1. Introduction
As a tyrosine kinase inhibitor, emodin
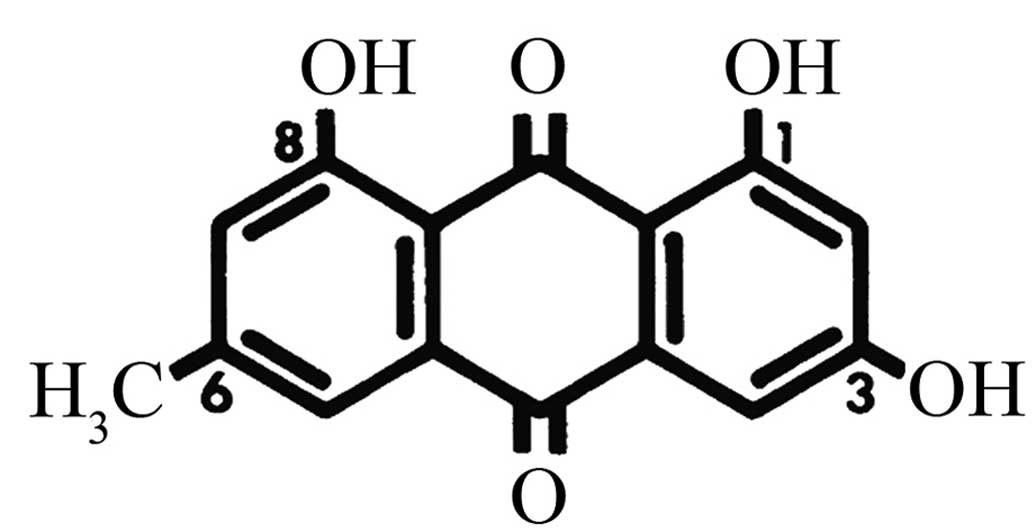
(1,3,8-trihydroxy-6-methyl-anthraquinone) (Fig. 1) is a natural anthraquinone
derivative found in the roots and rhizomes of numerous plants.
Pharmacological studies have demonstrated that emodin exhibits
various biological functions, such as anti-inflammatory,
anti-bacterial and anticancer activity. Studies have demonstrated
that emodin inhibits cell growth in several types of cancer cells
(1–3) and regulates genes related to the
control of cell apoptosis, oncogenesis, cell proliferation, cancer
cell invasion and metastasis (4–8).
Notably, previous studies have identified many types of drugs that
strengthen the therapeutic effect of gemcitabine on pancreatic
cancer (9–12). Similarly, our recent study also
concluded that emodin enhances the therapeutic effect of
gemcitabine in pancreatic cancer without additional toxic effects
(13). Although many researchers
have reported the antitumor effects of emodin in several types of
tumor cells, its specific molecular anticancer mechanisms have not
been fully elucidated. A recent resurgence of research into the
anticancer effects of emodin over the past 5 years has shown great
progress. Emodin was found to have antitumor effects in different
types of cancer, however, via different mechanisms. Therefore, the
exact antitumor mechanisms of emodin require further study. The
present review discusses the pharmacological activities of emodin
and the mechanisms that induce cell death in many types of human
cancer cells, both in vitro and in vivo. Research
findings on emodin-induced cytotoxicity and its protective effects
against cancer in different body systems as well as the antitumor
mechanisms involved are described below.
2. Effects of emodin on digestive system
cancer and the possible mechanisms involved
Human tongue squamous cancer
Lin et al(14) reported that emodin mediated
oxidative injury (DNA damage) based on reactive oxygen species
(ROS) production and endoplasmic reticulum (ER) stress based on the
levels of GADD153 and GRP78 that act as an early and upstream
change in the cell death cascade to caspase- and
mitochondria-dependent signaling pathways, triggers mitochondrial
dysfunction from Bcl-2 and Bax modulation, mitochondrial cytochrome
c release and caspase activation, consequently leading to
apoptosis in SCC-4 cells. Chen et al(15) also reported that emodin induced DNA
damage followed by the inhibition of expression of DNA
repair-associated genes, including ataxia telangiectasia mutated
(ATM), ataxia-telangiectasia and Rad3-related (ATR), 14-3-3σ,
breast cancer 1, early onset (BRCA1), DNA-dependent
serine/threonine protein kinase (DNA-PK) and
O6-methylguanine-DNA methyltransferase (MGMT) in SCC-4
human tongue cancer cells. Moreover, they found that emodin
inhibited the migration and invasion of SCC-4 cells and inhibited
the protein levels and activity of matrix metalloproteinase-2
(MMP-2), without affecting gene expression of MMP-2, however, it
inhibited the gene expression of MMP-9. MMP-9 (gelatinase-B) is
known to play an important role and is associated with tumor
migration, invasion and metastasis in various types of human
cancers. Furthermore, the authors demonstrated that emodin
decreased the levels of MMP-2 and urokinase plasminogen activator
(u-PA) in a concentration-dependent manner (16).
Gastric cancer and esophageal cancer
RhoA expression was found to be upregulated in
primary gastric carcinoma compared with normal gastric mucosa as
reported by Cai et al(17).
Emodin-induced generation of ROS was found to inhibit RhoA
activation to sensitize gastric carcinoma cells to anoikis
(17). Phosphatase of regenerating
liver-3 (PRL-3), a novel gene, has been found to play an important
role in the promotion of cellular proliferation as well as
inhibition of apoptosis in cancer cells. Sun and Bu (18) revealed that emodin inhibited cell
growth and induced apoptotic cell death in the SGC-7901 human
gastric carcinoma cell line via downregulation of PRL-3. Wang et
al(19) reported that human
esophageal carcinoma EC-109 cells treated with emodin underwent
rapid apoptosis. Emodin led to the growth inhibition of EC-109
cells in a time- and dose-dependent manner. Furthermore, the
intracellular pH (pHi) was found to decrease significantly by
0.47–0.78 units after treatment with emodin. Bursts of ROS took
place after the application of emodin (19). These results indicate that emodin
may be a strong anticancer drug against esophageal cancer by
causing various early events leading to tumor cell growth
inhibition, including the production of ROS and a decrease in pHi,
resulting in cellular apoptosis (19).
