Introduction
Laryngeal squamous cell carcinoma (LSCC), arises
from the larynx epithelium and is one of the most common head and
neck squamous cell carcinomas (HNSCCs) with a high rate of
metastasis and a poor prognosis (1,2).
Currently, LSCC accounts for almost 2% of all malignancies
worldwide (3,4). Although recent treatment outcomes are
improved, patients still have a poor prognosis with a 5-year
survival rate of ~60% (5). The main
cause of death is cancer metastasis (6) and the intrinsic microenvironment of
tumor cells has an important role in tumor metastasis (7). Therefore, a better understanding of
the underlying molecular metastasis-related mechanisms of LSCC is
essential for developing effective therapeutic strategies to
improve the survival rate and quality of life of these
patients.
Aurora kinase A (AURKA), a homologue of
aurora/Ipl1-related kinase, is located at chromosome 20q13.2
(8). It is an oncogene that causes
various epithelial malignant tumors, including pancreatic (9), ovarian (10), colorectal (11) and prostate cancer (12). We previously reported that the
expression of AURKA was elevated in human LSCC and promoted the
metastasis of LSCC (13,14). However, the underlying mechanism by
which AURKA enhances the metastasis of LSCC is still poorly
understood.
Recent accumulating evidence indicates that the
occurrence of epithelial-mesenchymal transition (EMT) may be the
principal mechanism of cancer metastasis, including bladder
(15), colorectal (16), pancreatic (17), breast (18), oral (19), thyroid (20) and prostate cancer (21), and LSCC (22). EMT is the process by which
epithelial cells de-differentiate into mesenchymal-like derivatives
(23). During the process,
epithelial cells acquire the characteristics of mesenchymal cells,
such as lack of polarization, increased motility and invasion,
decreased cell-cell junctions and chemotherapeutic resistance
(24–26). Recently, D'Assoro et al
reported that AURKA induced EMT to promote distant metastasis in
breast cancer (27). This indicates
an association between AURKA and EMT and cancer metastasis.
Therefore, in the present study, we investigated
whether AURKA is involved in EMT to promote the metastasis of LSCC.
The epithelial-related protein E-cadherin and the
mesenchymal-related proteins, N-cadherin, vimentin and slug, were
investigated either in AURKA-downregulated or parental Hep2 cells.
Effects of inhibition of the FAK/PI3K pathway on the
mesenchymal-like characteristics and cellular mobility, migration
and invasion ability were also studied in these Hep2 cells.
Materials and methods
Cell cultures
The human laryngeal cancer Hep2 cell line (Hep2
cells) was obtained from Shanghai Institutes of Biological
Sciences, Chinese Academy of Sciences. Hep2 cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine
serum (FBS) (both from Gibco, Bedford, MA, USA) and 1%
penicillin/streptomycin at 37°C under 5% CO2.
Cell proliferation assay
Cell proliferation was measured with the CCK-8 assay
kit (Gibco). Briefly, a total density of 3×103 Hep2
cells/well, growing in logarithmic phase, in 100 µl of DMEM
were plated into the 96-well plates. Then, 10 µl of CCK-8
was added into every well at the specific time points. After
culture for 2 h at 37°C under 5% CO2, OD450
absorbance values were monitored. In the present study, the
proliferation of T-Hep2 cells following treatment with the
selective inhibitor of AURKA (VX680) (Selleck Chemicals, Houston,
TX, USA), which was dissolved in dimethyl sulfoxide (DMSO), was
measured at 0, 24 and 48 h for 2 days.
Plate colony-formation assay
Hep2 cells treated with VX680 for 48 h were the
experimental group and the cells treated with DMSO or without any
treatment were considered the controls. Cells at a density of
2×103 were layered into 6-well plates. After culture for
2 weeks with DMEM containing 10% FBS, the cells were washed with
phosphate-buffered saline (PBS) 3 times and fixed with crystal
violet for 30 min at room temperature. The formed colonies were
counted in every well.
Immunofluorescence staining
Hep2 cells treated with VX680 for 48 h or without
any treatment, at the density of 5×104, were seeded into
Millicell® EZ Slides (Millipore, Bedford, MA, USA). The
cells were fixed with 4% paraformaldehyde (PFA) for 30 min when
they adhered to the wall of the slides. Then, the slides were
rinsed 3 times with 1X PBS for 5 min each time, followed by
blocking with primary cocktail (Cell Signaling Technology, Danvers,
MA, USA) (diluted 1:100 as indicated on the datasheet in antibody
dilution buffer) to examine the expression of EMT markers, vimentin
and E-cadherin. Following overnight incubation at 4°C, the blocking
solution was aspirated and the slides were washed 3 times with 1X
PBS for 5 min each time and were then incubated with Alexa
Fluor® 555 and Alexa Flour® 488 (Cell
Signaling Technology) (diluted 1:100 as indicated on the datasheet
in antibody dilution buffer) for 1 h at room temperature in the
dark. Then, the slides were rinsed 3 times with 1X PBS for 5 min
each. For immunofluorescence staining of p-AURKA (anti-p-AURKA;
1:100; Cell Signaling Technology), blocking with 5% BSA in PBS
containing 0.05% Triton for 1 h at room temperature was the only
difference in the protocol. Vimentin and E-cadherin were analyzed
using fluorescence microscopy magnification, ×10 (U-ULS100HG;
Olympus Optical Co., Ltd., Tokyo, Japan) (magnification, ×10).
