Introduction
The advent of the next-generation sequencing (NGS)
technology has revolutionized the current approaches in molecular
diagnostics of individuals with severe intellectual disabilities
(ID), developmental delay (DD), autism spectrum disorders (ASD) and
multiple congenital abnormalities (MCA). Recently, >1,000 genes
have been identified as the cause of ID and other
neurodevelopmental disorders (1).
A previous study showed that >30% of cases of
idiopathic ID and 13% of cases of severe ID (IQ<50) can be
explained with a specific molecular diagnosis using a targeted NGS
approach (2). Moreover, the
current molecular diagnostic method uses whole-exome sequencing
(WES), which is a highly powerful and cost-effective part of the
first-tier standard diagnostic approach, with a diagnostic yield of
up to 55% (3).
Bainbridge-Ropers syndrome (BRS; OMIM #615485) is a
rare congenital disorder characterized by delayed neuronal, motor
and growth development, severe ID accompanied by absent or poor
speech, muscular hypotonia, feeding difficulties and facial
dimorphism (4). It was first
described by Bainbridge et al (5), who identified rare de novo truncating
sequence variants in the additional sex-combs like 3 (ASXL3)
gene in a group of 4 unrelated affected children presenting similar
phenotypic features. The pathogenic sequence variants in the
ASXL3 gene lead to the reduced expression of the gene. The
haploinsuffiency of the ASXL3 gene with high penetrance is
the most likely the underlying and causative mechanism of the
disease (6).
In the present study, the case of a child (female,
born in 2013) with global developmental delay, central hypotonia,
microcephaly and poor speech is described. She was examined using a
multi-step molecular diagnostics algorithm, including karyotype and
array-comparative genomic hybridization (CGH) analyses. However, no
pathogenic chromosomal rearrangements or copy-number variants
(CNVs) that could explain the phenotype of the patient were
identified. Therefore, the patient and her healthy parents took
part in a pilot study using targeted NGS with a commercially
available gene-rich panel. This panel contained 2,742 genes
catalogued in the Online Mendelian Inheritance in Man (OMIM)
database whose pathogenic sequence variants are associated with
human inherited diseases. The results of this approach were
subsequently validated using consecutive Sanger sequencing.
Materials and methods
Clinical characteristics of the
patient
The proband was born from the second pregnancy of
healthy, unrelated parents (mother and father both born in 1981).
The mother has history of reproductive problems and the couple
attempted to have children for 2 years (from age 28 to 30 years).
After hormonal stimulation therapy, the mother conceived
spontaneously. The proband was delivered by breech birth after
Cesarean section as a dizygotic twin at 34 weeks of gestation
(2,160 g/45 cm reaching Apgar score 10-10-10, and the brother 2,460
g/45 cm). The prenatal ultrasound and biochemical screening did not
show any apparent abnormalities. After delivery, the proband
experienced neonatal icterus followed by 2-day-long phototherapy,
and developed a poor sucking reflex. The proband was transferred to
specialized neonatal unit, where she spent 18 days in the neonatal
incubator. She was vaccinated according to the recommended
vaccination schedule.
The proband was breast-fed up 7 months of the age,
with persisting feeding difficulties. Since the neonatal period,
she had been diagnosed with central hypotonia, delayed psychomotor
development and microcephaly, with an occipitofrontal circumference
(OFC) of 34.5 cm at 7 weeks of age. To improve motor development,
the proband was stimulated using Vojta therapy (7,8). At
the age of 6 months, a brain ultrasound examination did not uncover
any abnormalities. The proband started to roll over at the age of
13 months, as well as crawling and standing on four limbs during
the following month, without forward motion. Brain magnetic
resonance imaging (MRI) and electroencephalography did not show any
abnormalities at the age of 18 months, and the proband had an OFC
of 42.8 cm. At present, the proband exhibits mild facial dimorphism
including convergent strabismus, palate malformations and severely
delayed milestones in physiological and intellectual developmental
stages. The proband does not currently display coordinated gross
motor skills (walking) due to the persistent central hypotonia and
feet malformations. Due to the hypotonia, her head often leans
backwards. The proband does not speak at present, only vocalizes.
At the age of 9 months, she developed short-term recurrent
infections of the upper respiratory tract, which were treated with
antibiotics. Regarding skeletal and skin abnormalities, the proband
has a 4-finger line on the right palm, and the first and third toes
are crossed under the second toe on both feet. Some autistic
traits, including unprovoked laughter, have been identified, but
social contacts have started to develop. The proband is under
multidisciplinary specialized medical supervision and is undergoing
rehabilitative therapy. In summary, her psychomotor development
corresponds to that of a 1-year-old child. Her dizygotic twin
brother is healthy, and her older brother (born in 2012) suffers
from a cleft lip and mild facial dimorphism (hypertelorism and
epicanthus).
Table I provides an
overview of the phenotypic features of the proband and a brief
summary of selected previously reported BRS patients with the
ASXL3 gene pathogenic variants.
 | Table I.Clinical features of the proband and
summary of selected previous patients with the additional sex-combs
like 3 pathogenic variants. |
Table I.
