Introduction
Telomeres are special structures consisting of a
stretch of very simple tandemly repeated sequences and telomere
structural proteins at the terminal of chromosomes (1). Their main function is to cap the
chromosome ends and prevent chromosomal instability, while the
erosion of telomeres can lead to genetic instability, a pivotal
mechanism in the neoplastic process (2,3).
Because of incomplete replication of the termini of linear DNA
molecules, telomeric DNA is progressively lost with each cell
division (4,5). Telomere shortening reaching a
critically short length can activate DNA damage checkpoints and
result in induction of cellular senescence (6). The first checkpoint in response to
telomere shortening is a p53-dependent, permanent cell cycle
arrest. p53 plays a key role in cellular senescence and/or
apoptosis associated with telomere dysfunction (7). It may prevent entry into mitosis with
uncapped telomeres (8), and intact
p53 signaling could be a prerequisite for induction of senescence
and/or apoptosis in response to critical telomere shortening
(9). When p53 is mutated or
deleted, p53-dependent responses to telomere dysfunction are
mitigated and chromosomal fusions are tolerated. This results in
chromosome breakage and genomic copy number alterations (CNAs) and
drives development of carcinomas (3,10).
These all position p53 as the guard against tumorigenesis caused by
telomere dysfunction.
TP53 is a tumor-suppressor gene, whose
mutations and loss of heterozygosity (LOH) are hallmarks of most
human cancers (11,12). Mutations in the coding sequence can
cause dramatic defects in p53 function, and some polymorphisms in
the TP53 locus might have phenotypic manifestations
(13,14). LOH has emerged as the second hit in
tumor initiation which serves to inactivate or eliminate the
wild-type allele at the tumor-suppressor gene locus (12,15,16).
These mutations, polymorphisms or allelic loss (or LOH) that may
change p53 function have a relationship with telomere erosion and
tumorigenesis.
Both breast and esophageal cancers are the most
common tumors. However, no study has previously investigated the
relationship between TP53 gene variants and telomere length
(TL) in breast tumor and esophageal cancer. The relationships
between TP53 mutations, polymorphisms, allelic loss and TL
are still largely undefined. The present study, which investigated
the TP53 gene and TL from 126 Chinese breast tumor patients
and 68 Chinese esophageal cancer patients, was aimed at
investigating the potential interaction between p53 functional
mutations, polymorphisms, allelic loss and telomere erosion. This
study may help us better understand the molecular mechanism of
tumorigenesis, which should lead to improved screening and
treatment of cancer.
Materials and methods
Study population
A total of 126 breast tumor patients and 68
esophageal cancer patients of Chinese ancestry were included in the
present study. All of the breast tumor samples, including 45 malign
and 81 benign breast tumor samples, were consecutively collected
from the Yunnan Province. Breast tumor tissue and a blood sample
from each patient were collected for genomic DNA extraction and
genotyping. Sixty-eight esophageal cancer specimens were
consecutively collected from the Henan Province. For esophageal
cancer patients, each cancer tissue and normal tissue were
collected for study. Written consent was obtained from all
participants, in accordance with protocols approved by the
institutional review board at each contributing center.
Telomere measurement
Genomic DNA was isolated from whole blood samples
and tissues by standard phenol/chloroform method. Relative telomere
length was measured on extracted DNA using real-time quantitative
PCR (17,18) with minor modifications. Standard
curves for TL and single-copy gene (reference gene) were used to
transform cycle threshold into nanograms of DNA. Triplicate PCR
reactions were performed in 20 μl reactions comprising 8 μl
template DNA, 2 μl primer mixture and 10 μl SYBR® Premix
Ex Taq™ (Takara, Dalian, China). The final telomere and 36B4 primer
concentrations were 0.2 and 0.3 μM. The primer sequences were as
previously described (18). The
reaction mixture was initially denatured at 95°C for 2 min followed
by 40 cycles of 95°C for 5 sec, 58°C for 10 sec, and 72°C for 40
sec for the 36B4 reaction, or 25 cycles of 95°C for 5 sec, 56°C for
10 sec, and 72°C for 60 sec for the telomere reaction. All PCRs
were performed using a 96-well formatted LightCycler®
480 Real-Time PCR system (Roche Applied Science), and results were
obtained and analyzed using the LightCycler® 480 onboard
software (version 1.5).
Mutational screening and genotyping of
TP53
According to the TP53 somatic mutations
database (IARC TP53 database, http://www-p53.iarc.fr, R15 release) (11), 96% of somatic mutations are located
in exons 3–9 of the gene. Thus, we sequenced parts of the
TP53 gene to discover somatic mutations and other
variations. Exons 3–9 and respective intron-exon boundaries were
included. Primers for PCR and sequencing are listed in Table I. PCR was carried out in 25 μl of
reaction containing 1X LA PCR Buffer II (Mg2+ Plus), 20
ng DNA, 0.5 μM each of the primers, 0.4 mM each of deoxynucleotide
triphosphate, and 1.25 U of LA Taq DNA polymerase (Takara). The
reaction mixture was denatured at 95°C for 5 min followed by 10
cycles of 1 min of denaturation at 94°C, 1 min of reannealing at
60-50°C (decreased by 1°C every cycle), and 4 min of extension at
72°C; 25 cycles of 1 min of denaturation at 94°C, 1 min of
reannealing at 50°C and 4 min of extension at 72°C. The PCR was
completed by a final extension at 72°C for 10 min. The products
were purified with gel extraction kits (Watson BioMedical Inc.,
Shanghai, China) and were subjected to direct DNA sequencing using
the BigDye® Terminator v3.1 Cycle Sequencing kit and ABI
PRISM 3730 sequencer (Applied Biosystems Inc., USA). Sequences were
aligned and analyzed with DNAStar software package (DNAStar Inc.,
Madison, WI, USA). For the malign breast tumors and esophageal
cancer patients, both tissues for each patient were sequenced. For
the benign breast tumor patients that had variations not included
in the known polymorphisms, whole blood samples were also
sequenced. All somatic mutations found by direct sequencing of PCR
products were confirmed by sequencing of a second, independent PCR
product. All sequences were submitted to GenBank (accession no.
JQ751320-JQ752243).
 | Table IPrimers for PCR and sequencing. |
Table I
Primers for PCR and sequencing.
| Primer name | Primer sequence
5′-3′ | Type |
Amplified/sequencing fragmenta (locus) |
|---|
| TP53F |
ACGACGAGTTTATCAGGA | Amplifying |
g.11066-g.14379 |
| TP53R |
GACCTATGGAAACTGTGAG | Amplifying | |
| TP53S1 |
ACGGCATTTTGAGTGTTAG | Sequencing | g.13294-g.14084
(exons 7–9) |
| TP53S2 |
GGATGGGTAGTAGTATGGAAG | Sequencing | |
| TP53R1 |
CCTGATTTCCTTACTGCCTCTT | Sequencing | |
| TP53R2 |
TGCTTGCCACAGGTCTCC | Sequencing | |
| TP53S3 |
TCAAATAAGCAGCAGGAGA | Sequencing | g.12338-g.12805
(exons 5–6) |
| TP53R3 |
TGCCGTCTTCCAGTTGCT | Sequencing | |
| TP53S4 |
GTGAAGAGGAATCCCAAAG | Sequencing | g.11114-g.11656
(exons 3–4) |
| TP53R4 |
CCTATGGAAACTGTGAGTGGA | Sequencing | |
All polymorphisms in each individual were detected
and confirmed by sequencing the corresponding regions in both
tissues. Allelic loss was determined by comparing tumor and normal
single nucleotide polymorphism (SNP) allele types. Linkage
disequilibrium (LD) coefficient (D′ and r2) and Hardy-Weinberg
equilibrium P-value were estimated by Haploview software version
4.2 package (http://www.broad.mit.edu/mpg/haploview/). For each
polymorphism, Hardy-Weinberg equilibrium (HWE) was tested by
comparing the observed to expected genotype frequencies. All SNPs
were consistent with HWE (P>0.05). Reconstruction of the
TP53 haplotypes incorporating the 7 SNPs was accomplished
using Phase software. Four distinct haplotypes were observed in the
study population with a frequency >1%. Each individual was
assigned the best pair of haplotypes estimated by Phase
software.