Western blot analysis
Hep2 cells, treated with VX680, the inhibitor of FAK
(TAE226), and the inhibitor of PI3K (omipalisib) (both from Selleck
Chemicals), respectively, were lysed with RIPA buffer containing 1%
protease inhibitor cocktail (100:1) and quantified by the BCA
protein assay kit (all from Pierce, Rockford, IL, USA) according to
the manufacturer's instructions. Harvested proteins were separated
with 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) for 2 h and transferred onto polyvinylidene difluoride
(PVDF) membranes (Millipore) for 2 h. Then, the membranes were
blocked with 5% non-fat milk dissolved in 1X TBST (150 mM NaCl,
0.05% Tween-20, 10 mM Tris-HCl, pH 8.0) for 2 h, and incubated with
the primary antibodies at 4°C overnight including anti-E-cadherin
(1:2,000), anti-N-cadherin (1:2,000), anti-vimentin (1:2,000),
anti-slug (1:2,000) (all from Proteintech Group, Inc., Rosemont,
IL, USA), anti-p-AURKA (1:2,000), anti-AURKA (1:2,000), anti-FAK
(1:2,000), anti-p-FAK (Tyr397, 1:2,000) (all from Cell Signaling
Technology), anti-PI3K (1:2,000), anti-p-PI3K (1:2,000) and GAPDH
(1:5,000) (all from Abcam). After overnight incubation, the PVDF
membranes were washed with 1X TBST 3 times and incubated with a
secondary antibody (1:5,000; Cell Signaling Technology) for 2 h at
room temperature. Lastly, the membranes were detected with an
enhanced chemiluminescence detection system (Amersham Biosciences,
Piscataway, NJ, USA).
Wound healing assay
Hep2 cells treated with TAE226 and omipalisib were
considered the experimental groups, and groups treated with DMSO or
without any treatment were the control groups. Hep2 cells at a
density of 1×106 cell/well were seeded into 6-well
plates. After overnight culture, the Hep2 cells were scratched with
20-µl pipette tips to form straight lines. The floating
cells were removed using 1X PBS 3 times. Finally, the wound lines
were photographed at 0, 24 and 48 h at a magnification of ×2.
Cell migration and invasion assays
A density of 2×105 cell/well treated with
TAE226, omipalisib, DMSO or without any treatment suspended in 200
µl of serum-free DMEM were placed into the top of Transwell
chambers (Corning, Tewksbury, MA, USA) and the lower chambers were
filled with 600 µl of DMEM with 10% FBS. After overnight
culture, non-migratory Hep2 cells were removed with a cotton swab
from the top surface of the chamber and the lower filter of the
chamber was stained with crystal violet solution for 30 min at
37°C. Then, the migratory Hep2 cells were photographed using
microscopy at magnification, ×20. Briefly, the experimental
produces of the cell invasion and the cell migration assays were
identical except that BD Matrigel™ (Becton-Dickinson Labware,
Bedford, MA, USA) covered the membrane of the top chamber.
Statistical analysis
Data of all experiments were analyzed by GraphPad
Prism 6 software (GraphPad, San Diego, CA, USA) and are shown as
mean ± SD. The Student's t-test was performed to assess the
differences between experimental groups and controls. The level of
significance was set at P<0.05, and a high level of significance
was set at P<0.01.
Results
Inhibition of AURKA with VX680 reduces
the proliferation of Hep2 cells
Our previous study showed that AURKA was
overexpressed in Hep2 cells and promoted the metastasis of LSCC
(13,14). We downregulated AURKA with VX680 (80
nm/ml) (28), a selective inhibitor
of AURKA, in the Hep2 cells for 24 and 48 h (Hep2/VX680). Hep2
cells treated with DMSO (Hep2/ctrl) and without any treatment
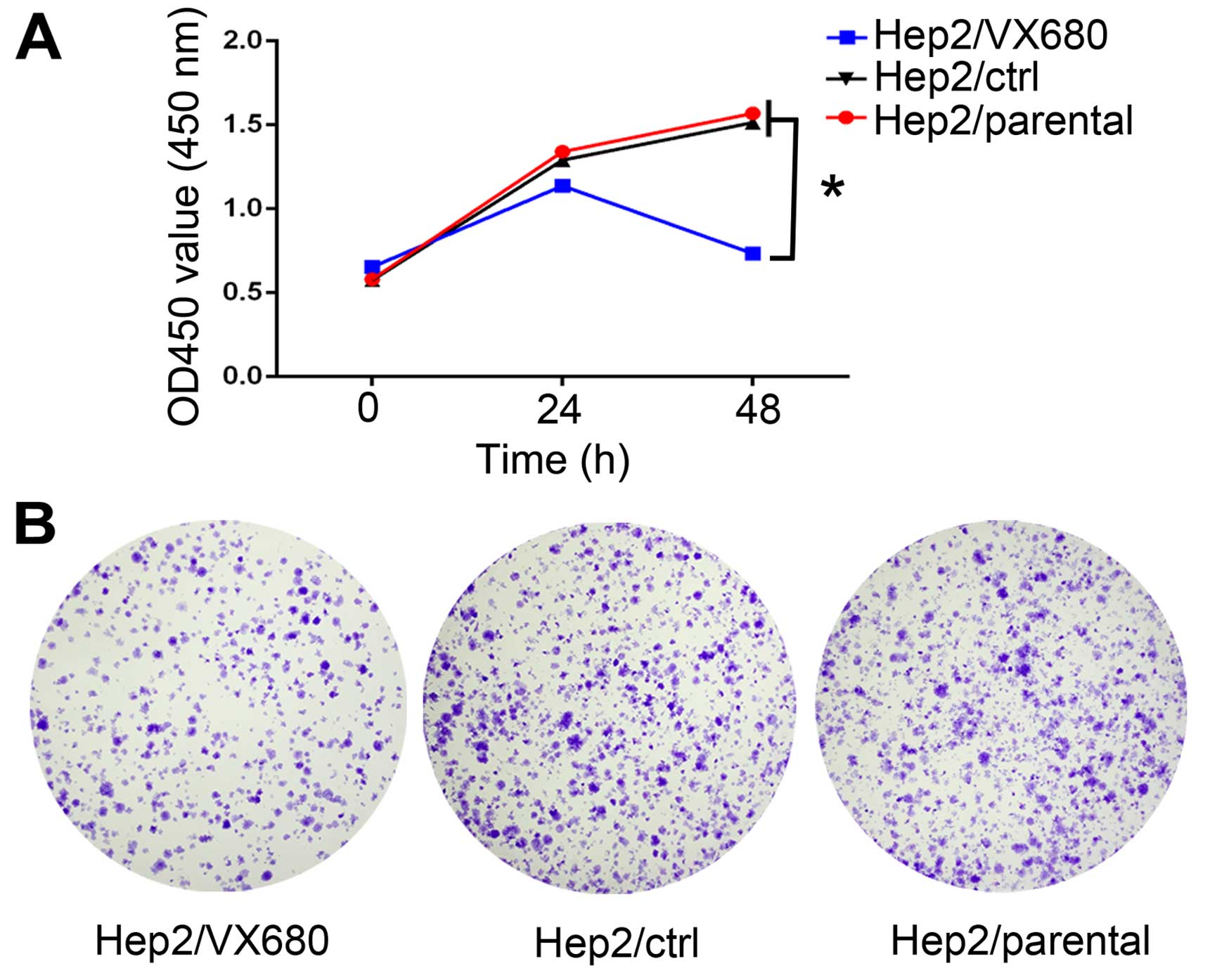
(Hep2/parental) were the control groups. The results of the CCK-8
assay showed less proliferation in the Hep2/VX680 cells compared
with the Hep2/ctrl and Hep2/parental cells (P<0.05; Fig. 1A). The plate colony-formation assay
showed parallel results in the Hep2/VX680 (404±18.95), Hep2/ctrl
(601±16.74) and Hep2/parental cells (607±24.11) (P<0.01;
Fig. 1B and C). These results
indicated that AURKA significantly promoted cell proliferation and
that VX680 effectively reduced the function of AURKA.