Clinical features of the proband and
summary of selected previous patients with the additional sex-combs
like 3 pathogenic variants.
|
|
|
|
|
|
|
|
| Craniofacial
features | Muscular
abnormalities |
|
|
|
|
|
|
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|---|
| Author, year | Sex (F/M
ratio) | Pregnancy
(normal/abnormal ratio) | Birth (weeks of
gestation) | Birth OFC (cm) | Birth weight
(kg) | Birth length
(cm) | Feeding
difficulties | Strabismus | Palate
abnormalities | Microcephaly | Hypotonia | Hypertonia | Motor delay | Free walking | Intellectual
disability | Speech
impairment | Autistic
features | Brain MRI
(normal/abnormal ratio) | EEG
(normal/abnormal ratio) | Skeletal
abnormalities | (Refs.) |
|---|
| Present study | F | AN | 34 | 34.5 | 2.16 | 45 | yes | yes | yes | yes | yes | no | yes | no | yes | yes | yes | N | N | no |
|
| Kuechler et
al, 2017 | 3/3 | 2/3 | 36–39 | 32-37.5 | 2.66–3.6 | 46–52 | 6/6 | 5/6 | 5/6 | 1/6 | 6/6 | 2/6 | 6/6 | 2/6 | 6/6 | 6/6 | 5/6 | 2/4 | 3/3 | 6/6 | 6 |
| Bainbridge | 2/2 | 1/3 | 38–40 | reduced | small | small | 3/4 | n.r. | 3/4 | 3/4 | 3/4 | 1/4 | 4/4 | no | 4/4 | 3/4 | 0/4 | 0/1 | n.r, | 3/4 | 5 |
| et al,
2013 |
|
|
| 3/4 | size 3/4 | size 3/4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Balasubramanian
et al, 2017 | 6/6 | 11/1 | 34-N | n.r. | m. red. | n.r. | 9/12 | 7/12 | 9/12 | 7/12 | 11/12 | 0/12 | m. del. | m. del. | 11/12 | 11/12 | 9/12 | 9/3 | n.r. | 7/12 | 45 |
| Srivastava et
al, 2016 | 1/2 | 2/1 | N | n.r. | 1/3 reduced | n.r. | 3/3 | 0/3 | n.r. | 0/3 | 3/3 | 0/3 | 3/3 | 0/3 | 3/3 | 3/3 | n.r. | 1/2 | 2/1 | 1/3 | 4 |
The proband was diagnosed at the Department of
Medical Genetics (University Hospital Brno) at the age of 18 months
in April 2015. The patient's parents provided written informed
consent. Peripheral blood samples were collected in sterile
heparinized tubes for cytogenetic analysis. Genomic DNA samples
were obtained from 1 ml peripheral blood in EDTA, according to the
standard DNA isolation process using the MagNaPure system (Roche
Diagnostics, Basel, Switzerland). Quality and quantity were checked
using a NanoDrop® ND-1000 (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and Qubit® 1.0 (Thermo Fisher
Scientific, Inc.).
G-banded karyotype analysis and
array-CGH technique
Cytogenetic analysis of the karyotype was performed
using a standard G-banding procedure as previously described
(9,10). Whole-genomic screening of
unbalanced chromosomal rearrangements by array-CGH was performed
using SurePrint G3 CGH Microarray 4×180 K (Agilent Technologies,
Inc., Santa Clara, CA, USA), following the manufacturer's
recommendations. The patient's DNA sample was matched with Human
Genomic DNA, Female reference (Promega Corporation, Madison, WI,
USA). The microarray slide was scanned with a DNA Microarray
Scanner (Agilent Technologies, Inc.). Data were obtained using
Agilent Feature Extraction software, version 12.0.2.2, and
visualized using Agilent Genomic Workbench Software, version
7.0.4.0 (both from Agilent Technologies, Inc.). Structural CNVs
were detected using the ADM-2 algorithm (11) with the following filters: >5
neighboring probes in genomic region; minimal size of 200 kb in
region; and minimal absolute average log ratio of 0.25 as cut-off.
All genomic positions were estimated on the human reference
sequence GRCh37/hg19. Microarray data are available in the Array
Express database (https://www.ebi.ac.uk/arrayexpress/) under the
accession number E-MTAB-7027.
Targeted NGS
High quality genomic DNA was used for the library
preparation. A total of 200 ng DNA was sheared using the Covaris
E-Series (Covaris, Inc., Woburn, MA, USA), and the size
distribution of fragments was evaluated using Agilent 2200
TapeStation (Agilent Technologies, Inc.). The DNA library was
processed using the SureSelectXT Target Enrichment
System and captured using ClearSeq Inherited DiseaseXT,
according to the manufacturer's recommendations (Agilent
Technologies, Inc.). This design included 2,742 genes involved in
the pathogenesis of human inherited diseases. Before the sequencing
run, the captured DNA library was checked for its quality (Agilent
2200 TapeStation; Agilent Technologies, Inc.) and quantity
(Qubit® 1.0; Thermo Fisher Scientific, Inc.). The
library was then sequenced on an Illumina MiSeq benchtop sequencer
following the manufacturer's recommendations (Illumina, Inc., San
Diego, CA, USA).
NGS data processing and data
analysis
The raw sequencing data were processed using a
multi-step advanced bioinformatics pipeline. The quality of the raw
sequencing files was checked using FastQC (version 0.11.5;
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
The presence of adapters was scanned using minion and swan (Kraken
package, version 15-065) (12).
The preprocessing of raw sequencing files was performed using
Cutadapt (version 1.11) (13).
Briefly, very low-quality ends were trimmed (Phred<5). Then, the
adapters from both reads of a pair were removed with a minimal
overlap of 3 bp and a maximum of 10% mismatch in a matched sequence
(removed adapters: R1, AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC; R2,
AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT).
