoptosis, 1×106
glutamate-treated HT22 cells were harvested using trypsin and
resuspended with PBS and stained with Annexin V-fluorescein
isothiocyanate (FITC)/propidium iodide (PI), according to the
manufacturer's protocol of the apoptosis detection kit (BD
Biosciences, Franklin Lakes, NJ, USA). The Annexin V-FITC/PI
positive cells were analyzed by flow cytometry on a BD FACSCalibur
(BD Biosciences). The results were analyses with Win MDI (version
2.9; BD Biosciences) software.
Cellular ROS detection
To determine the cellular ROS levels,
1×106 of the HT22 cells were treated with glutamate (0,
2, 4, 6, 8 and 10 mM) for 6 h. Subsequently, the cells were fixed
with 4% formaldehyde (room temperature, 15 min) and treated with
2.5% Triton X-100 for 5 min at room temperature, followed by
staining with 5 µM dihydroethidium (Beyotime Institute of
Biotechnology, Haimen, China) and 10 µg/ml DAPI (BIOSS, Beijing,
China) for 15 min at room temperature. Subsequently, the cellular
ROS levels were detected with a fluorescence microscope (5 field of
view, magnification, ×200) and the density of the fluorescence was
determined with ImageJ (K 1.45) software (National Institutes of
Health, Bethesda, MD, USA).
Western blot analysis
Cells were lysed using lysis buffer containing 20 mM
HEPES-OH (pH 7.0), 50 mM NaCl, 10% glycerol and 0.5% triton X-100
and proteinase inhibitors 0.5 µg/ml of leupeptin; 0.7 µg/ml of
pepstatin A; 0.1 mM of AEBSF [4-(2-aminonethly)-benzenesulfony
fluoride]; 2 µg/ml of aprotinin. Nuclear and cytosol proteins were
obtained using nuclear extraction kit (NXTRACT, Sigma-Aldrich,
Merck KGaA). The protein concentrations were analyzed with Bradford
assay. A total of 30 µg protein lysateswere separated on 12% SDS
gels and transferred onto nitrocellulose membranes (EMD Millipore).
Membranes were subsequently blocked with 5% skim milk
(Sigma-Aldrich, Merck KGaA) for 30 min at room tmeperature and
incubated with antibodies against Prx V (cat. no. SC-130337, Santa
Cruz Biotechnology, Inc., Dallas, TX, USA), Prx II (cat. no.
SC-515428), B-cell lymphoma-2 (Bcl-2; cat. no. SC-783),