Downregulation of AURKA causes less
mesenchymal-like characteristics in Hep2 cells
To investigate whether AURKA is involved in EMT to
promote the metastasis of LSCC, the EMT-related proteins
E-cadherin, N-cadherin, vimentin and slug were assessed in the Hep2
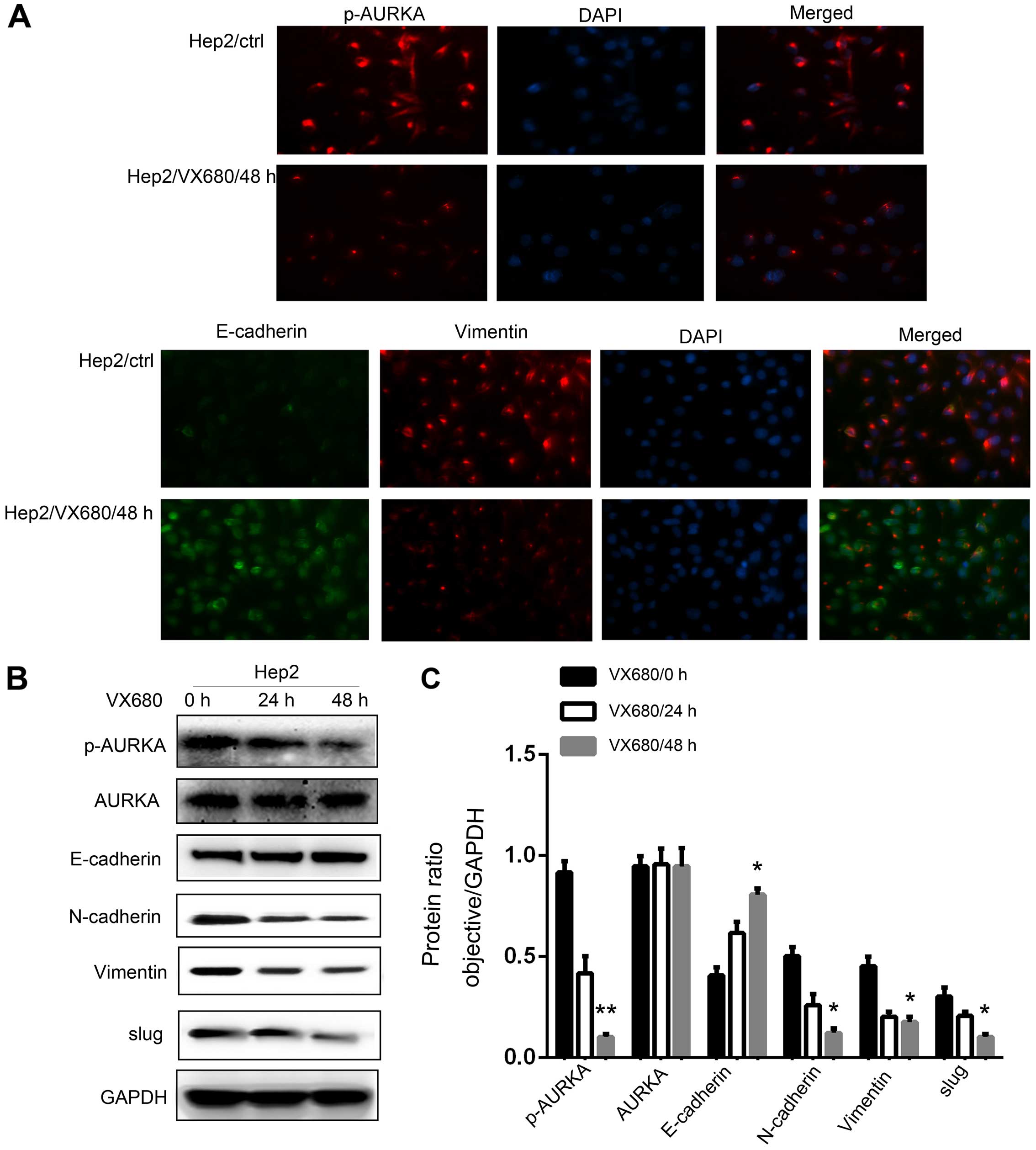
cells treated with VX680 at specific times. The results of
immunofluorescence staining showed that the expression of p-AURKA
and vimentin in the Hep2 cells was lower, while the expression of
E-cadherin was higher when compared with these levels in the
control group (Fig. 2A).
Subsequently, the results of western blotting also revealed that
the expression of p-AURKA was decreased almost 3-fold (P<0.01;
Fig. 2B and C) and the EMT-related
protein levels were altered in a time-dependent manner along with
the alteration of AURKA. Expression of E-cadherin was increased and
expression of N-cadherin, vimentin and slug was decreased (Fig. 2B and C). All of the aforementioned
observations indicated that AURKA may induce EMT to promote the
metastasis of LSCC.
