Introduction
Systemic lupus erythematosus (SLE) is an autoimmune
disease (1,2). The pathological mechanism comprises
the attack of numerous parts of healthy tissues in the body by the
patients' own immune system (3).
SLE patients present with painful and swollen joints, fever and
chest pain (4). The exact causes
of SLE have remained to be elucidated; however, hormonal,
environmental and genetic factors are thought to be associated with
the causes of SLE (5). At present,
no cure is available for SLE, and the life expectancy of SLE
patients is relatively short (6).
Therefore, further research is required in the field of SLE.
Understanding the exact causes of SLE will facilitate the therapy
and drug design for SLE.
In the past several years, numerous susceptibility
genes for SLE have been discovered, including interferon regulatory
factor (IRF)5, signal transducer and activator of transcription 4,
IKAROS family zinc finger 1, ETS proto-oncogene 1 and
deoxyribonuclease 1-like 3 (7–11).
Although the exact functional association between these genes and
SLE require further investigation, these results indicate a link of
genetic factors with SLE. Comparing the genomes of monozygotic
twins discordant for phenotypes is a tool to identify possible
mutations and copy number variations (CNVs) causing these
discordant phenotypes (12–14).
Kondo et al (15) studied a
monozygotic twin discordant for Van der Woude and popliteal
pterygium syndromes. They identified genetic differences in the
IRF6 gene that are associated with the phenotype discordance for
the monozygotic twin. In the present study, whole-genome sequencing
(WGS), a widely used research tool for studying genetic disease
(16–18), was applied to assess a monozygotic
twin discordant for SLE to identify the possibly responsible
mutation(s).
In the present study, a monozygotic twin discordant
for SLE was assessed by using WGS. Although the putative discordant
exonic variants between the twins were selected from the WGS data,
it was not possible to further verify these variants by
conventional Sanger sequencing. Of note, CNVs changes in SLE
pathway genes were detected in the twin discordant for SLE.
Therefore, CNVs changes in SLE genes may be associated with the
pathology of the twin discordant for SLE.
Materials and methods
Patients and genomic DNA
isolation
Whole blood samples of twin A and twin B were
obtained at the First People's Hospital of Yunnan Province
(Kunming, China) during their visit to the clinical center in
August 2016. The affected individual (twin A) was diagnosed with
SLE. The parents of the twins are healthy.
The present study was approved by the Ethics
Committee of the First People's Hospital of Yunnan Province
(Affiliated Hospital of Kunming University of Science and
Technology). Written informed consent was obtained from the
participants. Genomic DNA from the whole blood samples were
extracted using the E.Z.N.A.® Blood DNA Kit (cat. no.
D3392-02; Omega Bio-Tek, Inc., Norcross, GA, USA) according to the
manufacturer's protocol.
WGS and single nucleotide variant
(SNV)/insertions or deletions (indel) and CNV analysis
WGS was performed on the genomic DNA of the
leukocytes from each of the twins by using the HiSeq X Ten platform
(Macrogen, Inc., Seoul, South Korea). All of the experiments were
performed by using TruSeq Nano DNA kit (cat. no. 20015965;
Illumina, San Diego, CA, USA) based on the Illumina TruSeq Nano DNA
library preparation guide (Illumina). In brief, genomic DNA was
fragmented by Covaris sonicator system to obtain 300–400 bp
fragments. The DNA fragments were converted into blunt ends using
the End Repair mix (Illumina). A single ‘A’ nucleotide was added to
the 3′ ends of the blunted fragments to prevent them from ligating
to one another during the adapter ligation reaction. The ends of
the DNA fragments were ligated to multiple indexing adapters. The
DNA fragments were then sequenced using a HiSeq X Ten sequencer
(Macrogen, Inc.).