cleaved-caspase-3 (cat. no. SC-136219), AIF (cat. no. AC-5586),
laminB (all Santa Cruz Biotechnology, Inc.) and β-actin
(Sigma-Aldrich; Merck KGaA) at a dilution of 1:2,000 at 4°C
overnight. Membranes were then probed with horseradish
peroxidase-conjugated goat anti-mouse IgG (cat. no. SAB3701105,
Sigma-Aldrich; Merck KGaA) or anti-rabbit IgG (cat. no. SAB3700878,
Sigma-Aldrich; Merck KGaA) at a dilution of 1:5,000 for 1 h at room
temperature. Following the removal of excess antibody by washing
with TBS, specific binding was detected using a chemiluminescence
detection system with chemiluminescence detection kit (cat. no.
RPN2135; GE Healthcare, Chicago, IL, USA) according to the
manufacturer's protocol, and the densitometric analysis was
performed by ImageJ (K 1.45) software (National Institutes of
Health, Bethesda, MD, USA).
Statistical analysis
Statistical significance between experimental groups
was determined with mean ± standard deviation by a one-way analysis
of variance and a Tukey test. The data were analyzed by SPSS (19.0)
software (IBM Corp., Armonk, NY, USA). All experiments were done at
least 3 times independent. P<0.05 was considered to indicate a
statistically significant difference.
Results
Induction of neuronal cell death by
glutamate in HT22 cells
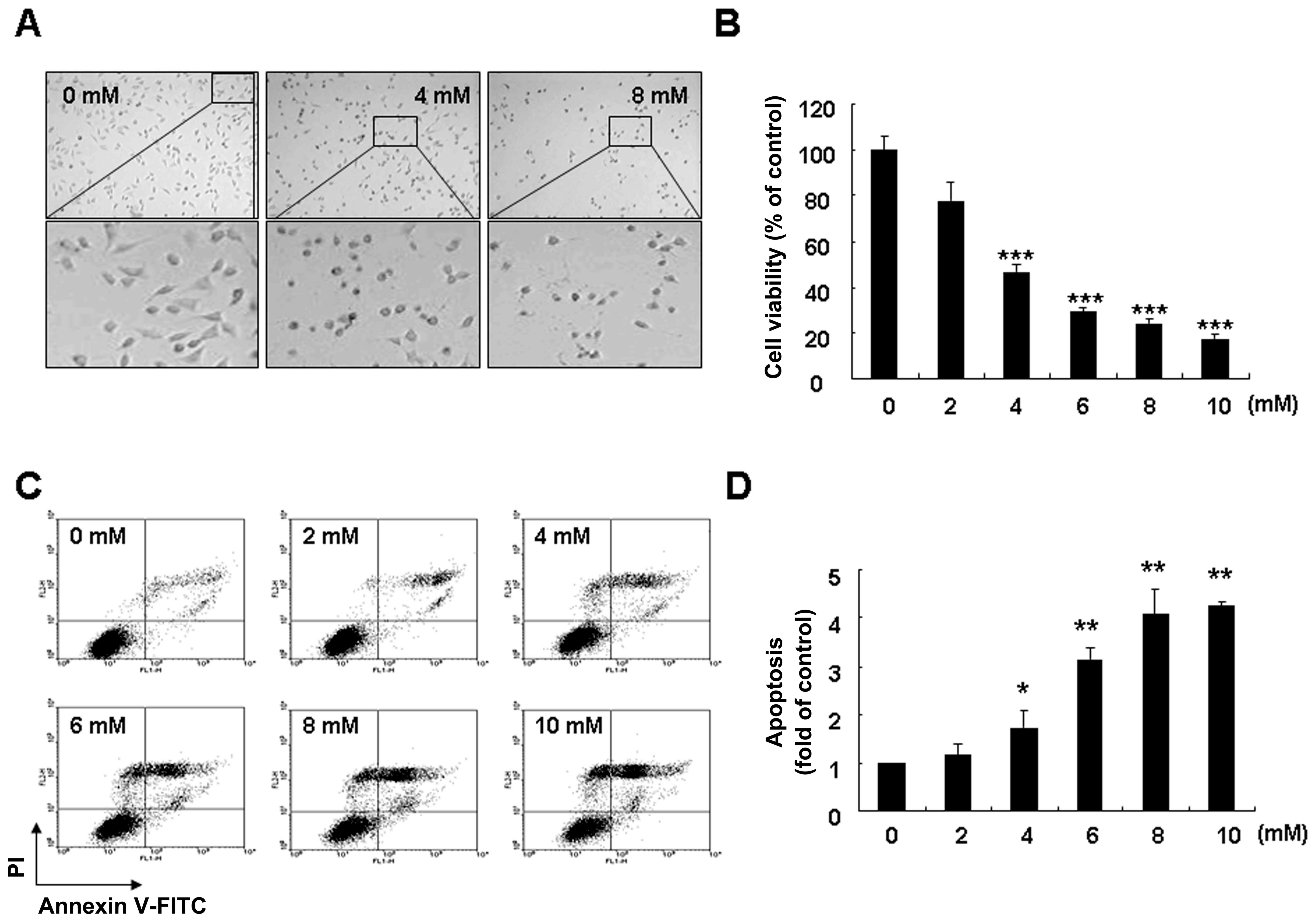
HT22 cells were treated with glutamate for 12 h, and
HT22 cells treated with glutamate for 12 h appeared to be less
viable compared with control cells (Fig. 1A). Furthermore, the present study
also determined the effect of glutamate on cell viability (Fig. 1B) by an MTT assay and cellular
apoptosis by fluorescence-activated cell sorting analysis (Fig. 1C and D) in HT22 cells. The results
demonstrated that treatment with glutamate significantly decreased
the cell viability and increased cell apoptosis in a
concentration-dependent manner in HT22 cells.
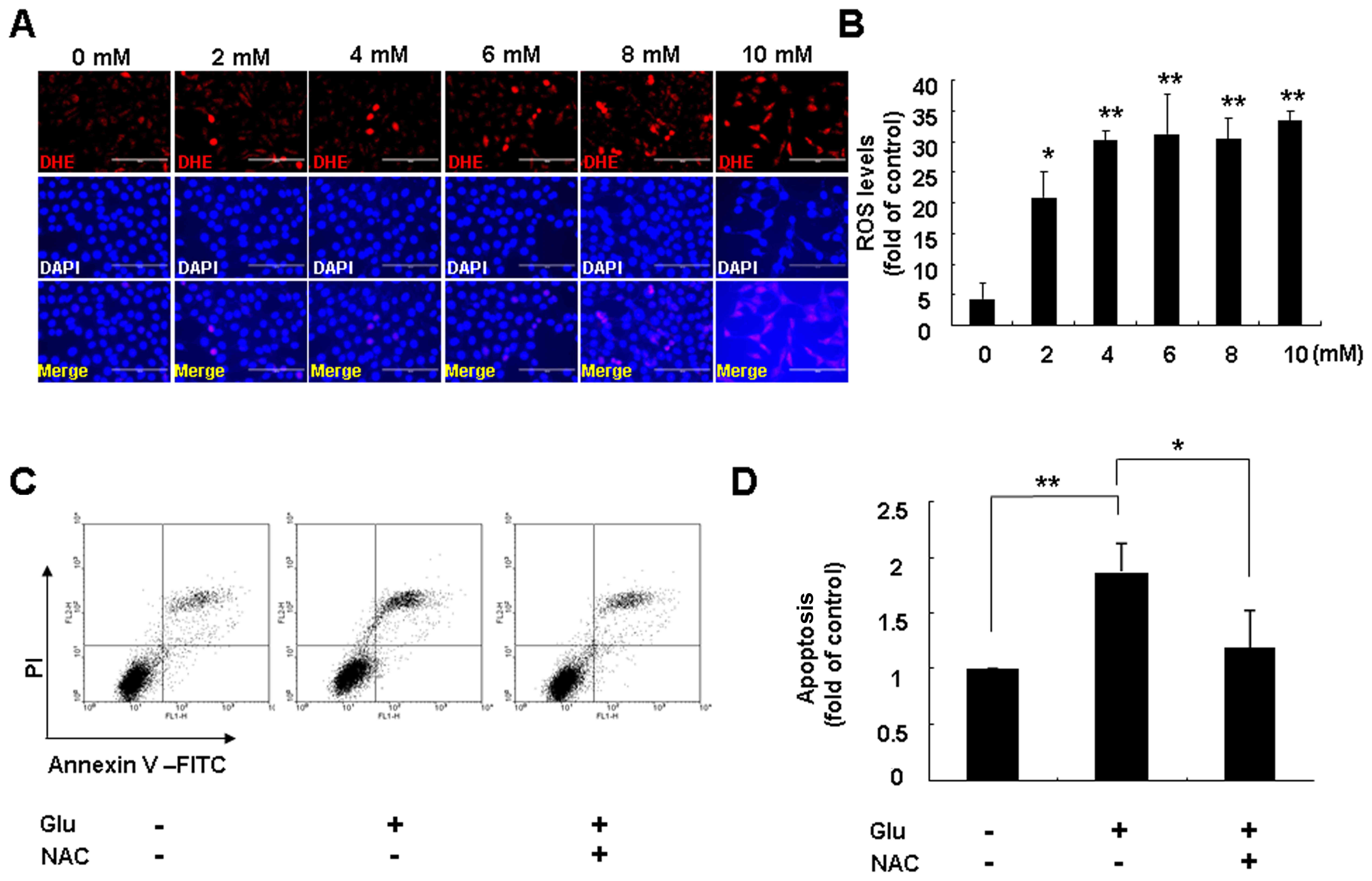
ROS is implicated in glutamate-induced
apoptosis in HT22 cells
It was previously reported that glutamate treatment
may increase the cellular ROS levels in HT22 cells (4,5,27).