AURKA promotes the metastasis of LSCC by
enhancing phosphorylation of the FAK/PI3K pathway
Given that the FAK pathway is regarded as a
regulatory factor in tumor metastasis (29–31)
and FAK is involved in PI3K-promoted EMT in lung cancer (32), we aimed to demonstrate that AURKA
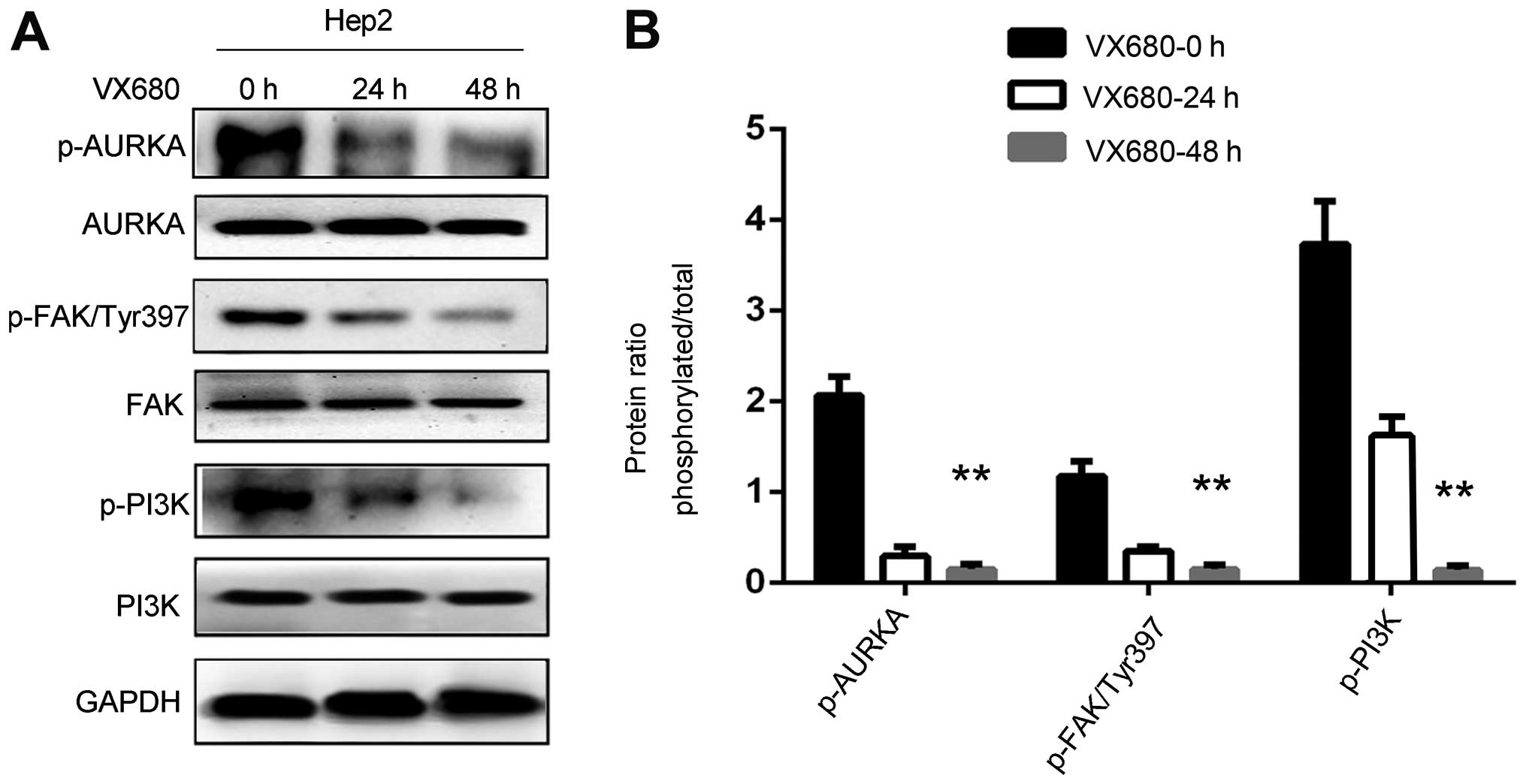
regulates those factors to induce EMT. As expected, the expression
of p-FAK (Tyr397) and p-PI3K was reduced following the diminution
of p-AURKA in the Hep2 cells (P<0.01; Fig. 3A and B), which demonstrated that FAK
and PI3K were regulated by AURKA and played a key role in
contributing to the tumorigenesis of LSCC.
Inhibition of the FAK/PI3K pathway causes
less mesenchymal-like characteristics
To explore whether the FAK/PI3K pathway plays a key
role in the induction of EMT in LSCC, we utilized the FAK inhibitor
TAE226 (2.1 µM/ml) (33),
and PI3K inhibitor omipalisib (500 nm/ml) (34) dissolved in DMSO. Treatment with
TAE226 altered the EMT-related proteins in a time-dependent manner.
The level of E-cadherin was increased, while N-cadherin, vimentin
and slug were decreased, indicating that FAK caused more
mesenchymal-like characteristics (Fig.
4A and B). p-AURKA, AURKA, p-FAK (Tyr397), FAK, p-PI3K and PI3K
in the Hep2/TAE226 cells were also examined. The expression of
p-FAK (Tyr397) was nearly decreased 2-fold after treatment with
TAE226 for 24 h (P<0.05; Fig. 4C and
D). In addition, the expression of p-PI3K was decreased
(P<0.01; Fig. 4C and D), whereas
p-AURKA, AURKA, FAK and PI3K were not changed, indicating that FAK
is a downstream factor of AURKA.
