Introduction
In recent years, due to the development of the
Chinese economy, the severity of problems associated with air
pollution has increased. Vehicle exhaust gas (EG) serves a
principal role in air pollution and may induce various
age-associated disorders, including cognitive dysfunction (1,2),
metabolic dysregulation (3,4),
cardiovascular disease (5,6) and cancer (7,8).
Numerous studies have investigated the mechanisms underlying
EG-associated disorders; DNA damage, epigenetic alterations,
inflammation and oxidative stress have been identified to serve a
role in these disorders (9–11).
Among the various chemical compounds identified in
vehicle EG, polycyclic aromatic hydrocarbons (PAHs) are a group of
chemicals containing at least two fused benzene rings without
heteroatoms (12). PAHs may be
released as a result of an incomplete combustion of derivatives of
coal, petroleum or organic polymers. However, in urban zones, PAHs
are primarily released from the engines of vehicles, suggesting
that vehicle EG is the major source of PAHs in air pollution
(13). The estimated concentration
of PAHs in EG ranges between 200 and 500 parts per million,
according to our previous study (data not published). Inhalation of
PAH-containing air may increase the risk of lung cancer in humans;
Osgood et al (14)
demonstrated that PAHs may induce inflammation and tumorigenesis in
mouse lung cells by activating extracellular signal-regulated
kinase 1/2, p38 mitogen-activated protein kinase (MAPK) and
inflammatory-associated genes, including cyclooxygenase 2 and
chemokine ligand 2. Eom et al (15) conducted a pilot nested case-control
study to examine the effects of exposure to PAH on lung
carcinogenesis and identified that oxidative stress induced by
exposure to PAH may be an important risk factor for lung cancer
development. Furthermore, Zhao et al (16) demonstrated that benzopyrene (BaP)
may promote lung cancer cell metastasis by activating the tumor
necrosis factor-α signaling pathway. Additionally, accumulating
evidence demonstrated that PAH may induce the methylation of genes
involved in the development of cancer; White et al (17) demonstrated that PAH exposure is
associated with hypomethylation and hypermethylation of various
promoter regions in breast cancer. Additionally, Kim et al
(18) identified that lipophilic
PAHs contribute to the pathogenesis of insulin resistance through
methylation-mediated suppression of the insulin receptor substrate
2 gene.
Zhu et al (19) and our previous studies (data not
published) identified three principal types of PAH in vehicle EG
derived from a gasoline internal combustion engine: Benzanthracene
(BaA), BaP and benzoperylene (BEP). Additionally, our previous
studies identified that exposure to vehicle EG may immunocompromise
BALB/C mice (data not published). However, whether exposure to
vehicle EG, particularly PAHs, is able to induce cell senescence
remains unknown.
In the present study, vehicle EG and pure PAHs were
used to simulate polluted air, and the cellular events following
long-term PAH exposure were investigated by analyzing mouse lung
fibroblast cells (mLFCs). PAHs were revealed to induce apoptosis of
mLFCs, promoting various apoptosis-associated factors. Notably,
PAHs induced cell senescence in mLFCs by activating the ATM
serine/threonine kinase (ATM)/H2A histone family member X (H2AX)
pathway. However, the epigenetic status of the promoter of
senescence-associated genes, including p16, was not affected by
exposure to PAHs. The present study may provide novel insights into
the underlying mechanism of vehicle EG and PAHs in promoting the
development of age-associated diseases.
Materials and methods
Animals and exposure model
A total of 24 BALB/C mice (12 male and 12 female,
6–8 weeks old, 16–18 g; Beijing Vital River Laboratory Animal
Technology Co., Ltd., Beijing, China) were exposed to EG (6 males
and 6 females) or clean air (6 males and 6 females). Mice were
housed at 25°C with 50–80% relative humidity under a 12:12-h
light/dark cycle, with access to food and water ad libitum.
The mice were placed in a cage in a sealed chamber (Fig. 1). Gas exposure (GE) was performed
for 5 h/day (between 9.00 a.m. and 2.00 p.m.), 5 consecutive days
per week, for 6 weeks in an exposure chamber (Fig. 1A). Following 30 days of treatment,
mice were sacrificed, and the lung tissues were collected for
further molecular experiments (Fig. 1B
and C). EG was collected from a gasoline internal combustion
engine and mixed with fresh air in a ratio of 1:10 using the
electromechanical injection system. Subsequently, the mixed gas was
injected into the chamber at a flow rate of 3 l/min. All animal
experiments were approved by The Animal Care and Use and Ethics
Committee of Jilin University (Changchun, China).
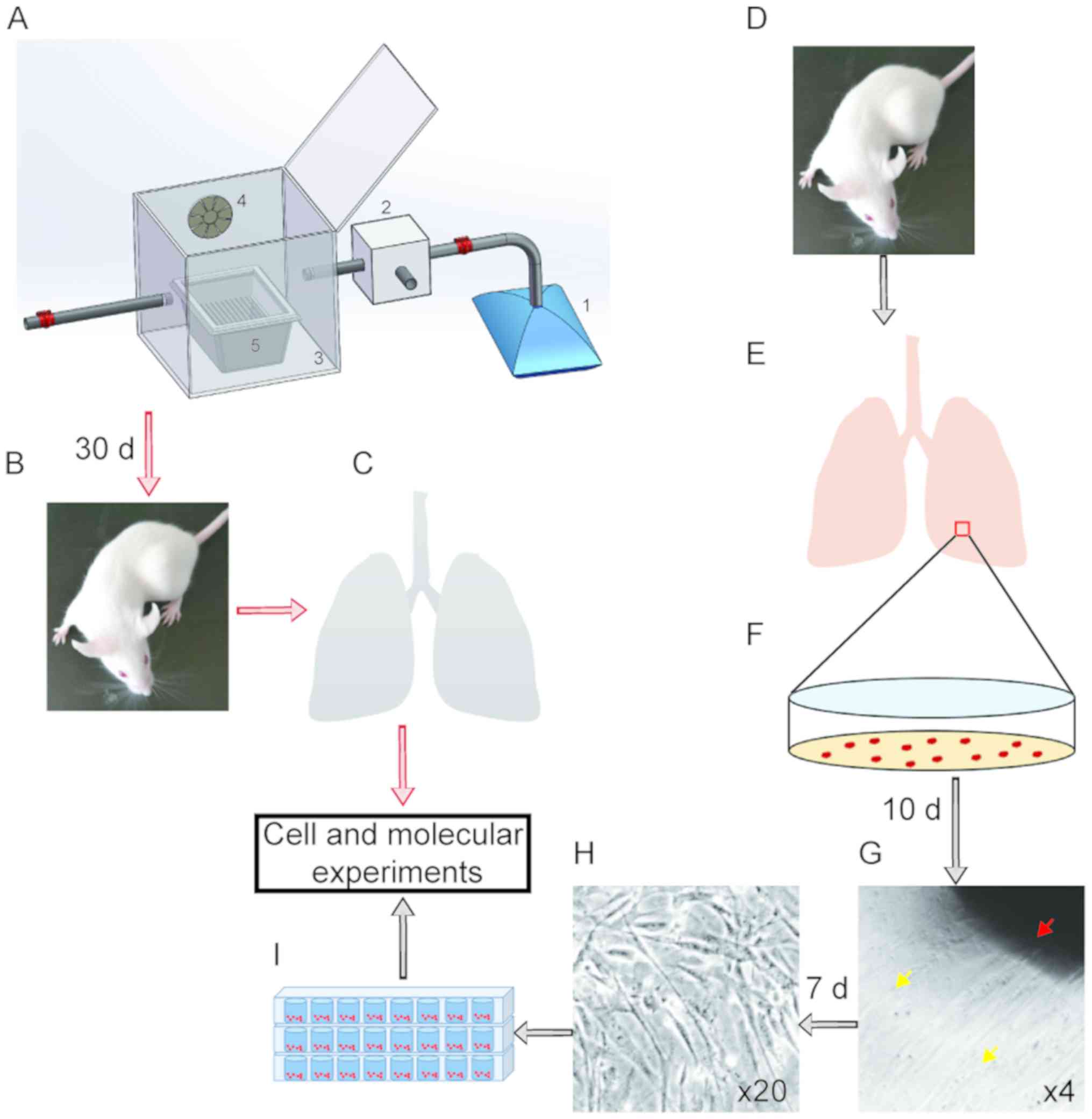 | Figure 1.Isolation of mLFCs and establishment
of EG and polycyclic aromatic hydrocarbons exposure model. (A)
Diagram of the exposure chamber, consisting of five principal
components: 1, Gas collecting bag; 2, controlled electromechanical
injection system; 3, inhalation chamber; 4, air-mixing fan; 5,
mouse cage. (B and C) BALB/C mice were exposed to EG for 30 days
and sacrificed. Lung tissues were collected for molecular