Candidate SNVs, indels and CNVs were extracted and
annotated by software including Isaac Aligner (19), Isaac Variant Caller (19), SnpEff (20), Control-FREEC (21) and Manta (22). The detailed steps of analyzing WGS
data are displayed in Table I. In
general, the non-synonymous, splicing, stop gained, stop loss and
frame shift variants were extracted from the total variants. The
SNVs/indels were further filtered by using the dbSNP (23), 1000 genome (24) and ESP6500 databases (http://evs.gs.washington.edu/EVS/) with a
threshold of minor allele frequency (MAF)<0.01. The candidate
discordant SNVs/indels between the twins were identified by
comparing the above variants of twin A (the affected individual)
with those of twin B. The discordant SNVs/indels observed in twin A
and twin B were determined as a candidate discordant SNVs/indels
set. These variants were further filtered by using SIFT score
<0.05 (25), Polyphen-2 score
>0.85 (26) and other criteria
[insufficient coverage (<5) and obvious false-positive
variants]. Integrative Genomics Viewer software (27,28)
was used to exclude the obvious false-positive variants by
visualization of alignment data from the two twins. After the above
filtrations, the putative discordant SNVs/indels were selected out
and validated by conventional Sanger sequencing.
 | Table I.Sequencing results and exclusion
criteria for whole genome sequencing data analysis. |
Table I.
Sequencing results and exclusion
criteria for whole genome sequencing data analysis.
| Parameter | Twin A | Twin B |
|---|
| Total reads | 782,904,696 | 793,057,758 |
| Total yield
(Mbp) | 117,435 | 118,958 |
| Mappable yield
(Mbp) | 100,041 | 101,845 |
| Mappable mean depth
(X) | 35.00 | 35.60 |
| SNVs and Indels |
|
|
| No. of total
variants | 3,585,567 | 3,586,390 |
| No. of
non-synonymous, splicing, stop gained, stop loss and frame shift
variants | 22120 | 22125 |
| No. of variants after
dbSNP, 1,000 genome and ESP6500 databases filtering | 234 | 234 |
| (Minor allele
frequency<0.01) |
|
|
| No. of variants in
twin A but not in twin B and filtered by SIFT (<0.05),
polyphen-2 | 17 |
|
| (>0.85) and other
criteria[insufficient coverage (<5) and obvious false-positive
variants] |
|
|
| No. of putative
discordant exonic variants chosen for Sanger sequencing (Table II) | 8 |
|
| No. of chosen
putative discordant variants validated by Sanger sequencing | 0 |
|
| Copy no.
variants |
|
|
| Gains
(>2) | 554 | 572 |
| Losses
(<2) | 285 | 316 |
In the CNV analysis, the CNVs between twin A and
twin B were compared. The discordant CNVs between twin A and twin B
were respectively subjected to Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway enrichment analysis by using PERL and the R
programming language. R's phyper was used to calculate the P-value
in the KEGG pathway enrichment analysis.
Polymerase chain reaction (PCR)
amplification and sequencing
The fragments of candidate genes harboring the
discordant SNVs/indels (Table I)
were amplified using PCR. The primers used for amplification and
sequencing of zinc finger protein 595, ankyrin repeat domain 20
family member (ANKRD20)A2, ANKRD20A4, ArfGAP with GTPase domain,
Speedy/RINGO cell cycle regulator family member E2B,
γ-glutamyltransferase 1 (GGT1) and eukaryotic translation
initiation factor 3 gene fragments were obtained from the Beijing
Genomics Institute (Shenzhen, China). The sequences of the primers
are available on request. Amplification of the fragments was
performed in a volume of 25 µl containing 30 ng genomic DNA, 50 µM
deoxynucleoside triphosphate, 10X LA Taq™ PCR buffer, 2.5 units of
Takara LA Taq™ (Takara Bio Inc., Otsu, Japan) and 0.2 µM of each
forward and reverse primer. The PCR amplification for these gene
fragments was performed using a denaturation step of 94°C for 5
min, followed by 35 cycles of denaturation at 94°C for 30 sec,
annealing at 60°C and extension at 72°C for 30 sec, and ended with
a final extension step at 72°C for 7 min.
The PCR products were purified using a Genomic DNA
Purification kit (cat. no. DP204-02; Tiangen Biotech Co., Ltd.,
Tiangen, China) and were sequenced using sequencing primers
(available upon request) and the Big Dye Terminator v.3.1 Cycle
Sequencing kit (cat. no. 4337456; Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) on an ABI Prism 3730 DNA
sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Sequences were compared between the two twins to verify the
discordant variants.
Results
WGS
To identify a potential association of de
novo variants and CNVs with SLE, WGS was performed on the DNA
of leukocytes from the two twins. A total of 3,585,567 and
793,057,758 variants were identified in twin A and B, respectively.