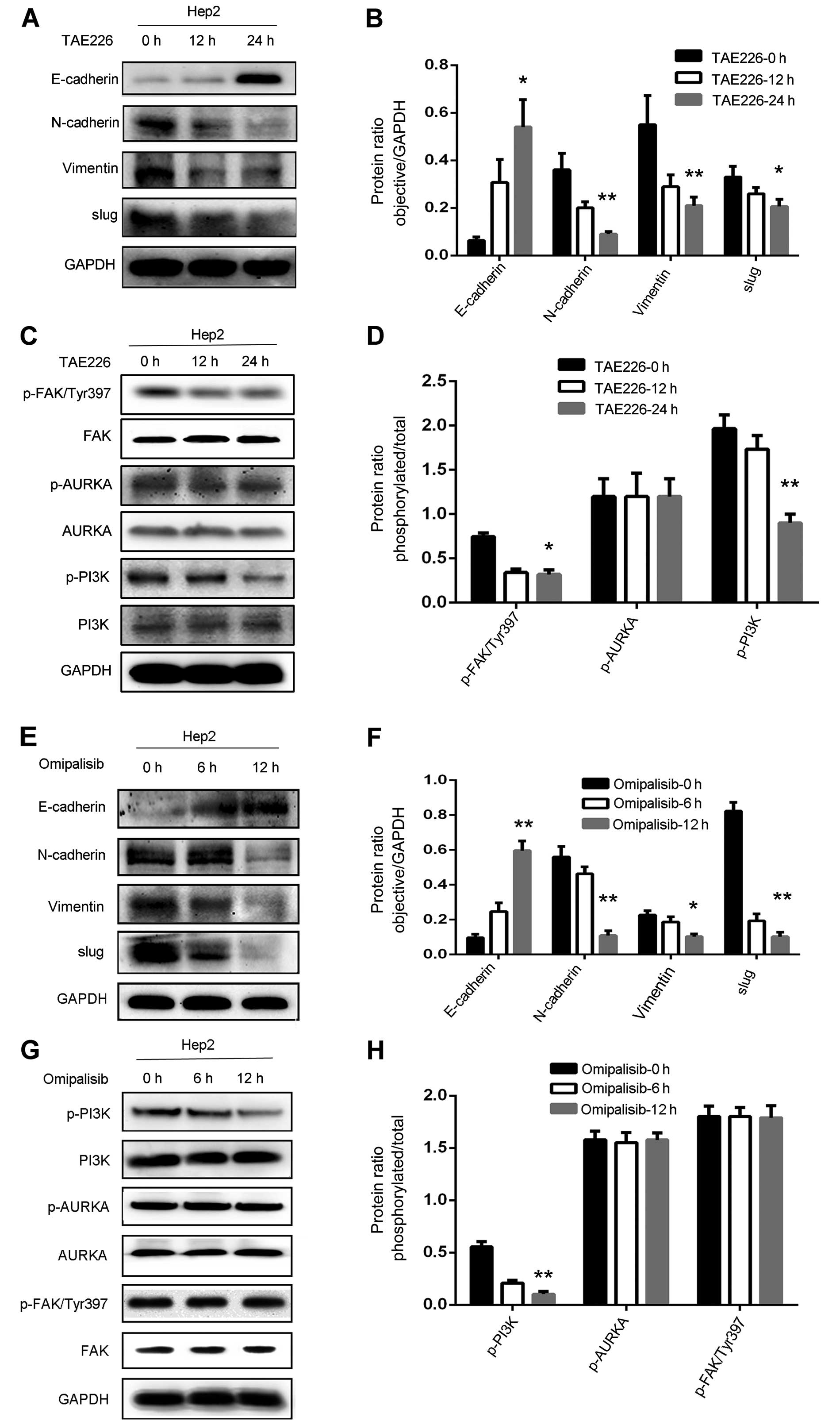 | Figure 4Effects of the inhibition of FAK/PI3K
using TAE226 or omipalisib in Hep2 cells. (A) Effects of TAE226 on
the expression of EMT-related proteins were analyzed by western
blotting. The expression of E-cadherin was increased, whereas
N-cadherin, vimentin and slug were decreased. (B) Quantification of
EMT-related protein ratio after treatment with TAE226
(*P<0.05, **P<0.01). (C) Effects of
TAE226 on the expression of p-AURKA, AURKA, p-FAK (Tyr397), FAK,
p-PI3K and PI3K were analyzed by western blotting. Expression of
p-FAK (Tyr397) and p-PI3K was decreased, while the others were not
changed. (D) Quantification of the protein ratio after treatment
with TAE226 (*P<0.05, **P<0.01). (E)
Effects of omipalisib on the expression of EMT-related proteins as
analyzed by western blotting. Expression of E-cadherin was
increased, whereas N-cadherin, vimentin and slug were decreased.
(F) Quantification of EMT-related protein ratio after treatment
with omipalisib (*P<0.05, **P<0.01).
(G) Effects of omipalisib on the expression of the regulatory
factors as analyzed by western blotting. Only p-PI3K was decreased.
(H) Quantification of related protein ratio after treatment with
omipalisib (**P<0.01). |
EMT-related proteins were also inhibited in the
Hep2/omipalisib cells. These observations showed that E-cadherin
was increased, while N-cadherin, vimentin and slug were decreased,
which also demonstrated that inhibition of PI3K caused less
mesenchymal-like characteristics (Fig.
4E and F). Moreover, p-AURKA, AURKA, p-FAK (Tyr397), FAK,
p-PI3K and PI3K were assessed after using omipalisib for 0, 6 and
12 h. From the results of the western blotting, p-PI3K was markedly
decreased (P<0.01; Fig. 4G and
H), whereas p-AURKA, AURKA, p-FAK (Tyr397), FAK and PI3K were
not changed, which illustratied that PI3K is a downstream factor of
AURKA/FAK (Fig. 4G and H).