Liver cancer
Hsu et al(20) demonstrated that emodin caused G2/M
arrest in several types of liver cancer cells (Huh7, Hep3B and
HepG2) indicating that emodin has a broad spectrum of activity
against human hepatoma cells. They revealed that 15 representative
genes were associated with the emodin treatment response in
hepatoma cell lines. These genes included CYP1A1, CYP1B1, GDF15,
SERPINE1, SOS1, RASD1 and MRAS which were all upregulated, while
NR1H4, PALMD and TXNIP were downregulated following treatment with
emodin for 6 h. Finally, levels of the TIPARP, SLC7A11 and CYR61
genes were increased while IGFBP3 levels decreased at 24 h
following emodin treatment. Yu et al(21) observed that emodin induced apoptosis
in HepG2 cells and caused a significant accumulation of cells in
the G1 phase. Emodin also caused an increase in cytochrome c
release into the cytosol from mitochondria and the activation of
caspase-9 and caspase-8. In addition, treatment with emodin
resulted in a dose-dependent accumulation of intracellular ROS.
Furthermore, the authors found that emodin increased the protein
expression level of p53 and decreased the protein level of
NF-κB/p65 in HepG2 cells, which may play a role in emodin-induced
apoptosis.
Gallbladder cancer
Gallbladder carcinoma is known to be resistant to
many forms of anticancer drugs. For this reason, new cancer therapy
strategies must be investigated for the future treatment and
management of this disease. The major platinum-containing drugs
including cisplatin, carboplatin and oxaliplatin are widely used
anticancer agents for the treatment of various solid tumors, but
have not been proven effective for treating gallbladder cancers
(22). Wang et al(23) reported that co-treatment of
cisplatin, carboplatin and oxaliplatin with emodin (an ROS
generator) effectively enhanced the chemosensitivity of gallbladder
carcinoma cell line SGC996 both in vitro and in vivo
in comparison with cisplatin, carboplatin or oxaliplatin treatment
alone. The mechanisms were attributed to emodin-induced reduction
in the glutathione level, and downregulation of multidrug
resistance-related protein 1 (MRP1) (a type of GS-X pump)
expression in SGC996 cells. Remarkably, emodin exhibited few
systemic toxic effects in vivo. This finding supports the
notion that ROS manipulation strategy could be selective between
cancerous and normal cells, as indicated by an increasing body of
evidence (24,25). The expression of survivin is
involved in the inhibition of apoptosis. Wang et al(26) reported that emodin potentiated the
antitumor effects of cisplatin (CDDP) on gallbladder cancer cells
by generating ROS and by downregulating the expression of survivin
without causing detectable toxic effects on normal tissues. Side
population (SP) cells are likely responsible for tumor metastasis
and recurrence since they exhibit enhanced potential for
proliferation, clonogenicity, tumorigenicity and invasion (27–29).
Li et al(30) isolated a
small population of stem-like SP cells from the gallbladder
carcinoma cell line, SGC-996. They found that SP cells displayed a
higher proliferation rate, a higher clonal-generating capability,
higher tumorigenic, migratory and invasive abilities than non-SP
cells. Notably, they found that emodin reduced the ratio, inhibited
colony formation and effectively eliminated sphere formation of SP
cells. Furthermore, emodin sensitized SP cells to cisplatin to
overcome chemo-resistance via inhibition of expression of
ATP-binding cassette subfamily G member 2 (ABCG2). In addition,
emodin/cisplatin co-treatment in vivo suppressed the tumor
growth derived from SP cells through downregulation of ABCG2
expression (31).
Colon cancer
Lu et al(32)
demonstrated that emodin inhibited cancer-cell growth by blocking
vascular endothelial growth factor (VEGF) receptor signaling and
revealed that emodin can be used as a potential inhibitor for colon
cancer angiogenesis. Ma et al(33) reported that emodin induced apoptosis
of LS1034 human colon cancer cells in vitro and inhibited
tumor xenografts derived from LS1034 cells in vivo. During
the in vitro study, emodin induced cell morphological
changes, decreased the percentage of viability, induced G2/M phase
arrest and increased ROS and Ca(2+) production as well as loss of
mitochondrial membrane potential (ΔΨm) in LS1034 cells.
Emodin-triggered apoptosis was also found to be
concentration-dependent. The protein levels of caspase-9,
cytochrome c and the ratio of Bax/Bcl-2 were increased in
LS1034 cells following emodin exposure. Their study suggests that
emodin induces the production of ROS and Ca(2+) release and alters
the levels of anti- and pro-apoptotic proteins, leading to
mitochondrial dysfunction and activation of caspase-9 and caspase-3
which in turn causes apoptosis in LS1034 cells. In the in
vivo study, emodin effectively suppressed tumor growth in tumor
xenografts bearing LS1034 cells but it was unknown whether emodin
caused tumor inhibition via mitochondrial dysfunction as in
vitro(33). Many colorectal
cancer (CRC) patients suffer from the unexpected development of
micrometastasis after curative resection of primary tumors, and
~50% develop liver metastasis at some time point during their
disease progression. PRL-3 is expressed at high levels in nearly
all metastatic lesions that are derived from CRC, regardless of the
metastatic site. Yet weak or no PRL-3 expression is observed in
normal colon and non-metastatic primary cancers (34). PRL-3 was found to promote metastasis
in a diverse array of cancers (35). Han et al(36) found that emodin strongly inhibited
phosphatase activity of PRL-3 in the colon cancer cell line DLD-1
and blocked PRL-3-induced tumor cell migration and invasion in a
dose-dependent manner. Emodin rescued the phosphorylation of ezrin,
which is a known PRL-3 substrate. The results of this study
indicate that emodin is a PRL-3 inhibitor and a lead molecule for
development of a selective PRL-3 inhibitor. MMP-2 and -9
(gelatinase A and B) are considered to be important biomolecules in
colon cancer progression. Nitric oxide (NO), a free radical that
plays an important role in signaling pathways, regulates MMP
expression. In contrast, tumor cell-derived NO promotes the
expression of angiogenic factors. Damodharan et al(37) demonstrated that emodin suppresses
the NO-mediated upregulation of MMP-2 and -9. Thus, emodin can be
selected as an effective antimetastatic agent in NO-induced tumor
progression. Therefore, elucidation of critical pathways in
metastasis, where emodin could exert its inhibitory effects, may
make it ‘a perfect fit’ for antitumor therapy. Finally, emodin may
be developed for the treatment of colon cancer in the future due to
its potential ability to inhibit tumor growth and metastasis.