The number of non-synonymous, splicing, stop gained, stop loss and
frame shift variants in twin A and B was 22,120 and 22,125,
respectively. The above potentially functional variants were
further filtered with the dbSNP (23), 1000 genome (24) and ESP6500 databases (http://evs.gs.washington.edu/EVS/), as well as
SIFT score <0.05 (19) and
Polyphen-2 score >0.85 (26),
as presented in Table I. At last,
8 putative variants in 7 genes were finally selected out for
subsequent Sanger sequencing (Table
II). Of the 7 genes, only the GGT1 gene has records in the
Online Mendelian Inheritance in Man database (29). The GGT1 gene has been reported to
be associated with diseases, including glutathionuria and
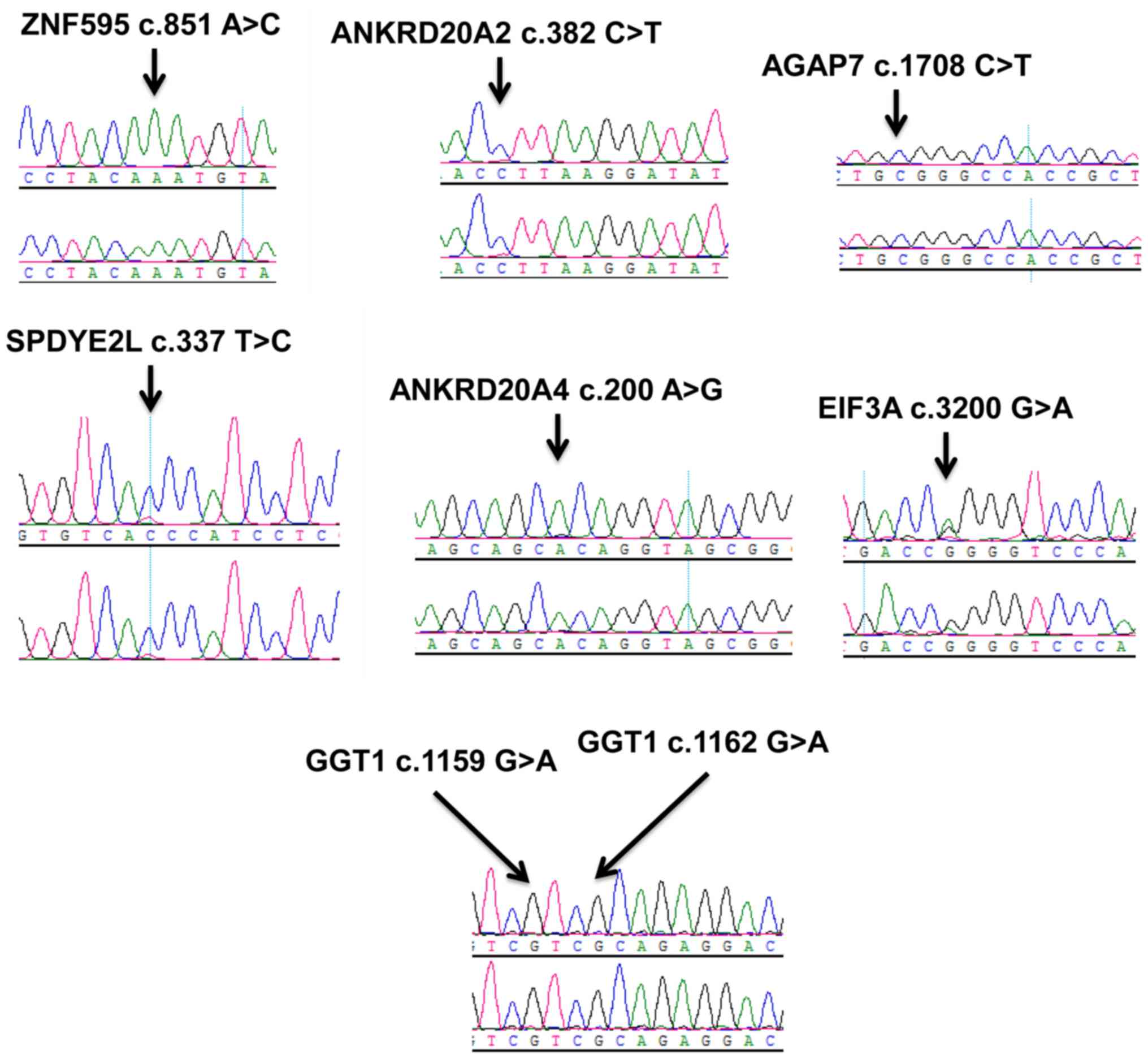
extrahepatic cholestasis. The 8 discordant variants were validated
by conventional Sanger sequencing. None of the above discordant
variants was successfully validated by Sanger sequencing (Fig. 1). To tell from the results, the
discordant variants from the WGS may have exhibited minor
differences between twin A and twin B.