experiments. (D-F) Mice were sacrificed, lung tissues were
collected and mLFCs were isolated. (G-I) mLFCs were
culture-expanded and used for subsequent experiments. (G)
Magnification, ×4; (H) magnification, ×20. Red arrow, mouse lung
tissue; orange arrows, mLFCs. EG, exhaust gas; d, days; mLFC, mouse
lung fibroblast cell. |
Cell culture
A male BALB/C mouse, housed under clean air
conditions (no GE), was sacrificed, and the fresh lung tissues were
cut into ~1 mm3 pieces (Fig. 1E and F). Subsequently, the lung
tissues were seeded in a 6-well plate and dried in a flow tissue
culture hood for 15 min until the tissues attached to the plate
surface. A total of 500 µl Dulbecco's modified Eagle's medium
(HyClone; GE Healthcare Life Sciences, Logan, MT, USA) supplemented
with 10% fetal bovine serum (HyClone; GE Healthcare Life Sciences)
and antibiotics (100 mg/ml streptomycin and 100 U/ml penicillin;
HyClone; GE Healthcare Life Sciences) was added to the culture
plates and the plates were incubated at 37°C in a humidified
incubator with 5% CO2 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The medium was replaced every other day until
mLFC proliferation was observed under a light microscope (ECLIPSE
Ts2; Nikon Corporation, Tokyo, Japan).
Cell proliferation assay
mLFCs (5×103) were treated with 3 µM BaA,
BaP or BEP (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 72
h, as previously described (20).
Subsequently, cell proliferation was determined using the
water-soluble tetrazolium salt (WST-1) cell proliferation reagent
(Beyotime Institute of Biotechnology, Haimen, China). Briefly, 20
µl WST-1 reagent was added to 200 µl cell culture medium and
incubated at 37°C in the dark for 2.5 h. The optical density (OD)
at 450 and 630 nm was measured using a microplate reader (Synergy
H1; BioTek Instruments, Inc., Winooski, VT, USA). Final OD values
were calculated using the following formula:
ODfinal=OD450-OD630-ODblank.
Cell apoptosis
Following treatment with 3 µM BaA, BaP or BEP for 72
h, cell apoptosis was determined by staining mLFCs with propidium
iodide (PI) and Annexin V-fluorescein isothiocyanate (FITC). Cells
(1×106) were washed with PBS and centrifuged at 200 × g
for 5 min at room temperature. The cellular pellet was suspended in
50 µl Annexin V solution containing 5 µl Annexin V-FITC and 5 µl PI
for 15 min at room temperature, provided in the Annexin V-FITC
Apoptosis Detection kit (BD Biosciences, Franklin Lakes, NJ, USA).
Data acquisition and data analysis were performed using a flow
cytometer (FACScan; Becton-Dickinson and Company, Franklin Lakes,
NJ, USA) with the FlowJo FACS analysis software (version 10.0;
FlowJo LLC, Ashland, OR, USA).
Protein extraction and western
blotting
Total protein was extracted from mLFCs treated with
BaA and BaP using a lysis buffer containing 50 mM Tris/acetate (pH
7.4), 1 mM EDTA, 0.5% Triton X-100, 150 mM sodium chloride and 0.1
mM phenylmethane sulfonyl fluoride. Total protein was quantified
using a Bradford protein assay kit (Beyotime Institute of
Biotechnology). Proteins (20 µg/lane) were separated by 8–10%
SDS-PAGE and subsequently transferred to polyvinylidene difluoride
(PVDF) membranes (EMD Millipore, Billerica, MA, USA). The PVDF
membranes were incubated in Tris-buffered saline buffer containing
0.5% Tween-20 (Sigma-Aldrich; Merck KGaA) and 5% skim milk at room
temperature for 1 h and subsequently incubated at 4°C overnight
with the following primary antibodies (all 1:1,000 dilution):
Anti-p53 (cat. no. ab26; Abcam, Cambridge, UK), anti-p21 (cat. no.
ab109199; Abcam), anti-cleaved caspase-3 (cat. no. 9661; Cell
Signaling Technology, Inc., Danvers, MA, USA), anti-cleaved
caspase-9 (cat. no. 9509; Cell Signaling Technology, Inc.),
anti-p16 (cat. no. ab189034; Abcam), anti-p27KIP1 (cat.
no. ab193379; Abcam), anti-ATM, (cat. no. ab78; Abcam), anti-H2AX
(cat. no. 7631; Cell Signaling Technology, Inc.) anti-
phosphorylated-H2AX (γH2AX; cat. no. 07-164, EMD Millipore) and
anti-β-actin (cat. no. sc-47778; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA). Following incubation with the primary antibodies,
membranes were incubated with horseradish peroxidase-conjugated
goat anti-rabbit (cat. no. sc-2004; Santa Cruz Biotechnology, Inc.)
or goat anti-mouse (cat. no. sc-2005; Santa Cruz Biotechnology,
Inc.) immunoglobulin G secondary antibodies at room temperature for
1 h (1:3,000 dilution). The immunoreactivity was detected using an
enhanced chemiluminescence detection kit (EMD Millipore). The
densitometric analysis was performed using Quantity One software
(version 4.6; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Senescence-associated β-galactosidase
(SA-β-Gal) staining
mLFCs were treated with 3 µM BaA or BaP for 72 h.
Cellular senescence was analyzed by measuring the activity of
SA-β-Gal. mLFCs were collected and fixed with 3% formaldehyde
(Beijing Chemical Works, Beijing, China) for 3 min at room
temperature. After washing with PBS, cell senescence was determined
with the Senescence β-Galactosidase Staining kit according to the
manufacturer's protocol (Beyotime Institute of Biotechnology).
Images of the cells were captured using a light microscope and
cells positive for β-Gal staining were subsequently counted.