Pancreatic cancer
Survivin, a member of the inhibitor of apoptosis
gene family, is involved in the control of cell division and
inhibition of apoptosis and is described as a β-catenin/Tcf/Lef
target gene. Guo et al(38)
reported that emodin not only downregulated the expression of
survivin and β-catenin but also decreased the translocation of
β-catenin to the nucleus in pancreatic cancer and, thus,
potentiated the antitumor effects of gemcitabine. A recent study by
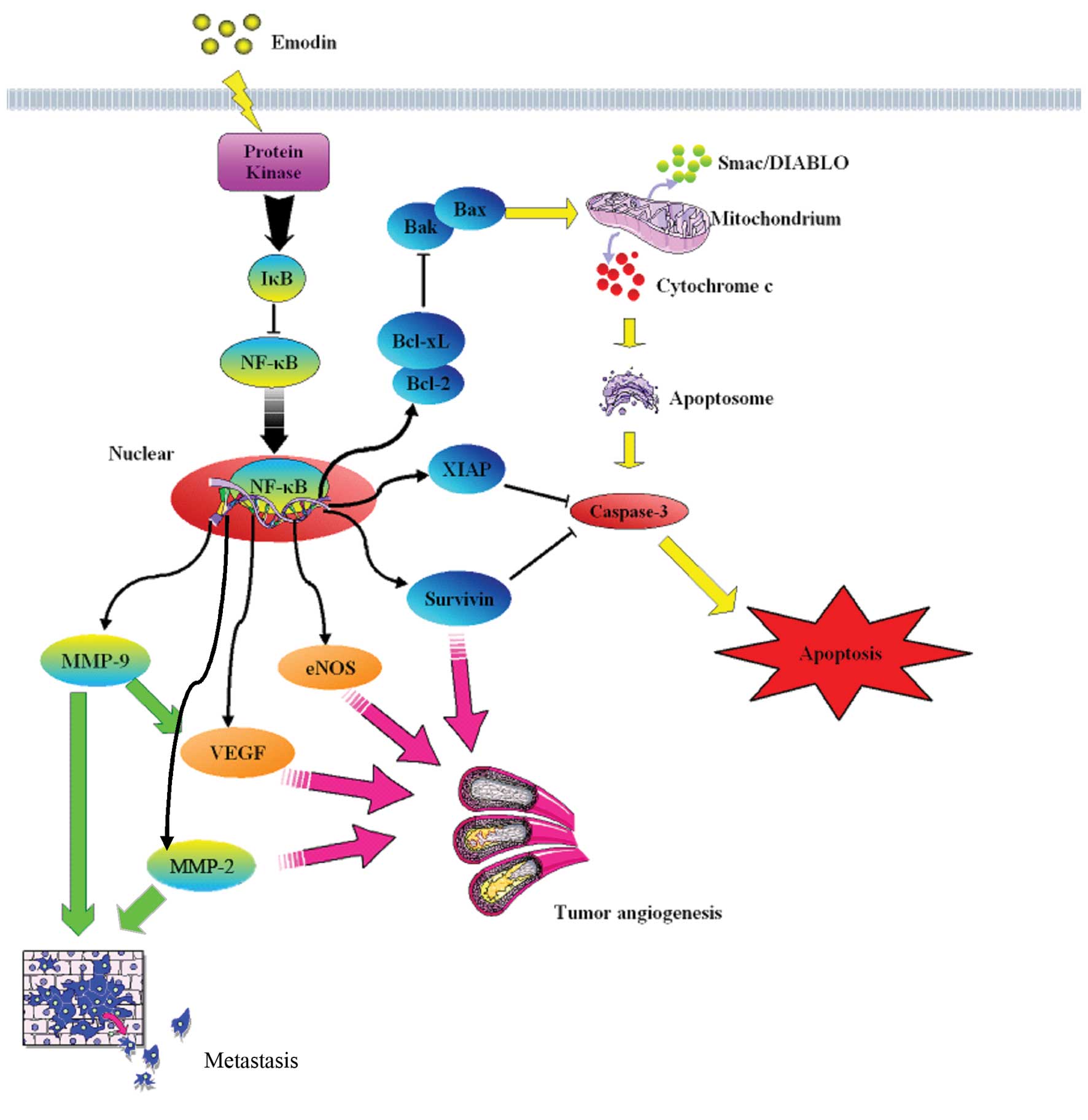
our research team suggests that emodin has a therapeutic effect on
pancreatic cancer through various antitumor mechanisms (Fig. 2). In Chen et al(39), we demonstrated that tumors in nude
mice subcutaneously injected with SW1990 cells and treated with a
combination of emodin (40 mg/kg) and gemcitabine (80 mg/kg) showed
a significant reduction in tumor volume, Ki-67 proliferation index
and expression of the Bcl-2/Bax ratio compared with tumors from
mice treated with sodium chloride, emodin alone (40 mg/kg) or
gemcitabine alone (125 mg/kg), and induced increased release of
CytC from the mitochondria to the cytoplasm and triggered caspase-3
activation leading to apoptosis. The results suggest that emodin
improves the antitumor effect of gemcitabine, even at a lower dose
of gemcitabine, so as to decrease the toxicity of chemotherapy on
transplanted tumors derived from the SW1990 cell line via the
enhancement of apoptosis induced by gemcitabine. This mechanism may
involve the downregulation of the Bcl-2/Bax ratio and the promotion
of CytC release from the mitochondria into the cytoplasm. In
another study, Liu et al(40) demonstrated that emodin induces
Panc-1 cell apoptosis mediated by a decrease in the mitochondrial
membrane potential. In addition, emodin at the dose of 40 mg/kg
improved the living state of model mice. In another study, Liu
et al(41) found that emodin
induced growth inhibition and apoptosis in the pancreatic cancer
cell line SW1990 and suppressed the migration and invasion of
SW1990 cells in a dose-dependent manner. Emodin significantly
downregulated NF-κB DNA-binding activity, survivin and MMP-9 in
SW1990 cells. They found that oral administration of emodin
significantly decreased tumor weight in vivo and metastasis
compared to a control. Furthermore, the expression of NF-κB,
survivin and MMP-9 was also suppressed in tumor tissues treated
with emodin. Their results indicated that emodin exerts
antiproliferative and antimetastatic activity on pancreatic cancer
both in vitro and in vivo, which may be related to
downregulation of NF-κB and its regulated molecules such as
survivin and MMP-9 proteins. Our previous study (42) demonstrated that treatment of
gemcitabine combined with emodin efficiently suppressed tumor
growth in mice inoculated with pancreatic tumor cells. This
treatment paradigm promoted apoptotic cell death and mitochondrial
fragmentation. Furthermore, it reduced the phosphorylated-Akt
(p-Akt) level, NF-κB activation, Bcl-2/Bax ratio, and increased
caspase-9/-3 activation and cytochrome c (CytC) release that
occurred in combination therapy or by emodin alone. Collectively,
emodin enhanced the activity of gemcitabine in tumor growth
suppression via inhibition of Akt and NF-κB activation, thus,
promoting the mitochondrial-dependent apoptotic pathway. In
addition, Wang et al(26)
reported that emodin enhanced the antitumor effect of gemcitabine
in SW1990 pancreatic cancer in vitro and in vivo,
which might be associated with downregulation of NF-κB expression,
thus inhibiting the expression of XIAP. Liu et al(43) reported that emodin promoted cell
apoptosis of the gemcitabine-resistant cell line SW1990/Gem in a
dose-dependent manner and enhanced the SW1990/Gem cell sensitivity
to gemcitabine in a time-dependent manner. They found that emodin
monotherapy or combination with gemcitabine decreased the gene and
protein expression levels of multi-drug resistance 1 (MDR-1), NF-κB
and Bcl-2 and inhibited the function of P-gp, but increased the
expression levels of Bax, cytochrome c (cytosol), caspase-9
and -3, thus promoting cell apoptosis. This study demonstrated that
emodin had a reversing effect on the gemcitabine-resistant cell
line SW1990/Gem, possibly via decrease in the function of P-gp and
activation of the mitochondrial apoptosis pathway. As members of
apoptosis inhibitors, survivin and XIAP play an important role in
chemotherapy resistance in pancreatic cancer. Guo et
al(44) reported that emodin
potentiates the antitumor effects of gemcitabine in PANC-1 cell
xenografts via downregulating expression of survivin and XIAP
leading to apoptosis. Recently, our study demonstrated that emodin