To confirm whether ROS was associated with glutamate-induced HT22
cell apoptosis, the cells were treated with the indicated
concentrations (0, 2, 4, 6, 8 and 10 mM) of glutamate for 6 h and
the cellular ROS levels were determined by dihydroethidium
staining. Stained cells were observed with a fluorescence
microscope. The results demonstrated that the cellular ROS levels
were dose-dependently increased by glutamate treatment (Fig. 2A and B). Furthermore, inhibition of
cellular ROS accumulation with NAC, an ROS scavenger, significantly
reduced glutamate-induced HT22 cell apoptosis (Fig. 2C and D), as observed by flow
cytometric analysis. These findings indicateda potential
involvement of ROS in glutamate-stimulated HT22 cell apoptosis.
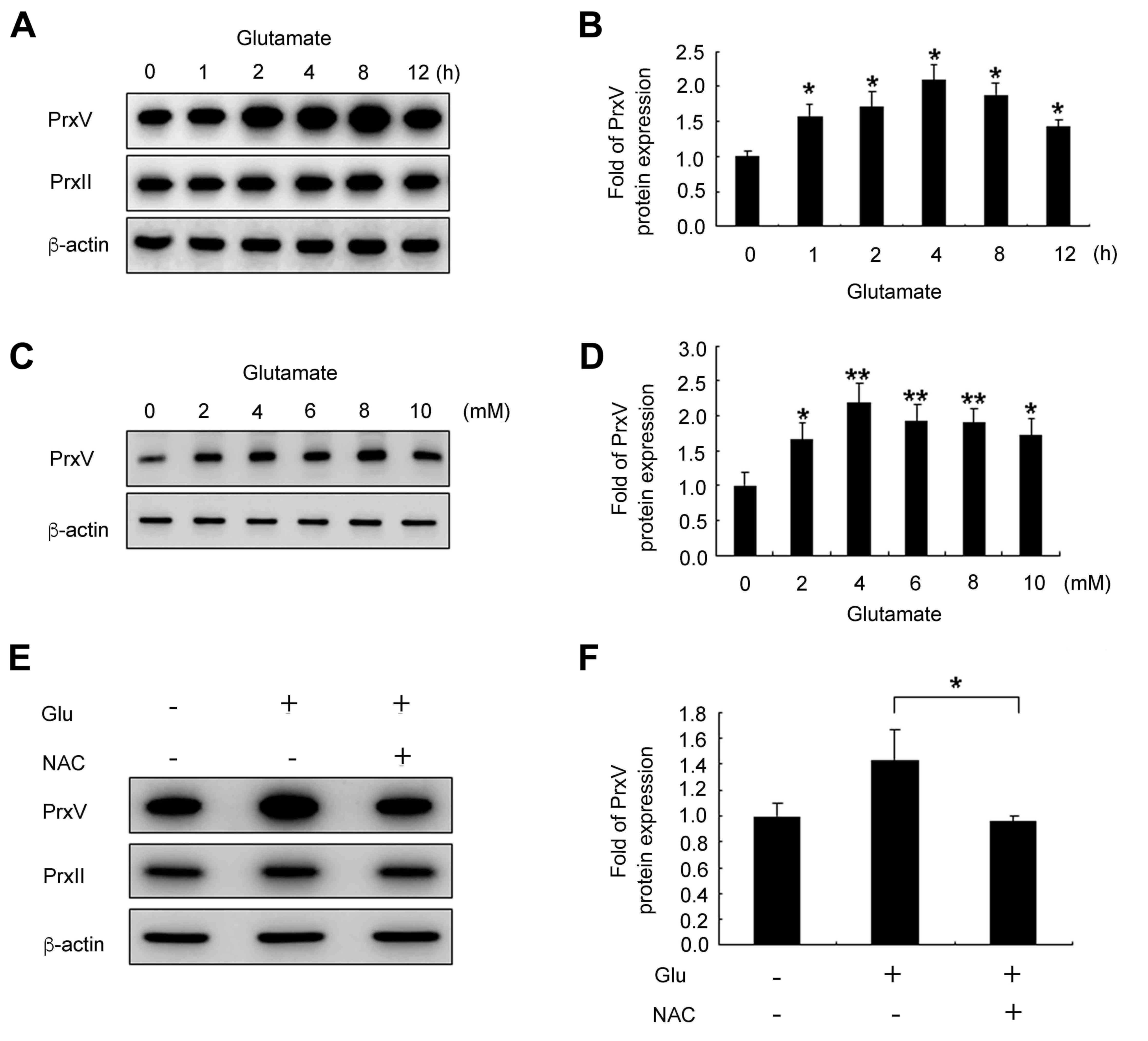
Glutamate treatment induces
upregulation of Prx V protein expression in HT22 cells
To investigate whether Prx V protein expression
levels are altered upon glutamate stimulation in HT22 cells, the
cells were stimulated for the indicated durations and
concentrations with glutamate and subjected to western blotting to
analyze Prx V protein expression. The protein expression of Prx V
by glutamate treatment was increased up to 4 h, and then decreases
at 8 and 12 h and up to 4 mM glutamate, then decreased at 6, 8 and
10 mM. (Fig. 3A-D), whereas Prx II
expression was not altered across any of the treatments. To verify
whether glutamate-induced Prx V protein upregulation was associated
with intercellular ROS accumulation, HT22 cells were pretreated
with NAC for 30 min following glutamate stimulation for 12 h. The
results demonstrated that treatment with NAC successfully reduced
Prx V protein expression levels in HT22 cells that also received
glutamate treatment (Fig. 3E and
F). These results indicated that Prx V may be involved in the
glutamate-induced HT22 cell apoptosis process that was associated
with intercellular ROS accumulation.
