Introduction
Hereditary sensory and autonomic neuropathy (HSAN),
also known as hereditary sensory neuropathy, is a rare
heterogeneous group of disorders with a wide range of clinical and
genetic diversity (1). HSAN is
traditionally classified into five subtypes (HSAN I–V) based on the
age of the patient at onset, the pattern of inheritance and
additional features such as degree of motor and sensory impairment,
autonomic dysfunction, particularly anhidrosis, and mental
retardation (2). HSAN type I (HSAN
I; MIM: 162400) is the most frequent HSAN subtype with autosomal
dominant inheritance characterized by marked distal sensory
impairment leading to painless ulceration, soft tissue infection
and osteomyelitis in the hands and feet (3). As the disease progresses, a variable
degree of distal muscle weakness and wasting with lancinating pain
may be observed.
Mutations in SPTLC1 (MIM: 605712), which
encodes the long-chain base subunit 1 of serine
palmitoyltransferase (SPT), are the major underlying causes of HSAN
I (1), and several mutations have
been reported to be relevant in HSAN I: p.C133W, p.C133Y, p.C133R,
p.V144D, p.S331F, pA310G, p.S331Y and p.A352V (2–13).
SPT catalyzes serine and palmitoyl coenzyme A (CoA), which is the
initial and rate-limiting step in the de novo biosynthesis
of sphingolipids. The age of onset for HSAN I is usually in the
second to fourth decades of life and motor involvement is variable
and limited to the distal limbs. In contrast to typical HSAN I,
three European cases of Ser331 mutations in SPTLC1 (two with
p.S331F and one with p.S331Y) demonstrated an early onset and a
severe phenotype (2,4–6).
In this study we report on the first known Korean
HSAN I patient to harbor a p.S331F mutation in SPTLC1, with
early onset and a severe phenotype.
Patients and methods
Patients
A Korean family with one HSAN I patient and three
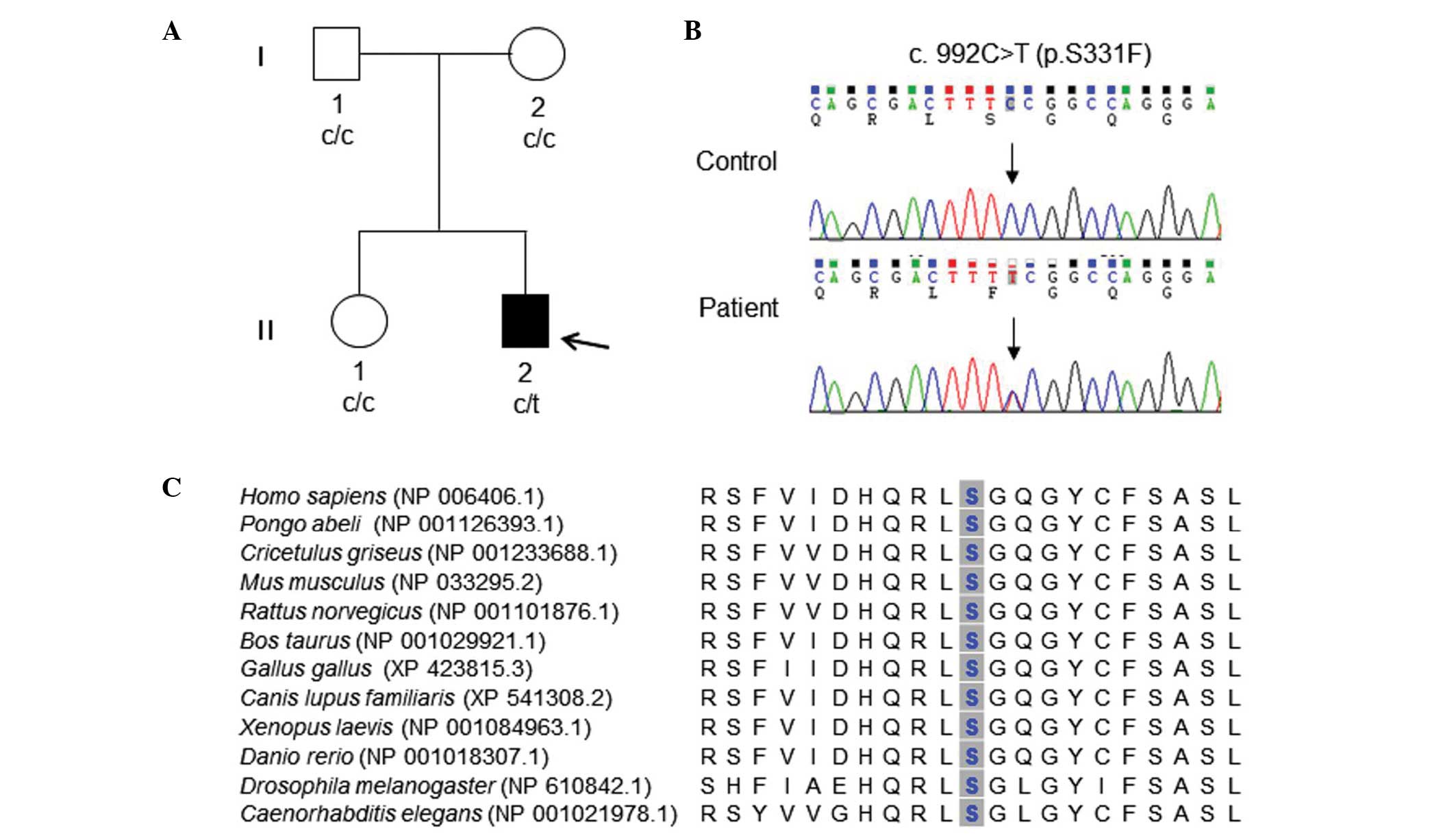
healthy individuals (ID: FC142, Fig.
1A) were enrolled in this study. Healthy Korean controls with
no familial history of neuromuscular disorders (n=300) were also
recruited for this study. Paternity was confirmed by the genotyping
of 15 microsatellites using a PowerPlex 16 system (Promega,
Madison, WI, USA). Written informed consent was obtained from all