inhibited tumor angiogenesis in vitro and in implanted
pancreatic cancer tissues, decreased the expression of
angiogenesis-associated factors (NF-κB and its regulatory factors
VEGF, MMP-2, MMP-9 and eNOS) and reduced eNOS phosphorylation. In
addition, we found that emodin had no effect on VEGFR expression
in vivo(13). In summary,
these results suggest that emodin has potential antitumor effect on
pancreatic cancer via its dual role in the promotion of apoptosis
and suppression of angiogenesis, probably through regulation of the
expression of NF-κB, NF-κB-regulated apoptosis-associated and
angiogenesis-associated factors. However, more intense research at
the animal level and clinical trials are required to confirm the
clinical antitumor efficacy.
3. Effects of emodin on respiratory system
cancer and the possible mechanisms involved
The recombinant protein Rad51 is essential for
homologous recombination repair, and Rad51 overexpression is
resistant to DNA double-strand break-inducing cancer therapies.
Chen et al(45) reported
that emodin enhanced the cytotoxicity induced by gefitinib in two
non-small cancer lung cancer (NSCLC) cell lines, A549 and H1650.
Emodin at low doses of 2–10 μM enhanced a gefitinib-induced
decrease in phospho-ERK1/2 and Rad51 protein levels by enhancing
Rad51 protein instability. Expression of constitutively active
MKK1/2 vectors (MKK1/2-CA) significantly rescued the reduced
phospho-ERK1/2 and Rad51 protein levels as well as cell viability
following gefitinib and emodin cotreatment. Blockage of ERK1/2
activation by U0126 (an MKK1/2 inhibitor) lowered Rad51 protein
levels and reduced cell viability in emodin-treated H1650 and A549
cells. ROS have been implicated in the phosphorylation of p53 that
is mediated by protein kinases, including p38MAPK,
ataxia-telangiectasia mutated (ATM) and ERK (46). Lai et al(47) demonstrated that emodin induced
mitochondria-dependent apoptotic cell death in human lung
adenocarcinoma A549 cells by activating a ROS-activated ATM-p53-Bax
signaling pathway. ERCC1 and Rad51 proteins are essential for
nucleotide excision repair and homologous recombination,
respectively. Furthermore, ERCC1 and Rad51 overexpression induces
resistance to DNA-damaging agents that promote DNA double-strand
breaks. Both ERCC1 and Rad51 protein levels as well as mRNA levels
were decreased in 4 different NSCLC cell lines after exposure to
emodin. These decreased levels were correlated with the
inactivation of the MKK1/2-ERK1/2 pathway (48). The most important chemotherapeutic
agents for patients with advanced NSCLC are platinum-containing
compounds such as cisplatin or carboplatin. Resistance to
platinum-based drugs is one of the major obstacles to cancer
treatment, and is often related to poor prognosis in patients with
NSCLC. Emodin was found to synergistically increase the
cytotoxicity induced by cisplatin in human NSCLC cell lines
(49). Exposure of human NSCLC
cells to emodin decreased cisplatin-elicited ERK1/2 activation and
ERCC1 protein induction by increasing instability of ERCC1 protein.
Su et al(50) reported that
exposure of the human NSCLC H1703 or A549 cell lines to emodin
decreased the antitumor antibiotic mitomycin C (MMC)-elicited
phosphorylated ERK1/2 and Rad51 levels. Moreover, emodin
significantly decreased the MMC-elicited Rad51 mRNA and protein
levels by increasing the instability of Rad51 mRNA and protein.
Thymidine phosphorylase (TP) is the rate-limiting enzyme for the
activation of capecitabine (a pro-drug of fluorouracil), and is a
useful predictor of tumor response to capecitabine-based
chemotherapy. Overexpression of Rad51 and ERCC1 induces resistance
to chemotherapeutic agents. Ko et al(51) reported that emodin enhanced
capecitabine-induced cytotoxic effects through ERK1/2 inactivation
and decreased the Rad51 and ERCC1 protein levels induced by
capecitabine. Enhancement of ERK1/2 signaling by constitutively
active MKK1/2 (MKK1/2-CA) resulted in increasing Rad51 and ERCC1
protein levels and cell viability in NSCLC cell lines treated with
emodin and capecitabine. Interestingly, emodin enhanced TP mRNA and
protein expression in capecitabine-treated NSCLC cell lines, and
depletion of the TP expression decreased the cytotoxic effects
induced by capecitabine and emodin. These findings suggest that the
enhanced cytotoxicity to capecitabine by emodin was mediated by
downregulation of the expression of Rad51 and ERCC1 and
upregulation of TP expression. He et al(52) found that emodin exerted a
suppressive effect on the proliferation of NSCLC in a
concentration-dependent manner. Protein and mRNA expression of
ERCC1 and Rad51 was also significantly decreased with the dose.