Detection of telomere length in
mLFCs
The relative average telomere length of mLFCs was
measured using total genomic mouse DNA as template, using
quantitative polymerase chain reaction (qPCR), as previously
described by Callicott and Womack (21), with certain modifications. Total
genomic DNA was extracted from control, and BaA- and BaP-treated
mLFCs using the Qiagen DNeasy Blood & Tissue kit (Qiagen China
Co., Ltd., Shanghai, China). The primers used for the analysis
were: Forward, 5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3′ and
reverse, 5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT-3′. Ribosomal
protein lateral stalk subunit P0 (RPLP0) gene was used as the
reference gene. The following primers were used to amplify RPLP0:
Forward, 5′-ACTGGTCTAGGACCCGAGAAG-3′ and reverse,
5′-TCAATGGTGCCTCTGGAGATT-3′. The qPCR reaction mixture to
investigate telomere length and RPLP0 comprised 12.5 µl 2×
SYBR-Green master mix (Roche Applied Science, Penzberg, Germany),
2.5 µl of each primer at a concentration of 300 nM and 20 ng
genomic DNA; double-distilled water was added to a total volume of
25 µl. The PCR thermocycling conditions conducted to determine
telomere length were as follows: Initial denaturation at 95°C for
10 min, followed by 30 cycles of 95°C for 15 sec and 56°C for 1
min, and final extension at 72°C for 5 min (ABI 7500; Applied
Biosystems; Thermo Fisher Scientific, Inc.). The PCR thermocycling
conditions for RPLP0 detection were as follows: Initial
denaturation at 95°C for 10 min, followed by 35 cycles of 95°C for
15 sec, 52°C for 20 sec and 72°C for 30 sec, and final extension at
72°C for 5 min. Telomere length was assessed by calculating the
relative ratio between telomeres and RPLP0. The 2−ΔΔCq
quantification method (22) was
used to quantify the qPCR results.
Detection of gene expression levels by
reverse transcription-qPCR (RT-qPCR) and semi-quantitative PCR
Total RNA was extracted from mLFCs using the Qiagen
RNeasy Mini kit (Qiagen China Co., Ltd.). Total RNA (800 ng) was
reverse transcribed to cDNA using Moloney murine leukemia virus
reverse transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The qPCR reaction mixture
contained: 2 µl cDNA, 4 µl 2X Taq PCR StarMix buffer (GenStar,
Beijing, China) and 2 µl of each primer. The semi-quantitative PCR
amplification was performed using the GeneQ cycler (Hangzhou Bori
Technology Co., Ltd., Hangzhou, China) and the thermocycling
conditions were as follows: Initial denaturation at 98°C for 3 min,
followed by 26–32 cycles of 95°C for 15 sec, 62°C for 15 sec and
72°C for 15 sec, and a final extension step at 72°C for 5 min. The
qPCR reaction was performed as follows: Initial denaturation at
95°C 10 min, followed by 40 cycles of 95°C for 10 sec and 60°C for
30 sec. The primer sequences used were as follows: p16, forward,
5′-GAGCCATCTGGAGCAGCATGGAG-3′ and reverse,
5′-GCCCATCATCATCACCTGAATC-3′; p27KIP1, forward,
5′-TGGGTTAGCGGAGCAGTGTC-3′ and reverse, 5′-CTCCACAGTGCCAGCGTTC-3′;
and β-actin, forward, 5′-CAGGTCATCACTATTGGCAACGAGC-3′ and reverse,
5′-CGGATGTCAACGTCACACTTCATGA-3′. For semi-quantitative PCR, the
reaction products were separated on a 3% agarose gel and visualized
by ethidium bromide. The densitometric analysis was performed using
Quantity One software (version 4.6; Bio-Rad Laboratories,
Inc.).
DNA methylation analysis
Mice were exposed to exhaust gas for 30 days, and
the CpG methylation state of the promoter of p16 was analyzed.
Genomic DNA was extracted from mouse lung tissues, and control and
3 µM BaP-treated mLFCs. In total, 1,000 ng was used for bisulfite
conversion with the QIAamp DNA Mini kit (Qiagen China Co., Ltd.)
according to the manufacturer's protocol. PCR was performed using
PrimeSTAR® HS DNA Polymerase mix (cat. no. R044A; Takara
Bio, Inc., Otsu, Japan). PCR thermocycling conditions were as
follows: Initial denaturation at 97°C for 10 min, followed by 35
cycles of 96°C for 30 sec, 60°C for 30 sec, 72°C for 30 sec, and a
final extension at 72°C for 5 min. The primers used for assessing
DNA methylation were as follows: Forward,
5′-GAGTTATTTGGAGTAGTATGGAG-3′ and reverse,
5′-ACCCATCATCATCACCTAAATC-3′. The PCR products were separated on 2%
agarose gel and purified with a DNA gel extraction kit (Tiangen
Biotech Co., Ltd., Beijing, China). Subsequently, the PCR products
were cloned into a CloneJET vector using a PCR Cloning kit (Thermo
Fisher Scientific, Inc.) and subjected to Sanger sequencing (Sangon
Biotech Co., Ltd., Shanghai, China) for analysis of the methylation
state of the p16 promoter.
Statistical analysis
SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA)
was used for statistical analysis. All experiments were repeated at
least three times, and data are presented as the means ± standard
deviation. Statistical significance was determined using Student's
t-test or one-way analysis of variance followed by Fisher's least
significant difference post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
mLFC harvesting
Extracted tissues were cut and cultured for 10 days,
after which, mLFCs began to proliferate (Fig. 1G). After 7 additional days, mLFCs
were trypsinized and transferred to a tissue culture plate for
subsequent experiments (Fig.
1H).
