Introduction
Bacteria stimulate the innate host immune system and
lead to the release of inflammatory molecules such as cytokines and
chemokines. Lipopolysaccharide (LPS) and peptidoglycan (PGN) are
the main cell wall components of Gram-negative and Gram-positive
bacteria, respectively. LPS and PGN stimulate different Toll-like
receptors (TLRs), TLR-4 and TLR-2 respectively, and activate a
number of inflammatory pathways, including the transcription
factor, nuclear factor (NF)-κB (1,2). NF-κB
is the key transcription factor involved in the inflammatory
responses, and also plays a critical role in tumor cell survival,
growth and migration. Many of the signal pathways implicated in
tumors converge to the NF-κB signal. Its activation aggravates
tumors, while its suppression inhibits tumor cell growth (3–5).
Therefore, the NF-κB signaling pathway has emerged as one of the
attractive targets for the treatment of many types of tumor,
including glioblastoma (6).
The endocannabinoid system, consisting of two types
of G-protein-coupled cannabinoid receptors (CB1 and CB2) and their
endogenous ligands (endocannabinoids) (7), has been implicated in a variety of
physiological and pathological conditions, including inflammation
and tumor progression. The CB1 receptor is abundantly expressed in
the CNS, while the CB2 receptor is predominantly expressed by
immune cells. Endogenous and exogenous cannabinoids exert
anti-inflammatory and antitumor effects (8–10).
However, the precise mechanisms underlying these actions are not
yet fully understood.
In the present study, we examined effects of
cannabinoids on PGN-induced NF-κB activation and cell growth using
U87MG human malignant glioma cells. We demonstrate that
cannabinoids suppress the PGN-induced phosphorylation of NF-κB and
cell growth in a CB1-dependent manner. Our findings provide a
mechanistic basis for opening up new therapeutic approaches of
treating or preventing brain tumors aggravated by inflammation.
Materials and methods
Chemicals
PGN, 2-arachidonoylglycerol (2-AG), WIN55,212-2 and
Dulbecco’s modified Eagle’s medium (DMEM) were obtained from Wako
Pure Chemical Industries, Ltd. (Osaka, Japan). AM281 (CB1 specific
antagonist) and AM630 (CB2 specific antagonist) were obtained from
Calbiochem (La Jolla, CA, USA). Fetal bovine serum (FBS) was
obtained from Invitrogen Corporation (Carlsbad, CA, USA).
Anti-phosphorylated (phospho)-specific NF-κB p65 (Ser536),
anti-β-actin and horseradish peroxidase (HRP)-linked anti-rabbit
IgG were purchased from Cell Signaling Technology, Inc. (Danvers,
MA, USA).
Cell culture
U87MG human malignant glioma cells were provided by
Dr Nakata. The cells were maintained in DMEM containing 10% FBS at
37°C in a 5% CO2 incubator.
NF-κB activity assay
To determine the effect of 2-AG on NF-κB activity,
we investigated the phospho-NF-κB level using western blot
analyses. An increase in the phospho-NF-κB level indicates the
NF-κB activation levels. The U87MG human malignant glioma cells
were incubated in DMEM containing serum for 24 h and pre-treated
with 2-AG (1 μM) for 10 min, and then treated with PGN (1–30 μg/ml)
for 16 h. Western blot analyses were performed using phospho-NF-κB
p65 (Ser536) antibody and anti-β-actin antibody.
Western blot analysis
Western blot analysis was performed as described
previously (11).
Reverse transcription-mediated polymerase
chain reaction (RT-PCR) analysis
To evaluate the expression patterns of CB1 and CB2
mRNA in the cells, RT-PCR was performed as follows: briefly, RNA
was extracted from the cells and reverse transcribed by using
reverse transcriptase ReverTra Ace (Toyobo, Tokyo, Japan).
PCR-based subtype-specific gene amplification for CB1, CB2 and
β-actin was performed with LA Taq (Takara, Tokyo, Japan) using the
following sets of primers: 5′-caggcctt cctaccacttcat-3′ and
5′-accccacccagtttgaacaga-3′ for CB1, 5′-aagccctcgtacctgttcat-3′ and
5′-acagaggctgtgaaggtcat-3′ for CB2, and 5′-atggtgggtatgggtcagaag-3′
and 5′-ctggggtgttgaagg tctcaa-3′ for β-actin.
Microscopic analysis
U87MG cells were seeded on 35-mm dishes at a density
of 1×103 cells/dish. After a 24-h incubation, the cells
in DMEM with serum were treated with 1 μM WIN55,212-2, a
combination of WIN55,212-2 (1 μM) and AM281 (1 μM) or a combination
of WIN55,212-2 (1 μM) and AM630 (1 μM) for 72 h. Morphological
changes of cells were assessed by light microscopy.