Statistical analysis
TL was analyzed as a continuous variable. The
Mann-Whitney U test or the Kruskal-Wallis H test was used as
appropriate to determine the differences in TL between different
groups. Correlation curves between age and TL were estimated by
linear regression model. A Chi-squared test or Fisher's exact test
was used as appropriate to assess differences in allelic loss
frequency between different groups and genotype distribution of
each tagSNP between malign and benign breast tumor patients. All
statistical analyses were performed using SPSS 13.0 (SPSS Inc.,
Chicago, IL, USA). Statistical significance was declared at
α=0.05.
Results
Breast tumors
TL and its association with somatic
p53 mutations
In 124 breast tumor samples, TL was determined by
real-time PCR. The mean level of TL in breast tumor tissues was
1.346 [standard error (SE) =0.039]. Mean TL of malign patients was
shorter than that of benign patients, although no significant
difference was observed (P=0.102). The mean TLs were 1.263
(SE=0.067) and 1.391 (SE=0.047) in malign breast tumors and benign
breast tumors, respectively. The TLs of the breast tumor tissues
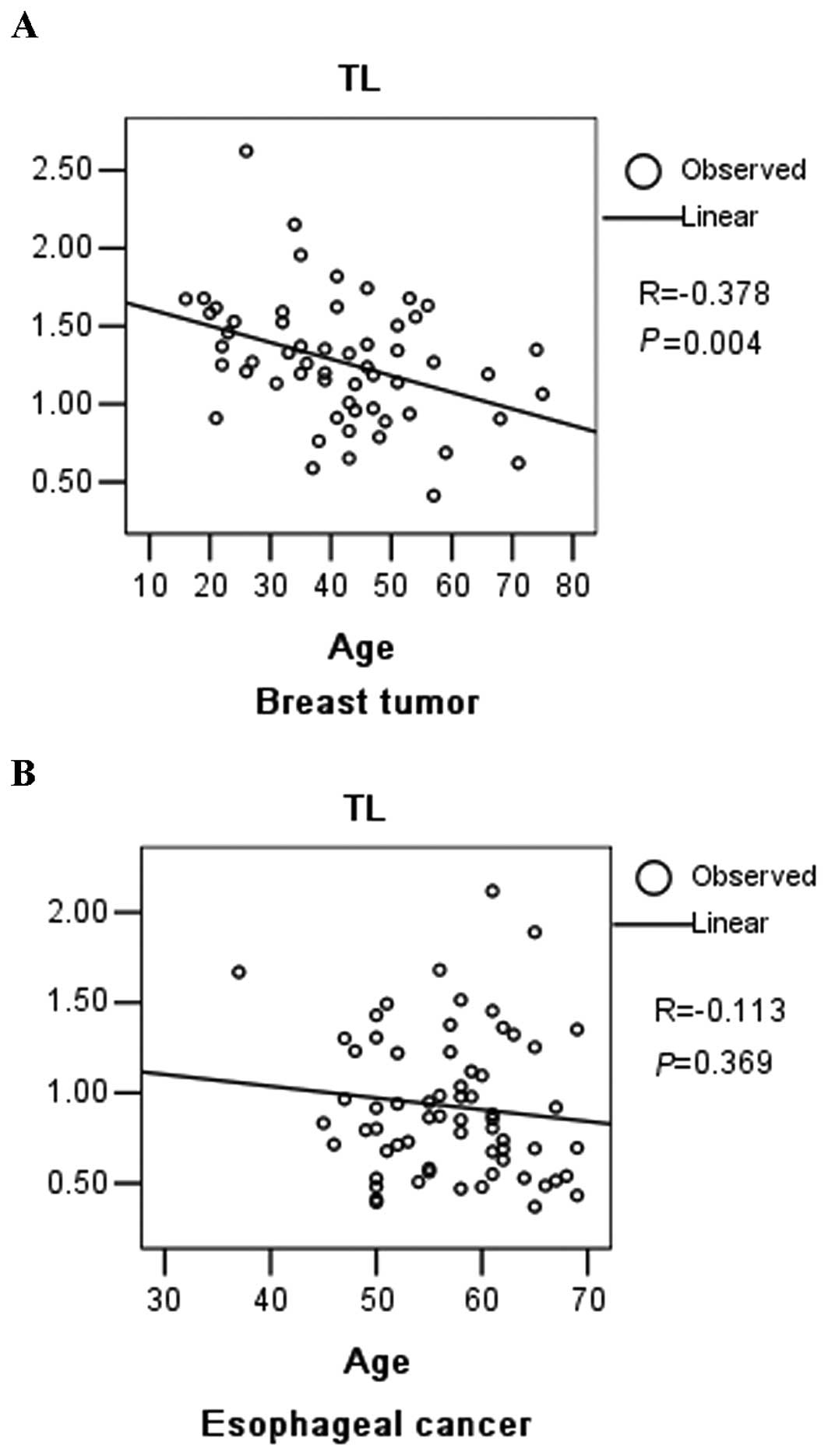
were plotted against patient age at sampling (Fig. 1A). The negative slope of the
best-fit line for breast tumor tissue indicated a decrease in the
TL with age in the breast tumor patients (R=−0.378, P=0.004).
Table II shows the
pattern and codon distribution of TP53 somatic mutations in
our patients. In the breast tumor patients, we found a total of 11
somatic mutations in 10 patients and 1 patient had double
mutations. Therefore, the frequency of TP53 gene somatic
mutations in breast cancer was 22.7% (10/44) in our study. The
proportions of different mutation types were 1/11 (9.1%) for
A:T→G:C, 2/11 (18.2%) for G:C→A:T, 3/11 (27.3%) for G:C→T:A, 1/11
(9.1%) for ins and 4/11 (36.4%) for del, respectively. All the
somatic mutations were found in the malign patients and located in
the coding region, including 6 missense mutations and 5 frameshift
mutations.
| Table IISomatic mutations detected in the
patients. |
Table II
Somatic mutations detected in the
patients.