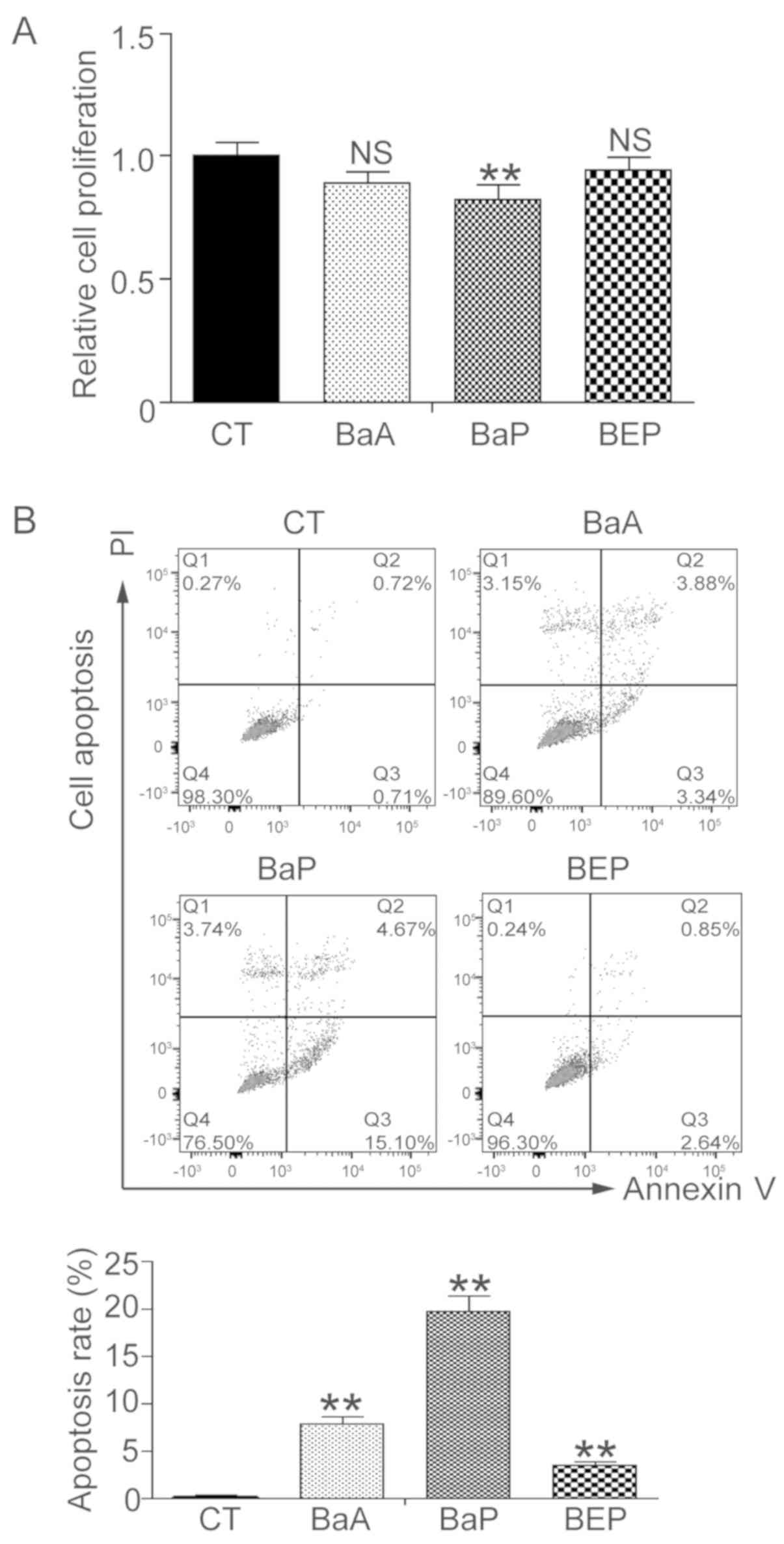
PAH induces mLFC apoptosis
mLFCs were treated with 3 µM BaA, BaP or BEP for 72
h, and cell proliferation was assessed using the WST-1 assay. mLFC
proliferation was significantly suppressed following treatment with
BaP, and proliferation was decreased by ~18% (P<0.01; Fig. 2A). Treatment with BaA decreased
proliferation by 11%; however, the difference was not significant
(P=0.13). Treatment with BEP did not alter cell proliferation. Cell
apoptosis was subsequently determined by flow cytometry. Compared
with the control group, cell apoptosis was increased by 7.8, 19.8
and 3.5% in the BaA, BaP and BEP groups, respectively (Fig. 2B). Treatment with BaP led to the
highest apoptosis rate.
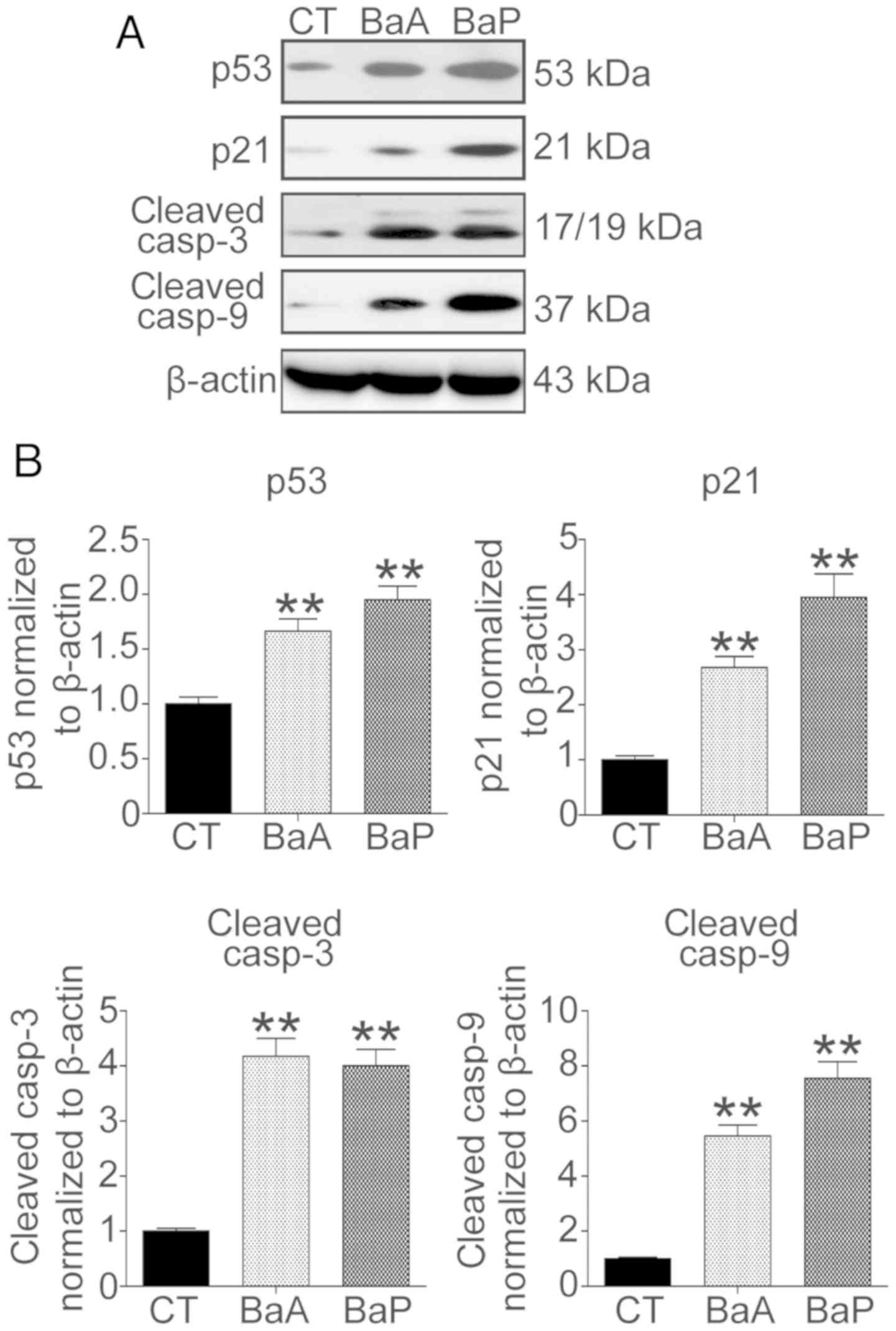
The protein expression levels of factors associated
with cell growth and apoptosis were analyzed by western blotting
(Fig. 3A). The results revealed
that following treatment with 3 µM BaA or BaP, the protein
expression levels of p53 and p21 were significantly increased. The
protein expression levels of p53 and p21 were increased 1.7- and
2.7-fold in the BaA group, respectively (Fig. 3B). Furthermore, following treatment
with BaP, the protein expression levels of p53 and p21 increased
1.9- and 3.9-fold, respectively (Fig.
3B). Additionally, the caspase-dependent cell apoptosis pathway
was activated. The protein expression levels of cleaved caspase-3
and cleaved caspase-9 were increased 4.2- and 5.4-fold in the BaA
group, and 4.0- and 7.5-fold in the BaP group, respectively
(P<0.01).