Cell proliferation assay
Cell proliferation was analyzed using the Cell
Counting kit 8 (Wako Pure Chemical Industries, Ltd.). U87MG human
malignant glioma cells were seeded in 96-well plates at a density
of 1×103 cells/well. After a 24-h incubation, the cells
were treated with PGN, WIN55,212-2 or AM281 for 72 h. The cells
were then incubated with 10 μl WST-8 for 2 h. The absorbance of the
colored formazan product produced by mitochondrial dehydrogenases
in metabolically active cells was recorded at 450 nm as the
background value. Cell proliferation was expressed as a percentage
of the absorbance obtained in the treated wells relative to that in
the untreated (control) wells.
Statistical analysis
Data are presented as the means ± SEM from at least
three independent experiments. Statistical analyses were performed
using ANOVA followed by Dunnett’s test and the results were
considered statistically significant when P<0.01.
Results
PGN enhances phosphorylation of NF-κB and
cell growth
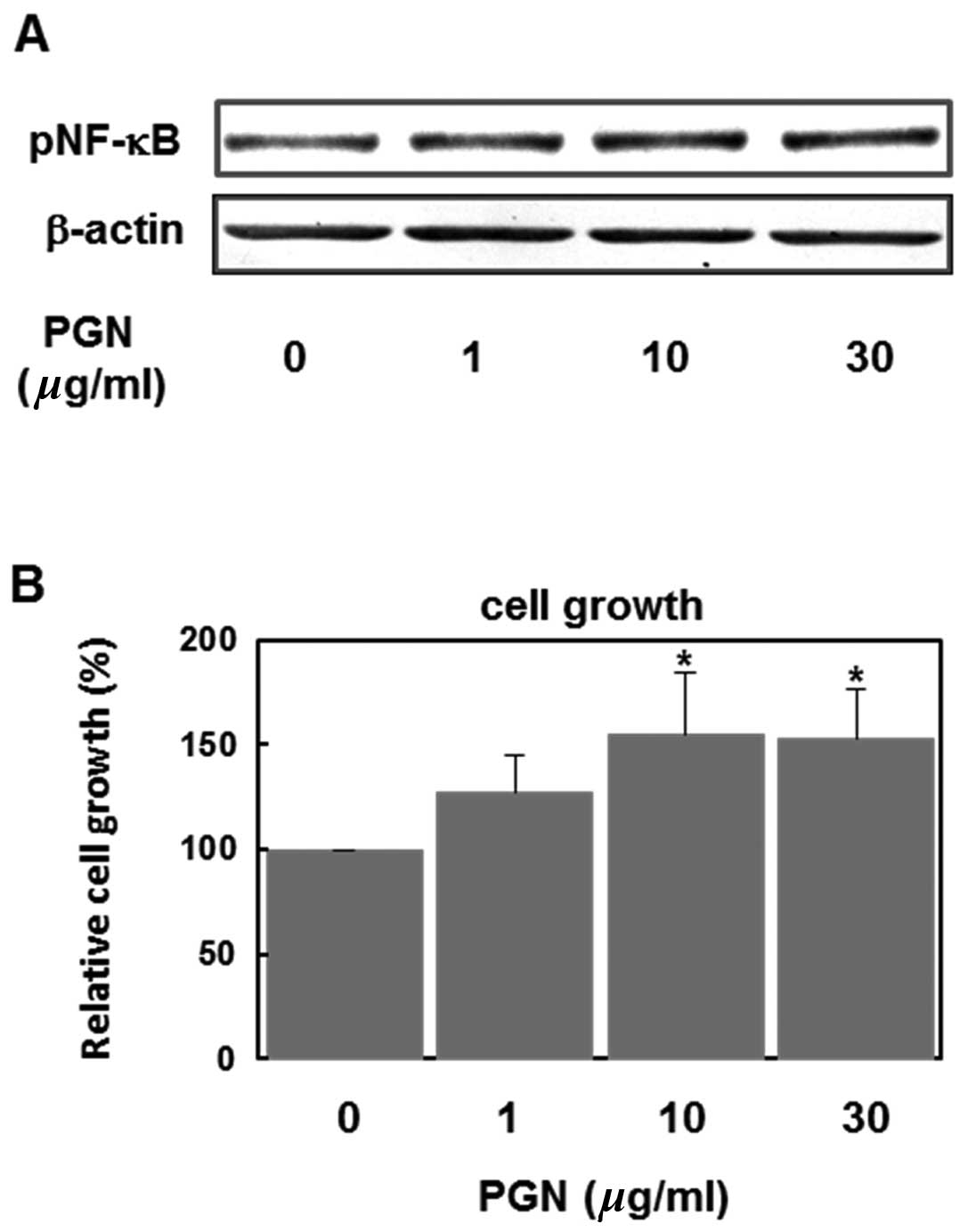
First, we examined effects of PGN, a major cell wall
component of Gram-positive bacteria, on NF-κB activation and cell
growth. As shown in Fig. 1A, PGN
(1–30 μg/ml) increased the level of phospho-NF-κB (p65) in a
concentration-dependent manner, indicating that PGN activates the
NF-κB signaling pathways in U87MG human glioblastoma cells. PGN
also accelerated the cell growth of these cells in a
concentration-dependent manner (Fig.