Knockdown of Prx V increases
glutamate-induced apoptosis in HT22 cells
As demonstrated in Fig.
4A, the Prx V protein expression levels were selectively
knocked down in the Prx V-shRNA lentivirus-infected cells (by
~80%), while the protein expression of Prx II was not affected. To
investigate the effect of Prx V shRNA on cell viability, mock and
Prx V shRNA-transfected HT22 cells were treated with glutamate and
cell viability was detected via an MTT assay. As demonstrated in
Fig. 4B, decreased cell viability
was observed in Prx V shRNA-transfected cells at 4 and 6 mM
compared with mock shRNA-transfected cells. Furthermore, the
apoptotic ratio between mock and Prx V shRNA-transfected HT22 cells
following glutamate treatment was analyzed by flow cytometry. The
results demonstrated that cellular apoptosis was significantly
increased in Prx V shRNA-transfected cells compared with mock cells
in HT22 cells treated with 4 mM glutamate, and increases in
apoptosis and decreases in cell viability were reversed by NAC
treatment (Fig. 4C-E).
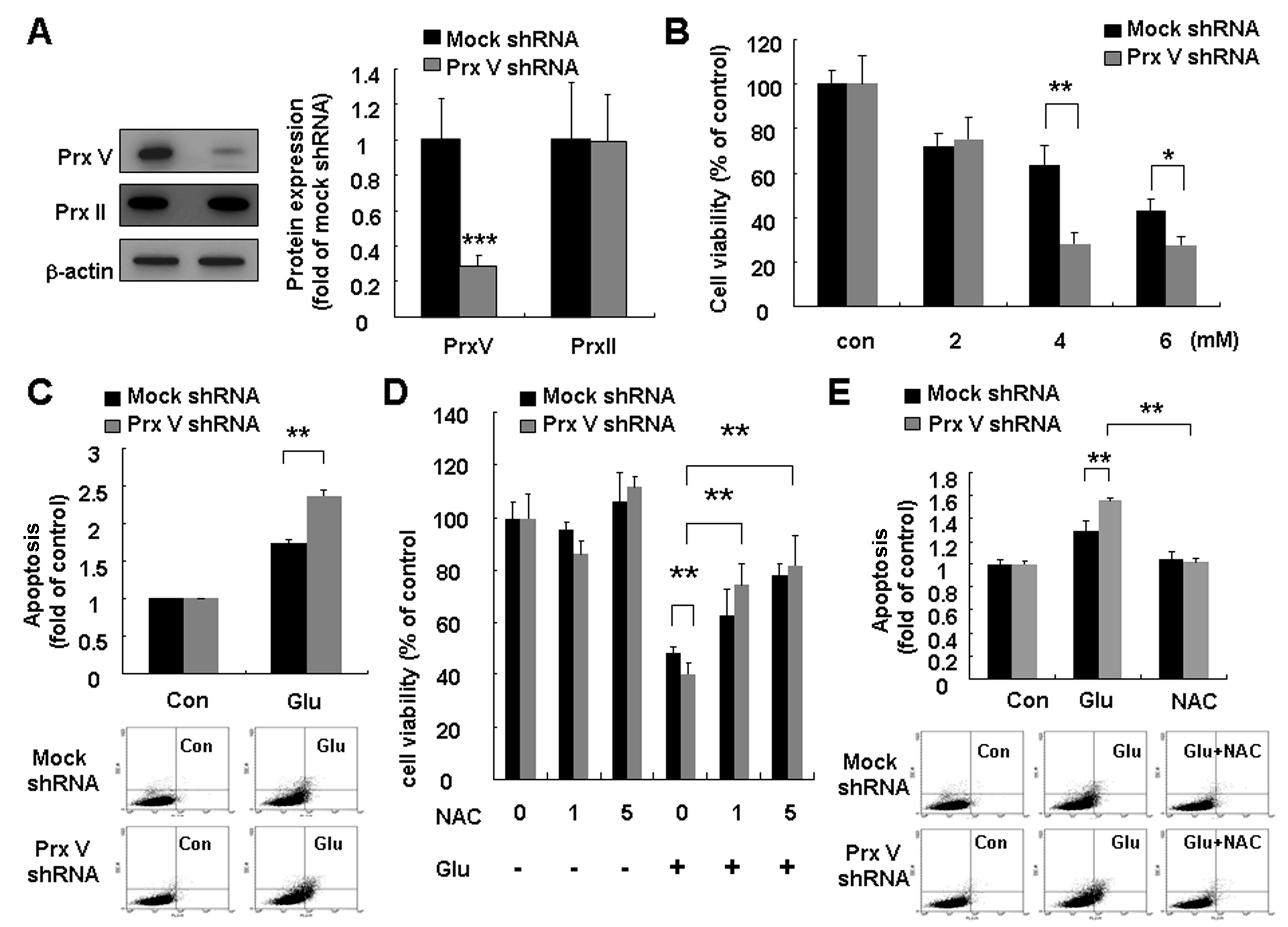 | Figure 4.Effect of Prx V on glutamate-induced
apoptosis in HT22 cells. (A) HT22 cells were stably transfected
with the Prx V shRNA or with a scrambled non-targeting shRNA
lentivirus vector, and western blot analysis was performed to
determine Prx V and Prx II protein expression levels. The results
were quantified by densitometric analysis. (B) Mock shRNA and Prx V
shRNA HT22 cells were stimulated with glutamate at the indicated
dosages and cell viability was determined via an MTT assay. (C)
Mock shRNA and Prx V shRNA HT22 cells were treated with 4 mM
glutamate and stained with Annexin V-FITC/PI. Cell apoptosis was
assessed by flow cytometry and the results were quantified and
statistically analyzed. (D) Mock shRNA and Prx V shRNA HT22 cells
were pretreated with NAC (1 and 5 mM) for 30 min followed by
glutamate treatment (4 mM), and cell viability was analyzed by an
MTT assay. (E) Mock shRNA and Prx V shRNA HT22 cells were
pretreated with NAC following treatment with 4 mM glutamate and
stained with Annexin V-FITC/PI. Cell apoptosis was assessed by flow
cytometry, and the results were quantified and statistically
analyzed. Data are presented as themean ± standard deviation (n=3).