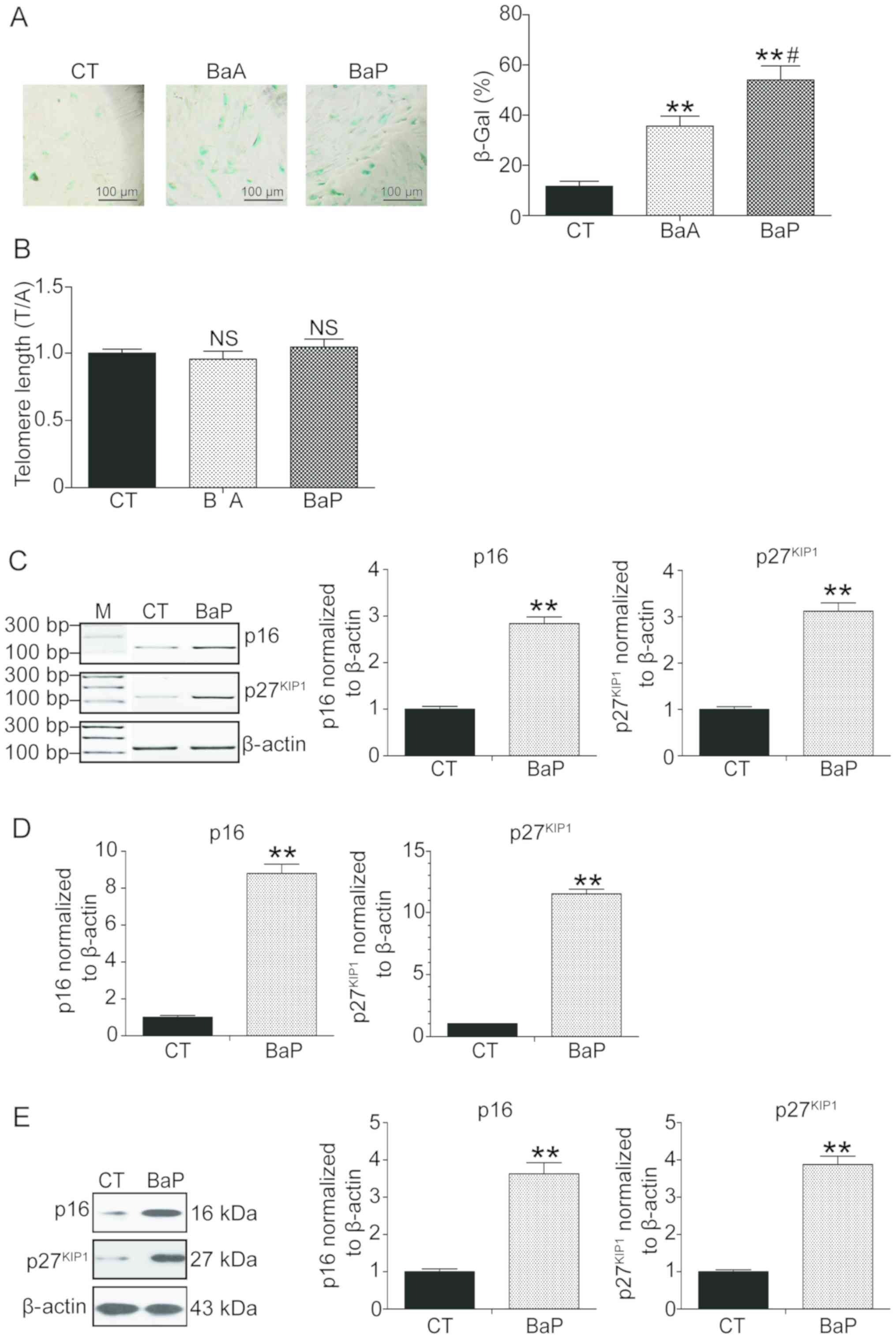
PAH induces premature senescence in
mLFCs
A previous study demonstrated that long-term
exposure to polluted air may cause biological aging and
age-associated diseases (9). In
the present study, PAH was hypothesized to induce premature
senescence. Therefore, mLFCs were exposed to PAH for 72 h and
senescence was examined. mLFCs treated with PAH were positive for
SA-β-Gal activity (Fig. 4A).
Compared with the control group, the number of cells positive for
SA-β-Gal in the BaA and BaP groups was increased 2.1- and 4.6-fold,
respectively (P<0.01; Fig. 4A).
To further investigate whether cell senescence was associated with
a decrease in telomere lengths, the telomeric regions were
investigated using qPCR. No significant differences in telomere
lengths between the control group and the samples exposed to PAH
were identified (P>0.05; Fig.
4B), suggesting that PAH may promote cell senescence via the
environment stress-induced premature senescence (SIPS) pathway and
not via the replicative senescence (RS) pathway (23,24).
The expression levels of two senescence-associated
factors, p16 and p27, were subsequently quantified (25–27).
BaP exhibted the most notable effect on the induction of premature
senescence (Fig. 4A); therefore,
it was selected as the pollutant for subsequent experiments. Using
semi-quantitative PCR, RT-qPCR and western blotting, the mRNA and
protein expression levels of p16 and p27 were revealed to be
significantly increased following treatment with BaP (P>0.01,
Fig. 4C-E).