1B).
Endocannabinoid 2-AG inhibits PGN-induced
phosphorylation of NF-κB
We then examined the effects of cannabinoids.
Anandamide and 2-AG are the two main endocannabinoids, and activate
both CB1 and CB2 cannabinoid receptors. Anandamide behaves as a
partial agonist at these cannabinoid receptors and also activates
the vanilloid receptor (TRPV1). 2-AG acts as a full agonist at
cannabinoid receptors and does not interact with TRPV1, suggesting
that 2-AG is a true natural ligand for cannabinoid receptors
(7). Therefore, to examine the
cannabinoid receptor-dependent effects we used 2-AG rather than
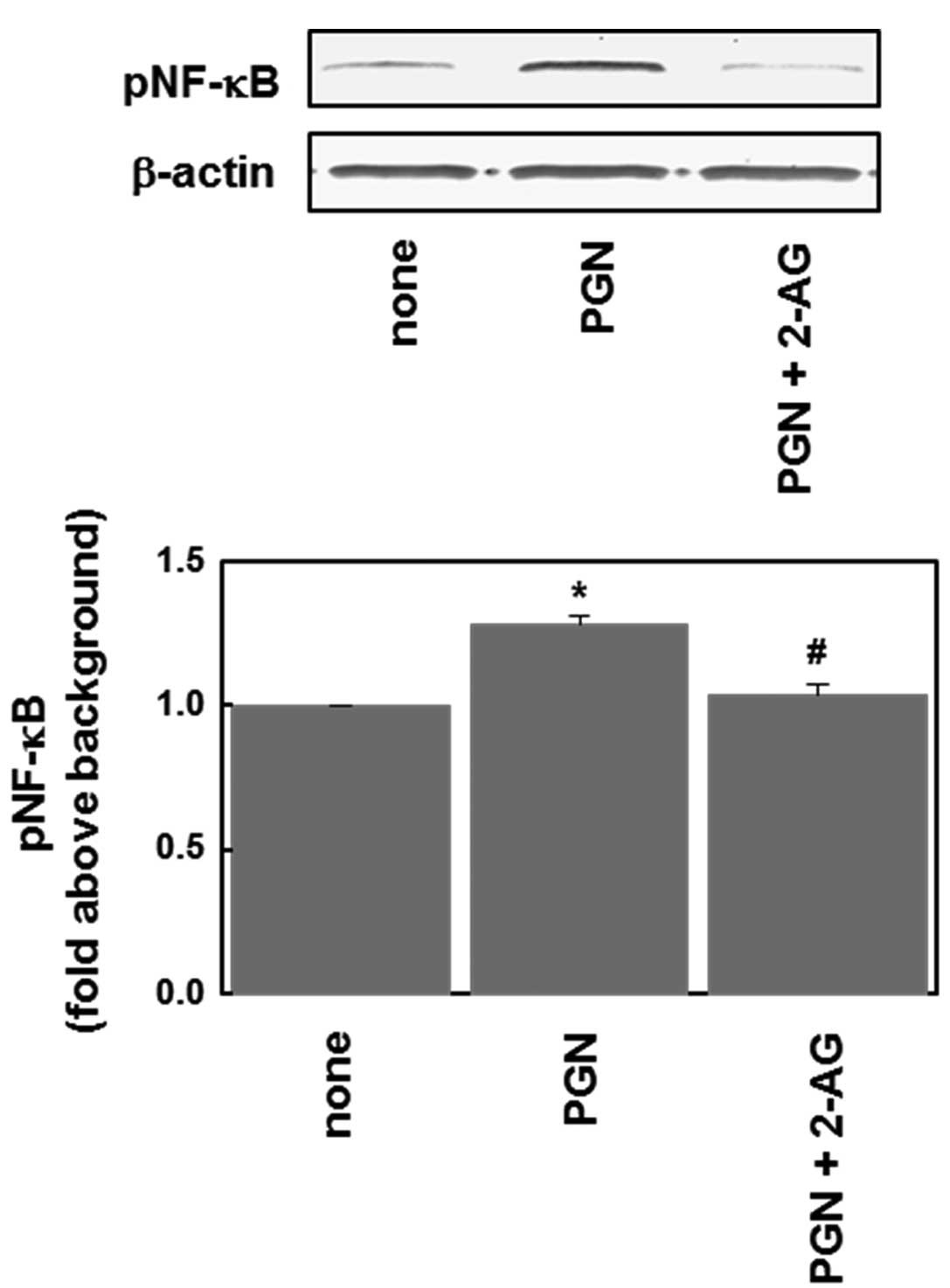
anandamide as an endocannabinoid. As shown in Fig. 2, the PGN (10 μg/ml)-induced
phosphorylation of NF-κB was significantly inhibited by 2-AG (1
μM).