Inhibition of the FAK/PI3K pathway
decreases cellular mobility, migration and invasion
To further demonstrate that activation of the
FAK/PI3K pathway may be crucial for AURKA-induced EMT in LSCC
metastasis, wound healing, migration and invasion assays were
performed. We down-regulated FAK and PI3K with TAE226 (Hep2/TAE226)
and omipalisib (Hep2/omipalisib), respectively. Similarly, Hep2
cells treated with DMSO (Hep2/ctrl) and without any treatment
(Hep2/parental) were considered the control groups. First the wound
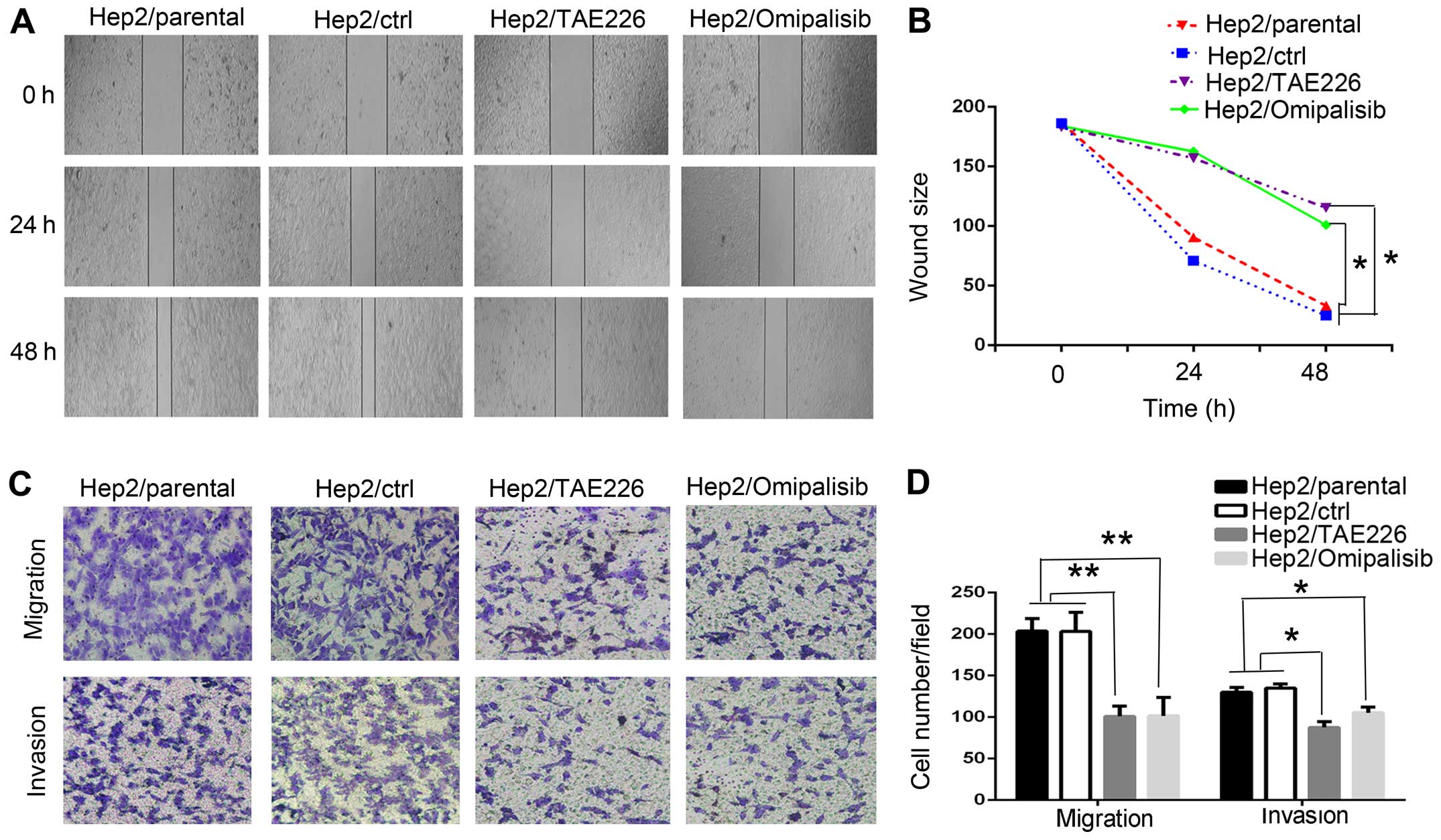
was scratched and observed for 0, 24 and 48 h. The results showed
that Hep2/TAE226 and Hep2/omipalisib cells moved slightly, while
Hep2/ctrl and Hep2/parental cells almost reached the middle of the
scratch, indicating that FAK and PI3K caused more mesenchymal-like
characteristics and markedly promoted Hep2 cell mobility
(P<0.05; Fig. 5A and B).
Similarly, the results of the migration assay
indicated that the numbers of Hep2/TAE226 (101±7.23) and
Hep2/omipalisib cells (101±12.7) migrating through the Transwell
chamber were significantly less than these numbers in the
Hep2/parental (203±8.76) and Hep2/ctrl cells (203±13.33). The
invasion assay also showed parallel results with Hep2/TAE226
(88±4.10), Hep2/omipalisib (105±3.93), Hep2/parental (130±3.46),
Hep2/ctrl cells (135±2.89) (Fig. 5C and
D). These observations indirectly illustrated that FAK and PI3K
acquired more mesenchymal-like characteristics and increased
mobility, migration and invasion.
Discussion
Cancer metastasis accounts for the majority of
tumor-related deaths worldwide. Understanding the potential
mechanism of metastasis has been long recognized as an essential
target in the treatment of cancer. However, the precise molecular
and cellular metastasis-related mechanisms are poorly understood
(35). Based on our previous
research showing that AURKA plays a key role in the tumorigenesis
and metastasis of LSCC in vitro (13) and in vivo (14), as well as growing evidence
suggesting that EMT may be the principal mechanism of cancer
metastasis (15–22), we explored the correlation between
AURKA and EMT in contributing to the metastasis of LSCC. To the
best of our knowledge, no previous study has been reported dealing
with this domain in LSCC.
AURKA, a member of the evolutionarily conserved
aurora serine/threonine kinase family, controls cell cycle
(36) by centrosome maturation,
mitotic entry, centrosome separation, bipolar spindle assembly,
chromosome alignment, cytokinesis and mitotic exit (37). Dysfunctional regulation during
mitosis leads to genetic instability and tumorigenesis. In the
present study, results of the CCK-8 and plate colony-formation
assays showed that VX680 inhibited the proliferation of Hep2 cells,
and that AURKA is a tumor-inducing gene in LSCC. Notably,
mesenchymal-related proteins, N-cadherin, vimentin and slug, which
endow cells with more migratory and invasive properties (26), were decreased in a time-dependent
manner following the reduction of AURKA, while the
epithelial-related protein E-cadherin was negatively correlated
with AURKA in the Hep2 cells. According to the aforementioned
observations, we demonstrated that AURKA may promote the metastasis
of LSCC by inducing EMT and may be regarded as a mesenchymal
marker.