Vacuolar degeneration was observed in A549 and SK-MES-1 cell lines
after emodin treatment by transmission electron microscopy. The
authors concluded that emodin may thus inhibit the cell
proliferation of NSCLC cells by downregulation of RCC1 and
Rad51.
4. Effects of emodin on reproductive system
cancer and the possible mechanisms involved
Breast cancer
Cheema et al(53) reported that the drug delivery and
efficacy of silk fibroin-coated liposomes (SF-ELP) encapsulating
the receptor tyrosine kinase inhibitor, emodin, inhibited both the
PI3K and MAPK pathways and reduced levels of phosphorylated
Her2/neu, which contribute to the enhanced growth inhibitory
effects of Her2/neu-overexpressing breast cancer cells. The present
study suggested that silk fibroin coating enhanced emodin delivery
in Her2/neu-overexpressing breast cancer cells thereby increasing
the overall efficacy of the drug. Huang et al(54) demonstrated that emodin induced
apoptosis through a decrease in the Bcl-2/Bax ratio and an increase
in cytoplasm cytochrome c concentration in human breast
cancer BCap-37 cells. Furthermore, Huang et al(55) demonstrated that emodin induced
alterations in gene expression profiling, but had no effects on
caspases. In addition, the p53 pathway may cooperate with the IGF-2
pathway, resulting in emodin-induced apoptosis through disruption
of the mitochondrial signaling pathway in BCap-37 cells. Wang et
al(56) prepared emodin-loaded
solid lipid nanoparticles (E-SLNs) by high pressure homogenization
(HPH) and evaluated their antitumor activity in vitro
against human breast cancer cell lines. The drug release of E-SLNs
in vitro lasted 72 h and exhibited a sustained profile,
which makes it a promising vehicle for oral drug delivery. Their
study showed that E-SLNs significantly enhanced the in vitro
cytotoxicity against human breast cancer cell lines MCF-7 and
MDA-MB-231 when compared to the emodin solution, while free emodin,
blank SLNs (B-SLNs) and E-SLNs all exhibited no significant
toxicity to human mammary epithelial MCF-10A cells. E-SLNs also
showed a highly significant cell cycle arrest effect in MCF-7 cells
compared to bulk emodin solution and induced higher apoptotic rates
in MCF-7 cells, indicating that cell cycle arrest and apoptosis may
be the underlying mechanism of the enhanced cytotoxicity. These
results suggest that the delivery of emodin by lipid nanoparticles
may be a promising approach for cancer therapy.
Cervical cancer
Treatment of eukaryotic cells with emodin, which is
described as an inhibitor of protein kinase CK2, was found to
induce apoptosis and the anti-apoptotic effect of CK2 is partially
mediated by targeting phosphorylation and upregulation of AKT by
CK2. Oslen et al(57)
further showed that the downregulation of AKT in the HeLa cell line
was partially due to the emodin-mediated target inhibition of
components of the PI3K pathway, which directly or indirectly affect
AKT activity. More studies are needed to demonstrate the effect of
emodin in cervical cancer.
Prostate cancer
Huang et al(58) found that emodin/cisplatin
co-treatment markedly elevated ROS levels and enhanced
chemosensitivity in DU-145 cells, when compared with cisplatin-only
treatment, but exerted little effect on non-tumor cells. The
mechanism may be associated with downregulation of MDR1 expression
and promotion of drug retention and suppression of transactivation
of HIF-1. Their results suggest that emodin may be recognized an an
effective adjunctive to improve the efficacy of cytotoxic drugs in
prostate cancer cells with over-activated HIF-1 and high MDR. Yu
et al(59) reported that
emodin caused a marked increase in apoptosis and a decrease in cell
proliferation in human prostate cancer LNCaP cells. The expression
of the androgen receptor (AR) and prostate-specific antigen (PSA)
was decreased and the expression of p53 and p21 was increased as
the emodin concentrations were increased. At the same time, emodin
induced apoptosis of LNCaP cells through the upregulation of
caspase-3 and -9, as well as the increase in the Bax/Bcl-2 ratio.
However, it did not involve modulation of Fas or caspase-8 protein
expression. This study concluded that emodin inhibits cell
proliferation by AR and p53-p21 pathways and induces apoptosis via
the mitochondrial pathway. Recent evidence indicates that the
CXCR4/CXCL12 axis is involved in promoting invasion and metastasis
in tumors. Thus, novel agents that downregulate CXCR4 expression
have therapeutic potential for suppressing cancer metastasis. Ok
et al(60) reported that
emodin downregulated the expression of both CXCR4 and HER2 without
affecting the cell viability of tumor cells. The suppression of
CXCR4 expression by emodin was found to correlate with the
inhibition of CXCL12-induced migration and invasion of both
prostate and lung cancer DU145 and A549 cells. Their findings
suggest that emodin is a novel blocker of CXCR4 expression and,
thus, has enormous potential as a powerful therapeutic agent for
metastatic cancer.
Ovarian cancer
The onset of drug resistance is a major impediment
toward the successful treatment of ovarian cancer. Li et
al(61) showed that emodin
alone induced apoptosis at a high concentration and increased
paclitaxel-induced apoptosis at a low concentration. It enhanced
the sensitivity of A2780/taxol cells to paclitaxel along with
downregulation of P-glycoprotein, XIAP and survivin. Their results
demonstrated a dual role for emodin in the inhibition of
drug-resistant ovarian tumor growth by increasing paclitaxel
cellular concentrations and re-sensitizing resistant cells to
paclitaxel. Their results suggest the possibility of an innovative
chemotherapeutic strategy that uses emodin in combination with
paclitaxel to increase the sensitivity of tumor cells. Human
ovarian A2780 cells were exposed first to emodin or DRB and then to
cisplatin alone, and the cytotoxic effects were monitored. The
cellular effects of cisplatin were significantly enhanced, whereas
simultaneous exposure did not enhance the cytotoxicity. The
increase in activity of cisplatin in the sequenced schedule was not
due to increases in intracellular levels of cisplatin or DNA
adducts, whereas the cytotoxic inhibition was related to a
significant decline in both intracellular platinum levels and DNA
adducts, which were ascribed to inhibition of cisplatin uptake.