2-AG inhibits PGN-induced phosphorylation
of NF-κB via CB1 receptor
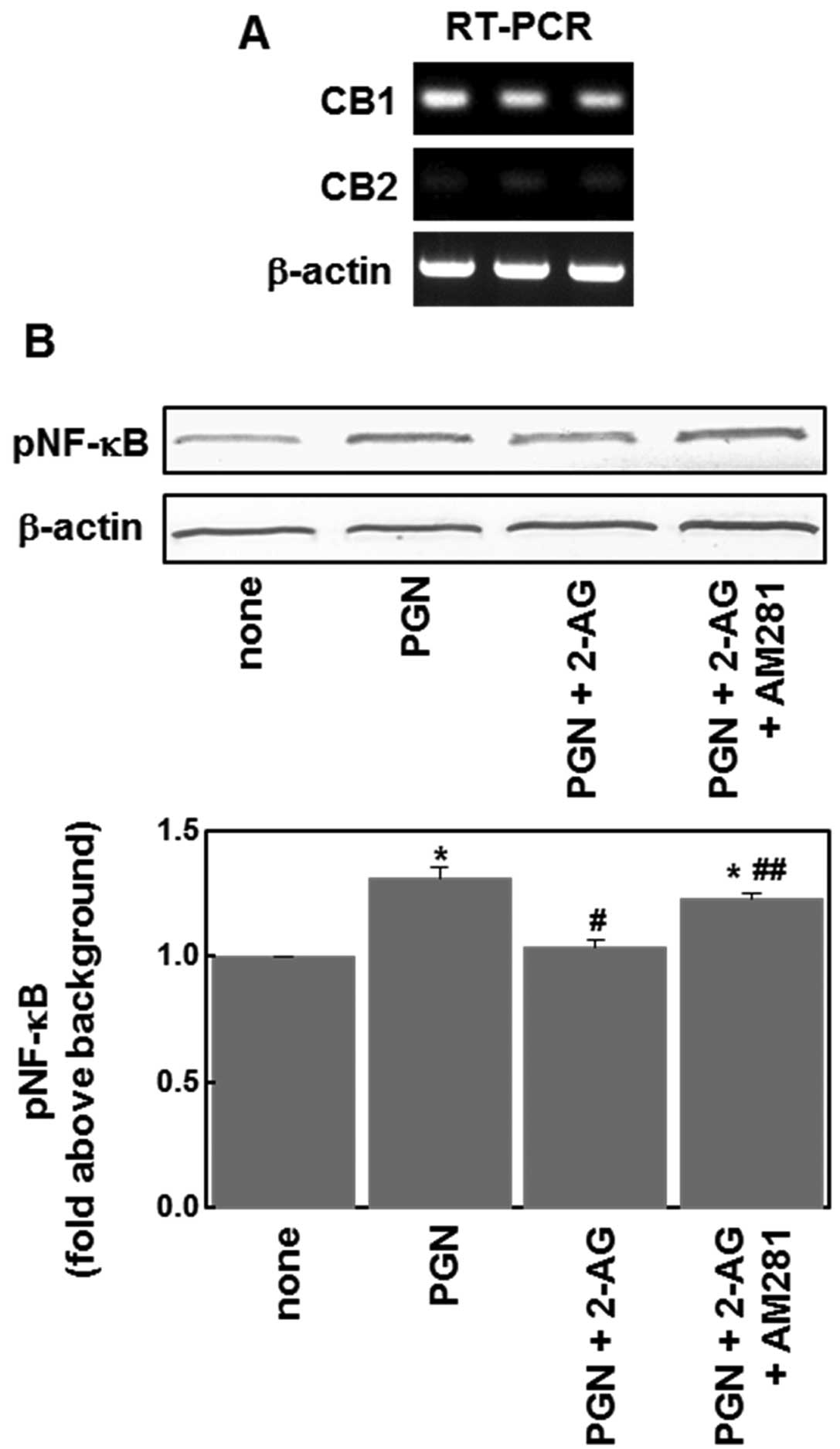
To examine whether cannabinoid receptors are
involved in the inhibitory effect of 2-AG, we first examined the
expression of CB1 and CB2 mRNAs in U87MG glioblastoma cells using
RT-PCR. The CB1 receptor expression was detectable in these cells,
but CB2 receptor expression was below the limit of detection
(Fig. 3A). We then examined effects
of the CB1 receptor antagonist, AM281, and found that AM281 (1 μM)
reversed the suppressive effect of 2-AG on PGN-induced NF-κB
phosphorylation (Fig. 3B). These
results indicate that CB1 receptors are involved in the effects of
2-AG on the NF-κB signaling pathway.