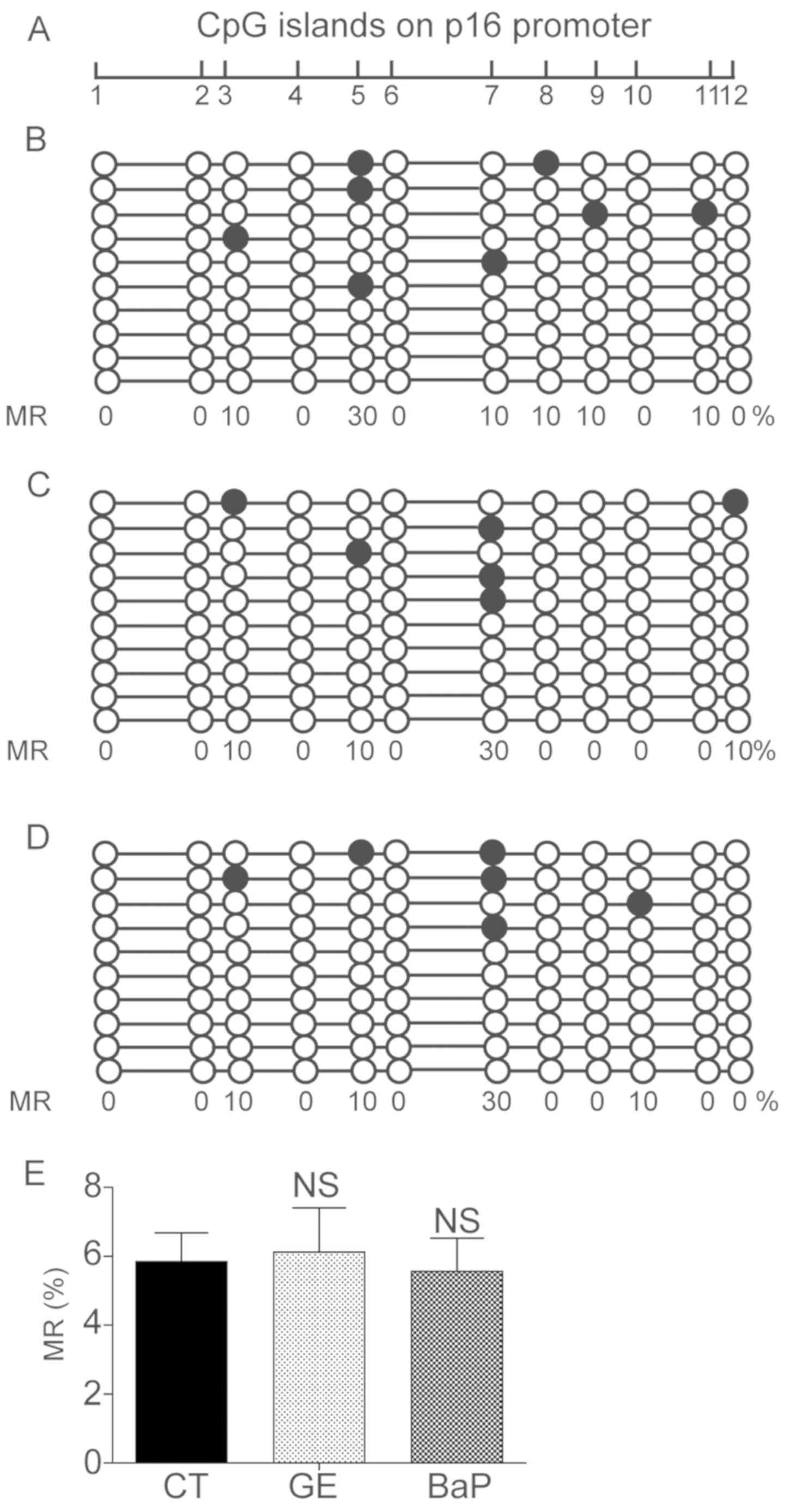
PAH does not influence the DNA
methylation state of the p16 promoter
Cellular senescence is associated with the
expression levels of p16, which is regulated by the methylation
state of its promoter region (28). Since the present results suggested
that PAHs may promote premature senescence and activate p16 in
mLFCs, the DNA methylation state of the promoter of p16 was
investigated following long-term exposure to EG or treatment with
BaP. However, DNA sequencing results suggested that the methylation
levels were similar between the control and the experimental groups
(Fig. 5). The present results
suggested that the premature senescence and the increase in the
expression levels of p16 induced by PAHs were not associated with
epigenetic alterations.
PAH exposure activates the ATM/H2AX
pathway
A previous study reported that ATM is involved in
senescence downstream to various stimuli, including
hyperproliferation and DNA damage (29). In response to DNA damage, ATM
induces H2AX phosphorylation at serine 139, resulting in the
phosphorylation of H2AX and an increase of γH2AX foci at the DNA
damage sites (30,31). In the present study, the ATM/H2AX
pathway was activated following exposure to PAHs. Western blot
analysis results suggested that exposure to EG and PAH induced a
significant upregulation of ATM and γH2AX (Fig. 6). The protein expression levels of
ATM were increased 2.5- and 3.6-fold in the GE group and PAH group,
respectively. Furthermore, the protein expression levels of γH2AX
were increased 1.5- and 2.0-fold in the GE group and PAH group,
respectively.
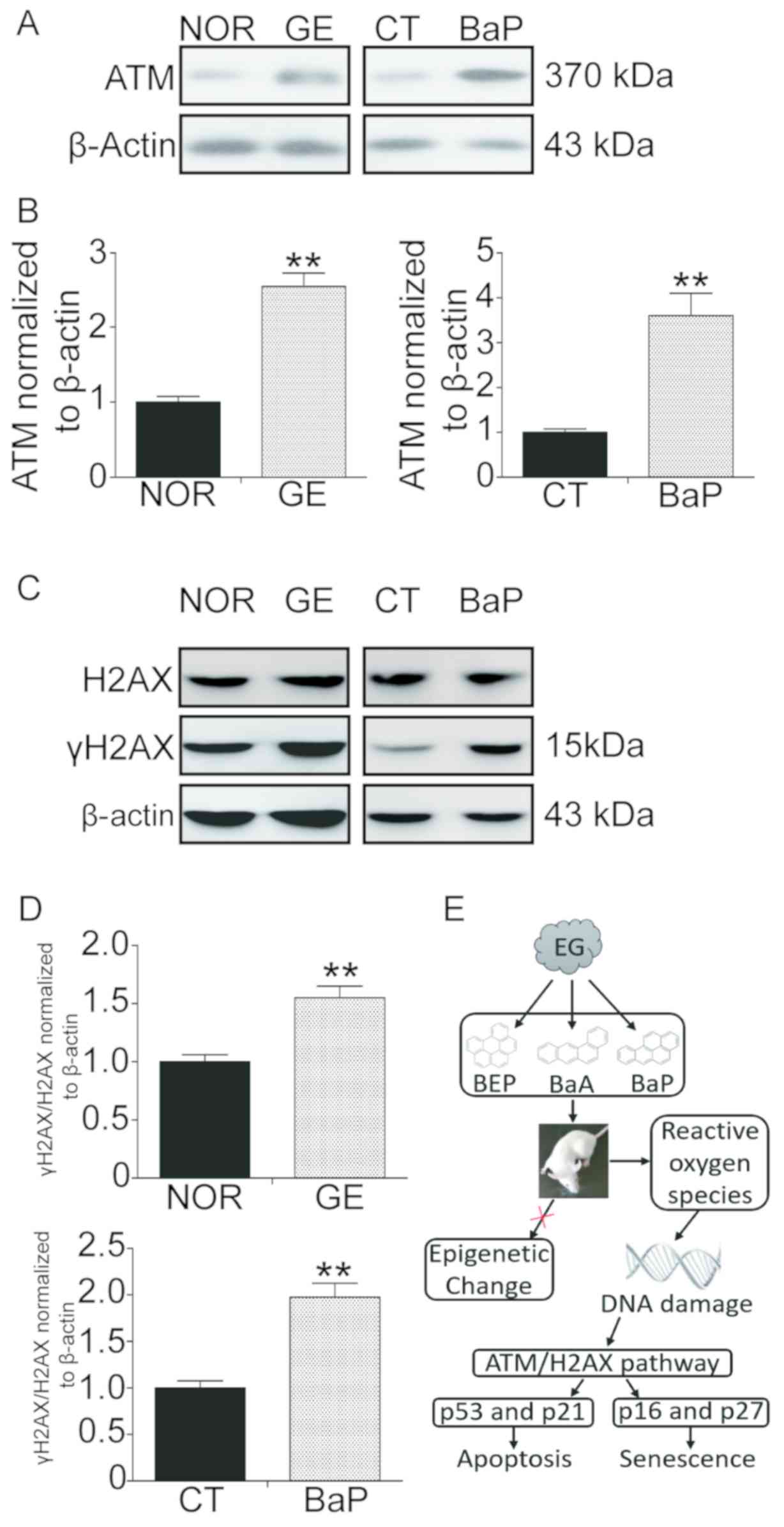 | Figure 6.GE and BaP exposure activates the
ATM/H2AX pathway in mouse lung tissues and lung fibroblast cells.