For part A, ***P<0.001 vs. mock shRNA; for parts B-E, *P<0.05
and **P<0.01, as indicated. Prx, peroxiredoxin; shRNA, short
hairpin RNA; FITC, fluorescein isothiocyanate; PI, propidium
iodide; PE, phycoerythrin; NAC, N-acetyl-L-cysteine; Con, control;
Glu, glutamate. |
Prx V regulates HT22 cell apoptosis
via ERK, MAPK and AIF signaling pathways
To understand the underlying molecular mechanism of
Prx V in glutamate-induced cell apoptosis of HT22 cells, three
types of MAPK inhibitors, including SB203580 (for p38 MAPK),
PD98059 (for ERK MAPK) and SP600125 (for JNK MAPK) were used in
combination with glutamate treatment. The increased apoptotic
ratios were analyzed between mock shRNA and Prx V shRNA-transfected
HT22 cells by flow cytometry. As demonstrated in Fig. 5A, increased apoptosis following
glutamate treatment was suppressed by PD98059 treatment, which
selectively inhibits the ERK signaling pathway, both in mock shRNA
and Prx V shRNA-transfected cells, while the other compounds
exhibited no effect. Furthermore, western blotting results
demonstrated that the protein expression levels of the proapoptotic
protein cleaved-caspase-3 and the antiapoptotic protein Bcl-2 were
not significantly different between mock shRNA and Prx V
shRNA-transfected cells (Fig. 5B).
In addition, the current study also investigated the
translocalization of AIF between the mock shRNA and Prx V
shRNA-transfected HT22 cells. As demonstrated in Fig. 5C and D, increased nuclear
translocalization of AIF was observed in Prx V shRNA-transfected
cells compared with mock shRNA-transfected cells following
treatment with glutamate.
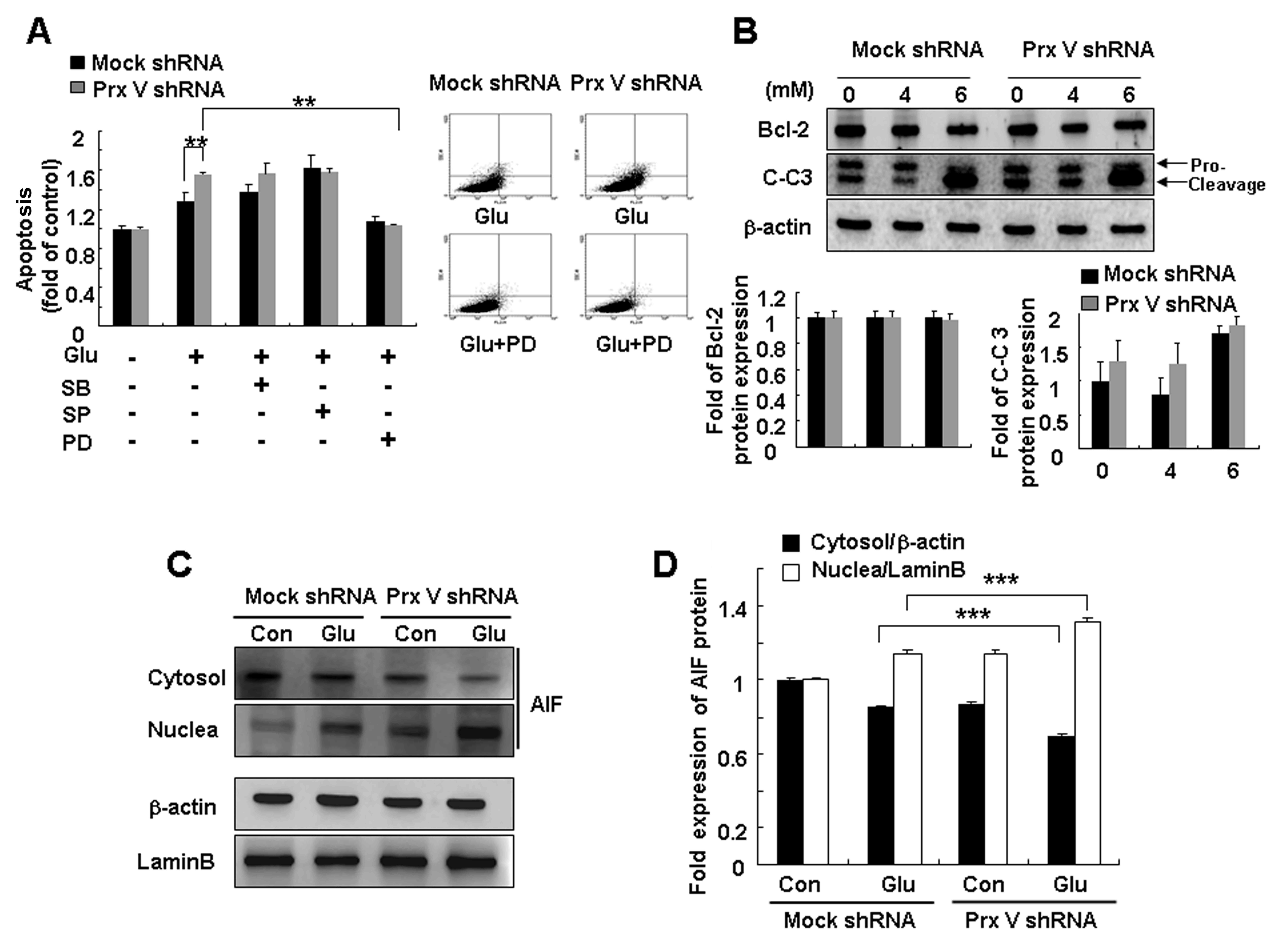 | Figure 5.Prx V regulates HT22 apoptosis via
ERK and AIF signaling pathways. (A) Mock shRNA and Prx V shRNA HT22
cells were pretreated with SB203580, PD98059 and SP600125, which
are inhibitors of p38, ERK and JNK mitogen-activated protein
kinases, respectively, for 30 min, followed by treatment with 4 mM
glutamate (12 h) and staining with Annexin V-FITC/PI. Cell
apoptosis was assessed by flow cytometry and the results were
quantified and statistically analyzed. (B) Western blot analysis
using Bcl-2 and cleaved-caspase-3 antibodies was performed in mock
shRNA and Prx V shRNA HT22 cells treated with 4 and 6 mM glutamate
(12 h). (C) Cytosolic and nuclear AIF protein levels were
determined by western blot analysis following glutamate treatment
(4 mM, 12 h) in mock shRNA and Prx V shRNA HT22 cells. (D)
Cytosolic and nuclear AIF protein levels were quantified by
densitometric analysis. Data are presented asthemean ± standard
deviation (n=3). **P<0.01 and ***P<0.001, as indicated. Prx,
peroxiredoxin; ERK, extracellular signal-regulated kinase; AIF,
apoptosis-inducing factor; shRNA, short hairpin RNA; JNK, c-Jun
N-terminal kinase; FITC, fluorescein isothiocyanate; PI, propidium