| Sample name | Telomere
length | Type of cancer | Mutation (PCR
product) | Genomic
descriptiona | Exon/intron
number | Mutational
type | Residue change
(Splice site) |
|---|
| 5C | 1.348 | Breast | 1856T→C | g.12524 | 5-exon | A:T→G:C | His179Arg |
| 31C | 0.412 | Breast | 1734G→T | g.12646 | 6-exon | G:C→T:A | His193Asn |
| 38C | 1.009 | Breast | 1665C→T | g.12715 | 6-exon | G:C→A:T | Val216Met |
| 39C | 0.827 | Breast | 1941del | g.12443 | 5-exon | del | Pro152NA |
| 41C | 1.559 | Breast | 2785-2786del | g.11596-11597 | 4-exon | del | Val122NA |
| 67C | 0.958 | Breast | 2823insCGGA | g.11560 | 4-exon | ins | Arg110NA |
| 72C | 0.688 | Breast | 547C→T | g.13833 | 8-exon | G:C→A:T | Glu285Lys |
| 94C | 1.371 | Breast | 1685-1686del | g.12694-12695 | 6-exon | del | Arg209NA |
| 97C | 0.561 | Breast | 570C→A | g.13810 | 8-exon | G:C→T:A | Cys277Phe |
| 98C | 0.916 | Breast | 348del | g.14032 | 9-exon | del | Lys320NA |
| 98C | 0.916 | Breast | 351C→A | g.14029 | 9-exon | G:C→T:A | Lys319Asn |
| C085 | 0.985 | Esophageal | 1001G→A | g.13379 | 7-exon | G:C→A:T at CpG | Arg248Trp |
| C090 | 0.679 | Esophageal | 562T→A | g.13818 | 8-exon | A:T→T:A | Arg280STOP |
| C091 | 1.361 | Esophageal | 582C→T | g.13798 | 8-exon | G:C→A:T at CpG | Arg273His |
| C093 | 0.729 | Esophageal | 2773C→T | g.11607 | 4-intron | G:C→A:T | NA (consensus
SD) |
| C094 | 0.780 | Esophageal | 582C→G | g.13798 | 8-exon | G:C→C:G | Arg273Pro |
| C095 | 0.880 | Esophageal | 567G→C | g.13813 | 8-exon | G:C→C:G | Pro278Arg |
| C097 | 2.118 | Esophageal | 1737G→A | g.12643 | 6-exon | G:C→A:T | Gln192STOP |
| C100 | 0.802 | Esophageal | 1641C→A | g.12739 | 6-exon | G:C→T:A | Glu224STOP |
| C100 | 0.802 | Esophageal | 1722C→G | g.12658 | 6-exon | G:C→C:G | Val197Leu |
| C101 | 1.681 | Esophageal | 1048A→G | g.13332 | 7-exon | A:T→G:C | Ile232Thr |
| C102 | 0.486 | Esophageal | 1021G→C | g.13359 | 7-exon | G:C→C:G | Ser241Cys |
| C104 | 0.507 | Esophageal | 1652T→C | g.12728 | 6-exon | A:T→G:C | Tyr220Cys |
| C107 | 0.479 | Esophageal | 1980C→G | g.12400 | 5-exon | G:C→C:G | Ala138Pro |
| C108 | 1.302 | Esophageal | 556G→A | g.13824 | 8-exon | G:C→A:T at CpG | Arg282Trp |
| C110 | 0.805 | Esophageal | 1652T→C | g.12728 | 6-exon | A:T→G:C | Tyr220Cys |
| C111 | 0.714 | Esophageal | 1652T→G | g.12728 | 6-exon | A:T→C:G | Tyr220Ser |
| C112 | 0.689 | Esophageal | 982-984del | g.13401-13403 | 7-exon | del | Ile255NA |
| C114 | 1.323 | Esophageal | 543insTT | g.13838 | 8-exon | ins | Glu287NA |
| C114 | 1.323 | Esophageal | 583G→A | g.13797 | 8-exon | G:C→A:T at CpG | Arg273Cys |
| C115 | 0.872 | Esophageal | 1001G→A | g.13379 | 7-exon | G:C→A:T at CpG | Arg248Trp |
| C116 | 1.306 | Esophageal | 1665C→A | g.12715 | 6-exon | G:C→T:A | Val216Leu |
| C120 | 0.979 | Esophageal | 2866insA | g.11514 | 4-exon | ins | Ser95NA |
| C121 | 0.481 | Esophageal | 1990A→C | g.12390 | 5-exon | A:T→C:G | Phe134Leu |
| C122 | 0.526 | Esophageal | 570C→A | g.13810 | 8-exon | G:C→T:A | Cys277Phe |
| C124 | 0.539 | Esophageal | 556G→A | g.13824 | 8-exon | G:C→A:T at CpG | Arg282Trp |
| C124 | 0.539 | Esophageal | 1875C→T | g.12505 | 5-exon | G:C→A:T | Val173Met |
| C125 | 0.916 | Esophageal | 313A→G | g.14067 | 9-intron | A:T→G:C | NA (consensus
SD) |
| C127 | 0.864 | Esophageal | 561C→A | g.13819 | 8-exon | G:C→T:A | Arg280Ile |
| C132 | 1.890 | Esophageal | 1865C→A | g.12515 | 5-exon | G:C→T:A | Cys176Phe |
| C134 | 0.432 | Esophageal | 583G→A | g.13797 | 8-exon | G:C→A:T at CpG | Arg273Cys |
| C134 | 0.432 | Esophageal | 1737G→A | g.12643 | 6-exon | G:C→A:T | Gln192STOP |
| C136 | 0.528 | Esophageal | 484G→A | g.13896 | 8-exon | G:C→A:T at CpG | Arg306STOP |