| Table II.Putative discordant variants chosen
for Sanger sequencing from whole genome sequencing data. |
Table II.
Putative discordant variants chosen
for Sanger sequencing from whole genome sequencing data.
| Chromosomal
location | Gene | Transcript ID | Amino acid
alteration | Ref | Alt | Quality | Zygosity | Read no. | Disease associated
with gene |
|---|
| chr4:86248 | ZNF595 | NM_182524.2 | K284T | A | C | 121 | Het | 24,15 |
|
| chr7:102294074 | SPDYE2L | NM_001166339.1 | S113P | T | C | 37 | Het | 10,5 |
|
| chr9:43129577 | ANKRD20A2 | NM_001012421.1 | L128F | G | A | 67 | Het | 9,8 |
|
| chr9:69382292 | ANKRD20A4 | NM_001098805.1 | H67R | A | G | 38 | Het | 13,5 |
|
| chr10:51464748 | AGAP7 | NM_001077685.2 | R570W | G | A | 129 | Het | 5,8 |
|
|
chr10:120801832 | EIF3A | NM_003750.2 | R1067Q | C | T | 136 | Het | 18,13 |
|
| chr22:25023537 | GGT1 | NM_001032364.2 | V387I | G | A | 46 | Het | 28,9 |
Glutathioninuria |
| chr22:25023540 | GGT1 | NM_001032364.2 | A388T | G | A | 36 | Het | 28,9 |
Glutathioninuria |
CNV analysis
As presented in Table
I, twin A as well as twin B displayed changes in CNVs. The
discordant CNVs in twin A and twin B were respectively subjected to
KEGG pathway enrichment analysis. Of all discordant CNVs in twin A
and twin B, only the genes with discordant copy number losses in
twin A (the affected individual) were significantly enriched in
pathways including SLE, alcoholism and olfactory transduction, as
presented in Table III. Enriched
SLE pathway genes with copy number losses in twin A were histone
cluster 2 H2A family member A3 (HIST2H2AA3), HIST2H2AA4, HIST2H3A,
HIST2H3C, HIST2H4A and HIST2H4B. The results suggested that CNV
changes may be associated with the phenotype of the monozygotic
twin discordant for SLE.
| Table III.Kyoto Encyclopedia of Genes and
Genomes pathway enrichment analysis of discordant copy no. losses
of twin A. |
Table III.
Kyoto Encyclopedia of Genes and
Genomes pathway enrichment analysis of discordant copy no. losses
of twin A.