In addition, the process of metastasis involves
numerous potential signaling pathways. Niu et al stated that
EMT could be correlated with the p38 MAPK/PI3K/Akt/mTOR pathway to
promote osteosarcoma metastasis (36), Kuo et al indicated that EMT
involves the NF-κB/ZEB1 pathway to promote lung cancer cell
migration and invasion (38). The
FAK-mediated PI3K/AKT pathway acts through EMT to enhance lung
cancer metastasis (31). In the
present study, down regulation of the expression of AURKA reduced
the expression of FAK and PI3K indicating that AURKA may act on FAK
and PI3K to facilitate tumor metastasis. Subsequently, TAE226 was
applied to block the expression of p-FAK (Tyr397). Notably, the
expression of p-PI3K was also decreased, whereas p-AURKA, AURKA,
FAK and PI3K were not altered, indicating that FAK is the
downstream factor of AURKA. p-PI3K was markedly decreased after
treatment with omipalisib for 12 h; inversely p-AURKA, AURKA, p-FAK
(Tyr397), FAK and PI3K were not altered, indicating that PI3K is a
downstream factor of AURKA/FAK. Finally, inhibition of FAK or PI3K
altered the expression of EMT markers which was the same as the
treatment of VX680 and allowed mesenchymal cells to acquire the
characteristics of epithelial cells to reduce mobility, migration
and invasion in Hep2 cells. All of these results imply that the
FAK/PI3K pathway is under the regulation of AURKA to promote the
process of EMT and enhance the metastasis of LSCC.
The present study suggests that VX680, TAE226 and
omipalisib, which are inhibitors and implicated in the
AURKA/FAK/PI3K signaling pathway, should not only be considered for
single use but also for combination therapy in the treatment of
patients with LSCC. Certainly, various doses and schedules of
targeted therapeutic drugs should be examined in clinical trials to
ensure the effectiveness and safety of LSCC therapy. Taken
together, our results demonstrated that AURKA may enhance
metastasis by inducing EMT via the FAK/PI3K pathway in LSCC. In
addition, in light of our observations of the therapeutic potential
in laryngeal cancer, the AURKA/FAK/PI3K pathway may present new
diagnostic biomarkers and potential therapeutic targets for
LSCC.
Acknowledgments
The present study was supported by grants from the
Science and Technology Commission of Shanghai Municipality (no.
12ZR1418700) and the Clinical Science and Technology Innovation
Project of Shenkang Hospital Development Center (SHDC12015114).
Abbreviations:
|
LSCC
|
laryngeal squamous cell carcinoma
|
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
AURKA
|
aurora kinase A
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Li D, Feng J, Wu T, Wang Y, Sun Y, Ren J
and Liu M: Long inter-genic noncoding RNA HOTAIR is overexpressed
and regulates PTEN methylation in laryngeal squamous cell
carcinoma. Am J Pathol. 182:64–70. 2013. View Article : Google Scholar
|
|
2
|
Bingol F, Yoruk O, Bingol BO, Erdemci B,
Ozkan O and Mazlumoglu MR: Estimation of the efficacy of
chemo-radiotherapy on tumor regression in the patients with
laryngeal cancer via computerized tomography using the Cavalieri
method. Acta Otolaryngol. 136:164–167. 2016. View Article : Google Scholar
|
|
3
|
Halec G, Holzinger D, Schmitt M,
Flechtenmacher C, Dyckhoff G, Lloveras B, Höfler D, Bosch FX and
Pawlita M: Biological evidence for a causal role of HPV16 in a
small fraction of laryngeal squamous cell carcinoma. Br J Cancer.
109:172–183. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu GP, Mehta V, Branovan D, Huang Q,
Hashibe M, Zhang ZF and Schantz SP: Improved survival among
patients with base of tongue and tonsil cancer in the United
States. Cancer Causes Control. 23:153–164. 2012. View Article : Google Scholar
|
|
5
|
Shen Z, Li Q, Deng H, Lu D, Song H and Guo
J: Long non-coding RNA profiling in laryngeal squamous cell
carcinoma and its clinical significance: Potential biomarkers for
LSCC. PLoS One. 9:e1082372014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mitra A, Mishra L and Li S: EMT, CTCs and
CSCs in tumor relapse and drug-resistance. Oncotarget.
6:10697–10711. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Amend SR and Pienta KJ: Ecology meets
cancer biology: The cancer swamp promotes the lethal cancer
phenotype. Oncotarget. 6:9669–9678. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marumoto T, Zhang D and Saya H: Aurora-A -
a guardian of poles. Nat Rev Cancer. 5:42–50. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rojanala S, Han H, Muñoz RM, Browne W,
Nagle R, Von Hoff DD and Bearss DJ: The mitotic serine threonine
kinase, Aurora-2, is a potential target for drug development in
human pancreatic cancer. Mol Cancer Ther. 3:451–457.
2004.PubMed/NCBI
|
|
10
|
Watanabe T, Imoto I, Katahira T, Hirasawa
A, Ishiwata I, Emi M, Takayama M, Sato A and Inazawa J:
Differentially regulated genes as putative targets of
amplifications at 20q in ovarian cancers. Jpn J Cancer Res.
93:1114–1122. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bischoff JR, Anderson L, Zhu Y, Mossie K,
Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, et
al: A homologue of Drosophila aurora kinase is oncogenic and
amplified in human colorectal cancers. EMBO J. 17:3052–3065. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xing Z, Gao S, Duan Y, Han H, Li L, Yang Y
and Li Q: Delivery of DNAzyme targeting aurora kinase A to inhibit
the proliferation and migration of human prostate cancer. Int J
Nanomedicine. 10:5715–5727. 2015.PubMed/NCBI
|
|
13
|
Zhang H, Chen X, Jin Y, Liu B and Zhou L:
Overexpression of Aurora-A promotes laryngeal cancer progression by
enhancing invasive ability and chromosomal instability. Eur Arch
Otorhinolaryngol. 269:607–614. 2012. View Article : Google Scholar :
|
|
14
|
Zhang H, Chen X, Liu B and Zhou L: Effects
of stable knockdown of Aurora kinase A on proliferation, migration,
chromosomal instability, and expression of focal adhesion kinase
and matrix metalloproteinase-2 in HEp-2 cells. Mol Cell Biochem.