Finally, very short (<35 bp) and unpaired reads after the
trimming were discarded. Preprocessed sequencing reads were mapped
to a reference genome (hg19; University of California Santa Cruz
Genome FTP; http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/)
(14,15) by BWA aln/sampe (version 0.7.15)
(16). Alignments were further
processed with Stampy (version 1.0.29) (17). Aligned bam files were sorted by
position and mate information was corrected using Samtools (version
1.3) (18). Since the library was
sequenced in two separate sequencing runs, bam files were merged
into one using Picard (version 2.1.0; http://picard.sourceforge.net). PCR duplicates were
marked and removed using Picard. Duplicate-clean bam files were
indel realigned using GenomeAnalysisTK (version 3.6) (19). Base Quality Score Recalibration
(BQSR) was performed in two steps using GenomeAnalysisTK, and dbSNP
(version 147) variants were used as a set of known variants
(https://www.ncbi.nlm.nih.gov/snp). The
coverage of targeted regions was explored using bedtools (version
2.23.0) (20). Additional quality
checks and statistics were obtained using Picard.
Raw variant calls were performed using VarScan2
(version 2.4.2) (21) with default
settings, except that the minimal variant frequency was set to 0.2,
and the VarScan2 P-value set to 0.05. The dbSNP ID was added if a
match between dbSNP and raw variants was found using SnpSift
(version 4.2) (22). Filtering of
the raw variants was performed using VarScan2 with the default
settings, except for the minimal P-value, which was set to 0.05.
SNPs in very close proximity to indels were removed from the calls.
The effect of the variants, ClinVar (downloaded on 10/18/2016)
(23) and dbSNP annotation was
added to the filtered variant calls using SnpEff (version 4.2)
(24). SnpEff also provided
putative variant impacts (HIGH, MODERATE, LOW, MODIFIER) to
categorize and prioritize variants.
Final variants were extracted from the filtered
variants by targeted regions and by association with genes of
interest using vcftools (version 0.1.15) and bcftools (version 1.3)
(25). Additional analyses and
annotations were performed using R (version 3.3.1; http://www.r-project.org/) with the data.table
(version 1.9.6; http://CRAN.R-project.org/package=data.table) and
VariantAnnotation (26) libraries.
The NGS data were manually checked and visualized using Integrative
Genomics Viewer (version 2.3.82) (27,28).
NGS data in .fastq and .bam format are available in the Array
Express database (https://www.ebi.ac.uk/arrayexpress/) under the
acces-sion number E-MTAB-7026.
The variants were classified using ACMG
recommendations (29) and detailed
information provided in databases OMIM (30), ClinVar (23), dbSNP (https://www.ncbi.nlm.nih.gov/snp),
UniProtKB/Swiss-Prot (31), ExAc
(32), 1000 Genomes (33) and relevant scientific literature.
The web-based application gene.iobio 3.0.5 (http://gene.iobio.io/) was used to assess the
variant's localization within the context of known pathogenic or
likely pathogenic variants from ClinVar (23). The in silico analysis was
performed using online tools: PROVEAN Tool (34), Polyphen-2 (35), MutationTaster2 (36) and VarSome (37).
Sanger sequencing and data
analysis
The pathogenic or likely pathogenic
single-nucleotide variants (SNVs) in the proband and parents were
validated using targeted Sanger sequencing. DNA primers were
designed using Primer3 (38,39),
Primer Blast (40), UCSC
In-Silico PCR (15) and
OligoAnalyzer 3.1 tools (https://eu.idtdna.com/calc/analyzer), and were
synthesized by Integrated DNA Technologies, Inc. (Coralville, IA,
USA). PCR was performed using the forward primer
5′-CAGAGCAACACAGCTTTGGA-3′ and a reverse primer
5′-GGAGACATTTCCAGGCCCTAT-3′, according to the manufacturer's
recommendations (Promega Corporation). PCR products were purified
using the Exonuclease I and FastAP Thermosensitive Alkaline
Phosphatase protocol (Thermo Fisher Scientific, Inc.). Then,
single-stranded DNA fragment libraries for direct Sanger sequencing
were prepared using the BigDye® Terminator v3.1 Cycle
Sequencing kit (Thermo Fisher Scientific, Inc.), according to the
manufacturer's recommendations. The sequencing reactions were run
on the DNA sequencer ABI 3130 (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The analysis was performed using
Sequencher® version 5.1 software (http://www.genecodes.com; Gene Codes Corporation, Ann
Arbor, MI, USA). The chromatograms of the proband and the parents
are stored in the Figshare online digital repository (doi:
10.6084/m9.figshare.6744224).
Results
Whole-genomic analyses and
verification
The proband was assessed to have a normal female
karyotype (46, XX) via cytogenetic analysis. Consequently, the
array-CGH analysis of the oligonucleotide DNA microarray 180 K CGH
exhibited the same result [arr(1–22,X)×2]. Due to the unexplained
severe pathological phenotype, which was suspected to have a
molecular genetic cause, the proband and her parents were involved
in a pilot project using a targeted NGS approach with capture
design ClearSeq Inherited disease (performed on proband DNA
samples), with consecutive Sanger sequencing verification
(performed on proband and parental DNA samples). In the course of
the NGS data analysis, the primary focus of the study was exonic
variants, and the data were filtered for non-synonymous exonic
variants (SNVs and indels) only. These variants were analyzed in
detail by searching through the ClinVar, OMIM, dbSNP and
UniProtKB/Swiss-Prot databases. Then, in silico tools were
used to predict the structural and functional impact of these
variants on the encoded protein (PROVEAN Tool, Polyphen-2,
MutationTaster2 and VarSome). Finally, the findings were correlated
to the patient phenotype, and were verified by independent analysis
of Sanger sequencing in the proband and unaffected parents.