| Pathway | Genes with CNV with
pathway annotation, n (%) | All genes with
pathway annotation, n (%) | Enrichment
factor | P-value | Q-value | Pathway ID | Names of genes with
CNV |
|---|
| Systemic lupus | 6 (15.00) | 133 (1.96) | 0.0451 | 0.000111 | 0.004227 | ko05322 | HIST2H2AA3,
HIST2H2AA4, HIST2H3A, |
| erythematosus |
|
|
|
|
|
| HIST2H3C, HIST2H4A,
HIST2H4B |
| Alcoholism | 6 (15.00) | 179 (2.63) | 0.0335 | 0.000558 | 0.010607 | ko05034 | HIST2H2AA3,
HIST2H2AA4, HIST2H3A, |
|
|
|
|
|
|
|
| HIST2H3C, HIST2H4A,
HIST2H4B |
| Olfactory
transduction | 6 (15.00) | 399 (5.87) | 0.015 | 0.027712 | 0.330570 | ko04740 | OR2G6, OR2T10,
OR2T11, OR2T27, OR2T29, |
|
|
|
|
|
|
|
| OR2T34 |
| Caffeine
metabolism | 1 (2.50) | 6 (0.09) | 0.1667 | 0.034797 | 0.330570 | ko00232 | CYP2A7 |
Discussion
At present, studies assessing monozygotic twins
discordant for SLE by using WGS are rare. Due to the possibility of
the existence of de novo variants and alterations of CNVs in
monozygotic twins (13,15,18,30),
the present study used WGS to explore the possible disease-causing
variants which are responsible for the discordance in monozygotic
twins regarding SLE. A total of 8 putative discordant variants in
the DNA of leukocytes were selected out for validation by Sanger
sequencing. However, no difference was identified in these variants
between the monozygotic twins. Of note, copy numbers in certain SLE
pathway genes exhibited alterations in monozygotic twins discordant
for SLE.
The invalidation of the selected discordant variants
in the present study was consistent with previous similar studies.
The studies by Baranzini et al (31), Chaiyasap et al (32) and Solomon et al (33) did not identify any discordant
variants in monozygotic twins discordant for multiple sclerosis,
congenital heart defect or vertebral defects, anal atresia, cardiac
defects, tracheo-esophageal fistula, renal anomalies and limb
abnormalities, respectively. By contrast, Reumers et al
(30) and Tang et al
(18) identified discordant
variants in the monozygotic twins discordant for schizophrenia. The
results of these studies suggested that the de novo
mutations in the monozygotic twin discordant for certain phenotypes
may be specific for certain types of disease. Furthermore, as for
the unsuccessful identification of discordant variants in the
present study and certain previous studies, the following
considerations were raised for using next-generation sequencing
(NGS) to study monozygotic twins discordant for certain phenotypes:
i) Although NGS technology is able to efficiently sequence a large
amount of genes at a time, it also has high error rates (34). The discordant variants obtained by
WGS in the present study may be due to sequencing errors. In
addition, the average sequencing depth in the present study was 30
X, so that not all genomic regions were completely sequenced. It is
possible that the sequencing coverage of the present study had no
sufficient power to identify de novo variants linked with
SLE. ii) DNA in leukocytes may not be an ideal sample for studying
monozygotic twins discordant for phenotypes (32,35).
In pregnancy, most monozygotic twins are monochorionic. As they
share the same blood circulation, it is possible that monozygotic
twins share the same hematopoietic systems. Tissue samples
displaying discordant phenotypes from monozygotic twins may be
better for studying the discordant variants, since they are
directly associated with the discordant phenotypes. iii) The
analyzing tool and standard for WGS data used in the present study
mainly focus on the functional variants, e.g. exonic missense
variants. It did not sufficiently assess the variants in non-coding
regions and may have missed the significant discordant variants of
the monozygotic twins.
SLE is an autoimmune disease in humans. The
mechanisms in monozygotic twins discordant for autoimmune disease
still remain to be fully elucidated. As for SLE, Javierre et
al (36) reported DNA
methylation pattern changes in a study of monozygotic twins
discordant for SLE. In the present study, CNVs changes of SLE
pathway genes were identified in monozygotic twins discordant for
SLE. The above results indicated that epigenetic changes and CNV
changes in SLE pathway genes may be responsible for monozygotic
twins discordant for SLE. The detailed mechanisms require further
investigation.
The present study employed the WGS technique to
reveal discordant variants and CNVs in monozygotic twins discordant
for SLE. The results suggested that CNV changes may be associated
with the occurrence of monozygotic twins discordant for SLE. The
results of the present study will be helpful in the future analysis
of mechanisms of monozygotic twins discordant for SLE.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Foundation of
Medical Discipline Leaders Program of the Health and Family
Planning Commission of Yunnan Province (grant no. D-201668), the
Personnel Training Project of Yunnan Province (grant no.
2017HB043), the National Natural Science Foundation of China (grant
nos. 81560126 and 81460424), the Joint Special Research Funds of
Kunming Medical University [grant no. 2017FE468(−010)] and the
Innovation Team of Yunnan Province (grant no. 2017HC009).