357:95–106. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rao Q, Chen Y, Yeh CR, Ding J, Li L, Chang
C and Yeh S: Recruited mast cells in the tumor microenvironment
enhance bladder cancer metastasis via modulation of ERβ/CCL2/CCR2
EMT/MMP9 signals. Oncotarget. 7:7842–7855. 2016.
|
|
16
|
Qin Y, Tang B, Hu CJ, Xiao YF, Xie R, Yong
X, Wu YY, Dong H and Yang SM: An hTERT/ZEB1 complex directly
regulates E-cadherin to promote epithelial-to-mesenchymal
transition (EMT) in colorectal cancer. Oncotarget. 7:351–631.
2016.
|
|
17
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Burnett JP, Korkaya H, Ouzounova MD, Jiang
H, Conley SJ, Newman BW, Sun L, Connarn JN, Chen CS, Zhang N, et
al: Trastuzumab resistance induces EMT to transform
HER2+ PTEN− to a triple negative breast
cancer that requires unique treatment options. Sci Rep.
5:158212015. View Article : Google Scholar
|
|
19
|
Attramadal CG, Kumar S, Boysen ME, Dhakal
HP, Nesland JM and Bryne M: Tumor budding, EMT and cancer stem
cells in T1-2/N0 oral squamous cell carcinomas. Anticancer Res.
35:6111–6120. 2015.PubMed/NCBI
|
|
20
|
Wang SC, Chai DS, Chen CB, Wang ZY and
Wang L: HPIP promotes thyroid cancer cell growth, migration and EMT
through activating PI3K/AKT signaling pathway. Biomed Pharmacother.
75:33–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Buczek ME, Miles AK, Green W, Johnson C,
Boocock DJ, Pockley AG, Rees RC, Hulman G, van Schalkwyk G,
Parkinson R, et al: Cytoplasmic PML promotes TGF-β-associated
epithelial-mesenchymal transition and invasion in prostate cancer.
Oncogene. 43:124–127. 2015.
|
|
22
|
Ota I, Masui T, Kurihara M, Yook JI,
Mikami S, Kimura T, Shimada K, Konishi N, Yane K, Yamanaka T, et
al: Snail-induced EMT promotes cancer stem cell-like properties in
head and neck cancer cells. Oncol Rep. 35:261–266. 2016.
|
|
23
|
Broster SA and Kyprianou N:
Epithelial-mesenchymal transition in prostatic disease. Future
Oncol. 11:3197–3206. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee SC, Kim OH, Lee SK and Kim SJ: IWR-1
inhibits epithelial-mesenchymal transition of colorectal cancer
cells through suppressing Wnt/β-catenin signaling as well as
survivin expression. Oncotarget. 6:27146–27159. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
D'Assoro AB, Liu T, Quatraro C, Amato A,
Opyrchal M, Leontovich A, Ikeda Y, Ohmine S, Lingle W, Suman V, et
al: The mitotic kinase Aurora-A promotes distant metastases by
inducing epithelial-to-mesenchymal transition in ERα+
breast cancer cells. Oncogene. 33:599–610. 2014. View Article : Google Scholar
|
|
28
|
Guan Z, Wang XR, Zhu XF, Huang XF, Xu J,
Wang LH, Wan XB, Long ZJ, Liu JN, Feng GK, et al: Aurora-A, a
negative prognostic marker, increases migration and decreases
radiosensitivity in cancer cells. Cancer Res. 67:10436–10444. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thiyagarajan V, Tsai MJ and Weng CF:
Antroquinonol targets FAK-signaling pathway suppressed cell
migration, invasion, and tumor growth of C6 glioma. PLoS One.
10:e01412852015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun L, Liu L, Liu X, Wang Y, Li M, Yao L,
Yang J, Ji G, Guo C, Pan Y, et al: MGr1-Ag/37LRP induces cell
adhesion-mediated drug resistance through FAK/PI3K and MAPK pathway
in gastric cancer. Cancer Sci. 105:651–659. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin Y, Rao J, Zha XL and Xu H:
Angiopoietin-like 3 induces podocyte F-actin rearrangement through
integrin αV/β3/FAK/PI3K pathway-mediated Rac1
activation. Biomed Res Int. 2013:1356082013.
|
|
32
|
Fu QF, Liu Y, Fan Y, Hua SN, Qu HY, Dong
SW, Li RL, Zhao MY, Zhen Y, Yu XL, et al: Alpha-enolase promotes
cell glycolysis, growth, migration, and invasion in non-small cell
lung cancer through FAK-mediated PI3K/AKT pathway. J Hematol Oncol.
8:222015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hochwald SN, Nyberg C, Zheng M, Zheng D,
Wood C, Massoll NA, Magis A, Ostrov D, Cance WG and Golubovskaya
VM: A novel small molecule inhibitor of FAK decreases growth of
human pancreatic cancer. Cell Cycle. 8:2435–2443. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Greger JG, Eastman SD, Zhang V, Bleam MR,
Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L and
Gilmer TM: Combinations of BRAF, MEK, and PI3K/mTOR inhibitors
overcome acquired resistance to the BRAF inhibitor GSK2118436
dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther.
11:909–920. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Evans EB and Lin SY: New insights into
tumor dormancy: Targeting DNA repair pathways. World J Clin Oncol.
6:80–88. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Niu NK, Wang ZL, Pan ST, Ding HQ, Au GH,
He ZX, Zhou ZW, Xiao G, Yang YX, Zhang X, et al: Pro-apoptotic and
pro-autophagic effects of the Aurora kinase A inhibitor alisertib
(MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the
activation of mitochondria-mediated pathway and inhibition of p38
MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Devel Ther.
9:1555–1584. 2015.PubMed/NCBI
|
|
37
|
Dar AA, Goff LW, Majid S, Berlin J and
El-Rifai W: Aurora kinase inhibitors - rising stars in cancer
therapeutics? Mol Cancer Ther. 9:268–278. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gu K, Li MM, Shen J, Liu F, Cao JY, Jin S
and Yu Y: Interleukin-17-induced EMT promotes lung cancer cell
migration and invasion via NF-κB/ZEB1 signal pathway. Am J Cancer
Res. 5:1169–1179. 2015.
|