| C138 | 0.562 | Esophageal | 2955del | g.11425 | 4-exon | del | Arg65NA |
| C141 | 0.410 | Esophageal | 1868C→T | g.12512 | 5-exon | G:C→A:T at CpG | Arg175His |
| C142 | 1.313 | Esophageal | 1677-1678del | g.12704-12705 | 6-exon | del | Phe212NA |
| C143 | 0.394 | Esophageal | 617-637del | g.13743-13763 | 8-exon | del
7-intron | Ser261NA |
| C143 | 0.394 | Esophageal | 1727A→G | g.12653 | 6-exon | A:T→G:C | Ile195Thr |
| C144 | 0.661 | Esophageal | 2868G→C | g.11512 | 4-exon | G:C→C:G | Ser94STOP |
| C145 | 0.628 | Esophageal | 2998C→A | g.11382 | 4-exon | G:C→T:A | Glu51STOP |
| C146 | 0.606 | Esophageal | 1638C→T | g.12742 | 6-intron | G:C→A:T | NA (consensus
SD) |
| C146 | 0.606 | Esophageal | 2871-2872del | g.11508-11509 | 4-exon | del | Leu93NA |
| C147 | 0.581 | Esophageal | 576C→A | g.13804 | 8-exon | G:C→T:A | Cys275Phe |
| C148 | 0.794 | Esophageal | 539C→T | g.13841 | 8-exon | G:C→A:T | Glu287Glu |
| C148 | 0.794 | Esophageal | 547C→T | g.13833 | 8-exon | G:C→A:T | Glu285Lys |
| C148 | 0.794 | Esophageal | 666C→T | g.13714 | 7-intron | G:C→A:T | NA |
| C149 | 0.550 | Esophageal | 568G→A | g.13812 | 8-exon | G:C→A:T | Pro278Ser |
| C150 | 0.369 | Esophageal | 484G→A | g.13896 | 8-exon | G:C→A:T at CpG | Arg306STOP |
| C151 | 0.514 | Esophageal | 1941G→A | g.12439 | 5-exon | G:C→A:T | Pro151Ser |
| C152 | 0.919 | Esophageal | 582C→T | g.13798 | 8-exon | G:C→A:T at CpG | Arg273His |
| C153 | 0.854 | Esophageal | 583G→A | g.13797 | 8-exon | G:C→A:T at CpG | Arg273Cys |
| C155 | 1.253 | Esophageal | 1041insG | g.13339 | 7-exon | ins | Asn235NA |
| C157 | 1.096 | Esophageal | 2877C→T | g.11503 | 4-exon | G:C→A:T | Trp91STOP |
| C158 | 0.738 | Esophageal | 1033A→T | g.13347 | 7-exon | A:T→T:A | Met237Lys |
| C159 | 0.692 | Esophageal | 1072T→G | g.13308 | 6-intron | A:T→C:G | NA (consensus
SA) |
| C160 | 0.674 | Esophageal | 618C→T | g.13762 | 7-intron | G:C→A:T | NA (consensus
SA) |
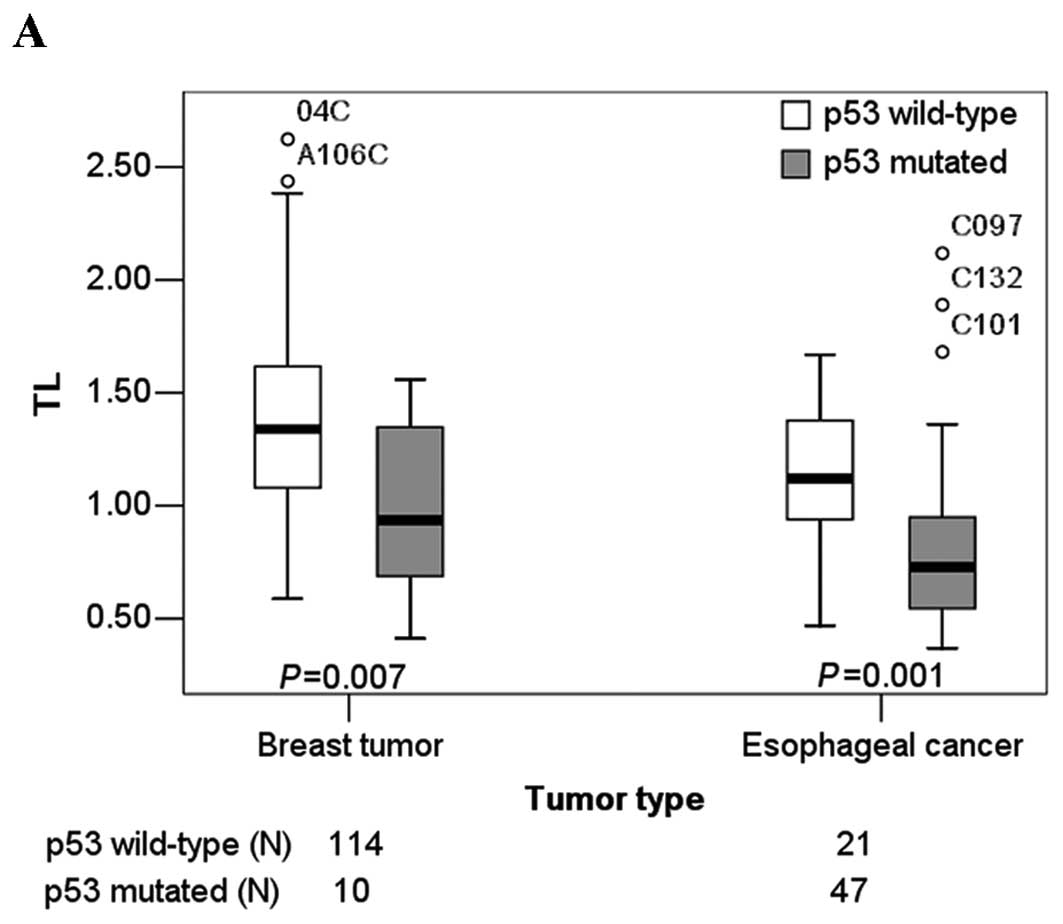
TLs were significantly shorter in patients with
somatic mutations when compared with patients with no mutation in
breast tumor tissues (P=0.007). Mean TLs of patients with and
without somatic mutation were 0.965 (SE=0.117) and 1.379 (SE=0.039)
respectively. The medians and the 25th, and 75th percentiles of TLs
in breast tumor patients with and without somatic mutations are
shown in Fig. 2A.
Relationship between TL and other
common p53 variants
Among the germline variants, four variants were
observed at low frequencies [minor allele frequency (MAF) <0.01]
in breast tumor patients. Variant 1621 (position in PCR product),
which was not included in the known SNPs, was detected in five
breast tumor patients both in leukocyte and breast tumor tissue.
The remnant common SNPs were rs12951053, rs12947788, rs1625895,
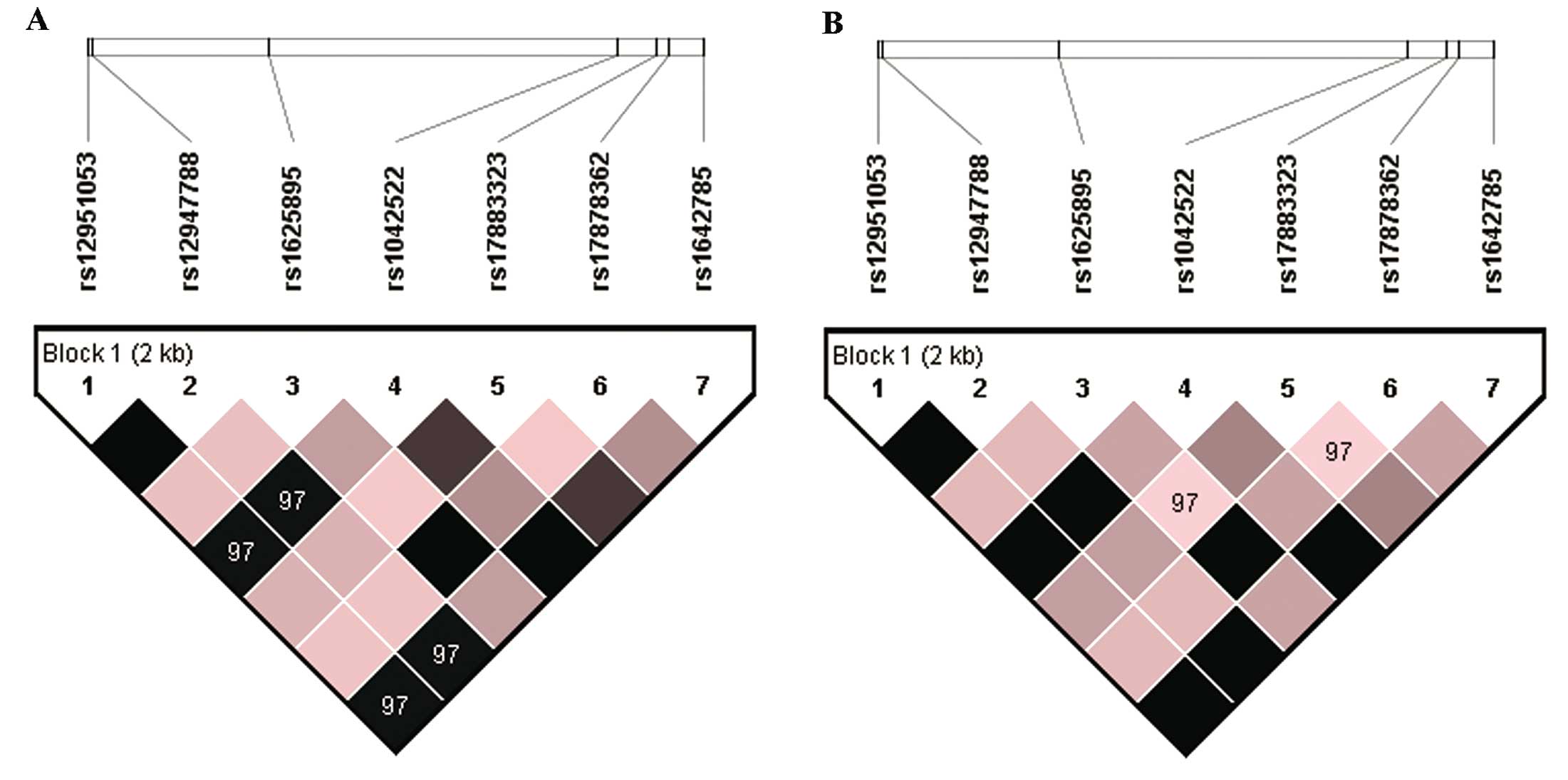
rs1042522, rs17883323, rs17878362, rs1642785. The locations of
polymorphisms ranged from 2-intron to 7-intron. LD coefficient (D′
and r2) and Hardy-Weinberg equilibrium P-value were estimated by
Haploview (Table III, Fig. 3A). For complete linkage SNPs, one
was selected for subsequent analysis. These were rs12951053,
rs1625895, rs1042522, rs17883323 and rs17878362. To obtain accurate
results, patients with somatic mutations were excluded of in the
association analysis of SNPs, haplotypes and TLs. In the group of
breast tumor patients, TLs of the different genotypes did not
achieve significant difference for all SNPs in the tumor tissue
(Table IV). There was also no
significant difference among the TLs of the different haplotypes
(data not shown).