Cannabinoid agonist WIN55,212-2 affects
the cell morphology via CB1 receptor
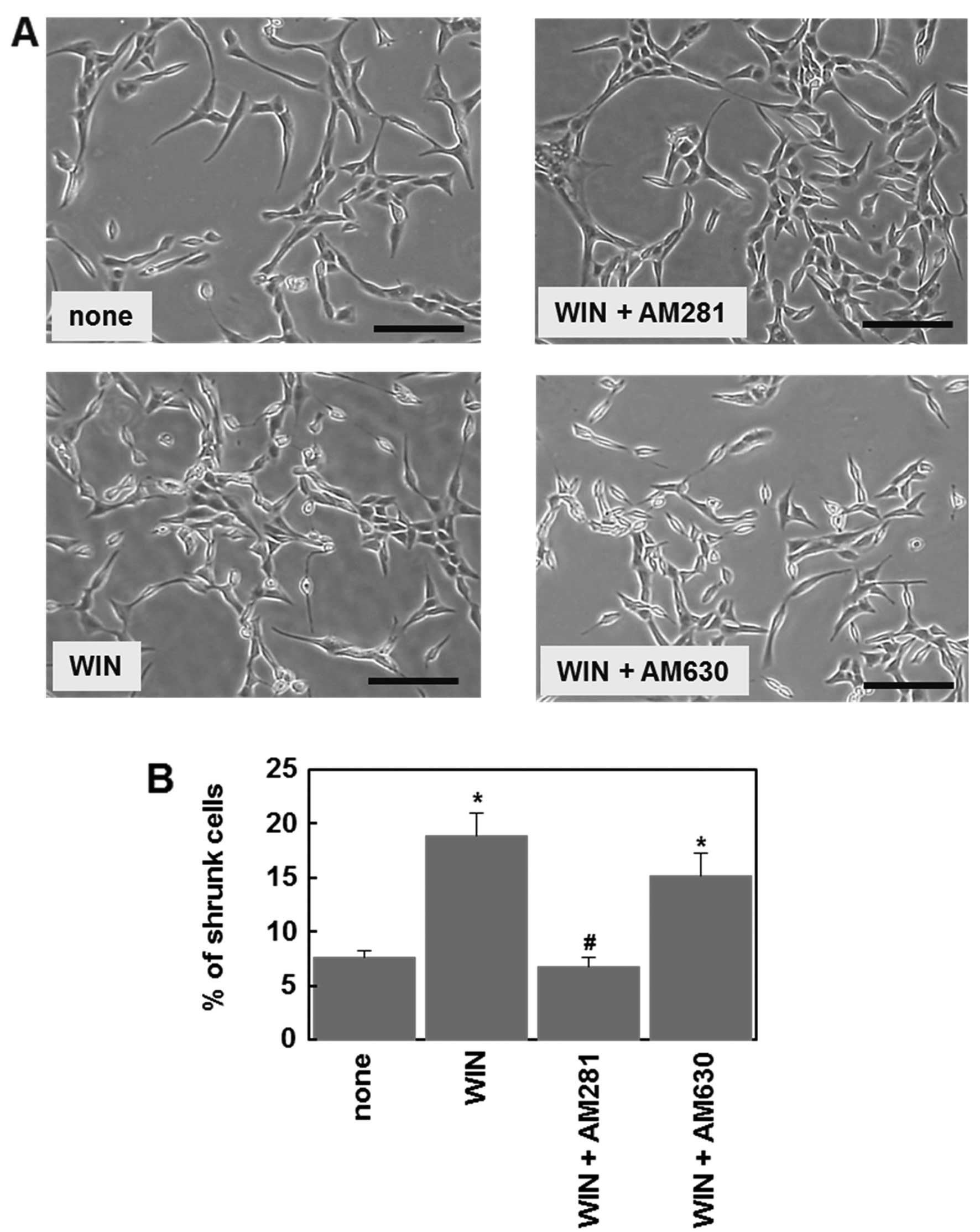
To further confirm the presence of functional CB1
receptors in U87MG cells, we examined effect of cannabinoid
receptor antagonists on cannabinoid-induced morphological changes.