Quality control of mapped reads to genomic targets
was performed. A total of >99% of reads were mapped to genomic
targets, with 30X coverage for >90% of bases. In the proband, a
total of 18,558 DNA sequence variants in targeted regions were
identified, including 17,560 variants (94.7%) classified with known
SNP identification numbers. In total, 16,794 SNVs, 887 insertions
and 877 deletions were identified. By performing NGS data analysis
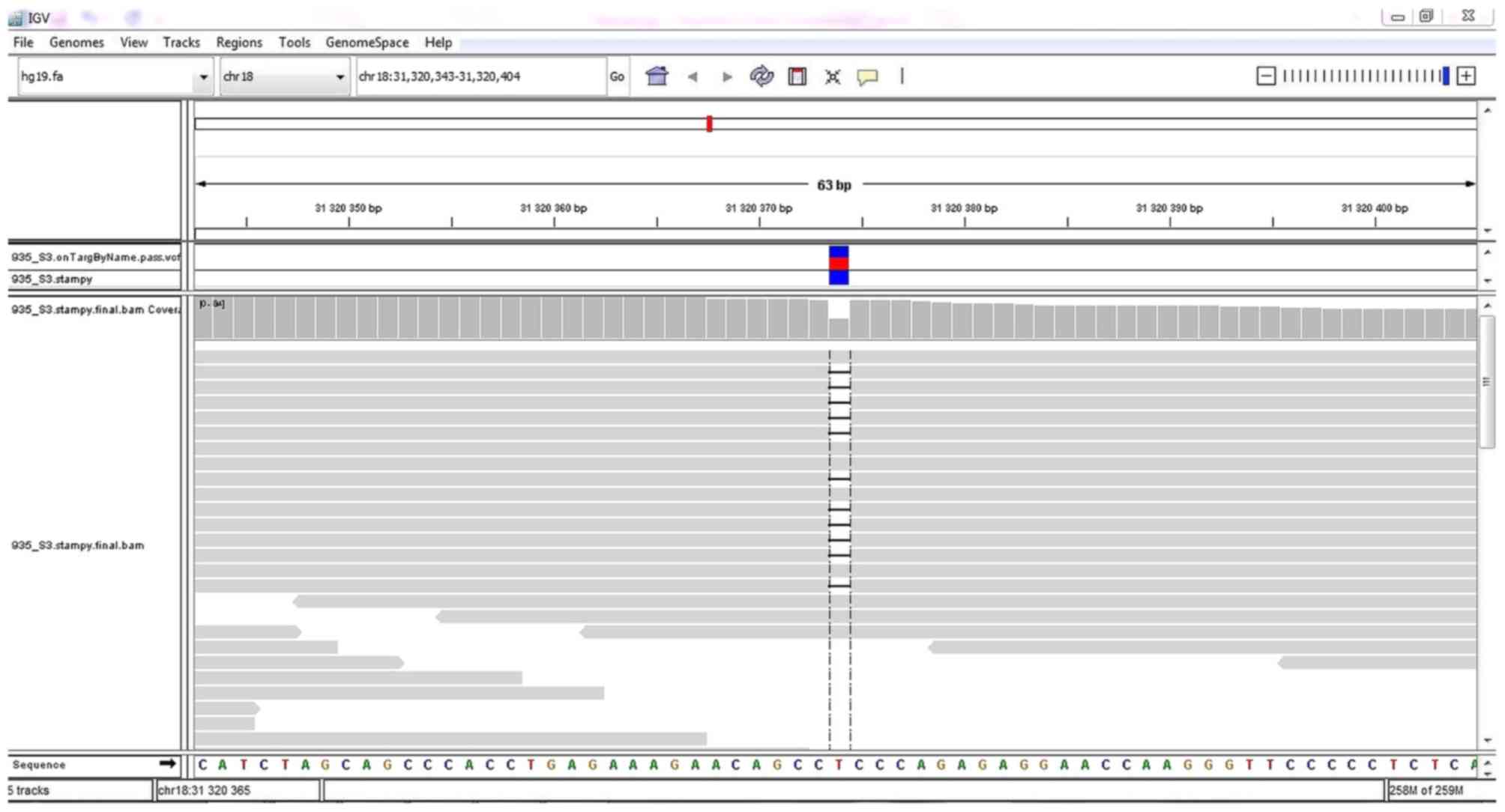
and variant filtering, a heterozygous 1-bp deletion variant
NC_000018.9:g.31320374delT (NM_030632.1:c.3006delT) affecting the
ASXL3 gene was identified, and it was present in 47.92% of
reads (23/48) covering this position (Fig. 1). No other pathogenic or likely
pathogenic variants related to the phenotype of the proband were
identified. This variant was not found in the ExAC or 1000 Genomes
databases. An in silico prediction analysis was performed
using the MutationTaster2 and VarSome tools to identify its impact
on the ASXL3 protein structure and function. This single-nucleotide
deletion in the ASXL3 gene results in a p.R1004Efs*21
frameshift leading to a premature termination codon. The protein
structure is likely to be affected due to the strong protein
truncation (>50% of protein length is missing). Using the
VarSome prediction tool, the p.R1004Efs*21 frameshift was
classified as likely pathogenic following the ACMG criteria
(29) (Fig. S1).