participants according to the protocol approved by the
Institutional Review Board for Ewha Womans University, Mokdong
Hospital (ECT 11-58-37; B.O. Choi).
Clinical and electrophysiological
assessments
The patient and the patient’s sister and parents
were examined for motor and sensory impairments, deep tendon reflex
abnormalities and muscle atrophy. The strength of the flexor and
extensor muscles was assessed manually using the Medical Research
Council scale (14). Physical
disabilities were assessed using the functional disability scale
(FDS) (15). Nerve conduction
studies (NCS) were performed on all family members of FC142 with a
surface electrode (Ambu, Ballerup, Denmark). Motor nerve conduction
velocities (MNCVs) of the median and ulnar nerves were determined
by providing stimulation at the elbow and wrist while recording the
compound muscle action potentials (CMAPs) over the abductor
pollicis brevis and adductor digiti quinti, respectively. In the
same manner, the MNCVs and CMAPs of the fibular and tibial nerves
were determined. Sensory nerve conduction velocities and sensory
nerve action potentials (SNAPs) of the median, ulnar and sural
nerves were obtained.
DNA preparation and pre-screening for
Charcot-Marie-Tooth (CMT) disease genes
DNA was purified from blood samples using a QIAamp
blood DNA purification kit (Qiagen, Hilden, Germany). The patient’s
DNA was pre-screened for a 1.4 Mbp length of 17p12
duplication/deletion, a major genetic cause of CMT, by using
hexaplex microsatellite polymerase chain reaction (16).
Exome sequencing and identification of
causative mutation
Exome sequencing was performed in the male patient
(II-2) according to the previous report (17). Exome capturing was achieved using a
SeqCap EZ, version 3.0 (Roche-NimbleGen, Madison, WI, USA), and
next generation sequencing was performed using a HiSeq 2000 Genome
analyzer (Illumina, San Diego, CA, USA). The UCSC assembly hg19
(NCBI build 37.1) was used as the reference sequence with BWA
aligner software (http://bio-bwa.sourceforge.net/). Variant calling was
achieved using a SAMtools program (http://samtools.sourceforge.net/). Single nucleotide
polymorphisms (SNPs) with a quality value >20 were considered as
candidates.