| Table IIISeven common SNPs identified by
sequencing. |
Table III
Seven common SNPs identified by
sequencing.
| | | Breast tumor | Esophageal
cancer | |
|---|
| | |
|
| |
|---|
| SNP | Position (PCR
product) | Locus | HWE P-value | MAF | HWE P-value | MAF | Alleles |
|---|
| rs12951053 | 869 | 7-intron | 0.897 | 0.381 | 1 | 0.353 | A:C |
| rs12947788 | 889 | 7-intron | 0.897 | 0.381 | 1 | 0.353 | G:A |
| rs1625895 | 1577 | 6-intron | 1 | 0.032 | 1 | 0.059 | C:T |
| rs1042522 | 2934 | 4-exon | 0.1973 | 0.488 | 0.8499 | 0.485 | C:G |
| rs17883323 | 3081 | 3-intron | 0.9608 | 0.075 | 0.9501 | 0.103 | G:T |
| rs17878362 | 3131 | 3-intron | 1 | 0.036 | 1 | 0.059 |
−:ccccagccctccaggt |
| rs1642785 | 3263 | 2-intron | 0.1973 | 0.488 | 0.8499 | 0.485 | C:G |
| Table IVAssociations of p53 common variants
in tumors without p53 somatic mutations and TLs in breast tumors
and esophageal cancer. |
Table IV
Associations of p53 common variants
in tumors without p53 somatic mutations and TLs in breast tumors
and esophageal cancer.
| Breast tumor | Esophageal
cancer |
|---|
|
|
|
|---|
| Genotype | N | TL (means ±
SE) | N | TL (means ±
SE) |
|---|
| rs12951053 |
| AA | 43 | 1.356±0.062 | 10 | 1.221±0.109 |
| AC | 52 | 1.376±0.057 | 7 | 1.139±0.105 |
| CC | 19 | 1.441±0.109 | 4 | 0.832±0.052 |
| P-value | | 0.958 | | 0.052 |
| rs1625895 |
| CC | 106 | 1.366±0.039 | 20 | 1.108±0.071 |
| CT | 8 | 1.551±0.229 | 1 | 1.351 |
| P-value | | 0.606 | | 0.509 |
| rs1042522 |
| CC | 33 | 1.377±0.064 | 7 | 1.106±0.129 |
| GC | 49 | 1.360±0.060 | 9 | 1.258±0.103 |
| GG | 32 | 1.411±0.085 | 5 | 0.889±0.070 |
| P-value | | 0.962 | | 0.079 |
| rs17883323 |
| GG | 99 | 1.408±0.042 | 18 | 1.071±0.071 |
| GT | 15 | 1.187±0.094 | 3 | 1.414±0.160 |
| P-value | | 0.108 | | 0.088 |
| rs17878362 |
| −/− | 105 | 1.361±0.039 | 20 | 1.108±0.071 |
|
−/ccccagccctccaggt | 9 | 1.591±0.206 | 1 | 1.351 |
| P-value | | 0.344 | | 0.509 |
Correlation of allelic loss with
somatic mutations and TL
In a comparative analysis of TP53 SNPs in
blood and tumor tissues of breast tumor patients, allelic loss was
detected in 11.3% (8/71) of tumors from heterozygous patients. Mean
TL of patients with allelic loss (1.170, SE=0.173) was shorter than
the mean TL of patients with no allelic loss (1.369, SE=0.054) with
a non-significant P-value (0.178). TP53 allelic loss was
detected in 60.0% (3/5) of breast tumor patients with somatic p53
mutations, which was more in comparison with individuals (7.6%,
5/66) without somatic p53 mutations (P=0.009). This suggests that
TP53 allelic loss was associated with TP53 mutations
among heterozygous breast tumor patients.
In the patients with TP53 allelic loss, all
of the heterozygous TP53 polymorphisms lost one allele,
which suggested that the loss type was large fragment loss. With
respect to every polymorphism included, the details of allelic loss
are shown in Table V. For the
famous rs1042522 (codon 72), 6 of 55 patients heterozygous for
codon 72 had allelic loss, including 1 (16.7%) loss of G allele
(Pro) and 5 (83.3%) loss of C allele (Arg).
| Table VDetails of the patients with allelic
loss. |
Table V
Details of the patients with allelic
loss.