iodide; PE, pychoerythrin; Bcl-2, B-cell lymphoma-2; Glu,
glutamate; SB, SB203580; SP, SP600125; PD, PD98059; c-c3,
cleaved-caspase-3; Con, control. |
Discussion
Previously, protective role of antioxidants in
glutamate-induced neuronal cell death has been investigated
extensively. Phosphoproteomic analysis demonstrated that 17 types
of proteins were up- or down-regulated in glutamate-treated HT22
cells, and overexpression of Prx II and SOD1 efficiently protected
neuron cell death induced by ischemia and oxidative insult, but not
glutamate excitotoxicity (28,29).
In addition, delivery of Prx VI into cells attenuated tumor
necrosis factor-α- and glutamate-induced retinal ganglion cell
death (30). These reports
suggested an association between Prxs and glutamate-induced
neuronal cell death; however, little is known regarding Prx V
expression and function in HT22 mouse hippocampal cells. We
previously reported high expression levels of Prx V in neuron cells
(31) and microglia Prx V in mouse
brains and primary cell could be up-regulated by lipopolysaccharide
(LPS) stimulation (20). The
results of the current study demonstrated that glutamate treatment
significantly reduced cell viability and increased apoptosis in a
concentration-dependent manner, which was associated with increased
ROS levels stimulated by glutamate.
In addition, in the current study, Prx V expression
levels were upregulated during glutamate-stimulated apoptosis in
HT22 cells, indicating a potential regulatory effect of Prx V in
HT22 cell apoptosis. Numerous reports have demonstrated that
antioxidant expression depends on oxidative conditions. For
example, Prx I and Prx VI may be effectively induced in response to
oxidative insults, such as tert-butyl hydroquinone and LPS, via
binding of nuclear factor erythroid 2-related factor 2 to
antioxidant response elements on their promoter regions (32–34).
The present study also challenged HT22 cells with the chemical
antioxidant NAC prior to stimulation with glutamate. The results
demonstrated that the Prx V protein expression levels were
downregulated compared with those treated with only glutamate,
which was accompanied by reduced cell apoptosis. These findings
indicated that ROS signaling pathways may be implicated in
glutamate-stimulated Prx V induction in HT22 cells.
According to the results of the present study, Prx V
protein expression was upregulated during glutamate-induced
apoptosis in HT22 cells. Knockdown of Prx V expression in HT22
cells enhanced glutamate-induced decreases in cell viability and
increased cellular apoptosis, which may be associated with
increases in the cellular ROS levels. The MAPK signaling pathway is
widely involved in cell proliferation, migration, apoptosis and
differentiation, and has been reported to be closely associated
with cellular ROS levels (35–37).