All variants occurring in CMT relevant genes (~60)
were initially selected. The functionally significant variants
(missense, nonsense, exonic indel (insertion and deletion) and
splicing site variants) were selected while the remaining variants
were filtered out. Sequencing variants were confirmed by the Sanger
sequencing method using an automatic genetic analyzer ABI3130XL
(Applied Biosystems, Foster City, CA, USA). A mutation was
considered to be an underlying cause when a candidate mutation was
only located in the affected member within the family, but was not
located in >200 control samples. The mutation nomenclature
recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/) were used to describe
the variants.
In silico analysis
The conservation pattern for the protein amino acid
sequence was performed using MEGA5 software, version 5.05
(http://www.megasoftware.net/) (18). Prediction of protein function
affection due to amino acid substitution was performed using the
online tools SIFT (http://sift.jcvi.org/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/)
and MuPro (http://mupro.proteomics.ics.uci.edu/).
Results
Identification of heterozygous missense
mutation in SPTLC1
The summary of whole exome sequencing data is
provided in Table I. The
sequencing yield was ~6.14 Gbp. Mappable reads and target coverage
(>10× reads) were 93.1 and 94.3%, respectively. Total observed
numbers of SNP and indels were 57,705 and 10,450, respectively. Of
these, coding SNPs and indels were 19,757 and 588,
respectively.
 | Table ISummary of exome sequencing for the
HSAN I patient (II-2). |
Table I
Summary of exome sequencing for the
HSAN I patient (II-2).
| Items | II-2 |
|---|
| Total yields | 6.14 Gbp |
| Mappable reads | 93.1% |
| Target coverage
(≥10×) | 94.3% |
| Total observed SNP
number | 57705 |
| Coding SNP
number | 19757 |
| Total observed indel
number | 10450 |
| Coding indel
number | 588 |
| Functionally
significant variants in CMT genesa | 32 |
In ~60 CMT relevant genes, 32 functionally
significant variants were identified (Table II). Of these, a heterozygous
missense mutation c.992C>T (p.S331F) was located in the
SPTLC1 gene, which was previously reported to cause HSAN I
(2), and was also reported in the
Inherited Peripheral Neuropathies Mutation Database (http://www.molgen.ua.ac.be/CMTMutations/). Capillary
sequencing for this variant demonstrated concordant results
(Fig. 1A and B). This mutation was
located in neither the parents nor the sister. This result suggests
that the SPTLC1 mutation in the patient occurred by de
novo mutation. Notably, this case and the case reported by
Rotthier et al (2) are
de novo. None of the 300 controls exhibited this mutation.
The SPTLC1 c.992C>T (p.S331F) variant is thus determined
to be the causative mutation for II-2. The mutation lies in the
aspartate aminotransferase superfamily (fold type I) domain of
pyridoxal phosphate-dependent enzymes.
| Table IIFunctionally significant variants
observed in CMT genes for the FC142 family (II-2). |
Table II
Functionally significant variants
observed in CMT genes for the FC142 family (II-2).