Cell morphology was assessed after a 72-h treatment with drugs. Due
to the long incubation time, we used the cannabinoid agonist,
WIN55,212-2, instead of 2-AG, which is easily degraded by
endogenous enzymes, such as monoacylglycerol lipase. Microscopic
analysis showed that treatment with WIN55,212-2 (1 μM) affected the
cell morphology and increased the number of shrunken cells
(Fig. 4A). The CB1 antagonist,
AM281 (1 μM), but not the CB2 antagonist, AM630 (1 μM), prevented
the WIN55,212-2-induced morphological change (Fig. 4). These results indicate that
functional CB1 receptors are expressed in U87MG cells, consistent
with the above observation (Fig.
3).
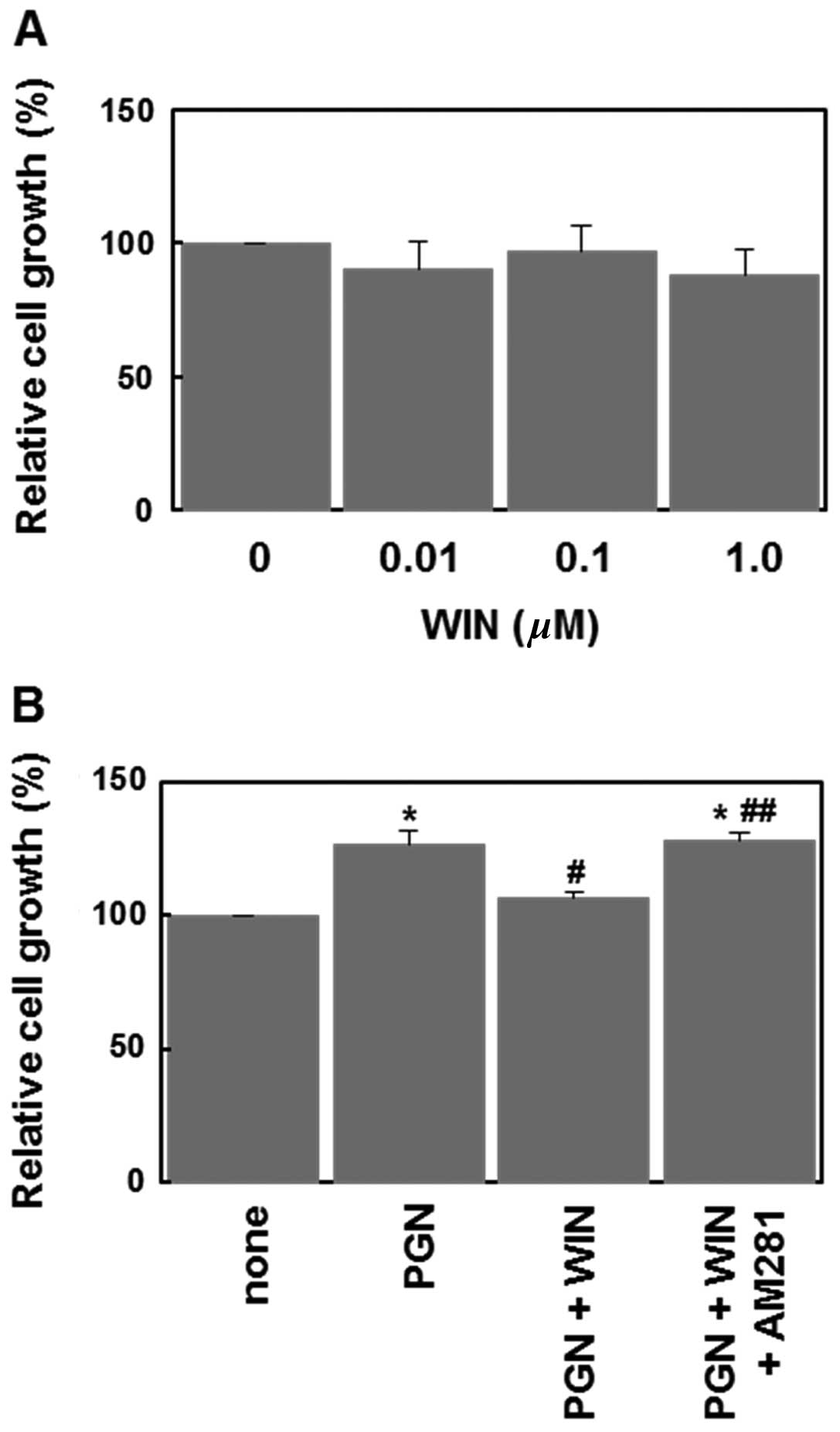
WIN55,212-2 inhibits PGN-induced cell
proliferation via CB1 receptor
Finally, we examined the ability of WIN55,212-2 to
inhibit PGN-induced cell proliferation. When applied alone,
WIN55,212-2 (0.01–1 μM) failed to significantly affect the cell
proliferation (Fig. 5A). However,
WIN55,212-2 (1 μM) significantly inhibited PGN-induced cell
proliferation (Fig. 5B). This
effect of WIN55,212-2 was reversed by AM281 (Fig. 5B). These results indicate that the
activation of CB1 receptors suppresses the PGN-induced cell
proliferation in U87MG human glioblastoma cells.