However, to the best of our knowledge, no previous evidence has
demonstrated an association between Prx V and MAPK in neuronal cell
apoptosis. It has been reported that Prx V may prevent amyloid-β
oligomer-induced neuronal cell death through inhibiting the ERK
signaling pathway in HT22 cells, indicating an association between
Prx V and MAPK signaling pathways in cellular apoptosis (38). In the current study, experiments
that employed various MAPK inhibitors demonstrated that increased
cell apoptosis in Prx V knockdown cells was reversed by ERK
inhibitor treatment, indicating that the regulatory effects of Prx
V on HT22 cell apoptosis may be regulated via ERK MAPK signaling
pathways. Additionally, it has previously been reported that
glutamate-stimulated apoptosis may be mediated via the
translocation of AIF from the cytosol to nuclei in HT22 cells
(10). Thus, the present study
also assessed the translocation of AIF from the cytosol to nuclei
and apoptosis-associated protein expression levels between mock and
Prx V shRNA-transfected cells following glutamate treatment. The
results demonstrated that increased translocation of AIF from the
cytosol to nuclei was observed in Prx V shRNA cells, whereas there
was no difference in the protein expression of Bcl-2 and
cleaved-caspase-3 in both transfection groups, indicating that AIF
translocation maybe a key process for Prx V in regulating HT22 cell
apoptosis. Although the findings of the current study may provide
insight for understanding the role of Prx V in neuronal cell death,
certain limitations exist. For example, the results in the current
study do not provide direct evidence of the mechanism by which Prx
V regulates AIF translocation and the ERK signaling pathway and
signaling pathways involved in glutamate-mediated Prx V induction
in HT22 cells remain unclear. However, according to the results of
the present study, it may be hypothesized that Prx V may serve a
protective role via the ROS/ERK/AIF signaling cascade, which was
induced by glutamate in HT22 cells. However, this mechanism
requires further investigation.
In conclusion, the results of the present study
indicated that glutamate-stimulated activation of ROS may provoke
the induction of Prx V, which has an antioxidative function in
glutamate-stimulated HT22 cell apoptosis, and PrxV may serve a role
in neural apoptosis. Identification of the mechanisms underlying
this may provide novel therapeutic targets for treatment of
oxidative stress induced neuronal cell death.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Research
Project of Heilongjiang Bayi Agricultural University (grant nos.
XA2014-04 and XYB2012-14) and the Scientific Research Foundation of
Heilongjiang Provincial Education Department of China (grant no.
1252HQ007).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GNS and LL performed the cell culture and wrote the
manuscript. LF, YJ and MHJ performed the data analysis. YHH, CHJ
and YZJ construct the shRNA cell lines and preformed image
analysis. DSL, THK and YDC were involved in drafting the manuscript
and data analysis. HNS was a major contributor in writing the
manuscript and made substantial contributions to conception and
design.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Reddy PH and Bal MF: Amyloid beta,
mitochondrial dysfunction and synaptic damage: Implications for
cognitive decline in aging and Alzheimer's disease. Trends Mol Med.
14:45–53. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
LaFerla FM, Green KN and Oddo S:
Intracellular amyloid-beta in Alzheimer's disease. Nat Rev
Neurosci. 8:499–509. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Selkoe DJ: Alzheimer's disease: Genes,
proteins, and therapy. Physiol Rev. 81:741–766. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maher P and Davis JB: The role of
monoamine metabolism in oxidative glutamate toxicity. J Neurosci.
16:6394–6401. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tan S, Wood M and Maher P: Oxidative
stress induces a form of programmed cell death with characteristics
of both apoptosis and necrosis in neuronal cells. J Neurochem.
71:95–105. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tan S, Schubert D and Maher P: Oxytosis: A
novel form of programmed cell death. Curr Top Med Chem. 1:497–506.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stanciu M, Wang Y, Kentor R, Burke N,
Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW
and DeFranco DB: Persistent activation of ERK contributes to
glutamate-induced oxidative toxicity in a neuronal cell line and
primary cortical neuron cultures. J Biol Chem. 275:12200–12206.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choi BH, Hur EM, Lee JH, Jun DJ and Kim
KT: Protein kinase Cdelta-mediated proteasomal degradation of MAP
kinase phosphatase-1 contributes to glutamate-induced neuronal cell
death. J Cell Sci. 119:1329–1340. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Suh HW, Kang S and Kwon KS: Curcumin
attenuates glutamate-induced HT22 cell death by suppressing MAP
kinase signaling. Mol Cell Biochem. 298:187–194. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Y and Bhavnani BR: Glutamate-induced
apoptosis in neuronal cells is mediated via caspase-dependent and
independent mechanisms involving calpain and caspase-3 proteases as
well as apoptosis inducing factor (AIF) and this process is
inhibited by equine estrogens. BMC Neurosci. 7:492006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fukui M, Song JH, Choi J, Choi HJ and Zhu
BT: Mechanism of glutamate-induced neurotoxicity in HT22 mouse
hippocampal cells. Eur J Pharmacol. 617:1–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fukui M and Zhu BT: Mitochondrial
superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical
role in protection against glutamate-induced oxidative stress and
cell death in HT22 neuronal cells. Free Radic Biol Med. 48:821–830.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rhee SG, Kang SW, Jeong W, Chang TS, Yang
KS and Woo HA: Intracellular messenger function of hydrogen
peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol.
17:183–189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Budanov AV, Sablina AA, Feinstein E,
Koonin EV and Chumakov PM: Regeneration of peroxiredoxins by
p53-regulated sestrins, homologs of bacterial AhpD. Science.
304:596–600. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ,
Stadtman ER and Rhee SG: Reversible oxidation and inactivation of
the tumor suppressor PTEN in cells stimulated with peptide growth
factors. Proc Natl Acad Sci USA. 101:16419–16424. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Evrard C, Capron A, Marchand C, Clippe A,
Wattiez R, Soumillion P, Knoops B and Declercq JP: Crystal
structure of a dimeric oxidized form of human peroxiredoxin 5. J
Mol Biol. 337:1079–1090. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Knoops B, Clippe A, Bogard C, Arsalane K,
Wattiez R, Hermans C, Duconseille E, Falmagne P and Bernard A:
Cloning and characterization of AOEB166, a novel mammalian
antioxidant enzyme of the peroxiredoxin family. J Biol Chem.