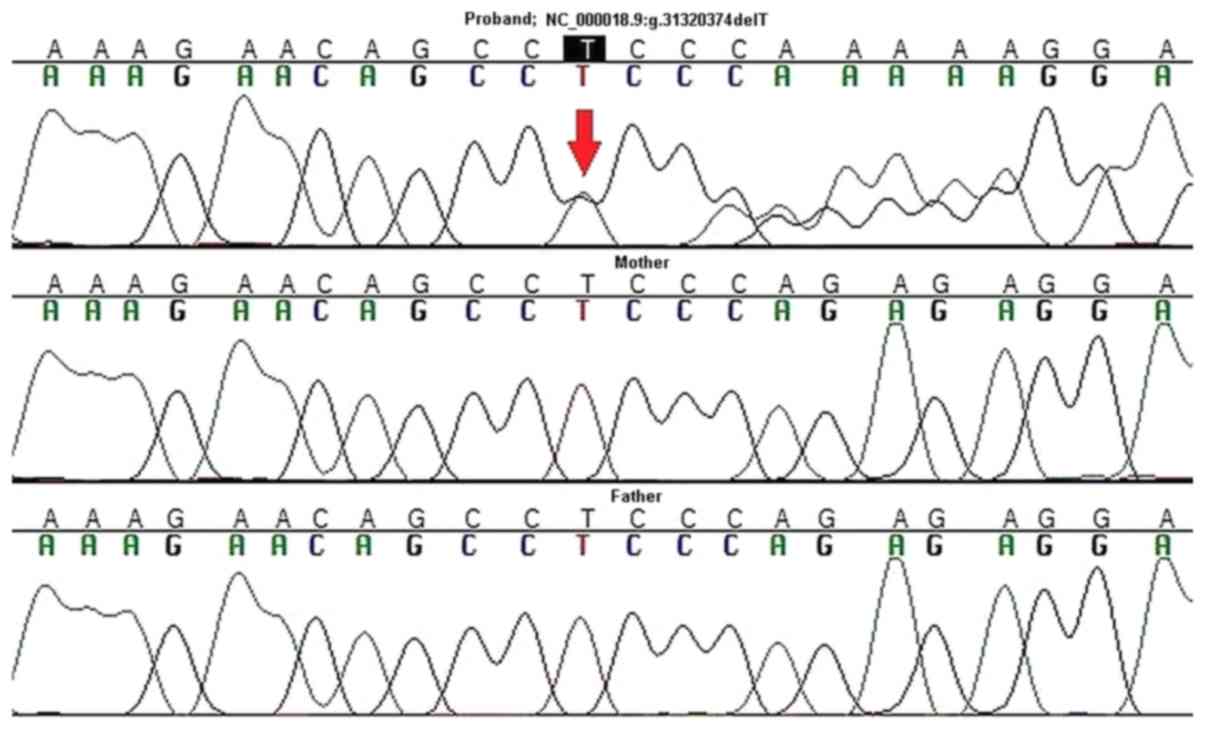
Consequently, targeted Sanger sequencing of DNA from
the proband and her parents was performed to validate the
ASXL3 variant and to assess its origin (de novo, or
paternal/maternal). A de novo origin and heterozygous state
was confirmed for p.R1004Efs*21 using this method (Fig. 2). This result provides strong
supporting evidence for its pathogenic or likely pathogenic
effect.
Using gene.iobio.io 3.0.5, it was determined that
the proband p.R1004Efs*21 variant is located in the same cluster as
other known pathogenic and likely pathogenic ASXL3 variants,
which enhances its clinical relevance (Fig. S2).
Discussion
The present study reports the case of a child with
severe psychomotor delay, hypotonia, microcephaly and facial
dimorphism. Using targeted NGS and following verification analysis
by Sanger sequencing, a novel de novo pathogenic sequence
variant in the ASXL3 gene, p.R1004Efs*21, was
identified.
The ASXL3 gene is located at the 18q12.1
chromosome region and encodes a member of the vertebrate ASX-like
protein family. ASXL family members represent epigenetic
scaffolding proteins that assemble specific epigenetic regulators
and transcription factors to specific genomic loci containing
histone modifications (41,42).
Polycomb protein ASX (ASX) was first identified in Drosophila
melanogaster as a part of the polycomb group of proteins
involved in embryonic development, maintaining HOX genes in a
transcriptionally repressive state (43,44).
The encoded protein ASXL3 contains 12 exons and has 2,248 amino
acids. Like other ASX family members, the ASXL3 protein has a
conserved domain structure: ASXN and ASXH domains in the N
terminus; ASXM1 and ASXM2 domains in the middle region; and the PHD
domain in the C terminus (41,42).
The ASXN and PHD zinc-finger domains play a role in the regulation
of gene transcription, representing putative DNA- or
histone-recognition sites. The region around the ASXH domain
creates protein-protein interaction sites for association with
epigenetic regulators. The ASXM1 and ASXM2 domains are involved in
protein-protein interactions. Between ASXH and ASXM1, there is the
5′ mutational cluster region (MCR), where truncated or splice
variants tend to cluster, giving rise to aberrant ASXL proteins
with intact ASXN and ASXH domains, while pathogenic sequence
variants in the 3′MCR lead to variants with changes between the
ASXM1 and ASXM2 domains (45).
Recent studies have highlighted important roles for
the ASXL3 gene in congenital disorders (45) and cancer (40) through the identification of
multiple ASXL3 pathogenic sequence SNVs and indels. Focusing
on congenital disorders, the pathogenic variants affecting the
ASXL3 gene have been identified as the cause of disorders in
patients sharing similar phenotypic features, including delayed
psychomotor development with missed milestones, microcephaly,
severe persisting feeding difficulties, poor growth, poor or absent
speech and dimorphic facial features (4,5,6). As
presented, the proband exhibits most of the typical BRS-related
phenotypic features (abnormal pregnancy, preterm birth and reduced
birth parameters, feeding difficulties, craniofacial abnormalities,
psychomotor delay and autistic features). Brain MRI scans in
patients with BRS usually indicate mild white matter loss and mild
corpus callosum hypoplasia, or mild cerebellar vermis hypoplasia.