| | | Genotype:
blood/normal tissue→tumor | |
|---|
| | |
| |
|---|
| Patient no. | Type of tumor | LOH type | rs12951053 | rs12947788 | rs1625895 | rs1042522 | rs17883323 | rs17878362 | rs1642785 | TP53
mutation |
|---|
| 20 | Breast cancer | Partial loss | AC→A- | AG→G- | - | - | GT→T- | - | - | − |
| 38 | Breast cancer | Partial loss | - | - | - | GC→G- | GT→T- | - | GC→G- | + |
| 64 | Breast cancer | Partial loss | AC→A- | AG→G- | - | GC→C- | - | - | GC→C- | − |
| 67 | Breast cancer | Complete loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| 94 | Breast cancer | Partial loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| A18 | Breast tumor | Complete loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | − |
| A71 | Breast tumor | Complete loss | AC→C- | AG→A- | - | - | GT→G- | - | - | − |
| A98 | Breast tumor | Partial loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | − |
| 85 | Esophageal
cancer | Complete loss | - | - | CT→C- | - | GT→T- | A1A2→A1- | - | + |
| 90 | Esophageal
cancer | Complete loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| 91 | Esophageal
cancer | Complete loss | AC→A- | AG→G- | CT→T- | - | - | A1A2→A2- | - | + |
| 93 | Esophageal
cancer | Partial loss | AC→A- | AG→G- | - | GC→C- | - | - | GC→C- | + |
| 94 | Esophageal
cancer | Complete loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| 95 | Esophageal
cancer | Complete loss | - | - | - | GC→C- | GT→G- | - | GC→C- | + |
| 100 | Esophageal
cancer | Complete loss | - | - | - | GC→G- | GT→T- | - | GC→G- | + |
| 101 | Esophageal
cancer | Complete loss | - | - | - | GC→C- | GT→G- | - | GC→C- | + |
| 102 | Esophageal
cancer | Complete loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| 104 | Esophageal
cancer | Complete loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| 107 | Esophageal
cancer | Complete loss | AC→A- | AG→G- | - | GC→C- | - | - | GC→C- | + |
| 111 | Esophageal
cancer | Complete loss | AC→A- | AG→G- | - | GC→C- | - | - | GC→C- | + |
| 112 | Esophageal
cancer | Partial loss | AC→A- | AG→G- | - | GC→C- | - | - | GC→C- | + |
| 115 | Esophageal
cancer | Complete loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| 116 | Esophageal
cancer | Complete loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| 127 | Esophageal
cancer | Partial loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| 136 | Esophageal
cancer | Partial loss | AC→A- | AG→G- | - | GC→C- | - | - | GC→C- | + |
| 141 | Esophageal
cancer | Complete loss | AC→A- | AG→G- | - | - | GT→T- | - | - | + |
| 142 | Esophageal
cancer | Complete loss | AC→C- | AG→A- | - | - | GT→G- | - | - | + |
| 144 | Esophageal
cancer | Complete loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| 145 | Esophageal
cancer | Partial loss | AC→A- | AG→G- | CT→T- | - | - | A1A2→A2- | - | + |
| 150 | Esophageal
cancer | Complete loss | AC→A- | AG→G- | CT→T- | - | - | A1A2→A2- | - | + |
| 151 | Esophageal
cancer | Complete loss | AC→A- | AG→G- | - | GC→C- | - | - | GC→C- | + |
| 153 | Esophageal
cancer | Complete loss | AC→C- | AG→A- | - | GC→G- | - | - | GC→G- | + |
| 158 | Esophageal
cancer | Complete loss | - | - | - | GC→G- | GT→T- | - | GC→G- | + |
| 160 | Esophageal
cancer | Complete loss | AC→A- | AG→G- | - | GC→C- | - | - | GC→C- | + |
Association of common SNPs and
susceptibility to malignant transformation in breast tumor
Associations between common SNPs of TP53 and
the susceptibility to tumor maligning in breast tumors were listed
in Table VI. No significant
difference was observed between malign and benign tumor patients in
5 common tagSNP genotypes and allele frequencies. This result
implies that all of the polymorphisms confer no effect on the risk
of tumor maligning in our breast tumor patients. The distribution
of haplotypes in benign and malign breast tumor patients were not
significantly different (data not shown).
| Table VIGenotype frequencies of common SNPs
and their association with susceptibility to malignant
transformation in breast tumors. |
Table VI
Genotype frequencies of common SNPs
and their association with susceptibility to malignant
transformation in breast tumors.
| SNP | Group | Genotype frequency
n, (%) | | |
|---|
| | AA | AC | CC |
| |
|
| rs12951053 | Benign | 27 (33.3) | 43 (53.1) | 11 (13.6) |
| Malign | 22 (48.9) | 15 (33.3) | 8 (17.8) |
| P-value | | | | 0.101 |
| | CC | CT | |
| |
|
| rs1625895 | Benign | 77 (95.1) | 4 (4.9) | |
| Malign | 41 (91.1) | 4 (8.9) | |
| P-value | | | | 0.455 |
| | CC | CG | GG |
| |
|
| rs1042522 | Benign | 22 (27.2) | 38 (46.9) | 21 (25.9) |
| Malign | 15 (33.3) | 17 (37.8) | 13 (28.9) |
| P-value | | | | 0.600 |
| | GG | GT | |
| |
|
| rs17883323 | Benign | 70 (86.4) | 11 (13.6) | |
| Malign | 37 (82.2) | 8 (17.8) | |
| P-value | | | | 0.528 |
| | −/− |
−/ccccagccctccaggt | |
| |
|
| rs17878362 | Benign | 76 (93.8) | 5 (6.2) | |
| Malign | 41 (91.1) | 4 (8.9) | |
| P-value | | | | 0.720 |
Esophageal cancer
TL and its association with somatic
p53 mutations
Of the 68 patients with esophageal cancer
investigated in this study, 55 TP53 gene somatic mutations
were found in 47 patients, and 7 patients had more than one
mutation. The frequency of TP53 gene somatic mutations in
esophageal cancer was therefore 69.1% (47/68) in our study. All of
the 47 patients had at least one mutation causing a amino acid
change or located in the splice-site. Among the 55 somatic
mutations identified, there were 31 missense mutations, 9 nonsense
mutations, 8 frameshift mutations, 5 splice-site mutations, 1
silent mutation and 1 intronic mutation. The proportions of
different mutational types were 5/55 (9.1%) for A:T→G:C, 12/55
(21.8%) for G:C→A:T at CpG, 12/55 (21.8%) for G:C→A:T, 2/55 (3.6%)
for A:T→T:A, 6/55 (10.9%) for G:C→C:G, 7/55 (12.7%) for G:C→T:A,
3/55 (5.5%) for A:T→C:G, 3/55 (5.5%) for ins and 5/55 (9.1%) for
del, respectively. Transitions were predominant (29/55, 52.7%),
followed by transversions (18/55, 32.7%).
In 68 esophageal cancer samples, TL was determined
by real-time PCR. The mean level of TL in esophageal cancer tissues
was 0.923 (SE=0.047). TLs were plotted against patient age at
sampling (Fig. 1B). No correlation
was found between age and TL in esophageal cancer tissue. TLs were
significantly shorter in patients with somatic mutations compared
with patients with no mutation in esophageal cancer tissues
(P=0.001). Mean TLs of patients with and without somatic mutations
were 0.835 (SE=0.057) and 1.120 (SE=0.069), respectively. The
medians, the 25th and the 75th percentiles of TLs in the esophageal
cancer patients with and without somatic mutations are shown in
Fig. 2A.
Relationship between TL and other
common p53 variants
Among the germline variants, two variants were
observed at low frequencies (MAF <0.01) in esophageal cancer
patients. The remnant common SNPs were rs12951053, rs12947788,
rs1625895, rs1042522, rs17883323, rs17878362 and rs1642785. Linkage
disequilibrium coefficient (D′ and r2) and Hardy-Weinberg
equilibrium P-value were estimated by Haploview (Table III, Fig. 3B). The SNPs selected for subsequent
analysis were the same as for the breast tumors.
In the group of esophageal cancer patients, TLs of
patients with minor genotype CC of rs12951053 and GG of rs1042522
were significantly shorter than patients with other genotypes of
this SNP in esophageal cancer tissue. Mean TLs of patients with
genotypes CC and AA&AC of rs12951053 were 0.832 (SE=0.052) and
1.187 (SE=0.076) respectively (P=0.020). Mean TLs of patients with
genotypes GG and CC&GC of rs1042522 were 0.889 (SE=0.070) and
1.192 (SE=0.080), respectively (P=0.032). The medians, the 25th and
the 75th percentiles of TLs in the esophageal cancer tissues
according to genotypes of rs12951053 and rs1042522 are shown in
Fig. 2B and C. For other SNPs, TLs
of different genotypes did not achieve significant difference
(Table IV). Haplotypes of 7 common
SNPs were estimated using the Phase software. Patients with
haplotype CACGG-G (rs12951053, rs12947788, rs1625895, rs1042522,
rs17883323, rs17878362, rs1642785) had a significantly shorter TL
than patients with the other haplotypes (P=0.009). Mean TLs of
patients with haplotype CACGG-G and the other haplotypes were 0.975
(SE=0.065) and 1.200 (SE=0.061), respectively.