274:30451–30458. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Seo MS, Kang SW, Kim K, Baines IC, Lee TH
and Rhee SG: Identification of a new type of mammalian
peroxiredoxin that forms an intramolecular disulfide as a reaction
intermediate. J Biol Chem. 275:20346–20354. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kropotov A, Gogvadze V, Shupliakov O,
Tomilin N, Serikov VB, Tomilin NV and Zhivotovsky B: Peroxiredoxin
V is essential for protection against apoptosis in human lung
carcinoma cells. Exp Cell Res. 312:2806–2815. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun HN, Kim SU, Huang SM, Kim JM, Park YH,
Kim SH, Yang HY, Chung KJ, Lee TH, Choi HS, et al: Microglial
peroxiredoxin V acts as an inducible anti-inflammatory antioxidant
through cooperation with redox signaling cascades. J Neurochem.
114:39–50. 2010.PubMed/NCBI
|
|
21
|
Plaisant F, Clippe A, Vander Stricht D,
Knoops B and Gressens P: Recombinant peroxiredoxin 5 protects
against excitotoxic brain lesions in newborn mice. Free Radic Biol
Med. 34:862–872. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou Y, Kok KH, Chun AC, Wong CM, Wu HW,
Lin MC, Fung PC, Kung H and Jin DY: Mouse peroxiredoxin V is a
thioredoxin peroxidase that inhibits p53-induced apoptosis. Biochem
Biophys Res Commun. 268:921–927. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Walbrecq G, Wang B, Becker S, Hannotiau A,
Fransen M and Knoops B: Antioxidant cytoprotection by peroxisomal
peroxiredoxin-5. Free Radic Biol Med. 84:215–226. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma Y, Li R, Zhang Y, Zhou L and Dai Y:
Knockdown of peroxiredoxin 5 inhibits the growth of osteoarthritic
chondrocytes via upregulating Wnt/β-catenin signaling. Free Radic
Biol Med. 76:251–260. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
De Simoni S, Linard D, Hermans E, Knoops B
and Goemaere J: Mitochondrial peroxiredoxin-5 as potential
modulator of mitochondria-ER crosstalk in MPP+-induced cell death.
J Neurochem. 125:473–485. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lu JL, Vallat JM, Pollard JD, Knoops B and
Ouvrier R: Expression of the antioxidant enzyme peroxiredoxin 5 in
the human peripheral nervous system. J Peripher Nerv Syst.
11:318–324. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lu J, Luo JH, Pang J, Cao JZ, Wu RH, Tong
ZT, Chen W and Xie D: Lentivirus-mediated RNA interference of
clusterin enhances the chemosensitivity of EJ bladder cancer cells
to epirubicin in vitro. Mol Med Rep. 6:1133–1139. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kang TH, Bae KH, Yu MJ, Kim WK, Hwang HR,
Jung H, Lee PY, Kang S, Yoon TS, Park SG, et al: Phosphoproteomic
analysis of neuronal cell death by glutamate-induced oxidative
stress. Proteomics. 7:2624–2635. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boulos S, Meloni BP, Arthur PG, Bojarski C
and Knuckey NW: Peroxiredoxin 2 overexpression protects cortical
neuronal cultures from ischemic and oxidative injury but not
glutamate excitotoxicity, whereas Cu/Zn superoxide dismutase 1
overexpression protects only against oxidative injury. J Neurosci
Res. 85:3089–3097. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fatma N, Kubo E, Sen M, Agarwal N,
Thoreson WB, Camras CB and Singh DP: Peroxiredoxin 6 delivery
attenuates TNF-alpha- and glutamate-induced retinal ganglion cell
death by limiting ROS levels and maintaining Ca2+ homeostasis.
Brain Res. 1233:63–78. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Murphy TH, Miyamoto M, Sastre A, Schnaar
RL and Coyle JT: Glutamate toxicity in a neuronal cell line
involves inhibition of cystine transport leading to oxidative
stress. Neuron. 2:1547–1558. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jin MH, Lee YH, Kim JM, Sun HN, Moon EY,
Shong MH, Kim SU, Lee SH, Lee TH, Yu DY and Lee DS:
Characterization of neural cell types expressing peroxiredoxins in
mouse brain. Neurosci Lett. 381:252–257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ishii T, Itoh K, Takahashi S, Sato H,
Yanagawa T, Katoh Y, Bannai S and Yamamoto M: Transcription factor
Nrf2 coordinately regulates a group of oxidative stress-inducible
genes in macrophages. J Biol Chem. 275:16023–16029. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chowdhury I, Mo Y, Gao L, Kazi A, Fisher
AB and Feinstein SI: Oxidant stress stimulates expression of the
human peroxiredoxin 6 gene by a transcriptional mechanism involving
an antioxidant response element. Free Radic Biol Med. 46:146–153.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun Y, Liu WZ, Liu T, Feng X, Yang N and
Zhou HF: Signaling pathway of MAPK/ERK in cell proliferation,
differentiation, migration, senescence and apoptosis. J Recept
Signal Transduct Res. 35:600–604. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou Y, Liang Y, Wei J, Chen J and Tang Q:
Lentiviral-mediated p38 MAPK RNAi attenuates aldosterone-induced
myocyte apoptosis. Mol Med Rep. 8:493–498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao WX, Tang SS, Jin X, Zhang CM, Zhang
T, Wang CC, Sun Y and Xiao XL: Olaquindox-induced apoptosis is
suppressed through p38 MAPK and ROS-mediated JNK pathways in HepG2
cells. Cell Biol Toxicol. 29:229–238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim B, Park J, Chang KT and Lee DS:
Peroxiredoxin 5 prevents amyloid-beta oligomer-induced neuronal
cell death by inhibiting ERK-Drp1-mediated mitochondrial
fragmentation. Free Radic Biol Med. 90:184–194. 2016. View Article : Google Scholar : PubMed/NCBI
|