With the exception of the brain MRI findings, these features are
different to those previously described as a consequence of
pathogenic variants in the ASXL1 gene, causing Bohring-Opitz
syndrome (BOS) (46). A recent
study identified a group of six unrelated patients having de
novo heterozygous truncating variants in the ASXL2 gene
(47). These patients shared
specific clinical features, some of which are also present in
patients with the ASXL1 gene (BOS) and ASXL3 gene
(BRS) truncating or splicing pathogenic variants (developmental and
intellectual impairment, facial dimorphism, feeding difficulties in
the neonatal period and hypotonia). Since 2013, ~30 patients with
ASXL3 pathogenic truncating or splicing pathogenic variants
causing BRS have been identified in scientific publications, to the
best of our knowledge. The variants are clustered predominantly in
two previously reported separated MCRs in exon 11 and exon 12,
respectively (45).
In the proband, a novel truncating variant,
p.R1004Efs*21, located in the terminal part of exon 11 outside the
reported MCRs, was identified. The mutation is located in an amino
acid coding region conserved among vertebrates; thus, the
MutationTaster2 and VarSome tools predicted a deleterious effect of
this variant on protein structure and function due to truncation.
Moreover, as mRNAs containing a premature stop codon can undergo
degradation through nonsense-mediated decay (NMD) (4), the truncating variants may have a
loss-of-function (LoF) impact. Functional studies have suggested
that all ASXL3 nonsense variants may be translated to
prematurely terminated proteins, which may consequently act in a
dominant-negative way (4,5). The correlation between the position
of the truncating variant and the severity of the phenotype has
also been debated: The disease severity may decrease as the variant
occurs further away from the 5′-end of exon 11 and towards the
3′-end (5). A recently described
case of a male child with atypical BRS also revealed the occurrence
of a novel heterozygous de novo variant, p.P1010Lfs*14, in
the ASXL3 gene (48). This
variant leads to an identically truncated ASXL3 protein as that
seen in the proband of the present study, but the phenotypic
features between the cases are different, namely the absence of
apparent structural brain abnormalities and less prominent facial
dimorphism in the proband.
As an exceptional case of two compound heterozygous
variants in the ASXL3 gene, there was a reported case of
another patient harboring a missense variant, p.R989G, of maternal
origin, and a missense variant, p.K1026N, of paternal origin
(49). The patient experienced
BRS-like features and primary insulin-like growth factor 1
deficiency. That study predicted that the phenotype was the product
of the synergistic or additive effect of these missense variants in
the LoF of the ASXL3 gene. The ASXL3 gene has also
been previously identified as an ASD risk gene, particularly
missense variants of this gene (50). In the ClinVar database (23), there are no reported cases of
patients with a BRS phenotype having heterozygous de novo
pathogenic missense variants in the ASXL3 gene.
Unfortunately, BRS is not a common clearly
recognizable syndromes, especially due to the absence of specific
phenotypic features and the low total number of reported cases.
Among patients with BRS, it is possible to observe varying degrees
of phenotypic severity, especially varying in the degree of ID
(5). This range in severity may
have an early or late post-zygotic origin, leading to pathogenic
variants with somatic mosaicism, which will then have an incomplete
penetrance and/or variable phenotypic features (5). Two of the described variants occur at
the 3′-end of the gene (p.E1761fs, p. E1824K), indicating escape
from NMD and the retention of protein activity. These observations
also confirm that a broad allelic heterogeneity may be implicated
in the pathogenesis of BRS.
Based on previously reported cases, the penetrance
of truncated or spliced pathogenic variants seems to be high, and
these have a de novo origin since they are absent in healthy
individuals. In the ExAC database (32) there are four reported LoF variants
(three exonic truncating variants and one splicing variant), none
of which have an allelic frequency higher than 8.34×10−6
(exonic truncating variants) or 1.38×10−5 (splicing
variant), and the probability of LoF intolerance is estimated to be
1.00. This value predicts high intolerance of the gene to LoF
mutations (32). A total of
>670 missense variants have been reported at present, and their
allele frequency varies significantly (from 8.281×10−6
to 0.6029), occurring across the whole coding region of the
ASLX3 gene. This observation is indicative of the different
impact of missense variants on the ASXL3 protein function, from
benign polymorphisms to possibly damaging effects (which was
evaluated using Polyphen-2 in silico prediction) (35).
In the present study, a novel de novo
frameshift variant in the ASXL3 gene, which could lead to a
truncated protein in a young patient with severe psychomotor
developmental delays and microcephaly, was detected. Based on these
findings and previous reported cases, it was proposed that the
identified frameshift variant in the ASXL3 gene may have
significantly affected ASXL3 protein structure and function. These
observations may help in defining and diagnosing BRS in patients
with similar phenotypic features.
Supplementary Material
Supporting Data
Acknowledgements
The Core Facility Bioinformatics of CEITEC Masaryk
University is gratefully acknowledged for their help collecting
scientific data presented in this paper. The authors would like to
acknowledge the CF Genomics CEITEC MU, supported by the NCMG
research infrastructure (LM2015091, funded by MEYS CR), for their
support in obtaining the scientific data presented in this paper.
The authors would also like to thank the ‘Projects of Large
Research, Development, and Innovations Infrastructures’ (CESNET,
LM2015042) for allowing access to computing and storage facilities,
owned by parties and projects contributing to the National Grid
Infrastructure MetaCentrum.