Mice were exposed to EG or clean air for 30 days and sacrificed,
and the lung tissues were collected and protein was extracted.
mLFCs were treated with 3 µM BaP for 72 h and the protein was also
extracted. (A) Protein expression levels of ATM assessed by western
blotting. (B) Densitometric analysis of the protein expression
levels of ATM. (C) Protein expression levels of H2AX and γH2AX
assessed by western blotting. (D) Densitometric analysis of the
protein expression levels of γH2AX. The ratio between H2AX and
γH2AX was determined, and expression levels were normalized to
β-actin. (E) Diagram illustrating polycyclic aromatic
hydrocarbons-promoted stress-induced premature senescence.
**P<0.01 compared with the normal or control group. γH2AX,
phosphorylated-H2AX; ATM, ATM serine/threonine kinase; BaA,
benzanthracene; BaP, benzopyrene; BEP, benzoperylene; CT, control
mLFCs; EG, exhaust gas; GE, gas exposure; H2AX, H2A histone family
member X; mLFC, mouse lung fibroblast cell; NOR, normal (clean
air). |
Discussion
Vehicle EG contains various chemical compounds
including carbon dioxide, hydrocarbons and nitrogen oxide, and it
is important to investigate the biological effects of these
components. PAHs are a group of toxic pollutants present in vehicle
EG. Although PAHs are involved in the pathological progression of
various tumors (32), the role of
PAHs in cell senescence remains unclear. In the present study,
under laboratory conditions, the biological effects of PAHs were
investigated on mouse lungs and mLFCs. Cell proliferation was
examined following exposure to PAHs for 72 h, and BaP, a type of
PAH, significantly suppressed proliferation of mLFCs. Additionally,
BaA and BaP were able to induce apoptosis of mLFCs. The molecular
factors associated with apoptosis, including p53, p21, cleaved
caspase-3 and cleaved caspase-9, were significantly upregulated
following treatment with BaA and BaP. BEP did not significantly
alter cell proliferation compared with the control, and exhibited a
markedly reduced effect on apoptosis compared with BaA and BaP.
Therefore, BaA and BaP were selected for further molecular
experiments. A previous study suggested that the cytotoxic effects
of BaA, BaP and BEP are distinct due to their differential
potential to activate the aryl hydrocarbon receptor signaling
pathway (33).
Two types of pathways may regulate cellular
senescence: RS and SIPS. RS is induced by serial passage of normal
cells in culture, whereas SIPS is primarily induced by exposure to
environmental stimuli, including radiation and chemical toxicants
(24). RS is associated with
telomere shortening and epigenetic alterations, whereas SIPS is
associated with DNA damage and genetic mutations. In the present
study, it was demonstrated that BaA and BaP may promote cell
senescence by increasing the expression levels of the
senescence-associated factors p16 and p27 in mLFCs. Since telomere
shortening is an important marker of RS (34), the lengths of telomeres were
investigated in the PAH exposure group and in the control group.
However, no significant difference between the two groups was
observed.
PAHs may form reactive epoxides in cells, covalently
binding to the DNA. These epoxides may induce epigenetic
alterations, including cytosine methylation (35). Methylation of the p16 promoter is
used as a marker of genomic hypermethylation (36). Furthermore, the hypermethylation of
CpG islands in the p16 promoter has been identified to be an early
event in lung cancer development; particularly in patients with a
history of exposure to cigarette smoke (37). In the present study, using the
bisulfite sequencing method, it was suggested that exposure to EG
and PAH was not sufficient to affect the methylation status of the
p16 promoter. Therefore, these present results, in combination with
the results of the telomere length assay, suggested that PAH may
induce SIPS and not RS.
Since DNA damage is a marker of SIPS, activation of
the ATM/H2AX pathway was examined. The ATM/H2AX pathway is
activated by DNA damage and regulates DNA repair (38). The present western blotting results
suggested that exposure to EG and PAH upregulated the protein
expression levels of ATM and γH2AX in mLFCs. A previous study
demonstrated that the reactive oxygen species formed following
exposure to BaP generate detrimental oxidative effects on cell
proliferation and cell survival via an increase in membrane lipid
peroxidation and oxidative DNA damage (16). Barascu et al (29) reported that oxidative stress may
induce an ATM-independent senescence response via the p38 MAPK
pathway. Therefore, SIPS induced by PAHs in vivo and in
vitro may be associated with the oxidative stress-induced DNA
damage response.
Collectively, the present study suggested that
exposure to PAH may induce apoptosis of mLFCs and increase the
protein expression levels of various apoptosis-associated factors,
including p53, p21, caspase-3 and caspase-9. Additionally, exposure
to PAH was revealed to generate a SIPS response in mLFCs,
upregulating the expression levels of p16 and p27. Exposure to PAH
did not influence the epigenetic status of the promoter of p16;
however, PAH was identified to induce SIPS via activation of the
ATM pathway, which may be initiated by reactive epoxides and
oxidative effects of PAHs (Fig.
6E). The present study may provide novel insights into the
underlying mechanism of vehicle EG and PAHs in promoting the
development of age-associated diseases.
Acknowledgεments
Not applicable.
Funding
The present study was supported financially by The
National Natural Science Foundation of China (grant no.
51306070).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DQ contributed to the design of the study and wrote
the manuscript. FY and KY performed the experiments and analyzed
the data. YH contributed to the design of the study. JL was
involved in conducting the experiments. YA analyzed data, providing
constructive comments. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by The Animal
Care and Use and Ethics Committee on The Use of Animals of Jilin
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gatto NM, Henderson VW, Hodis HN, St John
JA, Lurmann F, Chen JC and Mack WJ: Components of air pollution and
cognitive function in middle-aged and older adults in Los Angeles.
Neurotoxicology. 40:1–7. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ailshire JA and Clarke P: Fine particulate
matter air pollution and cognitive function among U.S. older
adults. J Gerontol B Psychol Sci Soc Sci. 70:322–328. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thiering E, Cyrys J, Kratzsch J, Meisinger
C, Hoffmann B, Berdel D, von Berg A, Koletzko S, Bauer CP and
Heinrich J: Long-term exposure to traffic-related air pollution and
insulin resistance in children: Results from the GINIplus and
LISAplus birth cohorts. Diabetologia. 56:1696–1704. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park SK, Adar SD, O'Neill MS, Auchincloss
AH, Szpiro A, Bertoni AG, Navas-Acien A, Kaufman JD and Diez-Roux
AV: Long-term exposure to air pollution and type 2 diabetes
mellitus in a multiethnic cohort. Am J Epidemiol. 181:327–336.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Franklin BA, Brook R and Arden Pope C III:
Air pollution and cardiovascular disease. Curr Probl Cardiol.
40:207–238. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meo SA and Suraya F: Effect of
environmental air pollution on cardiovascular diseases. Eur Rev Med
Pharmacol Sci. 19:4890–4897. 2015.PubMed/NCBI
|
|
7
|
Sax SN, Zu K and Goodman JE: Air pollution
and lung cancer in Europe. Lancet Oncol. 14:e439–e440. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ding N, Zhou N, Zhou M and Ren GM:
Respiratory cancers and pollution. Eur Rev Med Pharmacol Sci.
19:31–37. 2015.PubMed/NCBI
|
|
9
|
Ward-Caviness CK, Nwanaji-Enwerem JC, Wolf
K, Wahl S, Colicino E, Trevisi L, Kloog I, Just AC, Vokonas P,
Cyrys J, et al: Long-term exposure to air pollution is associated
with biological aging. Oncotarget. 7:74510–74525. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mumaw CL, Surace M, Levesque S, Kodavanti
UP, Kodavanti PRS, Royland JE and Block ML: Atypical microglial
response to biodiesel exhaust in healthy and hypertensive rats.