| | | Variants | | | |
|---|
| | |
| | | |
|---|
| Gene | RefSeqa | Chr: position | Ntb | AA | dbSNP137 | 1000Gc | Description |
|---|
| NGF | NM_002506.2 | chr01:115829313 | c.104C>T | A35V | rs6330 | 0.31 | Polymorphism |
| OPA1 | NM_130834.2 | chr03:193334991 | c.473G>A | S158N | rs7624750 | 0.46 | Polymorphism |
| FIG4 | NM_014845.5 | chr06:110064928 | c.1090A>T | M364L | rs2295837 | 0.10 | Polymorphism |
| FIG4 | - | chr06:110107517 | c.1961T>C | V654A | rs9885672 | 0.37 | Polymorphism |
| NEFL | NM_006158.4 | chr08:24811071 | c.1407_1408insC | P471fs | rs11300136 | - | Polymorphism |
| SPTLC1 | NM_006415.2 | chr09:94809543 | c.992C>T | S331F | - | - | Causative |
| IKBKAP | NM_003640.3 | chr09:111659439 | c.2490A>G | I830M | rs2230794 | 0.08 | Polymorphism |
| IKBKAP | - | chr09:111659483 | c.2446A>C | I816L | rs2230793 | 0.29 | Polymorphism |
| IKBKAP | - | chr09:111660851 | c.2294G>A | G765E | rs2230792 | 0.28 | Polymorphism |
| IKBKAP | - | chr09:111665169 | c.1804C>T | R602W | rs200397694 | 0.00 |
Non-cosegregation |
| LRSAM1 | NM_138361.5 | chr09:130242166 | c.952A>G | N318D | rs1539567 | 0.74 | Polymorphism |
| SETX | NM_015046.5 |
chr09:135139901 | c.7759A>G | I2587V | rs1056899 | 0.51 | Polymorphism |
| SETX | - |
chr09:135173685 | c.5563A>G | T1855A | rs2296871 | 0.41 | Polymorphism |
| SETX | - |
chr09:135203530 | c.3455T>G | F1152C | rs3739922 | 0.10 | Polymorphism |
| SETX | - |
chr09:135205006 | c.1979C>G | A660G | rs882709 | 0.21 | Polymorphism |
| DHTKD1 | NM_018706.6 | chr10:12111090 | c.58T>C | F20L | rs1279138 | 0.98 | Polymorphism |
| DHTKD1 | - | chr10:12131081 | c.814T>G | Y272D | rs3740015 | 0.47 | Polymorphism |
| DHTKD1 | - | chr10:12143105 | c.1821C>G | I607M | rs2062988 | 0.72 | Polymorphism |
| SBF2 | NM_030962.3 | chr11:9853777 | c.3646C>G | Q1216E | rs12574508 | 0.10 | Polymorphism |
| IGHMBP2 | NM_002180.2 | chr11:68678962 | c.602T>C | L201S | rs560096 | 0.70 | Polymorphism |
| IGHMBP2 | - | chr11:68705674 | c.2636C>A | T879K | rs17612126 | 0.23 | Polymorphism |
| WNK1 | NM_213655.4 | chr12:990912 | c.3922A>C | T1308P | rs956868 | 0.85 | Polymorphism |
| WNK1 | - | chr12:994487 | c.5273G>C | C1758S | rs7955371 | 0.99 | Polymorphism |
| KARS | NM_005548.2 | chr16:75669878 | c.601T>C | Y201H | rs150529876 | 0.00 | Polymorphism |
| SEPT9 | NM_001113495.1 | chr17:75494705 | c.1390A>G | M464V | rs2627223 | 0.92 | Polymorphism |
| CTDP1 | NM_048368.3 | chr18:77473127 | c.1019C>T | T340M | rs2279103 | 0.11 | Polymorphism |
| CTDP1 | - | chr18:77474626 | c.1166C>T | A389V | rs144647072 | 0.02 | Polymorphism |
| DNMT1 | NM_001379.2 | chr19:10273372 | c.931A>G | I311V | rs2228612 | 0.18 | Polymorphism |
| PRX | NM_181882.2 | chr19:40900865 | c.3394G>A | G1132R | rs268674 | 0.96 | Polymorphism |
| PRX | - | chr19:40902710 | c.1549C>T | L517F | - | - |
Non-cosegregation |
| DMPK | NM_001081563.1 | chr19:46275976 | c.1297C>G | L433V | rs527221 | 0.12 | Polymorphism |
| ATP7A | NM_000052.5 | chrX:77298857 | c.4048G>A | E1350K | rs4826245 | 1.00 | Polymorphism |
Appreciable in silico results were attained
by using three tools: SIFT yielding a deleterious score of 0.02
(damaging score: ≤0.05), a PolyPhen score of 2.317 (damaging score:
>1.0), and a MUpro protein instability score of −0.1034
(negative value: decreased stability). Analysis of multiple
sequences for the conservation pattern of the STPLC1 protein
demonstrated marked conservation throughout different species
(Fig. 1C).