Discussion
Several lines of evidence have shown that
cannabinoids exert a variety of physiological and pharmacological
effects (7,8,12).
Notably, cannabinoids inhibit tumor growth and have been proposed
as potential antitumor agents (8,13,14).
However, the mechanisms by which cannabinoids prevent tumor
progression are not yet fully understood. Our data suggest the
possibility that the antitumor and protective action of
cannabinoids against PGN-induced inflammatory assaults might be
associated with their ability to suppress the phosphorylation of
NF-κB via CB1 receptor signaling.
PGN is a characteristic cell-wall component of
Gram-positive bacteria that can stimulate inflammatory signaling in
several types of cells. In the present study, we found that PGN
potently induced NF-κB phosphorylation in human glioma cells
(Fig. 1A) and enhanced cell growth
(Fig. 1B). The NF-κB signal is
involved in a variety of physiological and pathological events,
including inflammation, immune responses and apoptosis. Over the
last decade, it has become clear that the NF-κB signaling pathway
also plays a critical role in cancer development and progression
(4,5,15,16).
The activation of NF-κB has been reported in a wide variety of
tumor cells (16). The results from
our study showed that 2-AG, one of the endocannabinoids, had an
inhibitory effect on PGN-induced NF-κB phosphorylation (Figs. 2 and 3B), suggesting that cannabinoids prevent
tumor progression during inflammation. Indeed, the cannabinoid
agonist, WIN-55212-2, abolished PGN-induced cell proliferation
(Fig. 5B). These results suggest
the therapeutic potential of cannabinoids in inflammation-dependent
tumor progression.
TLRs have been established to play an essential role
in the activation of innate immunity by recognizing specific
patterns of microbial components, such as PGN, LPS, flagellin, CpG
DNA and dsRNA derived from viral sequences (1). LPS, an essential component of the cell
wall of Gram-negative bacteria, activates the NF-κB signaling
pathway through TLR-4. The effects of cannabinoids on the
LPS-induced activation of the NF-κB signaling pathway have been
investigated in a number of cell types. The prototypic cannabinoid,
Δ9-tetrahydrocannabinol (THC), has been shown to inhibit
LPS-induced NF-κB activation in the macrophage cell line, RAW264.7,
that expresses CB2 receptors (17),
but not in the microglial cell line, BV-2, that also expresses CB2
(18). In hippocampal tissue,
including both neurons and glial cells, 2-AG inhibits LPS-induced
phosphorylation of NF-κB via CB1 receptors (19). The present study demonstrates that
cannabinoids also inhibit the NF-κB activation induced by the TLR-2
agonist, PGN, through a mechanism involving CB1 receptors (Figs. 2 and 3). Since the TLR-2 and TLR-4 signaling
pathways for the activation of NF-κB are shared by other TLRs
(1), it is likely that the
TLR-mediated NF-κB activation may generally be sensitive to
cannabinoids. The mechanisms underlying the suppression of NF-κB
activation by cannabinoids are not yet fully understood. A recent
study demonstrated that the effect of 2-AG on LPS-induced NF-κB
phosphorylation is mediated by peroxisome proliferator-activated
receptor-γ in hippocampal neurons (20). Further studies are required to
determine the molecular mechanisms involved.