Funding
This work was supported by the Ministry of Health,
Czech Republic, Conceptual Development of Research Organization
(grant no. FNBr, 65269705) and by the Project of Faculty of
Science, Masaryk University, Brno, Czech Republic (grant no.
MUNI/A/0824/2017).
Availability of data and materials
The microarray data analyzed in the present study
are available in the Array Express database (https://www.ebi.ac.uk/arrayexpress/) in the .txt
format under the accession number E-MTAB-7027. The NGS data
analyzed in this study are available in the Array Express database
(https://www.ebi.ac.uk/arrayexpress/)
in .fastq and .bam format under the accession number E-MTAB-7026.
Sanger sequencing data, including the chromatograms of the proband
and the parents, are stored in the Figshare online digital
repository (doi: 10.6084/m9.figshare.6744224).
Authors' contributions
MW analyzed and interpreted the proband's microarray
and NGS data regarding her phenotype using bioinformatics databases
and in silico tools. MW was a major contributor in writing
the manuscript. JO processed raw NGS data using advanced
bioinformatics tools through a unique multi-step pipeline, and
wrote the part of the manuscript related to the NGS data
processing. JS participated in the analysis of microarray and NGS
data. EH and HF performed targeted Sanger sequencing analysis. EM
and PN analyzed the proband's karyotype. RB and RG provided
specialized genetic counseling for the proband and her family, and
interpreted the findings. PK contributed towards the interpretation
of data, performed general scientific supervision and general
critical revision of the manuscript.
Ethics approval and consent to
participate
The Ethical committee of the University Hospital
Brno approved the present study. Written informed consent for
participation in the study was obtained from the parents of the
proband.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kochinke K, Zweier C, Nijhof B, Fenckova
M, Cizek P, Honti F, Keerthikumar S, Oortveld MA, Kleefstra T,
Kramer JM, et al: Systematic phenomics analysis deconvolutes genes
mutated in intellectual disability into biologically coherent
modules. Am J Hum Genet. 98:149–164. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Redin C, Gérard B, Lauer J, Herenger Y,
Muller J, Quartier A, Masurel-Paulet A, Willems M, Lesca G,
El-Chehadeh S, et al: Efficient strategy for the molecular
diagnosis of intellectual disability using targeted high-throughput
sequencing. J Med Genet. 51:724–736. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rauch A, Wieczorek D, Graf E, Wieland T,
Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di
Donato N, et al: Range of genetic mutations associated with severe
non-syndromic sporadic intellectual disability: An exome sequencing
study. Lancet. 380:1674–1682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Srivastava A, Ritesh KC, Tsan YC, Liao R,
Su F, Cao X, Hannibal MC, Keegan CE, Chinnaiyan AM, Martin DM and
Bielas SL: De novo dominant ASXL3 mutations alter H2A
deubiquitination and transcription in Bainbridge-Ropers syndrome.
Hum Mol Genet. 25:597–608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bainbridge MN, Hu H, Muzny DM, Musante L,
Lupski JR, Graham BH, Chen W, Gripp KW, Jenny K, Wienker TF, et al:
De novo truncating mutations in ASXL3 are associated with a novel
clinical phenotype with similarities to Bohring-Opitz syndrome.
Genome Med. 5:112013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kuechler A, Czeschik JC, Graf E, Grasshoff
U, Hüffmeier U, Busa T, Beck-Woedl S, Faivre L, Rivière JB, Bader
I, et al: Bainbridge-Ropers syndrome caused by loss-of-function
variants in ASXL3: A recognizable condition. Eur J Hum Genet.
25:183–191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vojta V and Peters A: Das Vojta Prinzip.
3rd edition. Springer; Heidelberg: 2007,
|
|
8
|
Jung MW, Landenberger M, Jung T,
Lindenthal T and Philippi H: Vojta therapy and neurodevelopmental
treatment in children with infantile postural asymmetry: A
randomized controlled trial. J Phys Ther Sci. 29:301–306. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Silva M, de Leeuw N, Mann K, Schuring-Blom
H, Morgan S, Giardino D, Rack K and Hastings R: European guidelines
for constitutional cytogenomic analysis. Eur J Hum Genet. 27:1–16.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Howe B, Umrigar A and Tsien F: Chromosome
preparation from cultured cells. J Vis Exp. e502032014.PubMed/NCBI
|
|
11
|
Roy S and Motsinger Reif A: Evaluation of
calling algorithms for array-CGH. Front Genet. 4:2172013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Davis MP, van Dongen S, Abreu-Goodger C,
Bartonicek N and Enright AJ: Kraken: A set of tools for quality
control and analysis of high-throughput sequence data. Methods.
63:41–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martin M: Cutadapt removes adapter
sequences from high-throughput sequencing reads. EMBnet.journal.
17:10–12. 2011. View Article : Google Scholar
|
|
14
|
Lander ES, Linton LM, Birren B, Nusbaum C,
Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al:
Initial sequencing and analysis of the human genome. Nature.
409:860–921. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Speir ML, Zweig AS, Rosenbloom KR, Raney
BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS,
et al: The UCSC Genome Browser database: 2016 update. Nucleic Acids
Res. 44:D717–D725. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lunter G and Goodson M: Stampy: A
statistical algorithm for sensitive and fast mapping of Illumina
sequence reads. Genome Res. 21:936–939. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The Sequence Alignment/Map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M, et al: The Genome Analysis Toolkit: A MapReduce framework for
analyzing next-generation DNA sequencing data. Genome Res.
20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Quinlan AR and Hall IM: BEDTools: A
flexible suite of utilities for comparing genomic features.