Correlation of allelic loss with
somatic mutations and TL
In a comparative analysis of TP53 SNPs in
tumor and normal tissue of esophageal cancer patients, allelic loss
was detected in 57.8% (26/45) of tumors from heterozygous patients.
The mean TL of patients with allelic loss (0.789, SE=0.063) was
shorter than the mean TL of patients with no allelic loss (1.008,
SE=0.092) with borderline statistical significance (P=0.056). The
frequency of TP53 allelic loss was significantly higher in
heterozygous esophageal cancer patients with somatic mutations
compared with patients with no mutation (P<0.001). The
frequencies of TP53 allelic loss were 74.3% (26/35) for
mutation esophageal tumors and 0.00% (0/10) for no-mutation
esophageal tumors. These suggest that TP53 allelic loss was
associated with TP53 mutations among heterozygous esophageal
cancer patients.
In the patients with TP53 allelic loss, all
of the heterozygous TP53 polymorphisms lost one allele which
was the same as for breast tumors (Table V). For the famous rs1042522 (codon
72), 20 of 36 patients heterozygous for codon 72 had allelic loss,
including 9 (45.0%) loss of G allele (Pro) and 11 (55.0%) loss of C
allele (Arg).
Discussion
The most important function of telomeres is the
maintenance of genomic integrity and stability (1,2). TL of
human somatic cells is a biomarker of cumulative oxidative stress,
biologic age and life stress (19,20).
It shortens with each cell division (4,5).
Oxidative stress (21) and life
stress (22) also can accelerate
its shortening. We observed an inverse correlation between TL and
age in breast tumor tissue, demonstrating a significant age-related
telomere loss in these tissues. This correlation was not detected
in esophageal cancer tissue, which suggested that other factors,
such as oxidative stress and life stress, rather than age mainly
influence the TL in these tissues.
Mutation of the tumor suppressor p53 is an almost
universal feature of human cancer. In our present study, we
detected 11 somatic mutations in 10 breast tumor patients. All of
the mutations were located in the coding region and caused amino
acid changes. G:C→A:T mutations are very frequent in sporadic
breast cancers (IARC TP53 database, http://www-p53.iarc.fr, R15 release) (11). Compared with global TP53
mutations in sporadic breast cancer, mutations in our patients had
less G:C→A:T transitions (18.18 vs. 46.56%) and more deletions
(36.36 vs. 11.17%) and G:C→T:A transversions (27.27 vs. 8.90%). Of
the 68 patients with esophageal cancer investigated in this study,
55 TP53 gene somatic mutations were found in 47 patients.
The proportions of different mutational types in our esophageal
cancer patients are similar with global TP53 mutation in
esophageal cancer (11), with a
higher transition followed by transversion.
Previous research has shown that mutant p53 proteins
have a dominant negative effect on wild-type p53, and inhibit or
activate the function of other p53 family members (13). Inhibition of p53 function enables
continuous cell division and critical telomere shortening, a
phenomenon known as telomere crisis, which causes telomere fusion
and genome instability (3,10,23).
Our study showed that telomeres were statistically shorter in
tumor/cancer tissue from patients with TP53 somatic
mutations than those with wild-type. This finding suggests that
mutant p53 enables continuous cell division and critical telomere
shortening and combines telomere erosion driving tumor
formation.
Chromosomal instability (CIN) is a feature of most
human cancers, and one mechanism of CIN is though the loss of
telomeres (24). LOH is one of the
representations of chromosomal instability, and short telomeres
have been reported to contribute to LOH in renal cell carcinoma
(25). In our study, patients with
allelic loss had a shorter TL than patients with no allelic loss,
and TP53 allelic loss was associated with TP53
mutations among heterozygous patients in both tumor types. These
results suggest that large fragment TP53 allelic loss may be
one of the representations of chromosomal instability caused by
telomere dysfunction combined with p53 function inhibition.
Notably, the patients with p53 allelic loss had a high proportion
of mutant alleles (50–100%). LOH has emerged as the second hit in
tumor initiation which serves to inactivate or eliminate the
wild-type allele at the tumor-suppressor gene locus (12,15).
Thus, LOH at the p53 locus caused by chromosomal instability may
constitute one of the major mechanisms for inactivation of the
intact allele associated with a p53 mutation (16).
Combined with our results and previous studies, we
hypothesize that the mechanisms of tumorigenesis associated with
telomere dysfunction and p53 mutations are as follows. i) Telomere
DNA is progressively lost with each cell division (4,5). ii)
Telomere shortening reaching a critically short length activates
DNA damage checkpoints, and results in induction of cellular
senescence, and the first checkpoint in response to telomere
shortening is a p53-dependent, permanent cell cycle arrest
(6). iii) Exogenous carcinogens and
endogenous biological processes cause p53 mutations (26). iv) Mutant p53 proteins enable
continuous cell division and critical telomere shortening, a
phenomenon known as telomere crisis, which causes telomere fusion
and genome instability. v) LOH occurring by chromosomal instability
inactivates the intact allele associated with p53 mutation. vi)
Recurrence of the above steps occurs. vii) Tumorigenesis and
malignant transformation transpires.
Rs1042522, viz. codon R72P SNP, is in exon 4, the
segment of TP53 that encodes the polyproline domain, which
is essential for p53 to mount a full apoptotic response to stress
and inhibit tumorigenesis (14). It
has been reported that p53-P72 has a weaker apoptotic potential
than p53-R72 (27). In esophageal
cancer tissue, we detected that patients with a minor genotype GG
of rs1042522 had a shorter TL than those with genotypes CC&GC,
and patients with minor genotype CC of rs12951053 had a shorter TL
than those with genotypes AA&AC. Genotype GG and CC of
rs1042522 were corresponded to P72 and R72 in our study. Thus, the
minor genotype GG has a weaker apoptotic potential, and may enable
critical telomere shortening. Rs12951053 is in intron 7, which has
a strong linkage relationship with rs1042522 (Fig. 3). Its significant difference in TL
between genotypes may be caused by this. Patients with haplotype
CACGG-G have a significantly shorter TL than patients with the
other haplotypes. This haplotype exclusively contains C allele of
rs12951053 and G allele of rs1042522 simultaneously. The
above-mentioned differences did not exist in breast tumor patients,
which suggests that the function of SNPs may be tissue- or tumor
type-specific. For other SNPs, we found no evidence for an
association with TL. Their TLs of different genotypes did not show
a significant difference. Our results showed that SNPs of
TP53 may, depending on tissue or tumor type, specifically
have a very feeble effect on cellular senescence and/or apoptosis
associated with telomere dysfunction. Elucidation of this issue
requires investigation with a large sample size and the use of more
types of cancer.
Although the relationships between TP53
variants and TL in breast tumor and esophageal cancer have not been
directly studied previously, several similar studies exist. Two
studies found that TLs in the peripheral blood cells of germline
p53 mutation carriers of Li-Fraumeni syndrome were shorter than
that of normal individuals (28,29).
Another similar research by Radpour et al found that TL is
inversely correlated with the promoter methylation profile of p53
in breast cancer, which suggests that p53 may function as a
gatekeeper to prevent critical telomere shortening and genome
instability (30). From the above
findings, it is evident that all similar research obtained
consistent results consistent with ours suggesting that TL
shortening cannot drive tumorigenesis alone; it is combined with
defects in cellular senescence and/or apoptosis. p53 plays a key
role in this pathway. This may explain the inconsistent results of
previous research investigating TL and cancer risk (31–34).