Except for SPTLC1 c.992C>T, other variants
in CMT relevant genes were not considered to be causative, as they
were polymorphic (observed in the controls) or not cosegregated
with the affected member within the pedigree. Despite the presence
of several rare heterozygous variants in recessive genes, the
possibility of an underlying cause was ruled out due the occurrence
of the same mutation in either parent.
Clinical manifestations and
electrophysiological features
The male proband (Fig.
1A, II-2) was the second child of non-consanguineous, healthy
parents. Birth weight was 3.2 kg and motor milestones during the
first year were normal. The patient noticed frequent falls, lower
leg weakness and a hand tremor at age five. At seven years of age,
the patient used an ankle-foot device due to a progressively
impaired gait, and the diagnosis of hereditary motor and sensory
neuropathy was made. Three years later, the patient had a cataract
operation. Following adolescence, the voice of the patient became
hoarse. Disease progression was rapid and the patient exhibited
with respiratory problems and walker dependency at the age of 27
years. A neurological examination at 28 years of age revealed
muscle weakness and atrophy of the proximal limb and trunk muscles
(body mass index, 12.2 kg/m2), and hypotonia with
prominent weakness in the distal muscles of the upper and lower
limbs (FDS 6, walking with a walker). The vibration and position
senses were markedly more preserved compared with the pain and
temperature senses. Marked reductions in pain and temperature
sensation were noted. Bilateral hand tremors and severe scoliosis
were observed. Deep tendon reflexes were absent in all extremities,
but pathological reflexes were not identified.
NCS was performed at the ages of 12, 26 and 28
years. SNAPs of the median, ulnar and sural nerves were not evoked,
and CMAPs of peroneal and tibial nerves were also absent. The
median nerves revealed low CMAPs (range, 0.1–0.2 mV) and slow MNCVs
(range, 12.2–25.9 m/s). CMAPs of the ulnar nerves were low and the
range was similar to that of the median nerves (CMAP range, 0.1–0.4
mV; MNCV range, 17.5–27.0 m/s). However, when NCS was performed on
the other family members (I-1, I-2, II-1), the results were
completely normal.
Discussion
The present study reports on the early-onset of a
severe phenotype of HSAN I with a missense mutation of c.992C>T
(p.S331F) in SPTLC1. HSAN I is characterized by autosomal
dominant inheritance, juvenile or adulthood disease onset, and
marked distal pain and temperature sensory impairment leading to
distal ulceration and mutilating arthropathy with relative
preservation of motor and autonomic functions (19). Other subtypes of HSAN (HSAN types
II–V) have autosomal recessive inheritance with an earlier onset of
the disease and variable motor involvement. Therefore, the
phenotype of this patient was different to the usual pattern of
HSAN I, with the features of: i) A sporadic case rather than
autosomal dominant inheritance; ii) earlier age of onset; iii)
severe proximal and distal motor involvement with wasting; and iv)
additional features including cataracts at a young age, hoarseness
of the voice, tremor, scoliosis and respiratory insufficiency.
To date, numerous SPTLC1 mutations have been
identified at restricted amino acid positions (p.C133W, p.C133Y,
p.C133R, p.V144D, p.S331F, p.S331Y, p.A310G, and p.A352V) (2–9).
There have only been three previously reported cases with a
mutation at the Ser331 position (2,4–6).
Common findings of these three cases are a de novo mutation,
symptom onset prior to 10 years of age, muscle hypotrophy due to
prominent motor involvement in the polyneuropathy, foot ulcers,
amputations and cataracts at a young age. Two of the cases also
exhibited joint hypermobility, vocal cord paralysis and respiratory
difficulties (2,4–6).