Bioinformatics. 26:841–842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
VarScan 2: Somatic mutation and copy number alteration discovery in
cancer by exome sequencing. Genome Res. 22:568–576. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cingolani P, Patel VM, Coon M, Nguyen T,
Land SJ, Ruden DM and Lu X: Using Drosophila melanogaster as a
model for genotoxic chemical mutational studies with a new program,
SnpSift. Front Genet. 3:352012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Landrum MJ, Lee JM, Benson M, Brown G,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al
ClinVar, : Public archive of interpretations of clinically relevant
variants. Nucleic Acids Res. 44:D862–D868. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cingolani P, Platts A, Wang le L, Coon M,
Nguyen T, Wang L, Land SJ, Lu X and Ruden DM: A program for
annotating and predicting the effects of single nucleotide
polymorphisms, SnpEff: SNPs in the genome of Drosophila
melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6:80–92.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Danecek P, Auton A, Abecasis G, Albers CA,
Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST,
et al: The variant call format and VCFtools. Bioinformatics.
27:2156–2158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Obenchain V, Lawrence M, Carey V, Gogarten
S, Shannon P and Morgan M: VariantAnnotation: A Bioconductor
package for exploration and annotation of genetic variants.
Bioinformatics. 30:2076–2078. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thorvaldsdóttir H, Robinson JT and Mesirov
JP: Integrative Genomics Viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hamosh A, Scott AF, Amberger JS, Bocchini
CA and McKusick VA: Online Mendelian Inheritance in Man (OMIM), a
knowledgebase of human genes and genetic disorders. Nucleic Acids
Res. 33:D514–D517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
UniProt Consortium: The universal protein
resource (UniProt). Nucleic Acids Res. 36:D190–D195.
2008.PubMed/NCBI
|
|
32
|
Lek M, Karczewski KJ, Minikel EV, Samocha
KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ,
Cummings BB, et al: Analysis of protein-coding genetic variation in
60,706 humans. Nature. 536:285–291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
1000 Genomes Project Consortium, ; Auton
A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini
JL, McCarthy S, McVean GA and Abecasis GR: A global reference for
human genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choi Y, Sims GE, Murphy S, Miller JR and
Chan AP: Predicting the functional effect of amino acid
substitutions and indels. PLoS One. 7:e466882012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kopanos C, Tsiolkas V, Kouris A, Chapple
CE, Albarca Aguilera M, Meyer R and Massouras A: VarSome: The human
genomic variant search engine. Bioinformatics. 2018.
|
|
38
|
Untergasser A, Cutcutache I, Koressaar T,
Ye J, Faircloth BC, Remm M and Rozen SG: Primer3-new capabilities
and interfaces. Nucleic Acids Res. 40:e1152012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Koressaar T and Remm M: Enhancements and
modifications of primer design program Primer3. Bioinformatics.
23:1289–1291. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ye J, Coulouris G, Zaretskaya I,
Cutcutache I, Rozen S and Madden TL: Primer-BLAST: A tool to design
target-specific primers for polymerase chain reaction. BMC
Bioinformatics. 13:1342012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Katoh M and Katoh M: Identification and
characterization of ASXL3 gene in silico. Int J Oncol.
24:1617–1622. 2004.PubMed/NCBI
|
|
42
|
Katoh M: Functional and cancer genomics of
ASXL family members. Br J Cancer. 109:299–306. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sinclair DA, Milne TA, Hodgson JW,
Shellard J, Salinas CA, Kyba M, Randazzo F and Brock HW: The
Additional sex combs gene of Drosophila encodes a chromatin protein
that binds to shared and unique Polycomb group sites on polytene
chromosomes. Development. 125:1207–1216. 1998.PubMed/NCBI
|
|
44
|
Gaytán de Ayala Alonso A, Gutiérrez L,
Fritsch C, Papp B, Beuchle D and Müller J: A genetic screen
identifies novel polycomb group genes in Drosophila. Genetics.
176:2099–2108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Balasubramanian M, Willoughby J, Fry AE,
Weber A, Firth HV, Deshpande C, Berg JN, Chandler K, Metcalfe KA,
Lam W, et al: Delineating the phenotypic spectrum of
Bainbridge-Ropers syndrome: 12 new patients with de novo,
heterozygous, loss-of-function mutations in ASXL3 and review of
published literature. J Med Genet. 54:537–543. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bohring A, Silengo M, Lerone M, Superneau
DW, Spaich C, Braddock SR, Poss A and Opitz JM: Severe end of Opitz
trigonocephaly (C) syndrome or new syndrome? Am J Med Genet.
85:438–446. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shashi V, Pena LD, Kim K, Burton B, Hempel
M, Schoch K, Walkiewicz M, McLaughlin HM, Cho M, Stong N, et al: De
novo truncating variants in ASXL2 are associated with a unique and
recognizable clinical phenotype. Am J Hum Genet. 99:991–999. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chinen Y, Nakamura S, Ganaha A, Hayashi S,
Inazawa J, Yanagi K, Nakanishi K, Kaname T and Naritomi K: Mild
prominence of Sylvian fissure in a Bainbridge-Ropers syndrome
patient with a novel frameshift variant in ASXL3. Clin Case Rep.
6:330–336. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Giri D, Rigden D, Didi M, Peak M, McNamara
P and Senniappan S: Novel compound heterozygous ASXL3
mutation causing Bainbridge-Ropers like syndrome and primary IGF1
deficiency. Int J Pediatr Endocrinol. 2017:82017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
De Rubeis S, He X, Goldberg AP, Poultney
CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al:
Synaptic, transcriptional and chromatin genes disrupted in autism.
Nature. 515:209–215. 2014. View Article : Google Scholar : PubMed/NCBI
|