Thus, in future research concerning telomere dysfunction and cancer
risk, the effects of cellular senescence and apoptosis should also
be considered.
In conclusion, our study revealed that telomeres of
patients with TP53 somatic mutations were statistically
shorter than those with wild-type in both breast tumor tissue and
esophageal cancer tissue, and large fragment TP53 allelic
loss was significantly associated with somatic mutations. These
findings suggest that mutant p53 enables continuous cell division
and critical telomere shortening and combines telomere erosion
driving tumor formation. Large fragment TP53 allelic loss
may be one of the representations of chromosomal instability caused
by telomere dysfunction combined with p53 function inhibition. The
SNPs of TP53 depending on tissue or tumor-type may have a
feeble effect on cellular senescence and/or apoptosis associated
with telomere dysfunction. Investigation with a large sample size
using more types of cancers may elucidate this issue.
Acknowledgements
We are greatly indebted to the persons who
participated in this research. In addition, we thank Dr Chengye
Wang for his help in preparing the study. This study was supported
by the National Natural Science Foundation of China (NSFC) and The
Bureau of Science and Technology of Yunnan Province.
References
|
1
|
Blackburn EH: Structure and function of
telomeres. Nature. 350:569–573. 1991. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hackett JA, Feldser DM and Greider CW:
Telomere dysfunction increases mutation rate and genomic
instability. Cell. 106:275–286. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rudolph KL, Millard M, Bosenberg MW and
DePinho RA: Telomere dysfunction and evolution of intestinal
carcinoma in mice and humans. Nat Genet. 28:155–159. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levy MZ, Allsopp RC, Futcher AB, Greider
CW and Harley CB: Telomere end-replication problem and cell aging.
J Mol Biol. 225:951–960. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harley CB, Futcher AB and Greider CW:
Telomeres shorten during ageing of human fibroblasts. Nature.
345:458–460. 1990. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
von Figura G, Hartmann D, Song Z and
Rudolph KL: Role of telomere dysfunction in aging and its detection
by biomarkers. J Mol Med. 87:1165–1171. 2009.PubMed/NCBI
|
|
7
|
Cosme-Blanco W, Shen MF, Lazar AJ, et al:
Telomere dysfunction suppresses spontaneous tumorigenesis in vivo
by initiating p53-dependent cellular senescence. EMBO Rep.
8:497–503. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thanasoula M, Escandell JM, Martinez P, et
al: p53 prevents entry into mitosis with uncapped telomeres. Curr
Biol. 20:521–526. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brassat U, Balabanov S, Bali D, et al:
Functional p53 is required for effective execution of telomerase
inhibition in BCR-ABL-positive CML cells. Exp Hematol. 39:66–76.
e1–2. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Artandi SE and DePinho RA: Telomeres and
telomerase in cancer. Carcinogenesis. 31:9–18. 2009. View Article : Google Scholar
|
|
11
|
Petitjean A, Mathe E, Kato S, et al:
Impact of mutant p53 functional properties on TP53 mutation
patterns and tumor phenotype: lessons from recent developments in
the IARC TP53 database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lasko D, Cavenee W and Nordenskjold M:
Loss of constitutional heterozygosity in human cancer. Annu Rev
Genet. 25:281–314. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Goh AM, Coffill CR and Lane DP: The role
of mutant p53 in human cancer. J Pathol. 223:116–126. 2011.
View Article : Google Scholar
|
|
14
|
Whibley C, Pharoah PD and Hollstein M: p53
polymorphisms: cancer implications. Nat Rev Cancer. 9:95–107. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Knudson AG Jr: Mutation and cancer:
statistical study of retinoblastoma. Proc Natl Acad Sci USA.
68:820–823. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saeki H, Kitao H, Yoshinaga K, et al:
Copy-neutral loss of heterozygosity at the p53 locus in
carcinogenesis of esophageal squamous cell carcinomas associated
with p53 mutations. Clin Cancer Res. 17:1731–1740. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cawthon RM: Telomere measurement by
quantitative PCR. Nucleic Acids Res. 30:e472002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gil ME and Coetzer TL: Real-time
quantitative PCR of telomere length. Mol Biotechnol. 27:169–172.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Babizhayev MA, Savel'yeva EL, Moskvina SN
and Yegorov YE: Telomere length is a biomarker of cumulative
oxidative stress, biologic age, and an independent predictor of
survival and therapeutic treatment requirement associated with
smoking behavior. Am J Ther. 18:e209–226. 2011. View Article : Google Scholar
|
|
20
|
Baird DM: Telomere dynamics in human
cells. Biochimie. 90:116–121. 2008. View Article : Google Scholar
|
|
21
|
von Zglinicki T: Oxidative stress shortens
telomeres. Trends Biochem Sci. 27:339–344. 2002.PubMed/NCBI
|
|
22
|
Epel ES, Blackburn EH, Lin J, et al:
Accelerated telomere shortening in response to life stress. Proc
Natl Acad Sci USA. 101:17312–17315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Artandi SE, Chang S, Lee SL, et al:
Telomere dysfunction promotes non-reciprocal translocations and
epithelial cancers in mice. Nature. 406:641–645. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Murnane JP: Telomeres and chromosome
instability. DNA Repair. 5:1082–1092. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen M, Ye Y, Yang H, et al: Genome-wide
profiling of chromosomal alterations in renal cell carcinoma using
high-density single nucleotide polymorphism arrays. Int J Cancer.
125:2342–2348. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Greenblatt MS, Bennett WP, Hollstein M and
Harris CC: Mutations in the p53 tumor suppressor gene: clues to
cancer etiology and molecular pathogenesis. Cancer Res.
54:4855–4878. 1994.PubMed/NCBI
|
|
27
|
Dumont P, Leu JI, Della Pietra AC III,
George DL and Murphy M: The codon 72 polymorphic variants of p53
have markedly different apoptotic potential. Nat Genet. 33:357–365.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tabori U, Nanda S, Druker H, Lees J and
Malkin D: Younger age of cancer initiation is associated with
shorter telomere length in Li-Fraumeni syndrome. Cancer Res.
67:1415–1418. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Trkova M, Prochazkova K, Krutilkova V,
Sumerauer D and Sedlacek Z: Telomere length in peripheral blood
cells of germline TP53 mutation carriers is shorter than that of
normal individuals of corresponding age. Cancer. 110:694–702. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Radpour R, Barekati Z, Haghighi MM, et al:
Correlation of telomere length shortening with promoter methylation
profile of p16/Rb and p53/p21 pathways in breast cancer. Mod
Pathol. 23:763–772. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pooley KA, Sandhu MS, Tyrer J, et al:
Telomere length in prospective and retrospective cancer
case-control studies. Cancer Res. 70:3170–3176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gramatges MM, Telli ML, Balise R and Ford
JM: Longer relative telomere length in blood from women with
sporadic and familial breast cancer compared with healthy controls.
Cancer Epidemiol Biomarkers Prev. 19:605–613. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zheng YL, Ambrosone C, Byrne C, Davis W,
Nesline M and McCann SE: Telomere length in blood cells and breast
cancer risk: investigations in two case-control studies. Breast
Cancer Res Treat. 120:769–775. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Svenson U, Nordfjall K, Stegmayr B, et al:
Breast cancer survival is associated with telomere length in
peripheral blood cells. Cancer Res. 68:3618–3623. 2008. View Article : Google Scholar : PubMed/NCBI
|