Availability of data and materials
The analyzed data sets generated during the study
and primer sequences are available from the corresponding author on
reasonable request.
Authors' contributions
FC, ZL and RL performed the experiments and analyzed
the data. ZL and YL wrote the manuscript and designed the study.
The final version of the manuscript was read and approved by all
authors.
Ethical approval and consent to
participate
The present study was approved by the Ethics
Committee of the First People's Hospital of Yunnan Province
(Affiliated Hospital of Kunming University of Science and
Technology). Written informed consent was from the
participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mirabelli G, Cannarile F, Bruni C, Vagelli
R, De Luca R and Carli L: One year in review 2015: Systemic lupus
erythematosus. Clin Exp Rheumatol. 33:414–425. 2015.PubMed/NCBI
|
|
2
|
Petri M: Review of classification criteria
for systemic lupus erythematosus. Rheum Dis Clin North Am.
31(245–254): vi2005.
|
|
3
|
Petri M: Epidemiology of systemic lupus
erythematosus. Best Pract Res Clin Rheumatol. 16:847–858. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ahmad YA and Bruce IN: Genetic
epidemiology: Systemic lupus erythematosus. Arthritis Res.
3:331–336. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mak A and Tay SH: Environmental factors,
toxicants and systemic lupus erythematosus. Int J Mol Sci.
15:16043–16056. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mok CC, Kwok RC and Yip PS: Effect of
renal disease on the standardized mortality ratio and life
expectancy of patients with systemic lupus erythematosus. Arthritis
Rheum. 65:2154–2160. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bodaño A, Amarelo J, González A,
Gómez-Reino JJ and Conde C: Novel DNASE I mutations related to
systemic lupus erythematosus. Arthritis Rheum. 50:4070–4071. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leng RX, Wang W, Cen H, Zhou M, Feng CC,
Zhu Y, Yang XK, Yang M, Zhai Y, Li BZ, et al: Gene-gene and
gene-sex epistatic interactions of MiR146a, IRF5, IKZF1, ETS1 and
IL21 in systemic lupus erythematosus. PLoS One. 7:e510902012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang C, Sandling JK, Hagberg N, Berggren
O, Sigurdsson S, Karlberg O, Rönnblom L, Eloranta ML and Syvänen
AC: Genome-wide profiling of target genes for the systemic lupus
erythematosus-associated transcription factors IRF5 and STAT4. Ann
Rheum Dis. 72:96–103. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang W, Shen N, Ye DQ, Liu Q, Zhang Y,
Qian XX, Hirankarn N, Ying D, Pan HF, Mok CC, et al: Genome-wide
association study in Asian populations identifies variants in ETS1
and WDFY4 associated with systemic lupus erythematosus. PLoS Genet.
6:e10008412010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Remmers EF, Plenge RM, Lee AT, Graham RR,
Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, et
al: STAT4 and the risk of rheumatoid arthritis and systemic lupus
erythematosus. N Engl J Med. 357:977–986. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ketelaar ME, Hofstra EM and Hayden MR:
What monozygotic twins discordant for phenotype illustrate about
mechanisms influencing genetic forms of neurodegeneration. Clin
Genet. 81:325–333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maiti S, Kumar KH, Castellani CA, O'Reilly
R and Singh SM: Ontogenetic de novo copy number variations (CNVs)
as a source of genetic individuality: Studies on two families with
MZD twins for schizophrenia. PLoS One. 6:e171252011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Silva S, Martins Y, Matias A and
Blickstein I: Why are monozygotic twins different? J Perinat Med.
39:195–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kondo S, Schutte BC, Richardson RJ, Bjork
BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S,
Sander A, et al: Mutations in IRF6 cause Van der Woude and
popliteal pterygium syndromes. Nat Genet. 32:285–289. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sobreira NL, Cirulli ET, Avramopoulos D,
Wohler E, Oswald GL, Stevens EL, Ge D, Shianna KV, Smith JP, Maia
JM, et al: Whole-genome sequencing of a single proband together
with linkage analysis identifies a Mendelian disease gene. PLoS
Genet. 6:e10009912010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Petersen BS, Spehlmann ME, Raedler A,
Stade B, Thomsen I, Rabionet R, Rosenstiel P, Schreiber S and
Franke A: Whole genome and exome sequencing of monozygotic twins
discordant for Crohn's disease. BMC Genomics. 15:5642014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang J, Fan Y, Li H, Xiang Q, Zhang DF, Li
Z, He Y, Liao Y, Wang Y, He F, et al: Whole-genome sequencing of
monozygotic twins discordant for schizophrenia indicates multiple
genetic risk factors for schizophrenia. J Genet Genomics.