Following a review of the literature with regard to the
SPTLC1 mutation, the present case and the aforementioned
three cases carrying the Ser331 mutation resemble each other in
terms of the inheritance pattern and the atypical clinical features
(Table III).
| Table IIIClinical features of HSAN patients
with SPTLC1 mutation with literature review. |
Table III
Clinical features of HSAN patients
with SPTLC1 mutation with literature review.
| AA change |
Origin/Inheritance | AAO (years) | NCS (PN) | Weakness/muscle
atrophy | Foot ulcer,
amputation | Respiratory
problem | Juvanile cataract
age (years) | Others | Reference (Author,
year, no.) |
|---|
| p.S331F | German/IC | Early
childhood | SM | Yes | Yes | NM | Yes (9) | Joint
contracture | Huehne et
al, 2008 (6) |
| French
(Gypsy)/IC | Congenital | SM | Yes | Yes | Yes | Yes | Mental retardation,
vocal cord palsy, joint hyperlaxity | Rotthier et
al, 2009 (2) |
| Korean/IC | 5 | SM | Yes walker
(27)a | Yes | Yes | Yes (10) | Hoarseness, tremor,
scoliosis | This study |
| p.S331Y | NM/IC | 4 | SM | Yes wheelchair
(14)a | Yes | Yes | Yes (13) | Tremor,
fasciculation, joint hypermobility | Auer-Grumbach et
al, 2013 (4) |
| p.C133W | Australian
English/NM | 65 | NM | Weakness in
4/38 | Yes | NM | NM | NM | Dawkins et
al, 2001 (8) |
| Canadian/NM | 20–40 | NM | No | Yes | NM | NM | NM | Bejaoui et
al, 2001 (9) |
| Chinese/AD | 20’s | SM | No | Yes | NM | NM | NM | Bi et al,
2007 (10) |
| Canadian/AD | 12, 60’s | S | Peroneal m.
atrophy | Yes | NM | NM | NM | Klein et al,
2005 (11) |
| English/AD, IC | 12–29 | SM | Yes | Yes | NM | NM | NM | Houlden et
al, 2006 (3) |
| p.C133Y | German/NM | NM | NM | NM | NM | NM | NM | NM | Bejaoui et
al, 2001 (9) |
| Australian
German/NM | NM | NM | NM | NM | NM | NM | NM | Dawkins et
al, 2001 (8) |
| Portuguese/AD | 20’s, 10 | SM | No | No | NM | No | Foot pain, dry
skin | Geraldes et
al, 2004 (12) |
| p.C133R | German/AD | 50 | No | No | NM | NM | NM | NM | Rautenstrauss et
al, 2009 (13) |
| p.V144D | Australian
German/NM | NM | NM | NM | NM | NM | NM | NM | Dawkins et
al, 2001 (8) |
| p.A310G | English/IC | 50’s | S | No | Yes | NM | NM | NM | Davidson et
al, 2012 (1) |
| p.A352V | Austrian/IC | 16 | SM | Mild pes cavus,
distal LL, peroneal atrophy | NM | NM | NM | NM | Rotthier et
al, 2009 (2) |
It is hypothesized that reduced SPT activity with
resultant accumulation of neurotoxic deoxysphingoid bases (DSB) may
be the underlying pathomechanism of HSAN I with SPTLC1
mutations. Rotthier et al (5) demonstrated that mutant proteins
(C133W, S331F and A352V) are enzymatically defective; however,
failed to demonstrate a positive correlation between DSB levels and
phenotypic severity. These results imply that there may be an
additional factor determining the severity of the disease,
including genetic modifiers or other interacting proteins.
This is the first report of a Korean HSAN I patient
with the p.S331F mutation in SPTLC1. The patient exhibited
early onset and severe clinical manifestations, which are uncommon
features of HSAN I but are an almost identical to those in patients
with an Ser331 mutation. Therefore, HSAN I with a p.S331 mutation
in SPTLC1 may exhibit an early onset, severe sensory motor
deficits and various other features, including cataracts, vocal
cord palsy and respiratory problems.
Acknowledgements
This study was supported by a grant from the Korean
Health Technology R&D Project, Ministry of Health and Welfare,
Republic of Korea (grant no. A120182).
References
|
1
|
Davidson G, Murphy S, Polke J, et al:
Frequency of mutations in the genes associated with hereditary
sensory and autonomic neuropathy in a UK cohort. J Neurol.
259:1673–1685. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rotthier A, Baets J, De Vriendt E, et al:
Genes for hereditary sensory and autonomic neuropathies: a
genotype-phenotype correlation. Brain. 132:2699–2711. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Houlden H, King R, Blake J, et al:
Clinical, pathological and genetic characterization of hereditary
sensory and autonomic neuropathy type 1 (HSAN I). Brain.