Neurotoxicology. 59:155–163. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Serra DS, Evangelista JSAM, Zin WA,
Leal-Cardoso JH and Cavalcante FSÁ: Changes in rat respiratory
system produced by exposure to exhaust gases of combustion of
glycerol. Respir Physiol Neurobiol. 242:80–85. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moorthy B, Chu C and Carlin DJ: Polycyclic
aromatic hydrocarbons: from metabolism to lung cancer. Toxicol Sci.
145:5–15. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bostrom CE, Gerde P, Hanberg A, Jernström
B, Johansson C, Kyrklund T, Rannug A, Törnqvist M, Victorin K and
Westerholm R: Cancer risk assessment, indicators, and guidelines
for polycyclic aromatic hydrocarbons in the ambient air. Environ
Health Perspect. 110 (Suppl 3):S451–S488. 2002. View Article : Google Scholar
|
|
14
|
Osgood RS, Upham BL, Hill T III, Helms KL,
Velmurugan K, Babica P and Bauer AK: Polycyclic aromatic
hydrocarbon-induced signaling events relevant to inflammation and
tumorigenesis in lung cells are dependent on molecular structure.
PLoS One. 8:e651502013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eom SY, Yim DH, Moon SI, Youn JW, Kwon HJ,
Oh HC, Yang JJ, Park SK, Yoo KY, Kim HS, et al: Polycyclic aromatic
hydrocarbon-induced oxidative stress, antioxidant capacity, and the
risk of lung cancer: A pilot nested case-control study. Anticancer
Res. 33:3089–3097. 2013.PubMed/NCBI
|
|
16
|
Zhao G, Wang Z, Huang Y, Ye L, Yang K,
Huang Q, Chen X, Li G, Chen Y, Wang J and Zhou Y: Effects of
Benzoapyrene on migration and invasion of lung cancer cells
functioning by TNF-α. J Cell Biochem. 119:6492–6500. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
White AJ, Chen J, Teitelbaum SL,
McCullough LE, Xu X, Hee Cho Y, Conway K, Beyea J, Stellman SD,
Steck SE, et al: Sources of polycyclic aromatic hydrocarbons are
associated with gene-specific promoter methylation in women with
breast cancer. Environ Res. 145:93–100. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim YH, Lee YS, Lee DH and Kim DS:
Polycyclic aromatic hydrocarbons are associated with insulin
receptor substrate 2 methylation in adipose tissues of Korean
women. Environ Res. 150:47–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu L, Wang J, Du Y and Xu Q: Research on
PAHs fingerprints of vehicle discharges. Huan Jing Ke Xue.
24:26–29. 2003.(In Chinese). PubMed/NCBI
|
|
20
|
Gordon MW, Yan F, Zhong X, Mazumder PB,
Xu-Monette ZY, Zou D, Young KH, Ramos KS and Li Y: Regulation of
p53-targeting microRNAs by polycyclic aromatic hydrocarbons:
Implications in the etiology of multiple myeloma. Mol Carcinog.
54:1060–1069. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Callicott RJ and Womack JE: Real-time PCR
assay for measurement of mouse telomeres. Comp Med. 56:17–22.
2006.PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Farhat N, Thorin-Trescases N, Voghel G,
Villeneuve L, Mamarbachi M, Perrault LP, Carrier M and Thorin E:
Stress-induced senescence predominates in endothelial cells
isolated from atherosclerotic chronic smokers. Can J Physiol
Pharmacol. 86:761–769. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kural KC, Tandon N, Skoblov M,
Kel-Margoulis OV and Baranova AV: Pathways of aging: Comparative
analysis of gene signatures in replicative senescence and stress
induced premature senescence. Bmc Genomics. 17:213–224. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Drummond D, Baravalle-Einaudi M, Lezmi G,
Vibhushan S, Franco-Montoya ML, Hadchouel A, Boczkowski J and
Delacourt C: Combined effects of in utero and adolescent tobacco
smoke exposure on lung function in C57Bl/6J mice. Environ Health
Perspect. 125:392–399. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim SY, Lee JH, Kim HJ, Park MK, Huh JW,
Ro JY, Oh YM, Lee SD and Lee YS: Mesenchymal stem cell-conditioned
media recovers lung fibroblasts from cigarette smoke-induced
damage. Am J Physiol Lung Cell Mol Physiol. 302:L891–L908. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Furlong HC, Stampfli MR, Gannon AM and
Foster WG: Cigarette smoke exposure triggers the autophagic cascade
via activation of the AMPK pathway in mice. Biol Reprod. 93:932015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sasaki M, Kajiya H, Ozeki S, Okabe K and
Ikebe T: Reactive oxygen species promotes cellular senescence in
normal human epidermal keratinocytes through epigenetic regulation
of p16(INK4a.). Biochem Biophys Res Commun. 452:622–628. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Barascu A, Le Chalony C, Pennarun G, Genet
D, Imam N, Lopez B and Bertrand P: Oxidative stress induces an
ATM-independent senescence pathway through p38 MAPK-mediated lamin
B1 accumulation. EMBO J. 31:1080–1094. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Burma S, Chen BP, Murphy M, Kurimasa A and
Chen DJ: ATM phosphorylates histone H2AX in response to DNA
double-strand breaks. J Biol Chem. 276:42462–42467. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McManus KJ and Hendzel MJ: ATM-dependent
DNA damage-independent mitotic phosphorylation of H2AX in normally
growing mammalian cells. Mol Biol Cell. 16:5013–5025. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Silva GS, Fe LML, Silva MNP and Val V: Ras
oncogene and Hypoxia-inducible factor-1 alpha (hif-1α) expression
in the Amazon fish Colossoma macropomum (Cuvier, 1818) exposed to
benzo[a]pyrene. Genet Mol Biol. 40:491–501. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun Y, Miller CA III, Wiese TE and Blake
DA: Methylated phenanthrenes are more potent than phenanthrene in a
bioassay of human aryl hydrocarbon receptor (AhR) signaling.
Environ Toxicol Chem. 33:2363–2367. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aubert G: Telomere dynamics and aging.
Prog Mol Biol Transl Sci. 125:89–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Herbstman JB, Tang D, Zhu D, Qu L, Sjödin
A, Li Z, Camann D and Perera FP: Prenatal exposure to polycyclic
aromatic hydrocarbons, benzo[a]pyrene-DNA adducts, and genomic DNA
methylation in cord blood. Environ Health Perspect. 120:733–738.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tessema M, Yu YY, Stidley CA, Machida EO,
Schuebel KE, Baylin SB and Belinsky SA: Concomitant promoter
methylation of multiple genes in lung adenocarcinomas from current,
former and never smokers. Carcinogenesis. 30:1132–1138. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Belinsky SA, Nikula KJ, Palmisano WA,
Michels R, Saccomanno G, Gabrielson E, Baylin SB and Herman JG:
Aberrant methylation of p16(INK4a) is an early event in lung cancer
and a potential biomarker for early diagnosis. Proc Natl Acad Sci
USA. 95:11891–11896. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shiloh Y: The ATM-mediated DNA-damage
response: Taking shape. Trends Biochem Sci. 31:402–410. 2006.
View Article : Google Scholar : PubMed/NCBI
|