Emodin in combination with cisplatin inhibits platinum drug uptake
by impacting the hCtr1 transporter thereby reducing the
cytotoxicity of cisplatin (62).
5. Effects of emodin on blood system cancer
and the possible mechanisms involved
Muto et al(7)
demonstrated that emodin significantly induced cytotoxicity in
human myeloma cells through the elimination of myeloid cell
leukemia 1 (Mcl-1). Emodin inhibited interleukin-6-induced
activation of Janus-activated kinase 2 (JAK2) and phosphorylation
of signal transducer and activator of transcription 3 (STAT3),
followed by the decreased expression of Mcl-1. Activation of
caspase-3 and caspase-9 was triggered by emodin, while the
expression of other antiapoptotic Bcl-2 family members, except
Mcl-1, were not altered in the presence of emodin. Induction of
apoptosis by emodin was almost abrogated in Mcl-1-overexpressing
myeloma cells similar to the level in parental cells, which were
not treated with emodin, suggesting the importance of Mcl-1 in
emodin-induced apoptosis. The use of arsenic trioxide
(As2O3) has been shown to effectively treat
acute promyelocytic leukemia (APL) with >80% of patients
achieving complete remission. The combination of arsenic and
interferon has also shown promising results in the treatment of
adult T-cell leukemia (ATL). The requirement for slow dose
increases of arsenic and the time required to achieve a
pharmacologic active dose in patients are major obstacles as the
median survival of patients with ATL is approximately 6 months.
Brown et al(63) reported a
potent synergistic effect of the combination of arsenic trioxide
and interferon α (As/IFN-α) with emodin on cell-cycle arrest and
cell death of HTLV-I-infected cells via the generation of ROS and
inhibition of Akt and AP-1. Importantly, clinically achievable
doses of emodin allowed for reduced arsenic concentrations by
100-fold while still achieving a high toxic effect on tumor cells.
Wang et al(64) reported
that emodin treatment may result in apoptosis of human chronic
myelocytic leukemia K562 cells. Both in vitro and in
vivo, the apoptosis-related protein Bcl-2 was decreased and the
Bax was increased after emodin treatment. Moreover, activation of
caspase-3, -8 and -9 by emodin has been demonstrated.
6. Conclusions
Overall, based on analyses of data from in
vitro or in vivo laboratory experimental models, we
suggest that emodin is a potent anticancer agent for cancer
treatment particularly in digestive system cancers such as
pancreatic cancer by regulating multi-molecular targets involved in
tumor growth, invasion, angiogenesis and metastasis. Studies in
recent years indicate that emodin has an anticancer effect in
several types of cancer, yet the mechanism of action in each type
of cancer appears to vary. Promoting apoptosis through the
mitochondrial pathway may be a similar antitumor mechanism of
emodin in different types of cancer from different tissues. This
review provides information regarding the promising anticancer
actions of emodin in cancer treatment and aimed to broaden the
therapeutic potential to cancer patients in the future. However,
despite the encouraging results summarized here, further studies
concerning the antitumor mechanisms of emodin and clinical trials
are warranted and required to confirm the clinical antitumor
efficacy before the possibility of large-scale clinical
application.
Acknowledgements
We are grateful for the funding support from the
Administration of Traditional Chinese Medicine of Zhengjing
Province, China (grant no. 2011ZZ010), the Zhejiang Provincial
Science Fund for Distinguished Young Scholars (grant no.
LR12H280001) and the National Natural Science Foundation of China
(grant no. 81173606).
References
|
1
|
Chen YC, Shen SC, Lee WR, et al: Emodin
induces apoptosis in human promyeloleukemic HL-60 cells accompanied
by activation of caspase 3 cascade but independent of reactive
oxygen species production. Biochem Pharmacol. 64:1713–1724. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Su YT, Chang HL, Shyue SK and Hsu SL:
Emodin induces apoptosis in human lung adenocarcinoma cells through
a reactive oxygen species-dependent mitochondrial signaling
pathway. Biochem Pharmacol. 70:229–241. 2005. View Article : Google Scholar
|
|
3
|
Srinivas G, Anto RJ, Srinivas P,
Vidhyalakshmi S, Senan VP and Karunagaran D: Emodin induces
apoptosis of human cervical cancer cells through poly(ADP-ribose)
polymerase cleavage and activation of caspase-9. Eur J Pharmacol.
473:117–125. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang Q, Shen HM, Shui G, Wenk MR and Ong
CN: Emodin inhibits tumor cell adhesion through disruption of the
membrane lipid Raft-associated integrin signaling pathway. Cancer
Res. 66:5807–5815. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kwak HJ, Park MJ, Park CM, et al: Emodin
inhibits vascular endothelial growth factor-A-induced angiogenesis
by blocking receptor-2 (KDR/Flk-1) phosphorylation. Int J Cancer.
118:2711–2720. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cha TL, Qiu L, Chen CT, Wen Y and Hung MC:
Emodin down-regulates androgen receptor and inhibits prostate
cancer cell growth. Cancer Res. 65:2287–2295. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Muto A, Hori M, Sasaki Y, et al: Emodin
has a cytotoxic activity against human multiple myeloma as a
Janus-activated kinase 2 inhibitor. Mol Cancer Ther. 6:987–994.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim MS, Park MJ, Kim SJ, et al: Emodin
suppresses hyaluronic acid-induced MMP-9 secretion and invasion of
glioma cells. Int J Oncol. 27:839–846. 2005.PubMed/NCBI
|
|
9
|
Bader Y, Hartmann J, Horvath Z, et al:
Synergistic effects of deuterium oxide and gemcitabine in human
pancreatic cancer cell lines. Cancer Lett. 259:231–239. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakagawa T, Shimizu M, Shirakami Y, et al:
Synergistic effects of acyclic retinoid and gemcitabine on growth
inhibition in pancreatic cancer cells. Cancer Lett. 273:250–256.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee SH, Ryu JK, Lee KY, et al: Enhanced
anti-tumor effect of combination therapy with gemcitabine and
apigenin in pancreatic cancer. Cancer Lett. 259:39–49. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang SJ, Gao Y, Chen H, et al:
Dihydroartemisinin inactivates NF-κB and potentiates the anti-tumor
effect of gemcitabine on pancreatic cancer both in vitro and in
vivo. Cancer Lett. 293:99–108. 2010.PubMed/NCBI
|
|
13
|
Lin SZ, Wei WT, Chen H, et al: Antitumor
activity of emodin against pancreatic cancer depends on its dual
role: promotion of apoptosis and suppression of angiogenesis. PLoS
One. 7:e421462012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin SY, Lai WW, Ho CC, et al: Emodin
induces apoptosis of human tongue squamous cancer SCC-4 cells
through reactive oxygen species and mitochondria-dependent
pathways. Anticancer Res. 29:327–335. 2009.