129:411–425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Auer-Grumbach M, Bode H, Pieber TR, et al:
Mutations at Ser331 in the HSN type I gene SPTLC1 are associated
with a distinct syndromic phenotype. Eur J Med Genet. 56:266–269.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rotthier A, Penno A, Rautenstrauss B, et
al: Characterization of two mutations in the SPTLC1 subunit of
serine palmitoyltransferase associated with hereditary sensory and
autonomic neuropathy type I. Hum Mutat. 32:E2211–2225. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huehne K, Zweier C, Raab K, et al: Novel
missense, insertion and deletion mutations in the neurotrophic
tyrosine kinase receptor type 1 gene (NTRK1) associated with
congenital insensitivity to pain with anhidrosis. Neuromuscul
Disord. 18:159–166. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Verhoeven K, Coen K, De Vriendt E, et al:
SPTLC1 mutation in twin sisters with hereditary sensory neuropathy
type I. Neurology. 62:1001–1002. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dawkins JL, Hulme DJ, Brahmbhatt SB,
Auer-Grumbach M and Nicholson GA: Mutations in SPTLC1, encoding
serine palmitoyltransferase, long chain base subunit-1, cause
hereditary sensory neuropathy type I. Nat Genet. 27:309–312. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bejaoui K, Wu C, Scheffler MD, et al:
SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat
Genet. 27:261–262. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bi H, Gao Y, Yao S, Dong M, Headley AP and
Yuan Y: Hereditary sensory and autonomic neuropathy type I in a
Chinese family: British C133W mutation exists in the Chinese.
Neuropathology. 27:429–433. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klein CJ, Wu Y, Kruckeberg KE, et al:
SPTLC1 and RAB7 mutation analysis in dominantly inherited and
idiopathic sensory neuropathies. J Neurol Neurosurg Psychiatry.
76:1022–1024. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Geraldes R, de Carvalho M, Santos-Bento M
and Nicholson G: Hereditary sensory neuropathy type 1 in a
Portuguese family-electrodiagnostic and autonomic nervous system
studies. J Neurol Sci. 227:35–38. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rautenstrauss B, Neitzel B, Muench C, Haas
J, Holinski-Feder E and Abicht A: Late onset hereditary sensory
neuropathy type 1 (HSN1) caused by a novel p.C133R missense
mutation in SPTLC1. J Peripher Nerv Syst. 14:124–125. 2009.
|
|
14
|
Paternostro-Sluga T, Grim-Stieger M, Posch
M, et al: Reliability and validity of the Medical Research Council
(MRC) scale and a modified scale for testing muscle strength in
patients with radial palsy. J Rehabil Med. 40:665–671. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Birouk N, LeGuern E, Maisonobe T, et al:
X-linked Charcot-Marie-Tooth disease with connexin 32 mutations:
clinical and electrophysiologic study. Neurology. 50:1074–1082.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choi BO, Kim J, Lee KL, Yu JS, Hwang JH
and Chung KW: Rapid diagnosis of CMT1A duplications and HNPP
deletions by multiplex microsatellite PCR. Mol Cells. 23:39–48.
2007.PubMed/NCBI
|
|
17
|
Lee SS, Lee HJ, Park JM, et al: Proximal
dominant hereditary motor and sensory neuropathy with proximal
dominance association with mutation in the TRK-fused gene. JAMA
Neurol. 70:607–615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tamura K, Peterson D, Peterson N, Stecher
G, Nei M and Kumar S: MEGA5: molecular evolutionary genetics
analysis using maximum likelihood, evolutionary distance, and
maximum parsimony methods. Mol Biol Evol. 28:2731–2739. 2011.
View Article : Google Scholar
|
|
19
|
Auer-Grumbach M, De Jonghe P, Verhoeven K,
et al: Autosomal dominant inherited neuropathies with prominent
sensory loss and mutilations: a review. Arch Neurol. 60:329–334.
2003. View Article : Google Scholar : PubMed/NCBI
|