In conclusion, in this study, we demonstrate that
cannabinoids prevent PGN-induced pathological signal transduction
and cell proliferation in U87MG human malignant glioma cells. Our
results provide evidence for the possibility of cannabinoids as a
new therapeutic approach for inflammation-dependent tumor
progression.
Acknowledgements
This study was supported in part by Grants-in-Aid
for Science and Culture from the Ministry of Education, Culture,
Sports, Science and Technology of Japan.
References
|
1
|
Takeda K and Akira S: TLR signaling
pathways. Semin Immunol. 16:3–9. 2004. View Article : Google Scholar
|
|
2
|
Buchanan MM, Hutchinson M, Watkins LR and
Yin H: Toll-like receptor 4 in CNS pathologies. J Neurochem.
114:13–27. 2010.PubMed/NCBI
|
|
3
|
Chaturvedi MM, Sung B, Yadav VR, Kannappan
R and Aggarwal BB: NF-κB addiction and its role in cancer: ‘one
size does not fit all’. Oncogene. 30:1615–1630. 2011.
|
|
4
|
Basseres DS and Baldwin AS: Nuclear
factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic
initiation and progression. Oncogene. 25:6817–6830. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nogueira L, Ruiz-Ontanon P,
Vazquez-Barquero A, Moris F and Fernandez-Luna JL: The NFκB
pathway: a therapeutic target in glioblastoma. Oncotarget.
2:646–653. 2011.
|
|
7
|
Kano M, Ohno-Shosaku T, Hashimotodani Y,
Uchigashima M and Watanabe M: Endocannabinoid-mediated control of
synaptic transmission. Physiol Rev. 89:309–380. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guindon J and Hohmann AG: The
endocannabinoid system and cancer: therapeutic implication. Br J
Pharmacol. 163:1447–1463. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Centonze D, Finazzi-Agro A, Bernardi G and
Maccarrone M: The endocannabinoid system in targeting inflammatory
neurodegenerative diseases. Trends Pharmacol Sci. 28:180–187. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cabral GA and Griffin-Thomas L:
Cannabinoids as therapeutic agents for ablating neuroinflammatory
disease. Endocr Metab Immune Disord Drug Targets. 8:159–172. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Saito T, Sugimoto N, Ohta K, Shimizu T,
Ohtani K, Nakayama Y, Nakamura T, Hitomi Y, Nakamura H, Koizumi S
and Yachie A: Phosphodiesterase inhibitors suppress
Lactobacillus casei cell wall-induced NF-κB and MAPK
activations and cell proliferation through protein kinase A- or
exchange protein activated by cAMP-dependent signal pathway.
Scientific World Journal. 2012.748572:
|
|
12
|
Klein TW: Cannabinoid-based drugs as
anti-inflammatory therapeutics. Nat Rev Immunol. 5:400–411. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guzman M: Cannabinoids: potential
anticancer agents. Nat Rev Cancer. 3:745–755. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sarfaraz S, Adhami VM, Syed DN, Afaq F and
Mukhtar H: Cannabinoids for cancer treatment: progress and promise.
Cancer Res. 68:339–342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aggarwal BB: Nuclear factor-kappaB: the
enemy within. Cancer Cell. 6:203–208. 2004.PubMed/NCBI
|
|
16
|
Prasad S, Ravindran J and Aggarwal BB:
NF-kappaB and cancer: how intimate is this relationship. Mol Cell
Biochem. 336:25–37. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jeon YJ, Yang KH, Pulaski JT and Kaminski
NE: Attenuation of inducible nitric oxide synthase gene expression
by delta 9-tetrahydrocannabinol is mediated through the inhibition
of nuclear factor-kappaB/Rel activation. Mol Pharmacol. 50:334–341.
1996.PubMed/NCBI
|
|
18
|
Kozela E, Pietr M, Juknat A, Rimmerman N,
Levy R and Vogel Z: Cannabinoids Delta(9)-tetrahydrocannabinol and
cannabidiol differentially inhibit the lipopolysaccharide-activated
NF-kappaB and interferon-beta/STAT proinflammatory pathways in BV-2
microglial cells. J Biol Chem. 285:1616–1626. 2010. View Article : Google Scholar
|
|
19
|
Zhang J and Chen C: Endocannabinoid
2-arachidonoylglycerol protects neurons by limiting COX-2
elevation. J Biol Chem. 283:22601–22611. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Du H, Chen X, Zhang J and Chen C:
Inhibition of COX-2 expression by endocannabinoid
2-arachidonoylglycerol is mediated via PPAR-γ. Br J Pharmacol.
163:1533–1549. 2011.PubMed/NCBI
|