|
|
15
|
Chen YY, Chiang SY, Lin JG, et al: Emodin,
aloe-emodin and rhein induced DNA damage and inhibited DNA repair
gene expression in SCC-4 human tongue cancer cells. Anticancer Res.
30:945–951. 2010.PubMed/NCBI
|
|
16
|
Chen YY, Chiang SY, Lin JG, et al: Emodin,
aloe-emodin and rhein inhibit migration and invasion in human
tongue cancer SCC-4 cells through the inhibition of gene expression
of matrix metalloproteinase-9. Int J Oncol. 36:1113–1120.
2010.PubMed/NCBI
|
|
17
|
Cai J, Niu X, Chen Y, et al:
Emodin-induced generation of reactive oxygen species inhibits RhoA
activation to sensitize gastric carcinoma cells to anoikis.
Neoplasia. 10:41–51. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun ZH and Bu P: Downregulation of
phosphatase of regenerating liver-3 is involved in the inhibition
of proliferation and apoptosis induced by emodin in the SGC-7901
human gastric carcinoma cell line. Exp Ther Med. 3:1077–1081.
2012.PubMed/NCBI
|
|
19
|
Wang QJ, Cai XB, Liu MH, Hu H, Tan XJ and
Jing XB: Apoptosis induced by emodin is associated with alterations
of intracellular acidification and reactive oxygen species in
EC-109 cells. Biochem Cell Biol. 88:767–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsu CM, Hsu YA, Tsai Y, et al: Emodin
inhibits the growth of hepatoma cells: finding the common
anti-cancer pathway using Huh7, Hep3B, and HepG2 cells. Biochem
Biophys Res Commun. 392:473–478. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu JQ, Bao W and Lei JC: Emodin regulates
apoptotic pathway in human liver cancer cells. Phytother Res.
27:251–257. 2012.PubMed/NCBI
|
|
22
|
Kim MJ, Oh DY, Lee SH, et al:
Gemcitabine-based versus fluoropyrimidine-based chemotherapy with
or without platinum in unresectable biliary tract cancer: a
retrospective study. BMC Cancer. 8:3742008. View Article : Google Scholar
|
|
23
|
Wang W, Sun YP, Huang XZ, et al: Emodin
enhances sensitivity of gallbladder cancer cells to platinum drugs
via glutathion depletion and MRP1 downregulation. Biochem
Pharmacol. 79:1134–1140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: a radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang J and Yi J: Cancer cell killing via
ROS: to increase or decrease, that is the question. Cancer Biol
Ther. 7:1875–1884. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang W, Sun Y, Li X, et al: Emodin
potentiates the anticancer effect of cisplatin on gallbladder
cancer cells through the generation of reactive oxygen species and
the inhibition of survivin expression. Oncol Rep. 26:1143–1148.
2011.PubMed/NCBI
|
|
27
|
Moserle L, Ghisi M, Amadori A and
Indraccolo S: Side population and cancer stem cells: therapeutic
implications. Cancer Lett. 288:1–9. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu C and Alman BA: Side population cells
in human cancers. Cancer Lett. 268:1–9. 2008. View Article : Google Scholar
|
|
29
|
Tabor MH, Clay MR, Owen JH, et al: Head
and neck cancer stem cells: the side population. Laryngoscope.
121:527–533. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li XX, Wang J, Wang HL, et al:
Characterization of cancer stem-like cells derived from a side
population of a human gallbladder carcinoma cell line, SGC-996.
Biochem Biophys Res Commun. 419:728–734. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li XX, Ding Y, Wang W, et al: Emodin as an
effective agent in targeting cancer stem-like side population cells
of gallbladder carcinoma. Stem Cells Dev. 22:554–566.
2012.PubMed/NCBI
|
|
32
|
Lu Y, Zhang J and Qian J: The effect of
emodin on VEGF receptors in human colon cancer cells. Cancer
Biother Radiopharm. 23:222–228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma YS, Weng SW, Lin MW, et al: Antitumor
effects of emodin on LS1034 human colon cancer cells in vitro and
in vivo: roles of apoptotic cell death and LS1034 tumor xenografts
model. Food Chem Toxicol. 50:1271–1278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Saha S, Bardelli A, Buckhaults P, et al: A
phosphatase associated with metastasis of colorectal cancer.
Science. 294:1343–1346. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ooki A, Yamashita K, Kikuchi S, Sakuramoto
S, Katada N and Watanabe M: Phosphatase of regenerating liver-3 as
a convergent therapeutic target for lymph node metastasis in
esophageal squamous cell carcinoma. Int J Cancer. 127:543–554.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han YM, Lee SK, Jeong DG, et al: Emodin
inhibits migration and invasion of DLD-1 (PRL-3) cells via
inhibition of PRL-3 phosphatase activity. Bioorg Med Chem Lett.
22:323–326. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Damodharan U, Ganesan R and Radhakrishnan
UC: Expression of MMP2 and MMP9 (gelatinases A and B) in human
colon cancer cells. Appl Biochem Biotechnol. 165:1245–1252. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo Q, Chen Y, Zhang B, Kang M, Xie Q and
Wu Y: Potentiation of the effect of gemcitabine by emodin in
pancreatic cancer is associated with survivin inhibition. Biochem
Pharmacol. 77:1674–1683. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen H, Wei W, Guo Y, et al: Enhanced
effect of gemcitabine by emodin against pancreatic cancer in
vivo via cytochrome C-regulated apoptosis. Oncol Rep.