44:295–306. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Raczy C, Petrovski R, Saunders CT, Chorny
I, Kruglyak S, Margulies EH, Chuang HY, Källberg M, Kumar SA, Liao
A, et al: Isaac: Ultra-fast whole-genome secondary analysis on
Illumina sequencing platforms. Bioinformatics. 29:2041–2043. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cingolani P, Platts A, le Wang L, Coon M,
Nguyen T, Wang L, Land SJ, Lu X and Ruden DM: A program for
annotating and predicting the effects of single nucleotide
polymorphisms, SnpEff: SNPs in the genome of Drosophila
melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6:80–92.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boeva V, Popova T, Bleakley K, Chiche P,
Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O and
Barillot E: Control-FREEC: A tool for assessing copy number and
allelic content using next-generation sequencing data.
Bioinformatics. 28:423–425. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen X, Schulz-Trieglaff O, Shaw R, Barnes
B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S and Saunders CT:
Manta: Rapid detection of structural variants and indels for
germline and cancer sequencing applications. Bioinformatics.
32:1220–1222. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smigielski EM, Sirotkin K, Ward M and
Sherry ST: DbSNP: A database of single nucleotide polymorphisms.
Nucleic Acids Res. 28:352–355. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
1000 Genomes Project Consortium, . Auton
A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini
JL, McCarthy S, McVean GA and Abecasis GR: A global reference for
human genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thorvaldsdottir H, Robinson JT and Mesirov
JP: Integrative Genomics Viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amberger JS, Bocchini CA, Schiettecatte F,
Scott AF and Hamosh A: OMIM.org: Online Mendelian Inheritance in
Man (OMIM®), an online catalog of human genes and
genetic disorders. Nucleic Acids Res. 43:D789–D798. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reumers J, De Rijk P, Zhao H, Liekens A,
Smeets D, Cleary J, Van Loo P, Van Den Bossche M, Catthoor K, Sabbe
B, et al: Optimized filtering reduces the error rate in detecting
genomic variants by short-read sequencing. Nat Biotechnol.
30:61–68. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Baranzini SE, Mudge J, van Velkinburgh JC,
Khankhanian P, Khrebtukova I, Miller NA, Zhang L, Farmer AD, Bell
CJ, Kim RW, et al: Genome, epigenome and RNA sequences of
monozygotic twins discordant for multiple sclerosis. Nature.
464:1351–1356. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chaiyasap P, Kulawonganunchai S,
Srichomthong C, Tongsima S, Suphapeetiporn K and Shotelersuk V:
Whole genome and exome sequencing of monozygotic twins with trisomy
21, discordant for a congenital heart defect and epilepsy. PLoS
One. 9:e1001912014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Solomon BD, Pineda-Alvarez DE, Hadley DW,
Hansen NF, Kamat A, Donovan FX, Chandrasekharappa SC, Hong SK,
Roessler E and Mullikin JC: NISC Comparative Sequencing Program:
Exome sequencing and high-density microarray testing in monozygotic
twin pairs discordant for features of VACTERL association. Mol
Syndromol. 4:27–31. 2013.PubMed/NCBI
|
|
34
|
Nielsen R, Paul JS, Albrechtsen A and Song
YS: Genotype and SNP calling from next-generation sequencing data.
Nat Rev Genet. 12:443–451. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Erlich Y: Blood ties: Chimerism can mask
twin discordance in high-throughput sequencing. Twin Res Hum Genet.
14:137–143. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Javierre BM, Fernandez AF, Richter J,
Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, Berdasco M,
Fraga MF, O'Hanlon TP, Rider LG, et al: Changes in the pattern of
DNA methylation associate with twin discordance in systemic lupus
erythematosus. Genome Res. 20:170–179. 2010. View Article : Google Scholar : PubMed/NCBI
|