25:1253–1261. 2011.PubMed/NCBI
|
|
40
|
Liu JX, Zhang JH, Li HH, et al: Emodin
induces Panc-1 cell apoptosis via declining the mitochondrial
membrane potential. Oncol Rep. 28:1991–1996. 2012.PubMed/NCBI
|
|
41
|
Liu A, Chen H, Wei W, et al:
Antiproliferative and antimetastatic effects of emodin on human
pancreatic cancer. Oncol Rep. 26:81–89. 2011.PubMed/NCBI
|
|
42
|
Wei WI, Chen H, Ni ZL, et al: Antitumor
and apoptosis-promoting properties of emodin, an anthraquinone
derivative from Rheum officinale Baill, against pancreatic
cancer in mice via inhibition of Akt activation. Int J Oncol.
39:1381–1390. 2011.PubMed/NCBI
|
|
43
|
Liu DL, Bu H, Li H, et al: Emodin reverses
gemcitabine resistance in pancreatic cancer cells via the
mitochondrial apoptosis pathway in vitro. Int J Oncol.
40:1049–1057. 2012.PubMed/NCBI
|
|
44
|
Guo HC, Bu HQ, Luo J, et al: Emodin
potentiates the antitumor effects of gemcitabine in PANC-1
pancreatic cancer xenograft model in vivo via inhibition of
inhibitors of apoptosis. Int J Oncol. 40:1849–1857. 2012.PubMed/NCBI
|
|
45
|
Chen RS, Jhan JY, Su YJ, et al: Emodin
enhances gefitinib-induced cytotoxicity via Rad51 downregulation
and ERK1/2 inactivation. Exp Cell Res. 315:2658–2672. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu B, Chen Y and St Clair DK: ROS and
p53: a versatile partnership. Free Radic Biol Med. 44:1529–1535.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lai JM, Chang JT, Wen CL and Hsu SL:
Emodin induces a reactive oxygen species-dependent and ATM-p53-Bax
mediated cytotoxicity in lung cancer cells. Eur J Pharmacol.
623:1–9. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ko JC, Su YJ, Lin ST, et al: Suppression
of ERCC1 and Rad51 expression through ERK1/2 inactivation is
essential in emodin-mediated cytotoxicity in human non-small cell
lung cancer cells. Biochem Pharmacol. 79:655–664. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ko JC, Su YJ, Lin ST, et al: Emodin
enhances cisplatin-induced cytotoxicity via down-regulation of
ERCC1 and inactivation of ERK1/2. Lung Cancer. 69:155–164. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Su YJ, Tsai MS, Kuo YH, et al: Role of
Rad51 down-regulation and extracellular signal-regulated kinases 1
and 2 inactivation in emodin and mitomycin C-induced synergistic
cytotoxicity in human non-small-cell lung cancer cells. Mol
Pharmacol. 77:633–643. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ko JC, Tsai MS, Kuo YH, et al: Modulation
of Rad51, ERCC1, and thymidine phosphorylase by emodin result in
synergistic cytotoxic effect in combination with capecitabine.
Biochem Pharmacol. 81:680–690. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
He L, Bi JJ, Guo Q, Yu Y and Ye XF:
Effects of emodin extracted from Chinese herbs on proliferation of
non-small cell lung cancer and underlying mechanisms. Asian Pac J
Cancer Prev. 13:1505–1510. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cheema SK, Gobin AS, Rhea R,
Lopez-Berestein G, Newman RA and Mathur AB: Silk fibroin mediated
delivery of liposomal emodin to breast cancer cells. Int J Pharm.
341:221–229. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Huang Z, Chen G and Shi P: Emodin-induced
apoptosis in human breast cancer BCap-37 cells through the
mitochondrial signaling pathway. Arch Pharm Res. 31:742–748. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Huang Z, Chen G and Shi P: Effects of
emodin on the gene expression profiling of human breast carcinoma
cells. Cancer Detect Prev. 32:286–291. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang S, Chen T, Chen R, Hu Y, Chen M and
Wang Y: Emodin loaded solid lipid nanoparticles: preparation,
characterization and antitumor activity studies. Int J Pharm.
430:238–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Olsen BB, Bjorling-Poulsen M and Guerra B:
Emodin negatively affects the phosphoinositide 3-kinase/AKT
signalling pathway: a study on its mechanism of action. Int J
Biochem Cell Biol. 39:227–237. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Huang XZ, Wang J, Huang C, et al: Emodin
enhances cytotoxicity of chemotherapeutic drugs in prostate cancer
cells: the mechanisms involve ROS-mediated suppression of multidrug
resistance and hypoxia inducible factor-1. Cancer Biol Ther.
7:468–475. 2008. View Article : Google Scholar
|
|
59
|
Yu CX, Zhang XQ, Kang LD, et al: Emodin
induces apoptosis in human prostate cancer cell LNCaP. Asian J
Androl. 10:625–634. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ok S, Kim SM, Kim C, et al: Emodin
inhibits invasion and migration of prostate and lung cancer cells
by downregulating the expression of chemokine receptor CXCR4.
Immunopharmacol Immunotoxicol. 4:768–778. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li J, Liu P, Mao H, Wanga A and Zhang X:
Emodin sensitizes paclitaxel-resistant human ovarian cancer cells
to paclitaxel-induced apoptosis in vitro. Oncol Rep.
21:1605–1610. 2009.PubMed/NCBI
|
|
62
|
Kurokawa T, He G and Siddik ZH: Protein
kinase inhibitors emodin and dichloro-ribofuranosylbenzimidazole
modulate the cellular accumulation and cytotoxicity of cisplatin in
a schedule-dependent manner. Cancer Chemother Pharmacol.
65:427–436. 2010. View Article : Google Scholar
|
|
63
|
Brown M, Bellon M and Nicot C: Emodin and
DHA potently increase arsenic trioxide interferon-α-induced cell
death of HTLV-I-transformed cells by generation of reactive oxygen
species and inhibition of Akt and AP-1. Blood. 109:1653–1659.
2007.PubMed/NCBI
|
|
64
|
Wang C-G, Yang J-Q, Liu B-Z, et al:
Anti-tumor activity of emodin against human chronic myelocytic
leukemia K562 cell lines in vitro and in vivo. Eur J Pharmacol.
627:33–41. 2010. View Article : Google Scholar : PubMed/NCBI
|